
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLoading of DOX into a tetrahedral DNA nanostructure: the corner does matter†
Yao
Xu
 a,
Shu-wei
Huang
a,
Yu-qiang
Ma
*a and
Hong-ming
Ding
*b
a,
Shu-wei
Huang
a,
Yu-qiang
Ma
*a and
Hong-ming
Ding
*b
aNational Laboratory of Solid State Microstructures and Department of Physics, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, China. E-mail: myqiang@nju.edu.cn
bCenter for Soft Condensed Matter Physics and Interdisciplinary Research, School of Physical Science and Technology, Soochow University, Suzhou 215006, China. E-mail: dinghm@suda.edu.cn
First published on 7th December 2021
Abstract
With the rapid development of nanotechnology, various DNA nanostructures have been synthesized and widely used in drug delivery. However, the underlying mechanisms of drug molecule loading into the DNA nanostructure are still elusive. In this work, we systematically investigate the interactions of a tetrahedral DNA nanostructure (TDN) with the anti-cancer drug doxorubicin (DOX) by combining molecular docking and all-atom molecular dynamics simulations. It is found that there are five possible binding modes in the single TDN–DOX interactions, namely the outside-corner mode, the inside-corner mode, the major-groove mode, the minor-groove mode, and the intercalation mode, where the van der Waals (VDW) interaction and the electrostatic (ELE) interaction dominate in the case of unionized DOX and ionized DOX, respectively. Moreover, with the increase of the DOX number, some of the interaction modes may disappear and the inside-corner mode is the most energy-favorable mode. The present study enhances the molecular understanding of the role of TDN as the drug carrier, which may provide a useful guideline for the future design of DNA nanostructures.
Introduction
Nucleic acids play an irreplaceable role in biological inheritance since most forms of life use DNA as a carrier of genetic information. In nature, there are two typical forms of DNA structures, namely the single-stranded (ss) form and the double-strand (ds) form that comes from two ssDNA molecules using the hybridization principle. The ss-DNA or ds-DNA can further fold or assemble into 3D structures like nucleosomes, which determine their biological functions in living organisms. Besides the most common B-form dsDNA, the DNA molecule has a variety of other structures.With the growing understanding of the structure of DNA molecules, recently the programmability of the DNA primary structure and richness of the secondary structure have attracted more and more attention from researchers in nanoscience and biochemistry.1–6 The advent of various assembly methods like DNA origami or DNA tiles made it possible to compose complicated 3D structures like DNA origami nanotubes,7,8 nanoflasks,9 nanocages,10,11etc. In the meantime, the various types of DNA-based nanomaterials synthesized have been widely used in biomedical applications.12–19
Drug delivery could be one of the most intriguing applications, where the DNA nanostructure is used as a carrier to deliver the drug molecules in the human body.20–23 Apart from increasing the solubility of drugs, protecting the drugs from enzyme attack as well as fascinating cellular delivery of the drugs (which other nanocarriers may also have), the good biocompatibility24 and the low cytotoxicity25 make the DNA nanostructure an ideal candidate for drug delivery. In the meantime, thanks to its editable characteristics, modifications for targeting can be easily applied. For example, some delivery systems or detection platforms have been developed based on DNA nanocages including tetrahedrons26–29 and octahedrons.30–32
Despite the extensive research on the in vitro/in vivo delivery of drug molecules by DNA nanostructures in the experiments,19,33–37 it is still largely unknown how the DNA structures interact with the drug molecules, which requires a deeper understanding of their interactions at the molecular level. More importantly, due to the unique topological structure of the DNA nanostructures, more possible drug-binding sites may occur, which may be quite different from the interaction of a single dsDNA chain with drug molecules.38–43
In this work, we take the tetrahedral DNA nanostructure (TDN) and doxorubicin44–46 (DOX) as examples of the DNA nanostructure and drug molecule, respectively, and investigate their interactions under different conditions by combining molecular docking and all-atom molecular dynamics simulations. As shown below, there are at least five different binding modes in the TDN–DOX interaction, which are dependent on the ionization state and the concentration of the DOX. The binding energy analysis along with its energy decomposition is further used to reveal the underlying molecular mechanism of the different binding behaviors.
Model and methods
The atomistic model of TDN-17 was built using the Polygen program,47 where 17 indicates that each side of the triangle that makes up the tetrahedron consists of 17 nucleotides including 16 hybridized nucleotides and 1 free nucleotide (Table 1). The initial conformation of DOX was downloaded from the PubChem database (PubChem CID: 31703).48 Then the geometry optimization of the DOX molecule was calculated by using the B3LYP functional together with a 6-311G(d,p) basis set, and was carried out with the Gaussian 09 program.49| Face | Sequence |
|---|---|
| F1 | AAGGCGAGGAATTTTTCCTGAGGGTGCTACCTGTCGGGTGTTTAAAGTTAT |
| F2 | GAAAATCTCGGCCTCTTGCCCACAAATTTCAATGCTACGAGAGAAGTGGCA |
| F3 | CTTTTAGAGCCGGAGAGGTTACATCCAGGCTACTTTCCGCTCCTTAAAAAT |
| F4 | GACTCCCACGATGGACTCAATGTAGGTCCGATGTGATGCTCTCTTCACCGG |
The HDOCK webserver was used for molecular docking.50,51 The optimized DOX along with a stabilized TDN-17 was submitted to the webserver (http://hdock.phys.hust.edu.cn/), where a pre-equilibrated TDN-17 was used as the receptor molecule and the DOX was used as the ligand molecule. After the docking job was completed, the server gave 100 pre-generated binding models based on the docking score and the root-mean-square deviation (RMSD) of the ligand. The top 30 predictions were examined manually based on the score ranking and cluster analysis to evaluate all the possible binding modes.
