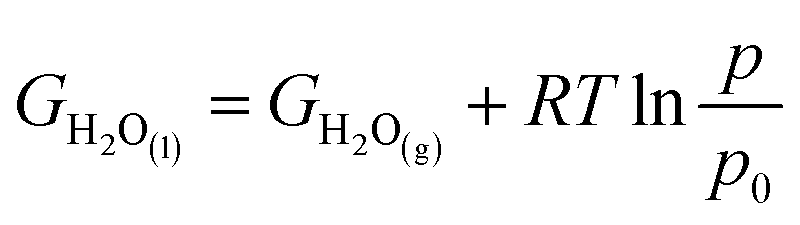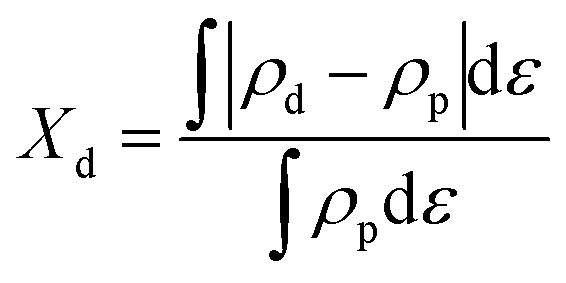DOI:
10.1039/D2NA00107A
(Paper)
Nanoscale Adv., 2022,
4, 2913-2921
First-principles design of hetero CoM (M = 3d, 4d, 5d block metals) double-atom catalysts for oxygen evolution reaction under alkaline conditions†
Received
15th February 2022
, Accepted 30th May 2022
First published on 31st May 2022
Abstract
As an extension of single-atom catalysts, the development of double-atom catalysts with high electrocatalytic activity for the oxygen evolution reaction (OER) is vital to facilitate hydrogen production and industrial applications. The CoM (M = 3d, 4d, 5d block metals) homo and double-atom catalysts supported on nitrogen-doped graphene (CoM/N4G) were prepared for electrochemical water oxidation under alkaline conditions, and the electrocatalytic activity was studied through density functional theory (DFT) calculations. The hetero CoCu/N4G double-atom catalyst indicated the highest OER activity with an onset potential of 0.83 V, while the homo Co2/N4G catalyst showed a higher onset potential of 1.69 V. The decoupled strain, dopant, and configurational effects based on the notable differences between the homo Co2/N4G and CoCu/N4G explained the enhanced OER activity, implying that the Cu dopant has a crucial impact on boosting the reactivity by reducing the affinity of reaction intermediates. The enhancement could also be understood from the perspective of the electron structure characteristic through d-orbital resolved density of states (ORDOS) (dz2, dxz, dyz, dxy, and dx2−y2) analysis. From the ORDOS analysis, we found an apparent alteration of the key orbitals between Co2/N4G (dz2, dxz, and dyz) and CoCu/N4G (dz2, dxz, dyz, and dxy) with a substantial change in the overlap ratio (Xd). This theoretical study offers beneficial insights into developing a strategy for efficient OER catalysts utilizing a double-atom structure.
