
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrathin CdS@BDC nanosheets derived from 2D metal–organic frameworks for enhanced photoinduced-stability and photocatalytic hydrogen production†
Jian
Li‡
ab,
Huaqiang
Wang‡
b,
Yihong
Gao
b,
Hao
Fang
b,
Peiyu
Chen
b and
Fangzhi
Huang
 *b
*b
aDepartment of Chemical and Chemical Engineering, Hefei Normal University, Hefei, 230601, P. R. China
bLab of Clean Energy & Environmental Catalysis, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Institute of Physical Science and Information Technology, School of Chemistry and Chemical Engineering, Anhui University, Hefei 230601, P. R. China. E-mail: huangfangzhi@163.com; Tel: +86-551-63861328
First published on 27th September 2022
Abstract
Two-dimensional metal–organic framework materials have attracted much attention due to their large surface area, porous structure and high carrier separation efficiency. Herein, we innovatively proposed a facile in situ vulcanization method, in which 2D Cd-MOF was used as the precursor and template to prepare a novel ultrathin CdS@1,4-benzenedicarboxylic (CdS@BDC) nanosheet in Na2S/Na2SO3 solution. The obtained CdS@BDC nanosheet has the overall morphology of a 1.46 nm average thickness nanosheet skeleton, and is uniformly in situ embedded with CdS nanoparticles with an average size of 6 nm. Due to the two-dimensional skeleton structure, the efficiency of capture, separation and migration of photogenerated carriers was significantly improved. The hydrogen evolution rate (HER) of ultrathin CdS@BDC nanosheet photocatalysts is up to 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 081 μmol h−1 g−1, which is 65 times higher than that of traditional CdS nanoparticles (199 μmol h−1 g−1). The excellent photocatalytic performance of CdS@BDC nanosheets can be attributed to the inherited structural and functional advantages of 2D Cd-MOF. For example, the efficiency of photo-generated carrier capture, separation and migration is significantly improved. For another example, the surface of the photocatalyst can adsorb a large amount of S2− ions, which not only consumes photogenerated holes as effective hole scavengers, but also accelerates the interfacial H2-production reaction due to the effective H+-capturing active sites. Thus, a tentative S2−-mediated mechanism for the enhanced photoinduced-stability of CdS@BDC nanosheets was also proposed. The present work provides a facile and fascinating strategy for the synthesis of highly efficient photocatalysts.
081 μmol h−1 g−1, which is 65 times higher than that of traditional CdS nanoparticles (199 μmol h−1 g−1). The excellent photocatalytic performance of CdS@BDC nanosheets can be attributed to the inherited structural and functional advantages of 2D Cd-MOF. For example, the efficiency of photo-generated carrier capture, separation and migration is significantly improved. For another example, the surface of the photocatalyst can adsorb a large amount of S2− ions, which not only consumes photogenerated holes as effective hole scavengers, but also accelerates the interfacial H2-production reaction due to the effective H+-capturing active sites. Thus, a tentative S2−-mediated mechanism for the enhanced photoinduced-stability of CdS@BDC nanosheets was also proposed. The present work provides a facile and fascinating strategy for the synthesis of highly efficient photocatalysts.
1. Introduction
With the excessive consumption of fossil fuels and the increasingly serious environmental pollution problem in the world, it is an important research topic to convert the inexhaustible solar energy into green chemical energy for the development of alternative energy.1–6 Since Honda and Fujishima pioneered photocatalytic water splitting on TiO2 electrodes in 1972,7 solar-driven splitting of water for hydrogen production has gradually become a promising environmental route to achieve renewable and clean hydrogen energy. To date, extensive efforts have been focused on semiconductor materials such as g-C3N4,8,9 BiVO4,10,11 Ta3N5,12,13 CNPs@PEI-Co,14 and CdS,15–18 which have been widely used to develop photocatalysts for hydrogen evolution. CdS has been recognized as one of the most attractive photocatalytic materials for hydrogen evolution among the various photocatalysts owing to its superior optical properties (Eg = 2.4 eV) for efficiently absorbing visible-light, and appropriate conduction band level for the proton reduction reaction.19,20 However, pure CdS still can't show gratifying photocatalytic activity due to its rapid photogenerated charge recombination and serious photocorrosion during the light irradiation reaction.21–23 In addition, the limited active sites on the surface of unmodified CdS also restrict its catalytic activity.24 To address the above shortcomings, the emergence of 2D nanostructures may be an effective way to inhibit the recombination of electron–hole pairs and provide more reactive sites.25,26 More importantly, photogenerated electrons and holes only need to overcome a short transmission distance to the surface of catalyst, and the separation efficiency can be greatly accelerated in 2D nanomaterials.Recently, two-dimensional (2D) metal–organic frameworks (MOFs), a new class of organic–inorganic hybrid materials constructed by metal ions/clusters and organic ligands have been vastly investigated in catalysis fields.27,28 Compared with other 2D nanomaterials such as graphene,29 transition metal dichalcogenides (TMDs),30 layered hydroxide31 and black phosphorus,32 the unique structural and compositional features of 2D MOFs, such as large surface area, abundant active sites, highly ordered porous structure and controllable composition, render them a promising template for creating 2D MOF-derived composite nanosheets.33–35 For example, Qiao et al.36 loaded CdS nanoparticles onto 2D Ni-based MOFs, which tremendously reduced the photocorrosion of CdS and the recombination of electrons and holes via electron transport from CdS to 2D Ni-based MOFs. Zhang et al.37 reported a facile approach for the synthesis of CuS/Cu-TCPP composite nanosheets, exhibiting a remarkably increased photocurrent compared to the pure Cu-TCPP nanosheets. Liu et al.38 synthesized 2D free-standing N-doped Ni-Ni3S2@carbon nanoplates by pyrolysis treatment from a precursor of Ni-based MOF nanoplates, which displayed a low overpotential of 284.7 mV at a current density of 10 mA cm−2 in alkaline solution. That is to say, metal sulfide associated with 2D MOF is a prospective strategy because of the following advantages: (i) the high surface area is capable of stabilizing the size and hindering the aggregation of nanoparticles; (ii) the ultrathin thickness results in a shorter transmission distance for photogenerated electrons reaching the surface of the catalyst, as well as reducing the recombination of electron–hole pairs; (iii) the large exposure of active sites is favorable for promoting the surface reduction of catalysts. However, to our knowledge, research on 2D MOFs in photocatalysis is still in its infancy. Based on various 2D MOFs, it remains a meaningful challenge to design and produce a more efficient system to enhance the photocatalytic performance by reducing the photocorrosion or producing more active sites of cadmium sulfide, etc.