All-atom molecular dynamics (MD) simulations were further performed based on the above-selected structures. Notably, each simulation system only considered one DOX monomer/aggregate and one TDN. All MD simulations were carried out by the GROMACS 2019.6 software package,52,53 and the Amber14sb_Parmbsc1 (ref. 54 and 55) force field was used to describe the properties of DNA and ions together with the TIP3P water model. NaCl was used to neutralize the system, and the concentration was set at 0.15 M, close to that under physiological conditions. The parameters for the DOX were generated by the general Amber force field56,57 (GAFF) with the restrained electrostatic potential58 (RESP) charges. The LINCS constraints59 were used for all bonds involving hydrogen atoms. The particle mesh Ewald (PME) method was used when calculating the long-range electrostatic interactions, and the Lennard–Jones (LJ) interactions were cut off at a distance of 1.0 nm. The periodic boundary conditions were adopted in all three directions.
In the simulation, the system was firstly energy-minimized by the steepest descent method until convergence was reached. Then each system was subjected to a 500 ps pre-equilibration process in the NVT and NPT ensembles, separately, with all the heavy atoms harmonically constrained by 1000 kJ mol−1 nm−2. Finally, the constraint was released and 100 ns free NPT simulations were performed. The temperature was controlled at 300 K by the V-rescale thermostat60 with a time constant of 0.2 ps and the pressure was kept at 1.0 bar by the Parrinello–Rahman barostat61 with a time constant of 2.0 ps. The typical snapshots of this work were all drawn using VMD software.62,63 The schematic diagrams of TDN–DOX interactions were drawn using PyMOL.64
In the binding energy calculation, the last 10 ns of each stabilized simulation trajectory was used with an interval of 100 ps, namely 100 frames for each run using the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) method.65–69 The gmx_mmpbsa script (https://jerkwin.github.io/gmxtool) together with the APBS70,71 program were used to calculate the binding energy and the corresponding energy decomposition term, namely the electrostatic (ELE) energy, the van der Waals (VDW) energy, and the solvation energy including the polar part (PB) and the non-polar part (SA) in this work.
Results and discussion
Binding of a single unionized DOX molecule to the TDN-17
A typical dsDNA molecule is formed by two helical strands of ssDNA. The nucleobases on the two strands complement each other to form the secondary structure of DNA. Since the double-helical structure is not spatially symmetric, there are two different grooves defined by spatial distance (i.e., the major groove and the minor groove, Fig. 1a), and the dsDNA may have two different binding sites for the DOX (Fig. 1b). Moreover, the TDN is a tetrahedron composed of 6 B-DNA double helices and 6 single-base staples. Thus, the TDN has a richer spatial structure than the regular dsDNA and may provide more binding sites for the DOX, which makes the TDN a promising drug carrier. As shown in Fig. 1c–g, five typical binding modes were observed by the molecular docking, namely the inside-corner mode, the outside-corner mode, the intercalation mode, the major-groove mode, and the minor-groove mode. Notably, the intercalation mode, the major-groove mode, and the minor-groove mode have been reported in previous DNA–DOX experiments.72,73 Moreover, a recent experimental study found that the DOX-loading ability of the TDN was much higher than that of the dsDNA with the same bps.74 Considering that the corner is the only difference between the TDN and dsDNA for the DOX loading, the efficient loading of DOX at the corner of the TDN should be the point, which gives indirect evidence for the two new modes (i.e., inside-corner and outside-corner modes) here.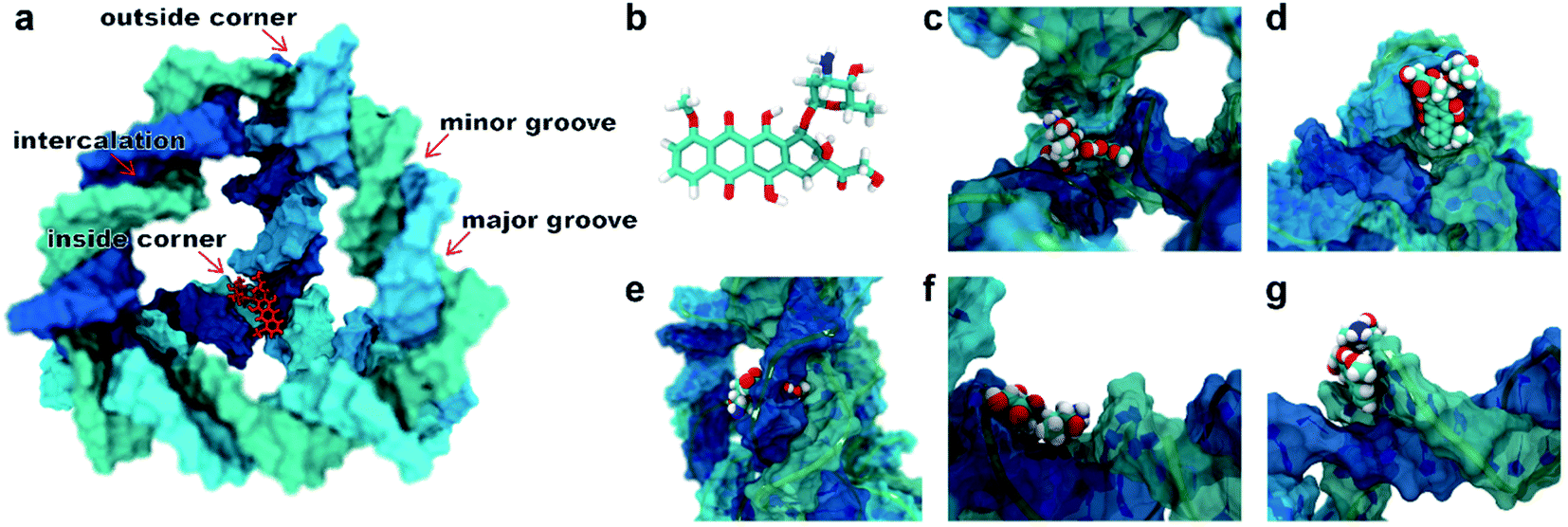 | ||
| Fig. 1 (a) Schematic illustration for a single DOX binding to the TDN-17 and (b) the structural formula of the DOX. The possible binding modes observed in the molecular docking: (c) the inside-corner mode, (d) the outside-corner mode, (e) the intercalation mode, (f) the major-groove mode, and (g) the minor-groove mode. | ||
To test the robustness of all the binding modes, we then performed all-atom molecular dynamics simulations for each complex above, respectively. As shown in Fig. 2, the root-mean-square deviation (RMSD) of TDN, the contact surface area (CSA) and the minimum distance between the DOX and TDN changed little with simulation time, indicating that the binding of DOX to TDN was relatively stable in all five modes.