Introduction
Water electrolysis using renewable energy is one of the most promising hydrogen production methods, because carbon dioxide (one of the main molecule for global warming) is not generated during the production and as a high purity of hydrogen can be obtained.1–4 However, in water electrolysis systems, the hydrogen evolution reaction (HER) at the cathode is limited by the oxygen evolution reaction (OER) at the anode because of the sluggish kinetics and large operation potential of the OER.5–8 Thus, novel metal-based catalysts, such as Ir or Ru oxides, are generally used as OER catalysts;9,10 however, their high cost and low atom-utilization efficiency make them difficult for industrial applications.11,12 Thus, the development of a cost-effective catalyst with high activity is needed for efficient hydrogen production. For this reason, many researchers have made efforts to enhance the catalytic activity through the particle size and morphology control of the catalysts or using a variety of support materials and dopants.13–20
Recently, the single-atom catalysts (SACs) have been extensively investigated due to their maximized atomic utilization with unique properties and a large number of active sites when the isolated single atoms are stabilized through a strong interaction with the support materials.21–28 Among those SACs, transition metal–nitrogen-doped carbon catalysts (M–N–C) are the most widely studied because of their good mechanical properties, excellent electrical conductivity, and high stability in acidic/alkaline conditions, showing superior catalytic activity for many electrochemical reactions, such as HER,29,30 oxygen reduction reaction (ORR),31,32 OER,33 and N2-reduction reaction (N2RR).34 Moreover, as an extension of SACs, double-atom catalysts (DACs) have also been vigorously studied for achieving better electrocatalytic performance than SACs with a modulated electronic structure of the active centers.35–40 For example, Yang et al. reported that transition-metal-doped double-atom Fe catalysts on graphene substrates (Fe-TMDA/GS) have more obvious advantages due to their lower Gibbs free energy variation for the potential determining step in the nitrogen-reduction reaction (NRR) compared to the single-atom Fe catalyst.41 Aside from double-atom Fe catalysts, diverse DACs such as Pt2/graphene, Ni–V/graphene, and Pd2/graphene have shown remarkable catalytic performance in the hydrolytic dehydrogenation of ammonia borane, CO oxidation, and formic acid dehydrogenation, respectively.42–44 Nevertheless, the study of DACs for the OER has not been much reported to date.
Herein, we constructed CoM (M = 3d, 4d and 5d block metals) double atoms supported on nitrogen-doped graphene (CoM/N4G), where the M metal atom was coordinated with the Co metal center, and then evaluated its structural stability. Then, the electrocatalytic activity of CoM/N4G for the OER was estimated through density functional theory (DFT) calculations. The CoCu/N4G catalyst with an onset potential of 0.83 V presented the best OER performance compared to the other CoM/N4G. To better understand the mechanism of the activity enhancement, the strain, dopant, and configurational effects on CoCu/N4G were decoupled and compared with those of the Co2/N4G catalyst so that we could clearly confirm the role of the Cu atom. In addition, we found the key descriptor representing the catalytic activity based on the 3d block-metal-introduced CoM/N4G catalyst and explored additional CoM/N4G catalysts employing 4d and 5d block metals. Finally, d-orbital resolved density of states (ORDOS) (dz2, dxz, dyz, dxy, and dx2−y2) analysis for the key intermediate adsorbed CoM/N4G system was performed to reveal the significant properties for the electronic structure of the active metal center related to the catalytic activity.
Computational details
All of the calculations here were performed on the basis of spin-polarized density functional theory (DFT) using the Vienna Ab initio Simulation Package (VASP).45 The generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) was used to approximate the exchange–correlation interaction.46–48 The projector-augmented wave (PAW) method was also employed to substitute the complicated interaction between the core ions and valence electrons.49 The plane wave functions were expanded with an energy cutoff of 400 eV for a reasonable number of plane waves. For all of the calculated models, the geometry optimization and electronic structure calculations were carried out with the Monkhorst–Pack mesh of 4 × 4 × 1 k-points to guarantee sufficient accuracy.50 Ionic relaxation was conducted until the residual force was less than 5 × 10−2 eV Å−1. In this study, the hetero CoM (M = 3d, 4d, and 5d block metals) double-atom catalysts were created by anchoring the CoM dimer into the di-vacancy site, which was associated with four N atoms (indicated by CoM/N4G), and the z-direction was fixed with the vacuum size of 20 Å to avoid undesirable interaction between periodic boundaries. When determining the optimized structure of CoM/N4G, nine types of geometry were considered. Then, the most stable geometry was determined by the formation energy calculation. The formation energy (ΔEf) from Co/N4G (a single Co atom) to CoM/N4G is described as follows:
| ΔEf = ECoM/N4G − ECo/N4G − EM |
where ECoM/N4G, ECo/N4G, and EM are the total energies of the CoM/N4G, Co/N4G, and single-M systems, respectively. Here, the choice of M atom for the formation energy calculation was related to the preparation method of the double-atom catalyst in which the sputtering or atomic layer deposition was accepted for the utilization of a single atom.51–53
The adsorption energy (ΔEads) was used to measure the adsorption strength between the intermediates and CoM/N4G surface, and the adsorption energy is defined by the following equation:
| ΔEads = Etot − Esur − Eint |
where the
Etot,
Esur, and
Eint mean the total energies of the adsorbed system, clean surface of the catalyst and the intermediate, respectively.