Hence, we use 2D Cd-MOF nanosheets (NSs) as the precursor and template, and report a method of the in situ synthesis of CdS@1,4-benzenedicarboxylic (CdS@BDC) nanosheet (NS) photocatalysts based on the reaction of cadmium ions in the Cd-MOF skeleton and sulfur ions in Na2S/Na2SO3 solution. In this study, the Na2S/Na2SO3 system can not only be used as a precursor of sulfur ions for the rapid synthesis of CdS, but also can act as a hole sacrificial agent to prevent the photocorrosion of CdS in the process of the photocatalytic reaction. More importantly, the ultrathin skeleton of CdS@BDC NSs not only successfully regulated the distribution and size of CdS nanoparticles, but also continued the advantage of 2D to promote its catalytic performance. Due to the porous structure and large surface area of the synthesized CdS@BDC NSs derived from the 2D Cd-MOF, a large number of S2− ions can be rapidly enriched to its surface, forming a S2−-mediated layer to prevent the photocorrosion. As expected, the synthesized CdS@BDC NSs system has excellent stability and reusability, and its photocatalytic activity is 65 times higher than that of traditional CdS nanoparticles. It not only provides a facile and fascinating strategy for the synthesis of highly efficient photocatalysts, but also opens up a new insight for the application of 2D MOF-based nanomaterials in photocatalysis.
2. Experimental sections
2.1 Chemicals and materials
Cadmium acetate dehydrate (Cd(CH3COO)2·2H2O), N,N-dimethylformamide (DMF) and 1,4-benzenedicarboxylic acid (H2BDC) were purchased from Shanghai Aladdin Biochemical Technology Co. Ltd. Sodium sulfide nonahydrate (Na2S·9H2O), sodium sulfite (Na2SO3), cadmium chloride (CdCl2·2.5H2O) and trisodium phosphate (Na3PO4·12H2O) were purchased from Shanghai Chemical Reagent Co. Ltd. All the reagents were of analytical grade and used without further purification. Ultrapure water with a resistivity of 18.2 MΩ cm was used throughout all the experiments.2.2 Catalyst preparation
For comparison, CdS nanoparticles (NPs) were synthesized using a modified method.39 Briefly, 30.0 mL of 0.1 M Na2S solution was added into 30.0 mL of 0.1 M CdCl2·2.5H2O solution under stirring conditions. After vigorously stirring for 10 min, the achieved suspension was added dropwise into 2.0 mL of 0.05 M Na3PO4·12H2O with stirring for 20 min. And then, the mixed solution was transferred into a 100 mL Teflon-lined autoclave and kept at 180 °C for 12 h. The product was collected by filtration, washed with absolute ethanol and deionized water separately, and finally dried under vacuum at 60 °C overnight.
2.3 Characterization
Scanning electron microscopy (SEM) images were acquired using a Zeiss Supra 40 scanning electron microscope with an acceleration voltage of 5 kV, combined with energy dispersive X-ray (EDX) spectroscopy. A JEM-2010-TEM electron microscope (JEOL, Japan) was used to obtain the elemental mapping, TEM and HRTEM images. Atomic force microscopy (AFM) images were recorded using a Dimension Edge instrument. Powder X-ray diffraction (PXRD) patterns were analyzed on a Philips X’Pert PRO SUPER X-ray diffractometer equipped with graphite-monochromated Cu Kα radiation (λ = 1.54056 Å). An ESCALab MKII XPS spectrometer system with Al Ka (1486.6 eV) as the X-ray source was used to carry out the XPS measurements. UV-vis spectra were measured using a spectrophotometer (Hitachi U-4100). The steady-state photoluminescence (PL) spectra for liquid samples were conducted through HitachF-4500 under an excitation wavelength of 425 nm. A FS5 fluorescence lifetime spectrometer (Edinburgh Instruments, UK) was used to obtain the time-resolved PL decay curves under the excitation of 425 nm and probed at 475 nm.2.4 Hydrogen evolution measurements
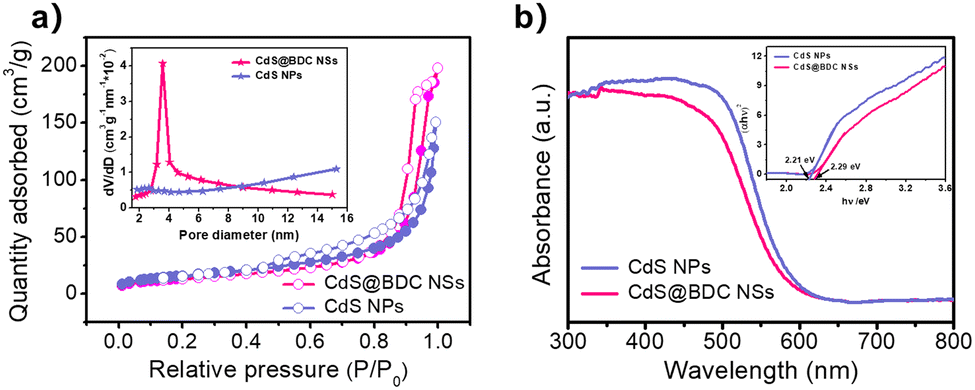
The photocatalytic H2 production experiments were performed in a 100 mL Pyrex flask connected to a closed gas circulation and evacuation system under a 300 W Xe lamp (CEL-HFX300, Beijing China Education Au-light Co., Ltd) equipped with an optical cut-off filter (λ > 380 nm). In a typical experiment (apart from CdS@BDC NSs sample), 5 mg of the photocatalyst was dispersed in 20.0 mL of an aqueous solution including Na2S (0.1 M) and Na2SO3 (0.1 M) as the sacrificial electron donors. Prior to irradiation, the suspension was stirred for 10 min and then bubbled with N2 for 30 min to completely remove the dissolved air. Subsequently, the photocatalytic reactor was irradiated by a Xe lamp with a UV cut off filter (>380 nm) at a fixed distance (10 cm). Throughout