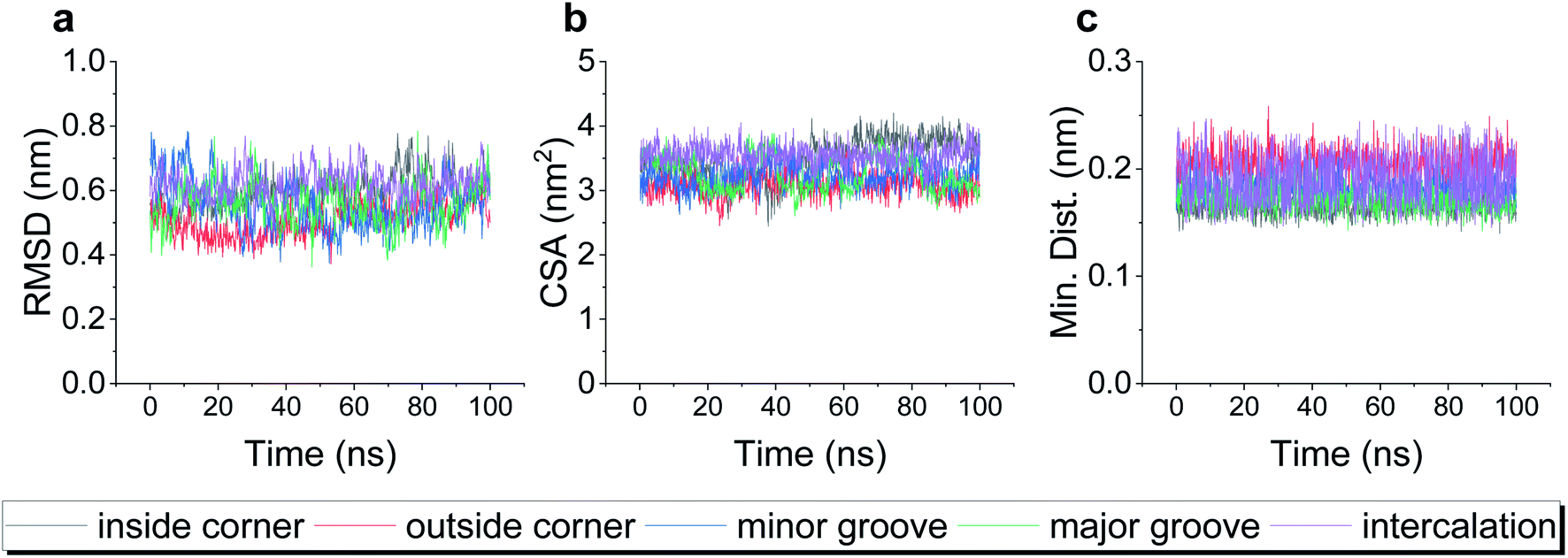 | ||
| Fig. 2 The root-mean-square deviation (RMSD) of TDN-17 (a), the contact surface area (CSA) between the TDN-17 and DOX (b), and the minimum distance between TDN-17 and DOX (c) as a function of time during the molecular dynamics simulations. | ||
Having demonstrated the stability of the observed modes, we then calculated the binding energy of TDN–DOX and analyzed the binding driving force in each mode (Fig. 3a). Among the five modes, the binding energy was the lowest in the intercalation mode (−182 kJ mol−1), followed by the outside-corner mode (−169 kJ mol−1), the inside-corner mode (−162 kJ mol−1), the minor-groove mode (−150 kJ mol−1), and the major-groove mode (−102 kJ mol−1). Notably, the VDW energy was very strong in the intercalation (−212 kJ mol−1) and outside-corner modes (−190 kJ mol−1) since there was an obvious π–π stacking between the adenine of DNA and an anthraquinone ring part of DOX (Fig. 3b and c). Notably, there were some unpacked nucleotides in the outside corner of the TDN, which can provide a more flexible region for π–π stacking. As a result, the DOX with aromaticity was more likely to adsorb on the outside of the TDN. In contrast, the electrostatic interaction was strong in the inside-corner, minor-groove, and the major-groove modes since there were some hydrogen bonds formed by the nitrogen atoms (blue) in DNA and oxygen atoms (red) in DOX (Fig. 3d–f). Nevertheless, there were many polar residues at the binding site, leading to a high solvation energy result, thus the binding energy was weak in these cases. In general, the VDW interaction was the dominant driving force for the binding of single unionized DOX to the TDN.