To predict the Gibbs free energy change (ΔG) depending on the individual reaction steps, we assumed an alkaline mechanism in which OH− is oxidized to H2O and O2 with electron (e−) release, and adopted the computational hydrogen electrode (CHE) approach.54,55 According to the CHE approach, the Gibbs free energy of (H+ + e−) can be calculated by the free energy of ½H2 in the gas phase, assuming the equilibrium,
under standard conditions [pressure (
pH2) = 1 bar, and temperature (
T) = 298.15 K]. In addition, the corrected Gibbs free energies of H
2O
(l) and O
2(g) (where the O
2 molecule is hard to describe by DFT) were obtained from the following equations:
| GO2(g) = 2GH2O(l) − 2GH2(g) + 4.92 |
where
R is the gas constant,
T corresponds to 298.15 K, and
p and
p0 are 0.035 bar and 1 bar, respectively (we assumed that H
2O
(l) at 298.15 K and 1 bar is considered to be in equilibrium with H
2O
(g) at 298.15 K and 0.035 bar). As reported in the CHE method, the only difference between acidic conditions and alkaline conditions for the Gibbs free energy calculation is related to the reference electrodes,
i.e., the standard hydrogen electrode and reversible hydrogen electrode (SHE and RHE).
55 Moreover, those two electrodes can be linked as per the following equation by adding the correction (described as
GpH):
GpH = kBT × pH × ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 10 |
| eURHE = eUSHE + kBT × pH × ln10 |
where
eURHE,
eUSHE,
kB, and
T are the electrode potential relative to RHE, SHE, Boltzmann constant, and temperature. To consider the alkaline environment of the solvent, the pH value is set to pH = 14. Thus, the equation for the Gibbs free energy change with the variation of the electrode potential (
USHE) can be expressed for both an acidic mechanism and alkaline mechanism by the equation:
| ΔG = ΔE − TΔS + ΔZPE − n(eUSHE + GpH) |
where Δ
E indicates the adsorption energy of a reaction intermediate; Δ
S, ΔZPE, and
GpH are the entropy, zero-point energy and free energy correction depending on pH condition, respectively; and
n denotes the number of electrons transferred in the reaction. The overlap ratio (
Xd) in this work means the quantitative degree of p–d orbital overlap between the d orbitals of CoM (M = Co, Cu) and the p orbital of the adsorbed O atom. That is, the fraction of p–d orbital overlap in the total p orbital can be calculated by the following equation:
where
Xd,
ρd,
ρp, and
ε are the overlap ratio, d density of state, p density of state, and energy
versus the Fermi energy, respectively.