the irradiation process, the above suspension was continuously stirred and kept at 6 °C through a flow of cooling water. The evolved H2 was measured via gas chromatography (Shimadzu GC-2014C, N2 as a carrier gas) using a thermal conductivity detector (TCD). For each evaluation of H2 generation, 200 μL of the headspace was injected into the GC and was quantified by a calibration plot to the internal hydrogen standard. During the long-time stability measurements, the reaction system used N2 to remove the produced hydrogen after every 4 h of irradiation, and the sacrificial agent remained unchanged.The quantum efficiency (QE) of H2 evolution was measured under conditions similar to the above photocatalytic reaction, except that the light source was equipped with a band-pass filter (420 nm). The photon flux of incident light was determined using a Ray virtual radiation actinometer (Apogee MQ-500, silicon ray detector, light spectrum, 389–692 nm; measurement range, 0–4000 mol−1 m−2 s−1). The following equation (1) was used to calculate the quantum efficiency (QE).40
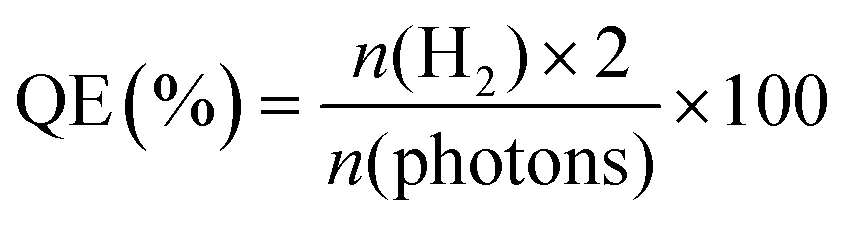 | (1) |
2.5 Photoelectrochemical measurements
The photoelectrochemical measurements were conducted on a CHI660E electrochemical workstation (Chenhua Instrument, Shanghai, China) in a conventional three-electrode system using the prepared FTO as the working electrode, Pt foil as the counter electrode, and Ag/AgCl (3 M KCl) as a reference electrode. A 0.1 M Na2S/Na2SO3 aqueous solution was employed as the electrolytes, and a 300 W Xe lamp (Beijing China Education Au-light Co., Ltd) with a UV-cutoff filter (>380 nm) served as the solar simulator. Amperometric I–t curves were obtained by applying a bias potential of −0.2 V under chopped light irradiation with an ON/OFF interval of 25 s. Electrochemical impedance spectroscopy (EIS) measurements were conducted by applying a Zahner IM6 (Zahner Elektrik, Kronach, Germany) in the dark within a frequency range of 10−1–104 Hz, and the amplitude sine wave potential was 10 mV in each case. Mott–Schottky plots were measured using the same three-electrode system in 0.2 M Na2SO4 aqueous solution. The above working electrode was prepared by mixing 5 mg photocatalyst with 5 μL Nafion and 0.4 mL ethanol to make slurry. Afterwards, the slurry (0.2 mL) was then coated onto the pretreated FTO glass (a 1.0 cm2 active area). Finally, the resulting electrode was dried in air and heated in an oven at 60 °C for 1 h. To convert the obtained potential (vs. Ag/AgCl) to RHE (NHE at pH = 0), the following relationship was used:| ERHE = E(Ag/AgCl) + 0.059pH + E0(Ag/AgCl), |
| E0(Ag/AgCl) = 0.1967 V. |
3. Results and discussion
The controlled fabrication process of CdS@BDC NSs was schematically presented in Fig. 1a. First, uniform ultrathin Cd-MOF NSs were obtained by a solvothermal reaction of Cd2+ ions with 1,4-benzenedicarboxylic acid (H2BDC), as shown in Fig. 1b. The corresponding AFM image and height profile intuitively exhibits their average thickness of ca. 1.46 nm (Fig. 1c). Then, the as-synthesized Cd-MOF NSs were transformed into CdS@BDC NSs via an in situ sulfidation method in Na2S/Na2SO3 solution. Intriguingly, compared with their Cd-MOF precursors, the overall morphology of the nanosheet framework remains basically unchanged as shown in Fig. 1d. Furthermore, the nanosheet framework was uniformly embedded with in situ CdS nanoparticles, and the average size of the CdS nanoparticles is about 6 nm (Fig. 1e and f). The corresponding lattice spacing value (Fig. 1g) measured as 0.34 nm corresponded to the (111) facet of cubic-structured CdS.41 The elemental mapping images (Fig. 1g–k) clearly depicted the uniform distribution of all elements (C, O, Cd, S) in their chemical composition, indicating that CdS@BDC NS is a homogeneous compound.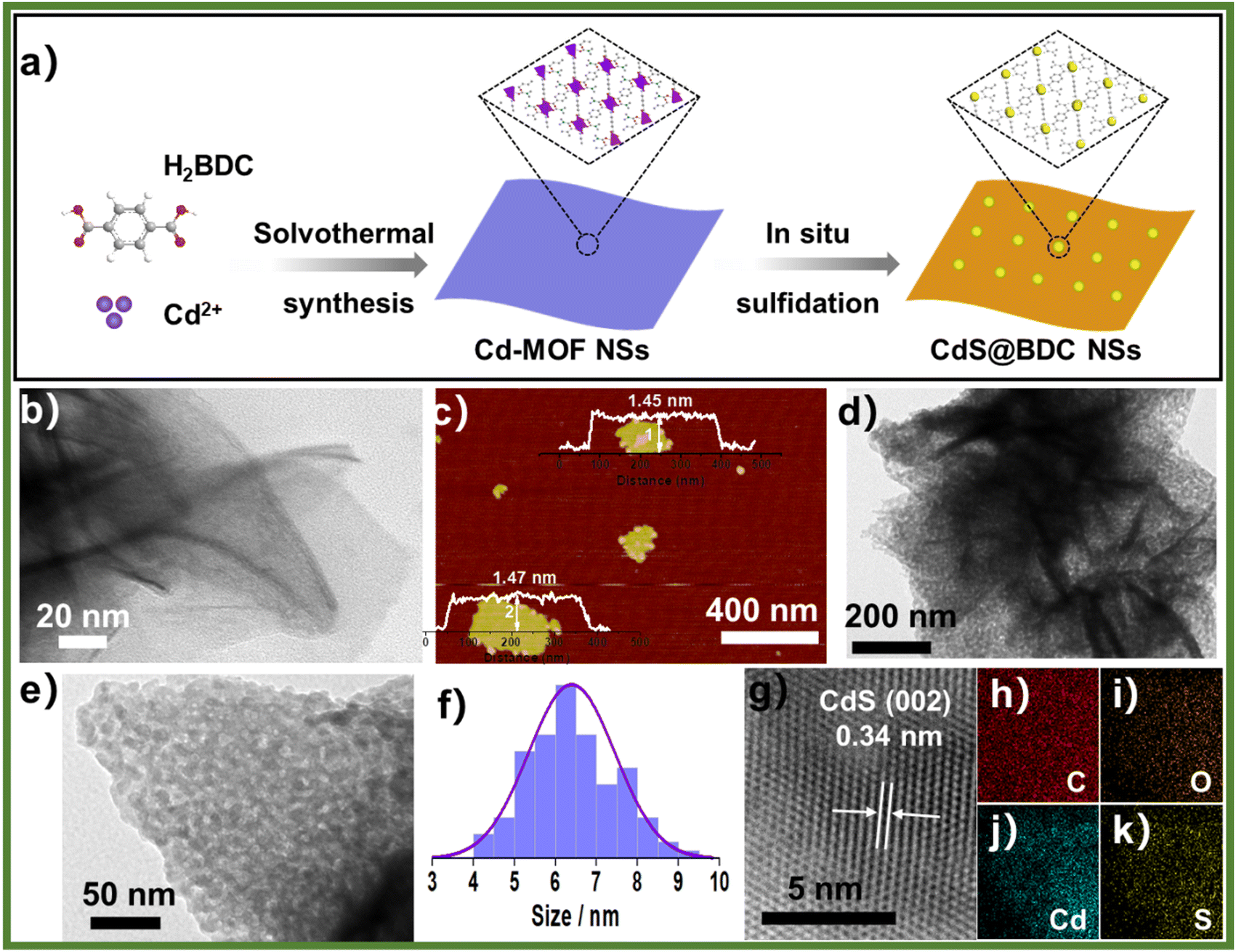 | ||
| Fig. 1 (a) Schematic illustration of the synthesis process of CdS@BDC NSs. (b) TEM image of Cd-MOF NSs. (c) AFM image of Cd-MOF NSs and the corresponding height profiles (inset). (d and e) TEM images and particle size distribution (f) of CdS@BDC NSs. (g) HRTEM graph of CdS@BDC NSs. (h–k) Elemental mapping images of C, O, Cd and S recorded from the same region of the CdS@BDC NSs. | ||
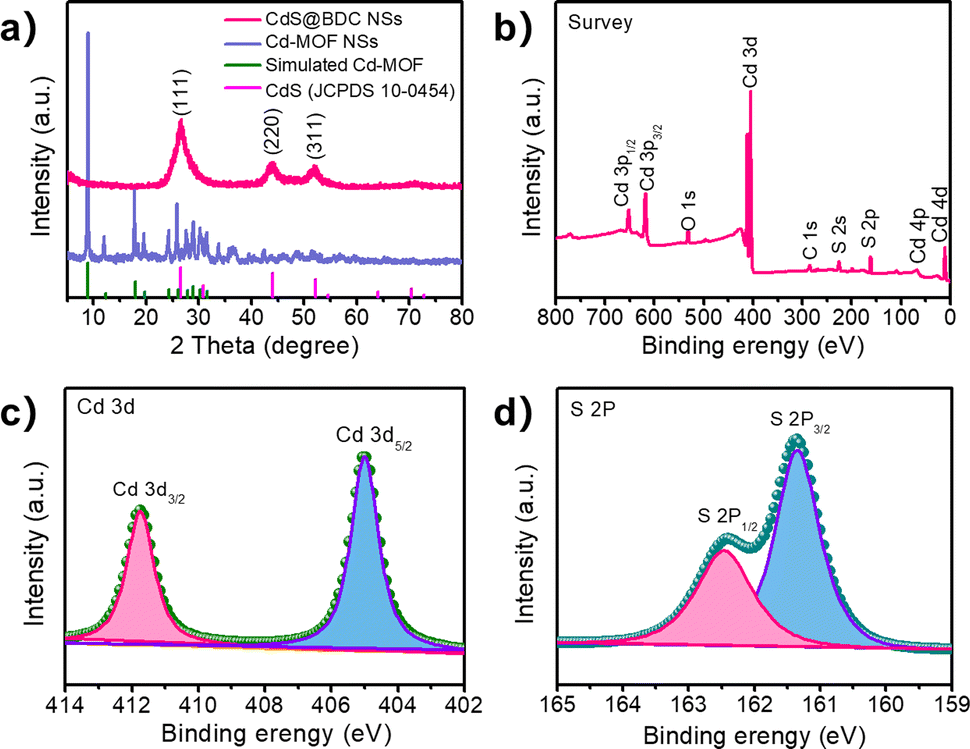