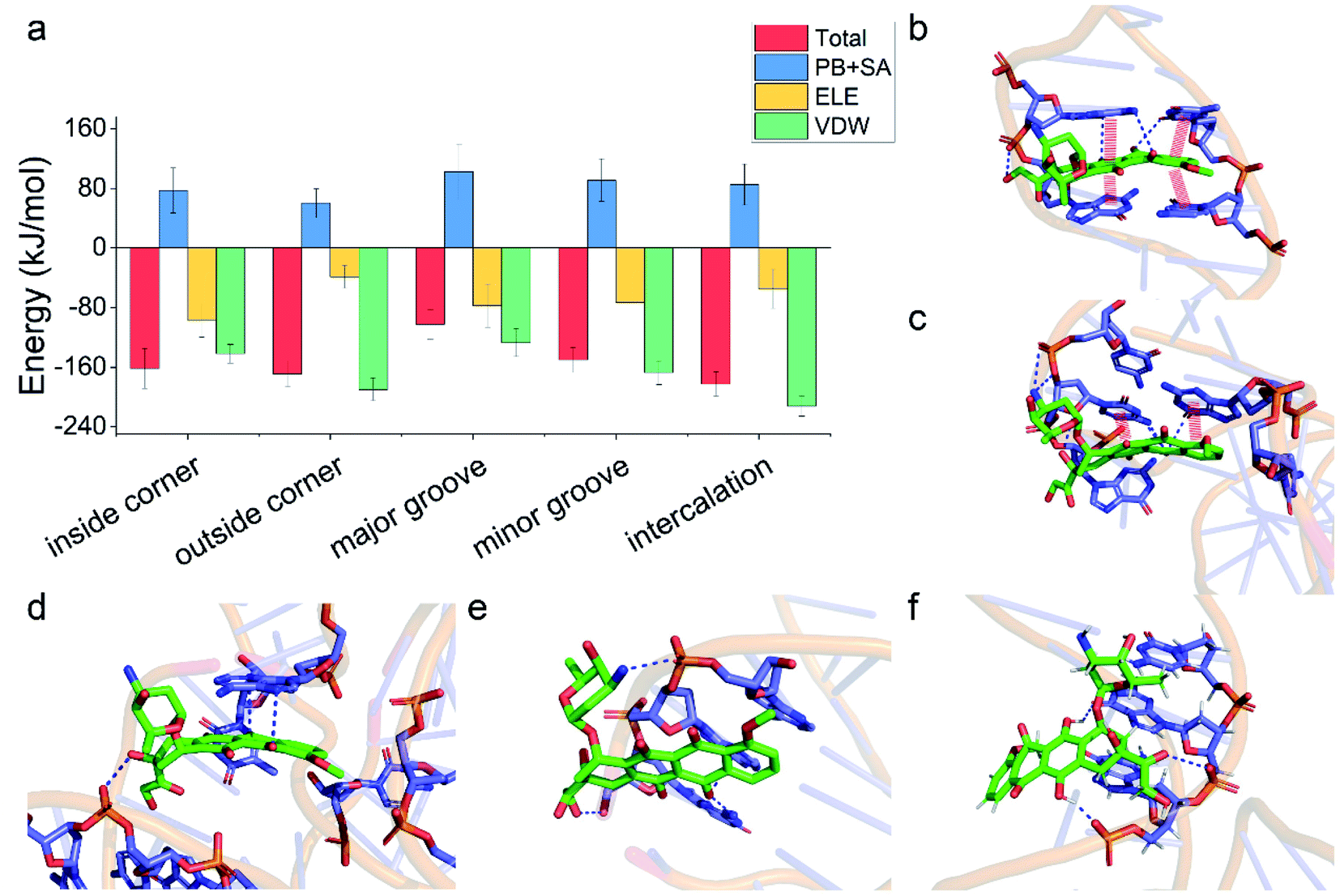 | ||
| Fig. 3 (a) The total binding energies and corresponding energy decomposition terms for the five binding modes. Structural depiction of key interfacial interactions in the five binding modes: (b) the intercalation mode, (c) the outside-corner mode, (d) the inside-corner mode, (e) the minor-groove mode, and (f) the major-groove mode. The red thick line indicates the π–π stacking and the blue dashed line represents the polar interaction. | ||
Binding of multiple unionized DOX molecules to the TDN-17
Due to its aromatic ring, the DOX molecules can aggregate with each other in water when the concentration of DOX is not very low. Could the aggregation of DOX affect its binding to the TDN?To answer this problem, we first simulated the self-assembly of two DOX molecules, four DOX molecules, and eight DOX molecules in water, respectively. After a 50 ns simulation, the DOX dimer, tetramer and octamer were obtained (Fig. S1†). Similar to the previous case, molecular docking was used to obtain the possible binding modes, followed by a 100 ns all-atom molecular dynamics simulation. The final snapshots for the possible binding modes are shown in Fig. 4a–c. Due to the formation of the aggregate, the size of the DOX became larger (than that of the single DOX), and there was not enough room for the DOX to insert into the intercalation space between two base pairs as well as the minor groove. As a result, the intercalation and the minor-groove modes can no longer be observed in this case. With a further increase of the DOX number, the outside-corner mode disappeared in the case of the DOX tetramer and octamer.