Result and discussion
Mechanism of the oxygen evolution reaction in a homo Co2 double-atom catalyst
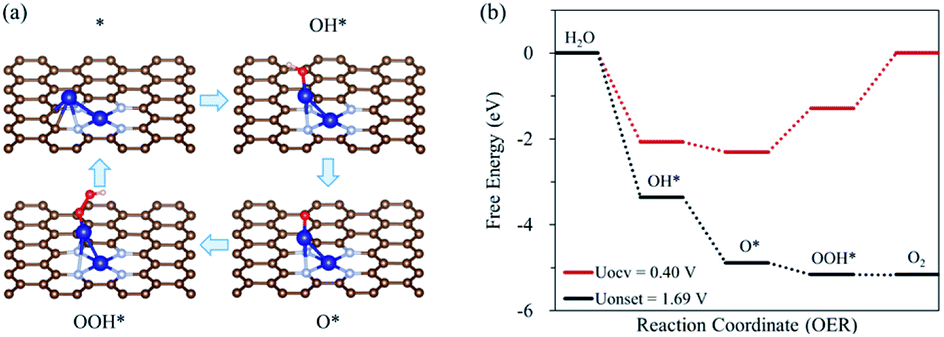
In this study, on a homo Co2 double-atom [indicated by Co2/N4G], we consider the following four steps for the oxygen evolution reaction (OER) [4OH− → O2(g) + 2H2O(l) + 4e−] under alkaline conditions.56,57
| OH* + OH− → O* + H2O(l) + e− |
| OOH* + OH− → * + O2(g) + H2O(l) + e− |
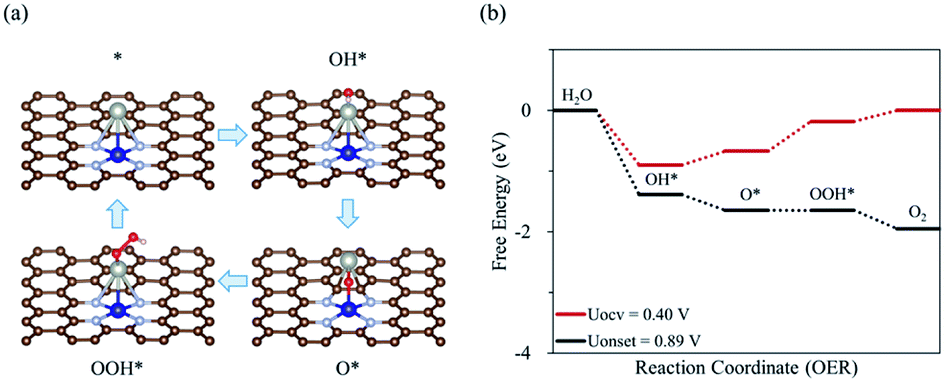
The first step for the OER is the adsorption of OH− on Co2/N4G, forming OH* (* denoted as the adsorbed state) and the release of an electron. Second, the adsorbed OH* reacts with OH−, activating the O–H bond dissociation and in turn liberating H2O, leaving O* and an electron behind. The third step is the reaction of adsorbed O* with OH−, leading to the formation of OOH* and an electron. The final step is the formation of O2, H2O, and electron via O–H scission by the reaction of adsorbed OOH* with OH−. On the other hand, the dissociative mechanism of OER (where the adsorbed O* directly forms O2 without OOH* formation) is not stated in this study. This is because the direct formation of O2 from O*, which is a chemical reaction, is a large endothermic reaction due to the strong O* adsorption on Co2/N4G (see Fig. S1†). Aside from Co2/N4G, all the CoM/N4G catalysts showed endothermic energy for the formation of O2 from O*.
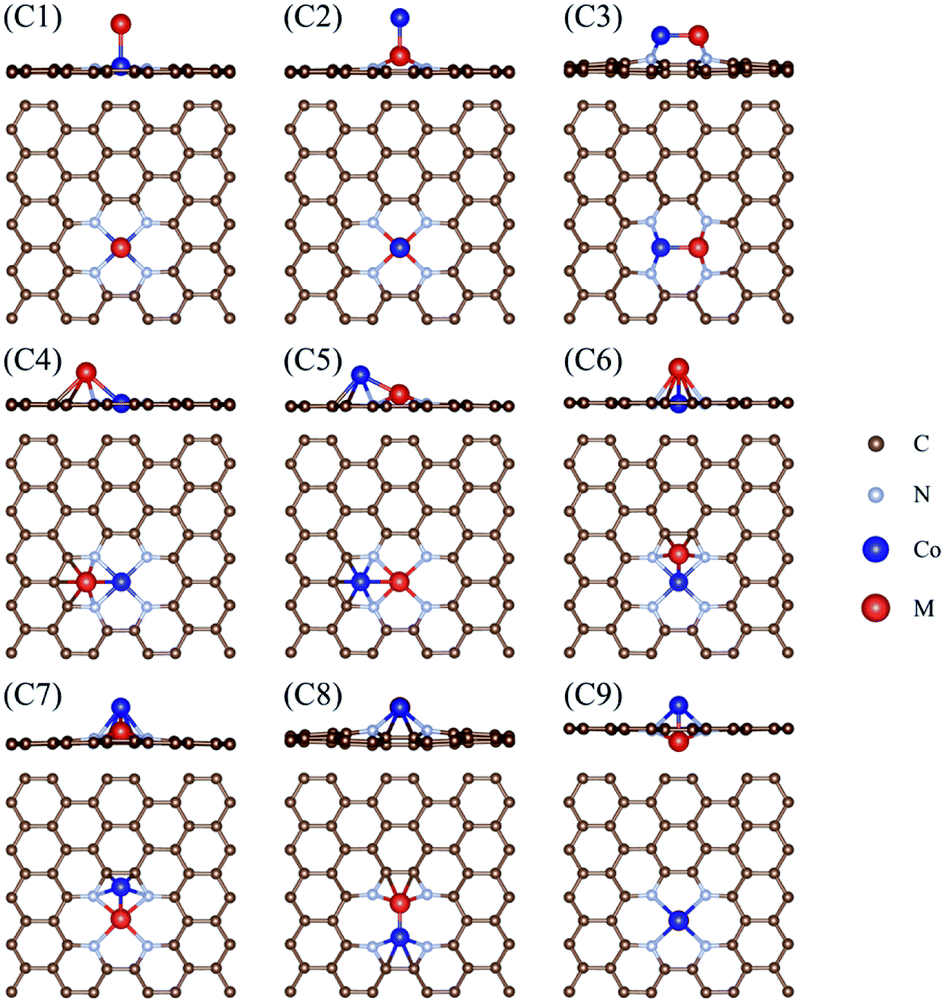
Fig. 1 shows the calculated free energy diagram for the OER and the molecular configurations of OER intermediates on a Co2/N4G catalyst. Here, we attempted to find the most stable structure of a homo Co2 double-atom supported on defective graphene by comparing the formation energies (ΔEf) of six possible configurations (denoted as C1, C3, C4, C6, C8, and C9) (see Table 1 and Fig. 2), which indicated that the homo Co2 double-atom was stabilized in the form of the C4 geometry (ΔEf = −1.63 eV) (where one Co atom is connected to a rectangular-shaped nitrogen, while the other Co atom is linked to a hexagonal-shaped nitrogen and carbons, resulting in the formation of the inclined Co2 structure).