The structure and chemical state of the as-synthesized CdS@BDC NSs were further measured by PXRD and XPS. As shown in Fig. 2a, all of the diffraction peaks in the pattern of the Cd-MOF sample agreed well with the simulated XRD pattern of Cd-MOF ([Cd(BDC)-(DMF)]n) with a space group P1(-)(2) on the basis of the reported crystal structure data.42 After in situ sulfidation treatment, the cubic-structured CdS phase was successfully obtained (JCPDS no. 10-0454).43 Meanwhile, the corresponding EDX spectroscopy measurement revealed that the molar ratio of Cd/S is 1:1.07 in CdS@BDC NSs (Fig. S1, ESI†). As displayed in Fig. 2b, the XPS survey spectrum of the CdS@BDC NSs indicated the existence of Cd, S, as well as C and O from the nanosheet framework. From the high-resolution XPS spectrum of Cd 3d (Fig. 2b), the binding energies of Cd 3d5/2 (405.1 eV) and Cd 3d3/2 (411.9 eV) peaks, demonstrating the valences of Cd were +2 in CdS@BDC NSs, while the S 2P core level spectra (Fig. 2c) consisted of a doublet located at a binding energy of 161.3 eV and 162.5 eV, ascribing to the characteristic peaks of S 2p1/2 and S 2p3/2 spin–orbit components of S2−, respectively.44 All these indicated and further confirmed the formation of a homogeneous CdS@BDC compound. According to the N2 adsorption/desorption isotherms (Fig. 3a), it is clearly seen that a sharp increase at P/P0 of 0.8–1.0, indicating typical type III isotherms,45 and the SBET of CdS@BDC NSs is 13.7 m2 g−1. The UV-vis diffuse reflectance spectra (DRS UV-vis, Fig. 3b) of CdS@BDC NSs and CdS NPs (Fig. S2, ESI†) indicate that their comparable absorption edges are very close, ranging from 300 nm to 650 nm. The corresponding band gap values of CdS@BDC NSs and CdS NPs are also similar, corresponding to 2.29 eV and 2.21 eV estimated from the (αhν)2versus photon energy (hν) plot (Fig. 3b, inset). The Mott–Schottky plots were also measured in 0.2 M Na2SO4 electrolyte with a pH value of 6.8. The positive slope of the C−2–E plot (Fig. S3, ESI†) revealed the n-type semiconductor property of CdS@BDC NSs and CdS NPs.46 The flat-band potential (Vfb) of CdS@BDC NSs and CdS NPs were −0.48 V and −0.41 V vs. Ag/AgCl (−0.21 V and −0.28 V vs. NHE) through extrapolating the intercept to the X-axis. For many n-type semiconductors, it is reported that the bottom of the conduction band (CB) potential is more negative by 0.1 V than the flat-band potential.47 Thus, the CBs of the CdS@BDC NSs and CdS NPs were calculated to be −0.38 V and −0.31 V vs. NHE, respectively, exhibiting that the CB potential of the CdS@BDC NSs is more negative than that of CdS NPs, which is helpful for photocatalytic H2 generation.
 | ||
| Fig. 2 (a) Powder XRD Profiles for Cd-MOF NSs and CdS@BDC NSs. (b) XPS survey spectra of the CdS@BDC NSs, the high-resolution XPS spectra of Cd 3d (c) and S 2p (d) XPS spectra of CdS@BDC NSs. | ||
 | ||
| Fig. 3 (a) Nitrogen adsorption/desorption isotherms of the CdS@BDC NSs and CdS NPs (the inset exhibits corresponding pore size distribution curves). (b) UV-vis diffuse absorption spectra and their calculated band gap (inset) of the CdS@BDC NSs and CdS NPs. | ||
The photocatalytic hydrogen evolution was conducted under visible-light irradiation (λ > 380 nm) using S2−/SO32− as sacrificial reagents without the use of precious co-catalysts of Pt. CdS nanoparticles (NPs) (Fig. S2, ESI†) and CdS quantum dots (QDs) (Fig. S4, ESI†) were also evaluated as photocatalysts for reference. From Fig. 4a, it can be found that CdS NPs and CdS QDs have a H2-evolution rate (HER) of 199 μmol h−1 g−1 and 760 μmol h−1 g−1, respectively. Prominently, the hydrogen production rate of CdS@BDC NSs is 13081 μmol h−1 g−1, and is 65 times and 17 times that of CdS NPs and CdS QDs, respectively. The excellent hydrogen production performance shows that CdS@BDC NS is one of the most promising noble-metal-free CdS-based photocatalysts (Table S1, ESI†). Photocatalytic quantum efficiency QE of the CdS-based photocatalyst and previously reported CdS-based photocatalyst is listed in Table S2 (ESI†). Therefore, the QE of CdS@BDC NS (4.61%) is higher than that of CdS NP (0.63%) and CdS QD (1.37%), respectively. In addition, the photocatalytic quantum efficiency of CdS@BDC NS also seems to be higher than that of some precious metal-free photocatalysts based on CdS (e.g., CdS/g C3N4, CdS/Cu7S4/g-C3N4and CdS/NiSe2). The corresponding photostability of the CdS@BDC NSs was investigated via repeating the H2-evolution experiments under the same experimental conditions, as illustrated in Fig. 4b. During four repeated photocatalytic experiments, no obvious reduction of hydrogen production was observed, implying its excellent photocatalytic stability. After four experimental cycles, the TEM image (Fig. S5a, ESI†) and PXRD patterns (Fig. S5b, ESI†) showed no significant changes in CdS@BDC NSs.