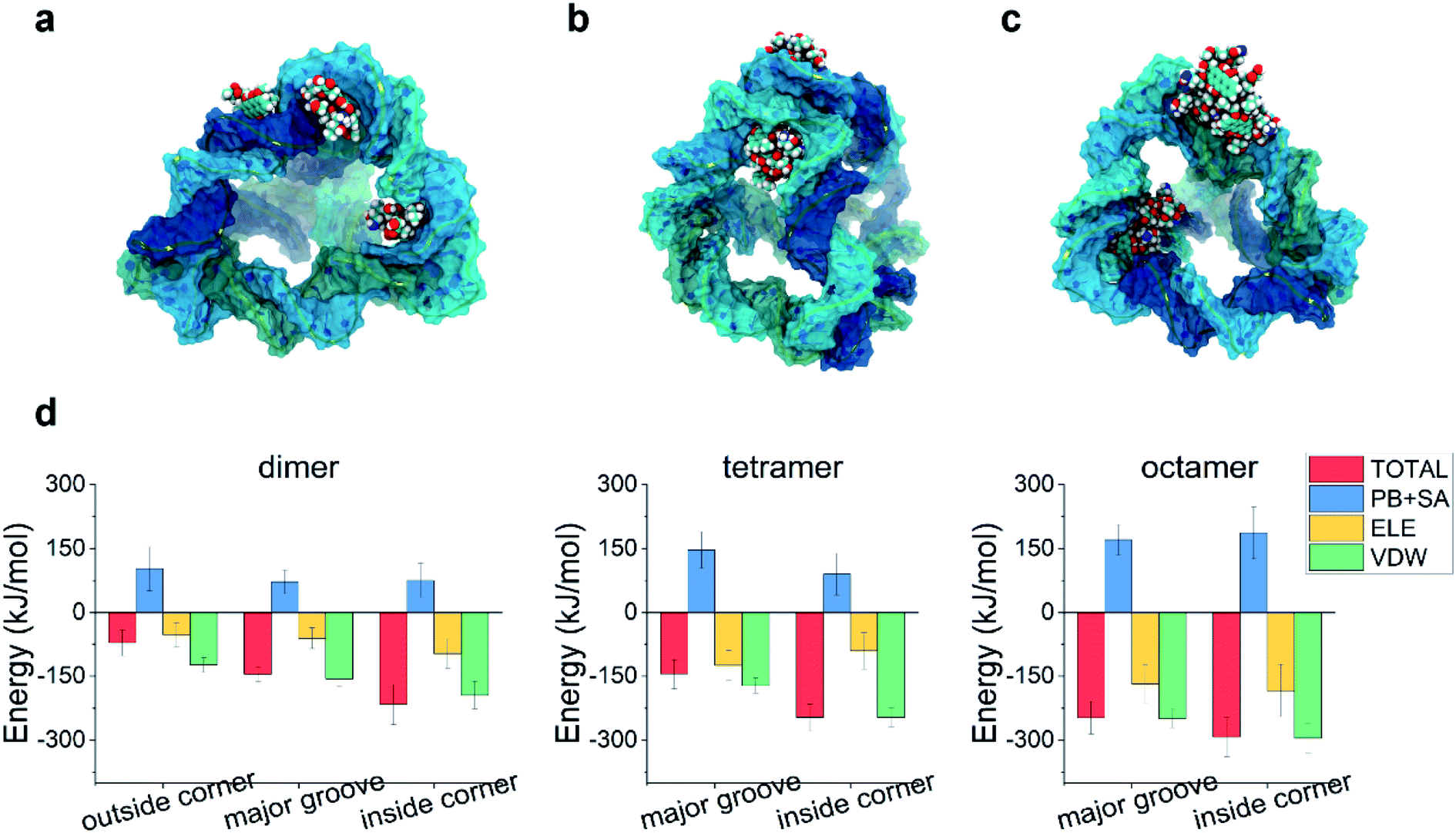 | ||
| Fig. 4 Final snapshots illustrating the typical modes in the multiple DOX–TDN-17 interaction: (a) DOX dimer, (b) DOX tetramer, and (c) DOX octamer. (d) The total binding energies and corresponding energy decomposition terms for the above binding modes. | ||
We then calculated the binding energies of TDN–DOX in these cases. As shown in Fig. 4d, in the case of the DOX dimer, the binding energy was the lowest in the inside-corner mode (−217 kJ mol−1), followed by the major-groove mode (−146 kJ mol−1) and the outside-corner mode (−72 kJ mol−1). Notably, the lowest binding energy here was a bit stronger than that in the intercalation mode of single DOX (−182 kJ mol−1). Moreover, the binding energy (−72 kJ mol−1) in the outside-corner mode here was even weaker than that in the case of single DOX (−169 kJ mol−1). With further increase of the DOX number, the lowest binding energy decreased gradually and was about −300 kJ mol−1 in the case of the DOX octamer. Actually, the formation of the DOX aggregates was mainly caused by the π–π stacking of the DOX molecules, which may shield some aromatic rings (from π–π stacking with the adenine in the TDN). As a result, the binding energy was not enhanced obviously. Moreover, the VDW interaction also dominated the binding process, which is similar to that in the single DOX.
Notably, the lowest binding energy of the multiple DOX–TDN interactions was observed in the inside-corner mode, which is the unique mode in the TDN (due to its 3D topological structure) and cannot be observed in the case of a single DNA chain. More importantly, the DOX was inside the TDN, which may protecte it from the undesired interactions during the in vivo cellular delivery. Notably, previous studies75,76 indicated that the corner-attack is an efficient way for DNA nanostructures to minimize electrostatic repulsion and translocate through the negatively charged cell membrane. Here, the corner of the TDN again exhibited its merit and the TDN was indeed a good nanocarrier for drug delivery.
Binding of ionized DOX molecules to the TDN-17
The pKa of doxorubicin is about 8.0, indicating that it can be partially ionized in the physiological environment (pH ∼ 7.4), namely, the amino group at the end of the DOX molecule is protonated into a positively charged one, leading to the whole molecule being positively charged. How did the ionization of DOX affect the binding behaviors?The possible binding modes along with their binding energies are listed in Fig. 5a and the corresponding final snapshots are shown in Fig. S2.† Similar to the case of unionized DOX, there still existed five, three, two, and two modes for the single ionized DOX, ionized DOX dimer, ionized DOX tetramer, and ionized DOX octamer, respectively. Nevertheless, the binding energies and the ranking of the binding modes became totally different. Since DNA is a highly negatively charged biomolecule and the ionized DOX carries one positive charge, there should be a strongly attractive electrostatic interaction between them. As a result, the binding energies were greatly enhanced. For example, the lowest binding energy of a single DOX–TDN was −398 kJ mol−1 and it was over twice as high as that in the unionized case (−182 kJ mol−1). Interestingly, the lowest binding energies were all observed in the inside-corner mode, which is probably due to the fact that the DOX molecules may contact more DNA bases inside the TDN.