 |
| | Fig. 1 (a) Schematic diagram of OER on Co2/N4G catalyst with the most favorable adsorption configuration of each intermediates, (b) free energy diagram depending on the OER pathway on Co2/N4G at OCV condition (UOCV = 0.401 V) and onset potential. | |
Table 1 Formation energies (ΔEf) (eV) of the considered CoM/N4G configurations (C1)–(C9)
| Catalyst |
(C1) |
(C2) |
(C3) |
(C4) |
(C5) |
(C6) |
(C7) |
(C8) |
(C9) |
| CoSc/N4G |
−2.64 |
−2.82 |
−1.82 |
−3.22 |
−2.15 |
−3.11 |
−2.47 |
— |
−2.62 |
| CoTi/N4G |
−2.35 |
−3.42 |
−2.03 |
−3.09 |
— |
−2.60 |
−3.02 |
−1.22 |
−2.10 |
| CoV/N4G |
−1.73 |
−2.76 |
−0.71 |
−2.00 |
−1.54 |
−1.91 |
−2.09 |
−0.12 |
−0.98 |
| CoCr/N4G |
−1.12 |
−0.53 |
1.18 |
— |
−0.05 |
— |
−0.08 |
2.04 |
— |
| CoMn/N4G |
−1.38 |
−0.04 |
1.13 |
— |
−0.31 |
— |
0.01 |
1.92 |
1.00 |
| CoFe/N4G |
−1.66 |
−0.86 |
0.10 |
−1.64 |
−1.30 |
— |
−1.25 |
0.96 |
0.37 |
| Co2/N4G |
−1.50 |
— |
0.47 |
−1.63 |
— |
−1.48 |
— |
1.21 |
0.35 |
| CoNi/N4G |
−1.59 |
−0.50 |
— |
−1.63 |
−1.12 |
−1.63 |
−0.78 |
— |
— |
| CoCu/N4G |
−1.40 |
2.22 |
— |
— |
1.43 |
— |
1.81 |
— |
— |
| CoZn/N4G |
−0.25 |
3.54 |
— |
— |
3.20 |
— |
3.58 |
— |
— |
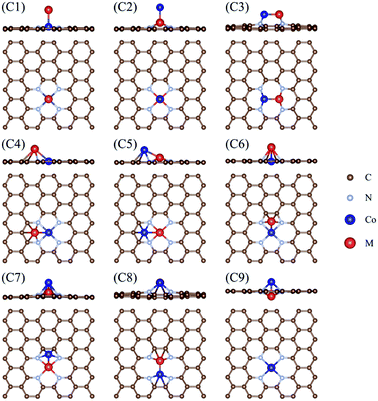 |
| | Fig. 2 Types of CoM/N4G catalysts considered: (C1) and (C2) have a vertical bonding configuration, and (C3)–(C9) have horizontally or diagonally combined features with a different coordination of N or C atoms. | |