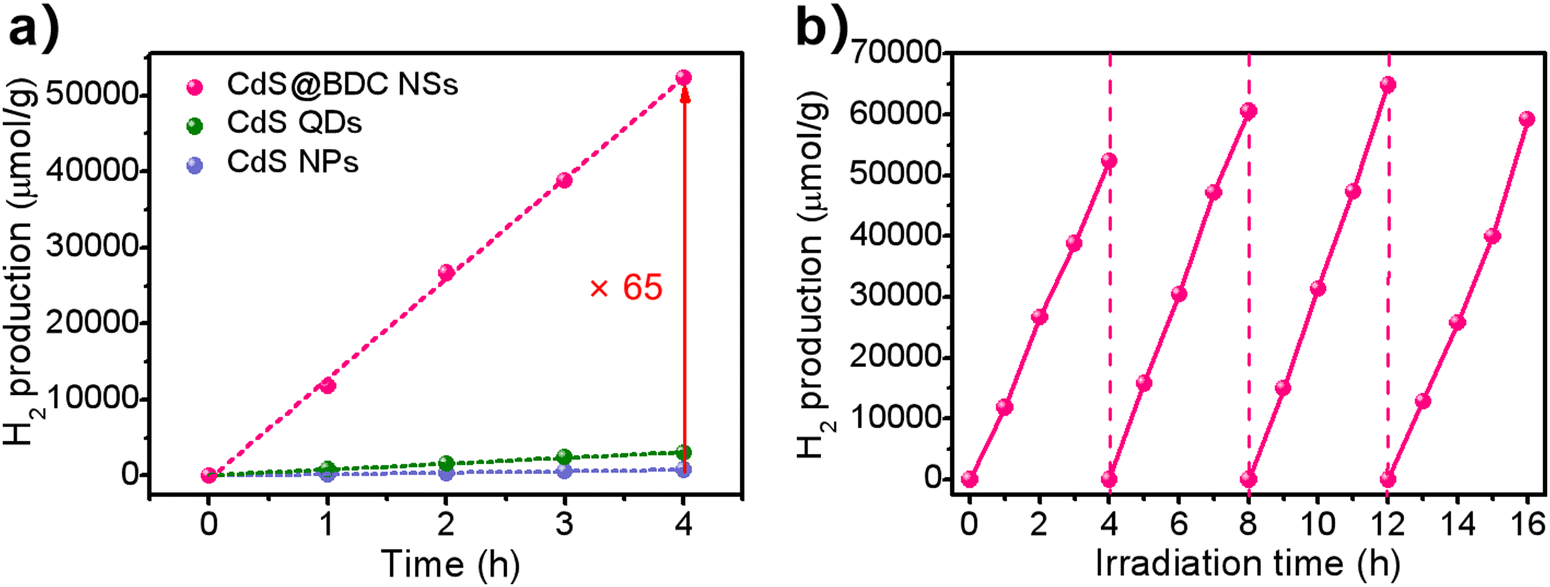 | ||
| Fig. 4 (a) Comparison of the photocatalytic H2-evolution activities of CdS@BDC NSs, CdS QDs and CdS NPs. (b) Cycling stability for the CdS@BDC NS photocatalysts. | ||
As we know, the efficiency of photogenerated carrier capture, separation and migration may play a crucial role in photocatalytic water splitting for H2 evolution. It can be seen from Fig. 5a that the peak intensity of photoluminescence (PL) of CdS@BDC NSs is much weaker than that of CdS NPs, indicating that the recombination of photoinduced electron–hole (e–h) pairs in CdS@BDC NSs is more efficiently repressed.48,49 Moreover, as displayed in the time-resolved PL spectra (Fig. 5b and Table S3, ESI†), the average photogenerated carrier lifetime of CdS@BDC NSs is 0.035 ns, which is longer than that of CdS NPs (0.021 ns), declaring that more photoelectrons can be used effectively for proton reduction.50,51 The transient photocurrent–time curves (Fig. 5c) also reveal that the photocurrent density of CdS@BDC NSs has been substantially improved compared with CdS NPs, suggesting faster charge separation and higher efficiency of photogenerated electron–hole pairs.36 In addition, as shown in Fig. 5d, the Nyquist plot of electrochemical impedance spectroscopy exhibits a smaller radius of CdS@BDC NSs than that of CdS NPs, corresponding to the lower charge-transport resistance.24,52 Evidently, CdS@BDC NSs have the above-mentioned excellent photoelectrochemical properties due to their special 2-dimensional lamellar structure, thus obtaining a high photocatalytic hydrogen production rate. What's more, when another lactic acid solution replaced Na2S/Na2SO3 solution as a hole sacrificing agent, the hydrogen production performance of CdS@BDC NSs was lower than that of conventionally synthesized CdS NPs (Fig. S6, ESI†). This strongly indicated that the presence of the Na2S/Na2SO3 system also had a crucial impact on the hydrogen production performance. According to the FT-IR spectra of CdS@BDC NSs (Fig. S7, ESI†), there are the characteristic peaks of S2− at 533 cm−1 and SO32− at 1440 cm−1. Meanwhile, the unwashed CdS NP sample was taken out of Na2S/Na2SO3 solution, and the two absorption peaks were very weak. Obviously, after separation of the suspensible CdS@BDC NS from sulfur-rich S2−/SO32− solution, the resultant sample shows the highest various absorption peaks including the adsorbed S2− and SO32−, and the selective adsorption of massive S2− ions on the CdS@BDC NSs surface.