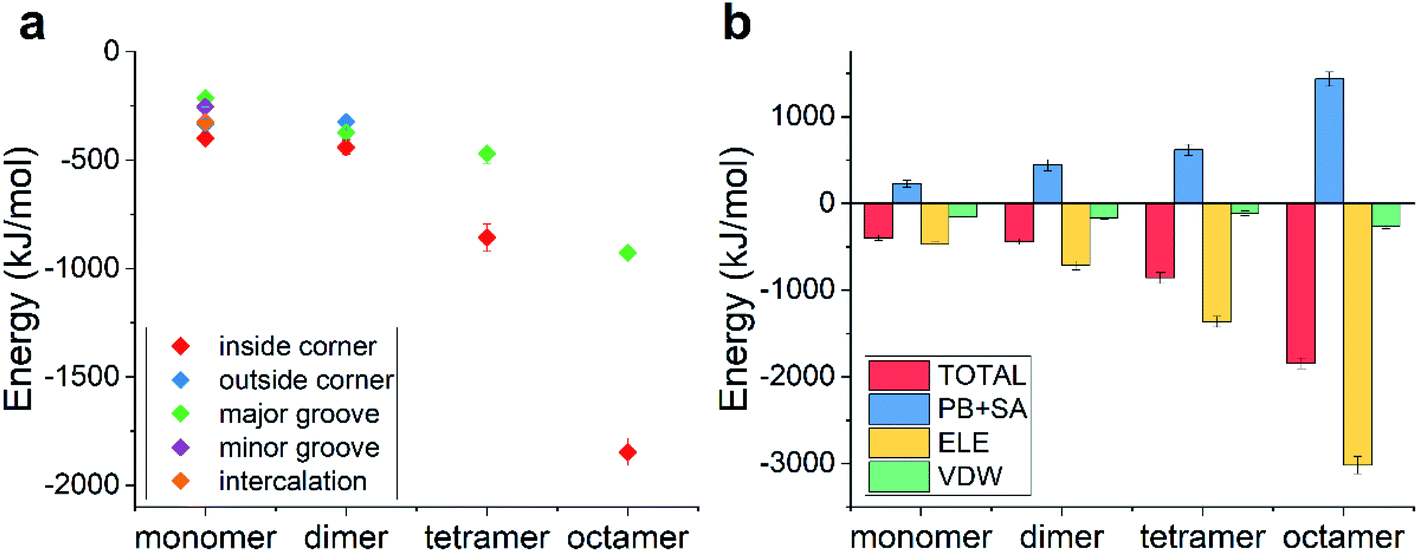 | ||
| Fig. 5 (a) The total binding energies as a function of the DOX number for the possible binding modes in the case of ionized DOX; (b) the energy decomposition terms as a function of the DOX number in the inside-corner binding mode. | ||
We further analyzed the energy terms of the inside-corner mode under different DOX numbers (Fig. 5b). Not surprisingly, the ELE energy dominated in all the cases and increased with the DOX number in a nearly linear manner. As a result, the binding energy increased obviously with the DOX number, which was different from the slow increase of the binding energy (with the DOX number) in the case of unionized DOX.
Conclusions
In summary, we have investigated the binding of DOX molecules to TDN-17 under different conditions by combining molecular docking and all-atom molecular dynamics simulations. Due to the unique structure and extra space of the TDN, five possible binding modes in the TDN–DOX interactions are observed, namely the outside-corner mode, the inside-corner mode, the major-groove mode, the minor-groove mode, and the intercalation mode. The binding energies of the binding modes and the ranking of the modes are dependent on the number and the ionization of the DOX. Moreover, it is found that the VDW interaction dominates in the TDN–unionized DOX interaction and the ELE interaction dominates in the TDN–ionized DOX interaction. Interestingly, the inside-corner mode is found to be the most energy-favorable mode in nearly all cases, which is due to the topological structure of the TDN. In general, the present study reveals the loading mechanism of the drug molecules into the TDN at the molecular level and may shed some light on the future development of functional DNA nanostructures in biomedical applications.Author contributions
Conceptualization: H. M. D. and Y. Q. M.; methodology: H. M. D. and Y. X.; formal analysis: Y. X.; investigation and visualization: Y. X. and S. W. H.; writing – original draft: H. M. D. and Y. X.; writing – review and editing: H. M. D. and Y. X.; funding acquisition: H. M. D. and Y. Q. M.; project administration: H. M. D. All authors approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 11874045, 12174184 and 11774147). We are grateful to the High Performance Computing Center (HPCC) of Nanjing University for performing the numerical calculations in this paper on its blade cluster system.References
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS PubMed.
- H. A. Day, P. Pavlou and Z. A. E. Waller, Bioorg. Med. Chem., 2014, 22, 4407–4418 CrossRef CAS PubMed.
- X. J. Yang, M. R. Cui, X. L. Li, H. Y. Chen and J. J. Xu, Chem. Commun., 2020, 56, 2135–2138 RSC.
- J. Li, L. Mo, C. H. Lu, T. Fu, H. H. Yang and W. Tan, Chem. Soc. Rev., 2016, 45, 1410–1431 RSC.
- H. M. Meng, H. Liu, H. Kuai, R. Peng, L. Mo and X. B. Zhang, Chem. Soc. Rev., 2016, 45, 2583–2602 RSC.
- F. Mo, K. Jiang, D. Zhao, Y. Wang, J. Song and W. Tan, Adv. Drug Delivery Rev., 2021, 168, 79–98 CrossRef CAS PubMed.
- E. Roth, A. Glick Azaria, O. Girshevitz, A. Bitler and Y. Garini, Nano Lett., 2018, 18, 6703–6709 CrossRef CAS PubMed.