For a homo Co2/N4G catalyst, our DFT calculations predicted that the OOH* + OH− → * + O2(g) + H2O(l) + e− reaction [defined as the potential limiting step (PLS)] determined the whole O2 evolution activity since that was the most energetically demanding step (requiring the largest Gibbs free energy change for the OER) among the reaction pathways of the OER, which led to an onset potential (as indicated by Uonset) of 1.69 V (over potential of 1.29 V). This was higher by 0.91 V than a previously reported Co oxide catalyst.19 Next, to further enhance the OER activity of a homo Co2/N4G catalyst, we replaced one Co atom in the Co2 double-atom by 3d block metals (M = Sc, Ti, V, Cr, Mn, Fe, Ni, Cu, and Zn).
Structure of the hetero double-atom catalysts
As displayed in Fig. 2, the hetero double-atom catalysts (indicated by CoM/N4G) could stably exist as one of nine possible configurations (denoted as C1–C9), whose stability could be quantitively understood by the calculation of the formation energy of the double atoms (ΔEf). Table 1 compares the ΔEf for the different configurations of the CoM/N4G catalysts. We found that the most stable ensemble structures of the CoM/N4G catalyst could be narrowed down to three geometries. That is, (1) the C1 geometry (we call the vertical Co double atom, where the Co atom of the CoM dimer was vertically bound to the rectangular-shaped four nitrogen atoms); CoCr (ΔEf = −1.12 eV), CoMn (ΔEf = −1.38 eV), CoFe (ΔEf = −1.66 eV), CoCu (ΔEf = −1.40 eV), and CoZn (ΔEf = −0.25 eV); (2) the C2 geometry (we called this the vertical M double atom, where the M atom of the CoM dimer was vertically associated with the rectangular-shaped four nitrogen atoms); CoTi (ΔEf = −3.42 eV), and CoV (ΔEf = −2.76 eV); (3) the C4 geometry (we called this the inclined hexagon M double atom, where the Co and M atoms were connected to the rectangular-shaped four nitrogen atoms and hexagonal-shaped carbon/nitrogen atoms, respectively); CoSc (ΔEf = −3.22 eV), and CoNi (ΔEf = −1.63 eV). The most stable ensemble geometries of the CoM/N4G catalysts are shown in Fig. S2 in ESI Section.† Next, we examined the OER activity on the surface of the most stable CoM/N4G catalysts.