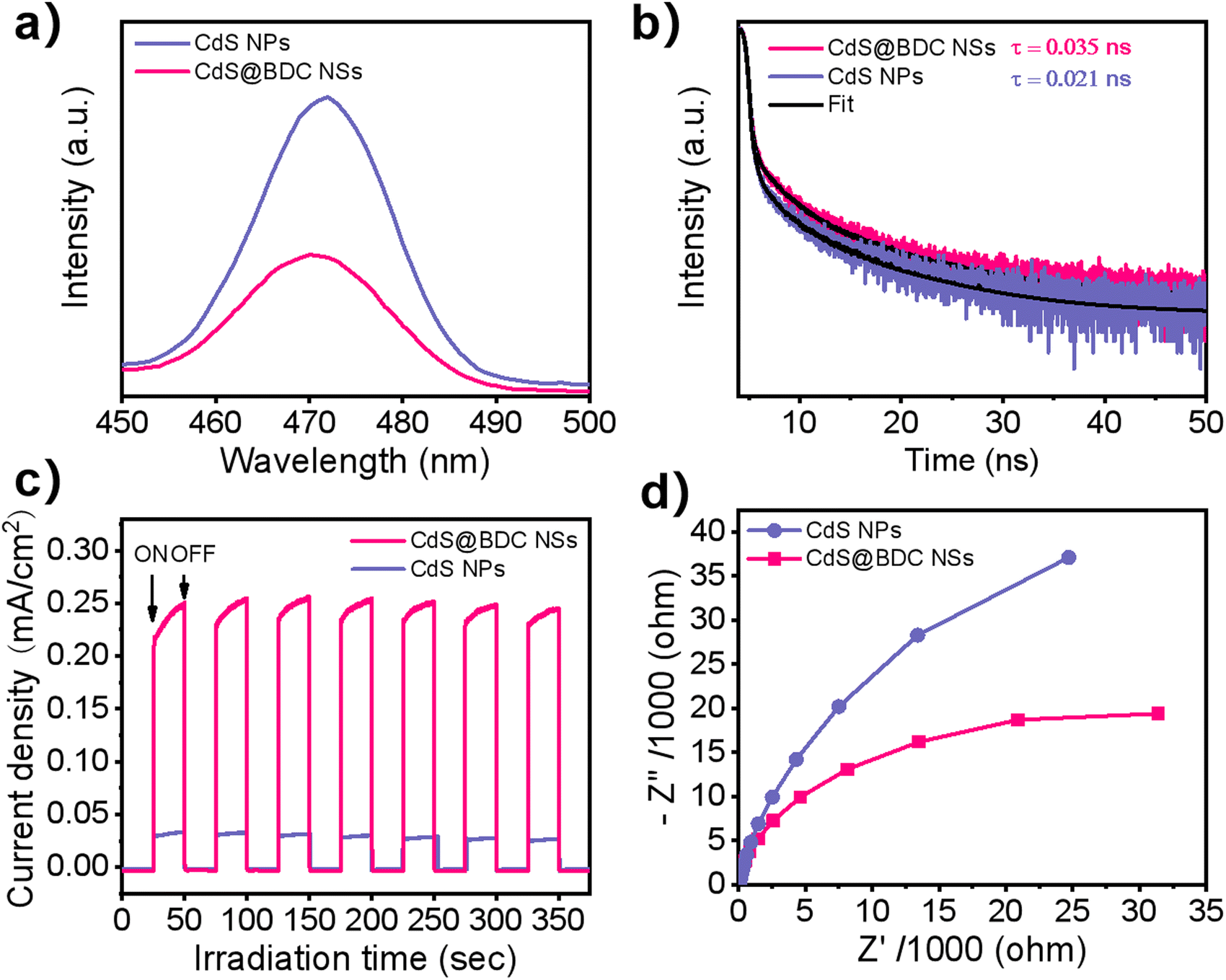 | ||
| Fig. 5 (a) Photoluminescence spectra. (b) Time-resolved photoluminescence decay spectra. (c) Photocurrent response. (d) Electrochemical impedance spectra for the CdS@BDC NSs and CdS NPs. | ||
On the basis of the above results, a tentative photocatalytic mechanism for the enhanced hydrogen production performance over CdS@BDC NSs was proposed in Fig. 6. In this work, a very important feature is that MOF-derived CdS@BDC NSs can efficiently adsorb a larger amount of S2−/SO32− compared with the traditional CdS NPs (Fig. S7, ESI† and Fig. 6A). Then, due to the two-dimensional skeleton structure on the surface of CdS@BDC NSs and a large number of exposed S2− ions, this supported the photo-excited electrons and holes to migrate to the spatially separated reduction center (at the small edge) and oxidation center (at the large base surface).53 On one hand, it is well known that S2−/SO32− has been widely used as an excellent sacrificial reagent for effectively consuming photogenerated holes in the photocatalytic H2 evolution of CdS photocatalysts, in which S2− ions can be oxidized into solid S0 by photogenerated holes at first, and then converted into S2O32− ions together with SO32− (Fig. 6B). The depletion of holes will inhibit the recombination of photo-generated electrons and holes, and as a result, more photoelectrons can be effectively used for proton reduction. This is also confirmed by the above photoluminescence spectrum and time-resolved luminescence decay spectrum analysis results shown in Fig. 5a and b. On the other hand, massive S2− adsorbed on the surface of ultrathin CdS@BDC NSs can also be exposed as interfacial hydrogen evolution active sites (also known as electronic co-catalysts), which can enrich H+ ions from sacrificial agent solution and subsequently reduce H+ to H2 through photoelectrons for promoting the hydrogen production reaction (Fig. 6C).54,55 Therefore, numerous S2−/SO32− ions adsorbed on the surface of CdS@BDC NSs can not only act as a hole-scavenger to promote the rapid capture and consumption of photogenerated holes, but also serve as an active site for catalytic reaction, and synergistically enhance the photocatalysis hydrogen production performance. That is to say, CdS in CdS@BDC NSs is the catalytic center, while BDC mainly plays an auxiliary role in the framework. Moreover, BDC with a porous structure assisted CdS to form an ultra-thin structure similar to complex, which made a large amount of S2−/SO32− adsorbed on the surface, thus removing a large amount of photo-generated holes and inhibiting the photo-corrosion of CdS. When another lactic acid solution replaces Na2S/Na2SO3 solution as the hole sacrificial agent, the hydrogen production performance of CdS@BDC NSs is lower than that of the conventionally synthesized CdS NPs (Fig. S6, ESI†). Because lactic acid molecules can't be enriched on the surface of CdS in CdS@BDC NSs due to the steric hindrance and the adsorption effect, which leads to a sharp decrease in performance, it helps to verify the structural function of CdS@BDC NSs.