- Y. X. Zhao, A. Shaw, X. Zeng, E. Benson, A. M. Nyström and B. Högberg, ACS Nano, 2012, 6, 8684–8691 CrossRef CAS PubMed.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS PubMed.
- Z. Ge, M. Lin, P. Wang, H. Pei, J. Yan, J. Shi, Q. Huang, D. He, C. Fan and X. Zuo, Anal. Chem., 2014, 86, 2124–2130 CrossRef CAS PubMed.
- Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nat. Commun., 2016, 7, 10619 CrossRef CAS PubMed.
- R. P. Goodman, I. A. T. Schaap, C. F. Tardin, C. M. Erben, R. M. Berry, C. F. Schmidt and A. J. Turberfield, Science, 2005, 310, 1661–1665 CrossRef CAS PubMed.
- J. Liu, L. Song, S. Liu, S. Zhao, Q. Jiang and B. Ding, Angew. Chem., Int. Ed., 2018, 57, 15486–15490 CrossRef CAS PubMed.
- H. Pei, X. Zuo, D. Zhu, Q. Huang and C. Fan, Acc. Chem. Res., 2014, 47, 550–559 CrossRef CAS PubMed.
- H. Li, N. Arroyo-Currás, D. Kang, F. Ricci and K. W. Plaxco, J. Am. Chem. Soc., 2016, 138, 15809–15812 CrossRef CAS PubMed.
- E. Kopperger, J. List, S. Madhira, F. Rothfischer, D. C. Lamb and F. C. Simmel, Science, 2018, 359, 296–301 CrossRef CAS PubMed.
- M. Lin, P. Song, G. Zhou, X. Zuo, A. Aldalbahi, X. Lou, J. Shi and C. Fan, Nat. Protoc., 2016, 11, 1244–1263 CrossRef CAS PubMed.
- J. Li, C. Fan, H. Pei, J. Shi and Q. Huang, Adv. Mater., 2013, 25, 4386–4396 CrossRef CAS PubMed.
- H. Lee, A. K. R. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS PubMed.
- Q. Zhang, Q. Jiang, N. Li, L. Dai, Q. Liu, L. Song, J. Wang, Y. Li, J. Tian, B. Ding and Y. Du, ACS Nano, 2014, 8, 6633–6643 CrossRef CAS PubMed.
- W. Cheng, J. Nie, L. Xu, C. Liang, Y. Peng, G. Liu, T. Wang, L. Mei, L. Huang and X. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 18462–18473 CrossRef CAS PubMed.
- Q. Sun, X. Sun, X. Ma, Z. Zhou, E. Jin, B. Zhang, Y. Shen, E. A. Van Kirk, W. J. Murdoch, J. R. Lott, T. P. Lodge, M. Radosz and Y. Zhao, Adv. Mater., 2014, 26, 7615–7621 CrossRef CAS PubMed.
- M. Liu, W. Ma, Q. Li, D. Zhao, X. Shao, Q. Huang, L. Hao and Y. Lin, Cell Proliferation, 2019, 52, 1–8 CAS.
- A. S. Walsh, H. Yin, C. M. Erben, M. J. A. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS PubMed.
- S. H. Ko, H. Liu, Y. Chen and C. Mao, Biomacromolecules, 2008, 9, 3039–3043 CrossRef CAS PubMed.
- N. Liu, X. Zhang, N. Li, M. Zhou, T. Zhang, S. Li, X. Cai, P. Ji and Y. Lin, Small, 2019, 15, 1–12 Search PubMed.
- T. Kato, R. P. Goodman, C. M. Erben, A. J. Turberfield and K. Namba, Nano Lett., 2009, 9, 2747–2750 CrossRef CAS PubMed.
- T. Tian, T. Zhang, T. Zhou, S. Lin, S. Shi and Y. Lin, Nanoscale, 2017, 9, 18402–18412 RSC.
- P. Wang, Z. Xia, J. Yan, X. Liu, G. Yao, H. Pei, X. Zuo, G. Sun and D. He, Nanoscale, 2015, 7, 6467–6470 RSC.
- Y. Zhang and N. C. Seeman, J. Am. Chem. Soc., 1994, 116, 1661–1669 CrossRef CAS.
- W. M. Shih, J. D. Quispe and G. F. Joyce, Nature, 2004, 427, 618–621 CrossRef CAS PubMed.
- C. L. P. Oliveira, S. Juul, H. L. Jørgensen, B. Knudsen, D. Tordrup, F. Oteri, M. Falconi, J. Koch, A. Desideri, J. S. Pedersen, F. F. Andersen and B. R. Knudsen, ACS Nano, 2010, 4, 1367–1376 CrossRef PubMed.
- F. Wang, Y. Zhou, S. Cheng, J. Lou, X. Zhang, Q. He, N. Huang and Y. Cheng, Nanoscale Res. Lett., 2020, 15, 150 CrossRef CAS PubMed.
- M. Liu, W. Ma, Q. Li, D. Zhao, X. Shao, Q. Huang, L. Hao and Y. Lin, Cell Proliferation, 2019, 52, 1–8 CAS.
- T. Zhang, T. Tian, R. Zhou, S. Li, W. Ma, Y. Zhang, N. Liu, S. Shi, Q. Li, X. Xie, Y. Ge, M. Liu, Q. Zhang, S. Lin, X. Cai and Y. Lin, Nat. Protoc., 2020, 15, 2728–2757 CrossRef CAS PubMed.
- J. Yan, J. Chen, N. Zhang, Y. Yang, W. Zhu, L. Li and B. He, J. Mater. Chem. B, 2020, 8, 492–503 RSC.
- R. Duangrat, A. Udomprasert and T. Kangsamaksin, Cancer Sci., 2020, 111, 3164–3173 CrossRef CAS PubMed.