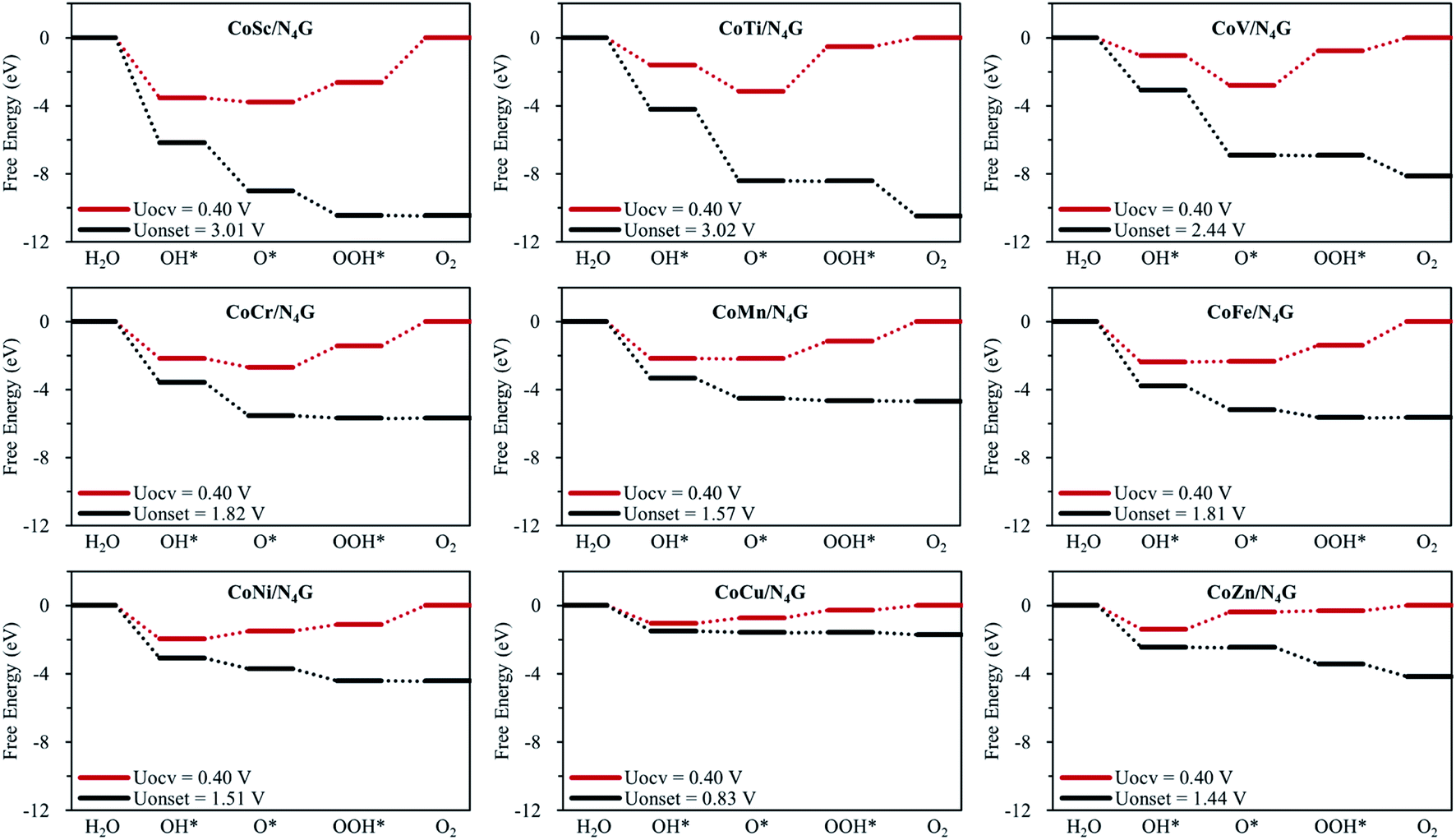
Electrocatalytic OER activity
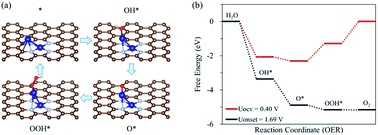
Fig. 3 shows the Gibbs free energy diagram for OER on the hetero CoM/N4G catalysts. We found that the hetero CoCu (Uonset = 0.83 V), CoMn (Uonset = 1.57 V), CoNi (Uonset = 1.51 V), and CoZn (Uonset = 1.44 V) double-atom catalysts had a lower onset potential (enhanced activity) for oxygen evolution than the homo Co2/N4G catalyst (Uonset = 1.69 V). Especially, the CoCu/N4G catalyst exhibited a significant improvement in OER activity compared to the other hetero double-atom cases. This enhancement was related to the large reduction in the affinity of the reaction intermediates (O*, OH*, and OOH*) [ΔEads(O*) = −4.27 eV, ΔEads(OH*) = −3.47 eV, and ΔEads(OOH*) = −2.06 eV] (see Table 2) on the CoCu/N4G catalyst than for the homo Co2 double-atom case [ΔEads(O*) = −5.86 eV, ΔEads(OH*) = −4.48 eV, and ΔEads(OOH*) = −3.06 eV], which resulted in a shift of the PLS from OOH* + OH− → * + O2(g) + H2O(l) + e− to O* + OH− → OOH* + e− and a decrease in the endothermicity of the PLS.