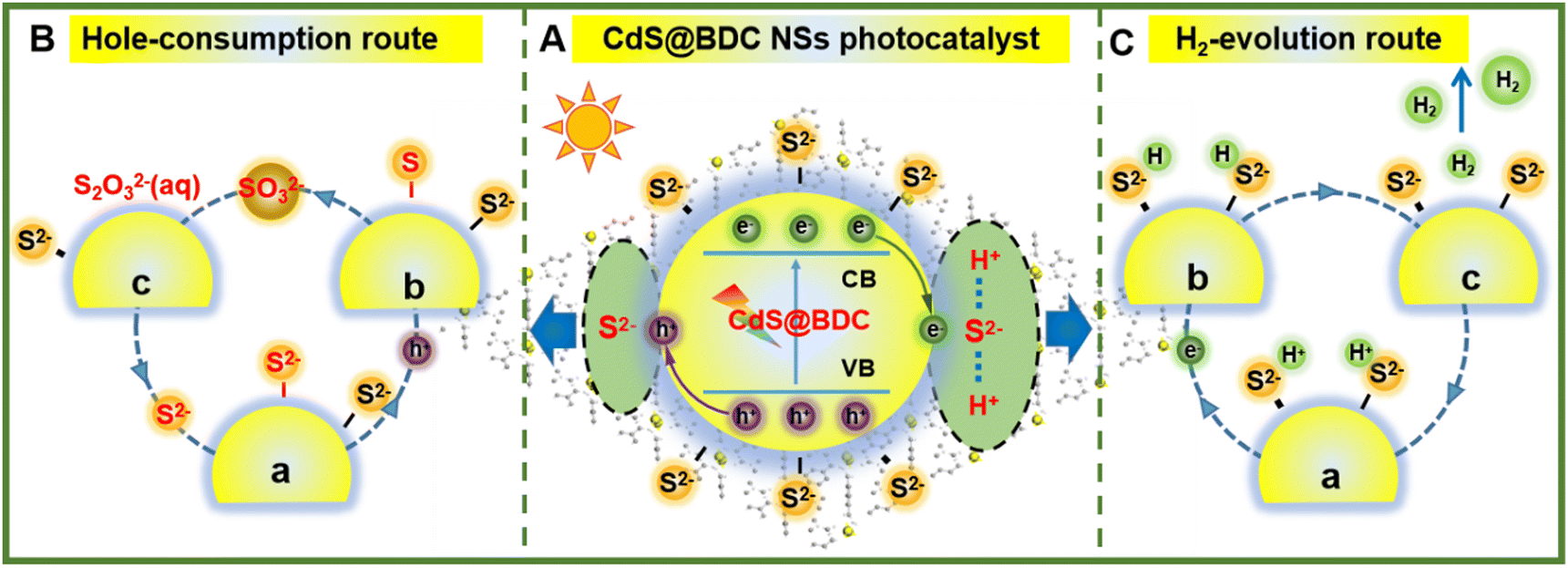 | ||
| Fig. 6 Schematic illustration of the S2−-mediated mechanism in CdS@BDC NSs: (A) S2−-mediated interfacial charge transfer and separation in the CdS@BDC NSs, (B) hole-consumption route by S2−/SO32− ions, and (C) H2-evolution reaction route on the adsorbed S2− ions as the catalytic active sites. | ||
In view of the above research on the synthesis method and hydrogen production performance of CdS@BDC NSs, it is speculated that this synthesis strategy and the corresponding photocatalytic enhancement mechanism are also universal in other 2D MOF-based nanomaterials. Therefore, we further synthesized Zn-MOF NSs and the corresponding ZnS@BDC NSs (Fig. S8a–d, ESI†). As a comparison, traditional ZnS NPs were also synthesized. As shown in Fig. S9 (ESI†), compared with traditional ZnS NPs (101 μmol h−1 g−1), ZnS@BDC NSs exhibit a substantial increase in photocatalytic hydrogen production activity of 2801 μmol h−1 g−1. Hence, our proposed synthesis method and the corresponding enhancement mechanism of photocatalytic hydrogen evolution can be used as an effective method to develop new efficient hydrogen production photocatalysts.
4. Conclusions
In summary, we have successfully prepared ultrathin CdS@BDC nanosheet photocatalysts via in situ sulfidation treatment from a precursor of 2D Cd-MOF nanosheets. In this case, due to the porous structure and larger surface area of the synthesized CdS@BDC NSs derived from the 2D Cd-MOF, a large number of S2− ions can be rapidly enriched to its surface, preventing light corrosion and acting as an active site of the surface catalytic reaction. It was proposed that the resultant CdS@BDC NS photocatalysts exhibited a perfect hydrogen generation activity of 13081 μmol h−1 g−1, which was about 65 times higher than that of CdS NPs. In addition, a tentative photocatalytic mechanism for the enhanced hydrogen production performance over CdS@BDC NSs was also proposed. Our work not only provides a facile and effective method for the synthesis of highly efficient photocatalysts, but also opens up new insight for the application of 2D MOF-based nanomaterials in photocatalysis.
Author contributions
Jian Li: conceptualization, investigation, formal analysis, data curation, validation, and writing – review and editing. Huaqiang Wang: conceptualization, investigation, formal analysis, data curation, methodology, and writing – original draft. Yihong Gao: theoretical calculation. Hao Fang: methodology and investigation. Peiyu Chen: formal analysis and performed the theoretical calculations. Fangzhi Huang: conceptualization, data curation, formal analysis, funding acquisition, resources, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21771001, 51872002, and 21371002), the key research and development projects in Anhui Province (202004a07020026), and the Key Lab of Photovoltaic and Energy Conservation Materials, Chinese Academy of Sciences, which is gratefully acknowledged by the authors.References
- S. B. Wang, B. Y. Guan and X. W. D. Lou, J. Am. Chem. Soc., 2018, 140, 5037–5040 CrossRef CAS PubMed.
- J. R. Ran, W. W. Guo, H. L. Wang, B. C. Zhu, J. G. Yu and S. Z. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed.
- J. R. Ran, B. C. Zhu and S.-Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 CrossRef CAS PubMed.
- K. Yang, X. X. Li, C. L. Yu, D. B. Zeng, F. Y. Chen, K. L. Zhang, W. Y. Huang and H. B. Ji, Chin. J. Catal., 2019, 40, 796–818 CrossRef CAS.
- Z. X. Qin, Z. X. Huang, M. L. Wang, D. Y. Liu, Y. B. Chen and L. J. Guo, Appl. Catal., B, 2020, 261, 118211 CrossRef CAS.
- J. Cao, H. Wang, Y. Zhao, Y. Liu, Q. Wu, H. Huang, M. Shao, Y. Liu and Z. Kang, J. Mater. Chem. A, 2020, 8, 3701–3707 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- N. Tian, Y. H. Zhang, X. W. Li, K. Xiao, X. Du, F. Dong, G. I. N. Waterhouse, T. R. Zhang and H. W. Huang, Nano Energy, 2017, 38, 72–81 CrossRef CAS.
- W. Wang, G. Li, T. An, D. K. L. Chan, J. C. Yu and P. K. Wong, Appl. Catal., B, 2018, 238, 126–135 CrossRef CAS.
- C. Lai, M. M. Zhang, B. S. Li, D. L. Huang, G. M. Zeng, L. Qin, X. G. Liu, H. Yi, M. Cheng, L. Li, Z. Chen and L. Chen, Chem. Eng. J., 2019, 358, 891–902 CrossRef CAS.
- K. K. Dey, S. Gahlawat and P. P. Ingole, J. Mater. Chem. A, 2019, 7, 21207–21221 RSC.
- T.-D. Pham, B.-K. Lee, V. N. Nguyen and V.-D. Dao, J. Catal., 2017, 352, 13–21 CrossRef CAS.
- M. Xiao, B. Luo, M. Lyu, S. Wang and L. Wang, Adv. Energy Mater., 2018, 8, 1701605 CrossRef.
- J. Wu, W. Xia, M. H. Lan, X. J. Xing, J. C. Hu, L. Huang, J. Liu, Y. Y. Ren, H. F. Liu and F. Wang, J. Mater. Chem. A, 2020, 8, 21690 RSC.
- X. M. Ning, Y. L. Wu, X. F. Ma, Z. Zhang, R. Q. Gao, J. Chen, D. L. Shan and X. Q. Lu, Adv. Funct. Mater., 2019, 29, 1902992 CrossRef.
- C. Zhu, C. Liu, Y. J. Zhou, Y. J. Fu, S. J. Guo, H. Li, S. Q. Zhao, H. Huang, Y. Liu and Z. H. Kang, Appl. Catal., B, 2017, 216, 114–121 CrossRef CAS.
- G. T. Sun, S. M. Mao, D. D. Ma, Y. J. Zou, Y. X. Lv, Z. H. Li, C. He, Y. H. Cheng and J.-W. Shi, J. Mater. Chem. A, 2019, 7, 15278–15287 RSC.