- H. Ijäs, B. Shen, A. Heuer-Jungemann, A. Keller, M. A. Kostiainen, T. Liedl, J. A. Ihalainen and V. Linko, Nucleic Acids Res., 2021, 49, 3048–3062 CrossRef PubMed.
- C. Pérez-Arnaiz, N. Busto, J. M. Leal and B. García, J. Phys. Chem. B, 2014, 118, 1288–1295 CrossRef PubMed.
- C. Wang, Y. Yu, M. Irfan, B. Xu, J. Li, L. Zhang, Z. Qin, C. Yu, H. Liu and X. Su, Small, 2020, 16, 1–9 CAS.
- H. Lei, X. Wang and C. Wu, J. Mol. Graphics Modell., 2012, 38, 279–289 CrossRef CAS PubMed.
- K. Butowska, K. Żamojć, M. Kogut, W. Kozak, D. Wyrzykowski, W. Wiczk, J. Czub, J. Piosik and J. Rak, Int. J. Mol. Sci., 2020, 21, 1–17 Search PubMed.
- Z. Shen, K. A. Mulholland, Y. Zheng and C. Wu, J. Mol. Model., 2017, 23, 256 CrossRef PubMed.
- C. Carvalho, R. Santos, S. Cardoso, S. Correia, P. Oliveira, M. Santos and P. Moreira, Curr. Med. Chem., 2009, 16, 3267–3285 CrossRef CAS PubMed.
- K. R. Kim, H. Y. Kim, Y. D. Lee, J. S. Ha, J. H. Kang, H. Jeong, D. Bang, Y. T. Ko, S. Kim, H. Lee and D. R. Ahn, J. Controlled Release, 2016, 243, 121–131 CrossRef CAS PubMed.
- O. Tacar, P. Sriamornsak and C. R. Dass, J. Pharm. Pharmacol., 2013, 65, 157–170 CrossRef CAS PubMed.
- C. Alves, F. Iacovelli, M. Falconi, F. Cardamone, B. Morozzo Della Rocca, C. L. P. De Oliveira and A. Desideri, J. Chem. Inf. Model., 2016, 56, 941–949 CrossRef CAS PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2021, 49, D1388–D1395 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, 2016.
- Y. Yan, D. Zhang, P. Zhou, B. Li and S. Y. Huang, Nucleic Acids Res., 2017, 45, W365–W373 CrossRef CAS PubMed.
- Y. Yan, H. Tao, J. He and S. Y. Huang, Nat. Protoc., 2020, 15, 1829–1852 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- T. E. Cheatham and D. A. Case, Biopolymers, 2013, 99, 969–977 CAS.
- I. Ivani, P. D. Dans, A. Noy, A. Pérez, I. Faustino, A. Hospital, J. Walther, P. Andrio, R. Goñi, A. Balaceanu, G. Portella, F. Battistini, J. L. Gelpí, C. González, M. Vendruscolo, C. A. Laughton, S. A. Harris, D. A. Case and M. Orozco, Nat. Methods, 2015, 13, 55–58 CrossRef PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- J. Stone, An Efficient Library for Parallel Ray Tracing and Animation, Master Thesis, University of Missouri-Rolla, 1998.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- W. L. Delano, The PyMOL Molecular Graphics System, 2002 Search PubMed.
- N. Homeyer and H. Gohlke, Mol. Inf., 2012, 31, 114–122 CrossRef CAS PubMed.
- C. Wang, D. Greene, L. Xiao, R. Qi and R. Luo, Front. Mol. Biosci., 2018, 4, 1–18 Search PubMed.
- F. Chen, H. Sun, J. Wang, F. Zhu, H. Liu, Z. Wang, T. Lei, Y. Li and T. Hou, RNA, 2018, 24, 1183–1194 CrossRef CAS PubMed.
- H. M. Ding, Y. W. Yin, S. Di Ni, Y. J. Sheng and Y. Q. Ma, Chin. Phys. Lett., 2021, 38, 018701 CrossRef CAS.
- Y. J. Sheng, Y. W. Yin, Y. Q. Ma and H. M. Ding, J. Chem. Inf. Model., 2021, 61, 2454–2462 CrossRef CAS PubMed.
- M. J. Holst and F. Saied, J. Comput. Chem., 1995, 16, 337–364 CrossRef CAS.
- E. Jurrus, D. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen, K. Liles, M. Chun, P. Li, D. W. Gohara, T. Dolinsky, R. Konecny, D. R. Koes, J. E. Nielsen, T. Head-Gordon, W. Geng, R. Krasny, G. W. Wei, M. J. Holst, J. A. McCammon and N. A. Baker, Protein Sci., 2018, 27, 112–128 CrossRef CAS PubMed.
- C. A. Frederick, L. D. Williams, G. Ughetto, G. A. van der Marel, H. J. van Boom, A. Rich and A. H. J. Wang, Biochemistry, 1990, 29, 2538–2549 CrossRef CAS PubMed.
- L. B. Liao, H. Y. Zhou and X. M. Xiao, J. Mol. Struct., 2005, 749, 108–113 CrossRef CAS.
- Z. Xia, P. Wang, X. Liu, T. Liu, Y. Yan, J. Yan, J. Zhong, G. Sun and D. He, Biochemistry, 2016, 55, 1326–1331 CrossRef CAS PubMed.
- H. Ding, J. Li, N. Chen, X. Hu, X. Yang, L. Guo, Q. Li, X. Zuo, L. Wang, Y. Ma and C. Fan, ACS Cent. Sci., 2018, 4, 1344–1351 CrossRef CAS PubMed.
- M. Li, H. Ding, M. Lin, F. Yin, L. Song, X. Mao, F. Li, Z. Ge, L. Wang, X. Zuo, Y. Ma and C. Fan, J. Am. Chem. Soc., 2019, 141, 18910–18915 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00753j |
| This journal is © The Royal Society of Chemistry 2022 |