- J. C. Hu, S. S. Sun, M. D. Li, W. Xia, J. Wu, H. F. Liu and F. Wang, Chem. Commun., 2019, 55, 14490 RSC.
- T. Xiong, H. Wang, Y. Zhou, Y. J. Sun, W. L. Cen, H. W. Huang, Y. X. Zhang and F. Dong, Nanoscale, 2018, 10, 8066–8074 RSC.
- P. Wang, Y. Sheng, F. Z. Wang and H. G. Yu, Appl. Catal., B, 2018, 220, 561–569 CrossRef CAS.
- D. P. Kumar, S. Hong, D. A. Reddy and T. K. Kim, Appl. Catal., B, 2017, 212, 7–14 CrossRef CAS.
- Y. J. Hao, S.-Z. Kang, X. Liu, X. Q. Li, L. X. Qin, J. Mu and A. C. S. Sustainable, Chem. Eng., 2016, 5, 1165–1172 Search PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- J.-D. Xiao and H.-L. Jiang, Small, 2017, 13, 1700632 CrossRef PubMed.
- J.-X. Lv, Z.-M. Zhang, J. Wang, X.-L. Lu, W. Zhang and T.-B. Lu, ACS Appl. Mater. Interfaces, 2018, 11, 2655–2661 CrossRef PubMed.
- D. D. Ma, J.-W. Shi, Y. J. Zou, Z. Y. Fan, X. Ji and C. M. Niu, ACS Appl. Mater. Interfaces, 2017, 9, 25377–25386 CrossRef CAS PubMed.
- H. X. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–474 CrossRef CAS PubMed.
- B. L. Zhu, B. Z. Lin, Y. Zhou, P. Sun, Q. R. Yao, Y. L. Chen and B. F. Gao, J. Mater. Chem. A, 2014, 2, 3819–3827 RSC.
- C. L. Tan and H. Zhang, Chem. Soc. Rev., 2015, 44, 2713–2731 RSC.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- S. K. Muduli, E. Varrla, Y. Xu, S. A. Kulkarni, A. Katre, S. Chakraborty, S. Chen, T. C. Sum, R. Xu and N. Mathews, J. Mater. Chem. A, 2017, 5, 24874–24879 RSC.
- D. D. Zhu, J. L. Liu, Y. Q. Zhao, Y. Zheng and S. Z. Qiao, Small, 2019, 15, 1805511 CrossRef PubMed.
- B. Han, X. W. Ou, Z. Q. Deng, Y. Song, C. Tian, H. Deng, Y.-J. Xu and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 16811–16815 CrossRef CAS PubMed.
- Y. Huang, M. T. Zhao, S. K. Han, Z. C. Lai, J. Yang, C. L. Tan, Q. L. Ma, Q. Q. Lu, J. Z. Chen, X. Zhang, Z. C. Zhang, B. Li, B. Chen, Y. Zong and H. Zhang, Adv. Mater., 2017, 29, 1700102 CrossRef PubMed.
- J. R. Ran, J. T. Qu, H. P. Zhang, T. Wen, H. L. Wang, S. M. Chen, L. Song, X. L. Zhang, L. Q. Jing, R. K. Zheng and S. Z. Qiao, Adv. Energy Mater., 2019, 9, 1803402 CrossRef.
- Q. P. Lu, M. T. Zhao, J. Z. Chen, B. Chen, C. L. Tan, X. Zhang, Y. Huang, J. Yang, F. F. Cao, Y. F. Yu, J. F. Ping, Z. C. Zhang, X. J. Wu and H. Zhang, Small, 2016, 12, 4669–4674 CrossRef CAS PubMed.
- Y. F. Lin, G. Chen, H. Wan, F. S. Chen, X. H. Liu and R. Z. Ma, Small, 2019, 15, 1900348 CrossRef PubMed.
- R.-B. Wei, Z.-L. Huang, G.-H. Gu, Z. Wang, L. X. Zeng, Y. B. Chen and Z.-Q. Liu, Appl. Catal., B, 2018, 231, 101–107 CrossRef CAS.
- W. C. Wang, Y. Tao, J. C. Fan, Z. P. Yan, H. Shang, D. L. Phillips, M. Chen and G. S. Li, Adv. Funct. Mater., 2022, 32, 2201357 CrossRef CAS.
- W. L. Zhen, X. F. Ning, B. J. Yang, Y. Q. Wu, Z. Li and G. X. Lu, Appl. Catal., B, 2018, 221, 243–257 CrossRef CAS.
- A. D. Burrows, K. Cassar, T. Düren, R. M. W. Friend, M. F. Mahon, S. P. Rigby and T. L. Savarese, Dalton Trans., 2008, 2465–2474 RSC.
- X. F. Ning, S. G. Meng, X. L. Fu, X. G. Ye and S. F. Chen, Green Chem., 2016, 18, 3628–3639 RSC.
- G. D. Yang, B. L. Yang, T. C. Xiao and Z. F. Yan, Appl. Surf. Sci., 2013, 283, 402–410 CrossRef CAS.
- X. X. Zhao, J. R. Feng, J. Liu, W. Shi, G. M. Yang, G. C. Wang and P. Cheng, Angew. Chem., Int. Ed., 2018, 57, 9790–9794 CrossRef CAS PubMed.
- Y. J. Xiao, Y. Qi, X. L. Wang, X. Y. Wang, F. X. Zhang and C. Li, Adv. Mater., 2018, 30, 1803401 CrossRef PubMed.
- Y. Matsumoto, J. Solid State chem., 1996, 126, 227–234 CrossRef CAS.
- S. Thaweesak, M. Q. Lyu, P. Peerakiatkhajohn, T. Butburee, B. Luo, H. J. Chen and L. Wang, Appl. Catal., B, 2017, 202, 184–190 CrossRef CAS.
- M. Ou, W. G. Tu, S. M. Yin, W. N. Xing, S. Y. Wu, H. J. Wang, S. P. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 130, 13758–13762 CrossRef.
- F. Raziq, L. Q. Sun, Y. Y. Wang, X. L. Zhang, M. Humayun, S. Ali, L. L. Bai, Y. Qu, H. T. Yu and L. Q. Jing, Adv. Energy Mater., 2018, 8, 1701580 CrossRef.
- J. Bian, Y. Qu, X. L. Zhang, N. Sun, D. Y. Tang and L. Q. Jing, J. Mater. Chem. A, 2018, 6, 11838–11845 RSC.
- M. Zhou, S. B. Wang, P. J. Yang, C. J. Huang and X. C. Wang, ACS Catal., 2018, 8, 4928–4936 CrossRef CAS.
- N. F. Wang, S. Cheong, D. E. Yoon, P. Lu, H. J. Lee, Y. K. Lee, Y. S. Park and D. C. Lee, J. Am. Chem. Soc., 2022, 144(37), 16974–16983 CrossRef CAS PubMed.
- H. G. Yu, W. Zhong, X. Huang, P. Wang and J. G. Yu, ACS Sustain. Chem. Eng., 2018, 6, 5513–5523 CrossRef CAS.
- W. Zhong, X. Huang, Y. Xu and H. G. Yu, Nanoscale, 2018, 10, 19418–19426 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00866a |
| ‡ These authors contribute equally to this paper. |
| This journal is © The Royal Society of Chemistry 2022 |