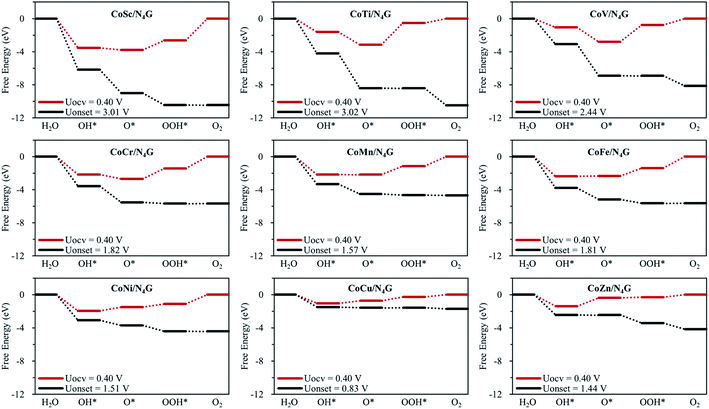 |
| | Fig. 3 Free energy diagrams for CoM/N4G (M = 3d block metals) at OCV condition (UOCV = 0.401 V) and onset potential. | |
Table 2 Adsorption energies (ΔEads) of the intermediates in units of [eV], and the onset potential (Uonset) for the CoM/N4G catalysts
| Catalyst |
ΔEads(O*) |
ΔEads(OH*) |
ΔEads(OOH*) |
U
onset (V) |
| CoSc/N4G |
−7.33 |
−5.95 |
−4.38 |
3.01 |
| CoTi/N4G |
−6.71 |
−4.01 |
−2.30 |
3.02 |
| CoV/N4G |
−6.37 |
−3.44 |
−2.55 |
2.44 |
| CoCr/N4G |
−6.26 |
−4.57 |
−3.19 |
1.82 |
| CoMn/N4G |
−5.74 |
−4.56 |
−2.94 |
1.57 |
| CoFe/N4G |
−5.90 |
−4.79 |
−3.18 |
1.81 |
| Co2/N4G |
−5.86 |
−4.48 |
−3.06 |
1.69 |
| CoNi/N4G |
−5.04 |
−4.37 |
−2.87 |
1.51 |
| CoCu/N4G |
−4.27 |
−3.47 |
−2.06 |
0.83 |
| CoZn/N4G |
−3.92 |
−3.82 |
−2.06 |
1.44 |
On the other hand, for the CoSc (Uonset = 3.01 V), CoTi (Uonset = 3.02 V), CoV (Uonset = 2.44 V), and CoFe (Uonset = 1.81 V) cases, the onset potential increased (OER activity decreased) due to the substantial increase in the affinity of the reaction intermediates (particularly, O*). Note the adsorption energies for CoSc [ΔEads(O*) = −7.33 eV, ΔEads(OH*) = −5.95 eV, and ΔEads(OOH*) = −4.38 eV], CoTi [ΔEads(O*) = −6.71 eV, ΔEads(OH*) = −4.01 eV, and ΔEads(OOH*) = −2.30 eV], CoV [ΔEads(O*) = −6.37 eV, ΔEads(OH*) = −3.44 eV, and ΔEads(OOH*) = −2.55 eV], and CoFe [ΔEads(O*) = −5.90 eV, ΔEads(OH*) = −4.79 eV, and ΔEads(OOH*) = −3.18 eV] (see Table 2). The most favorable adsorption configurations of CoM/N4G are shown in Fig. S3.†
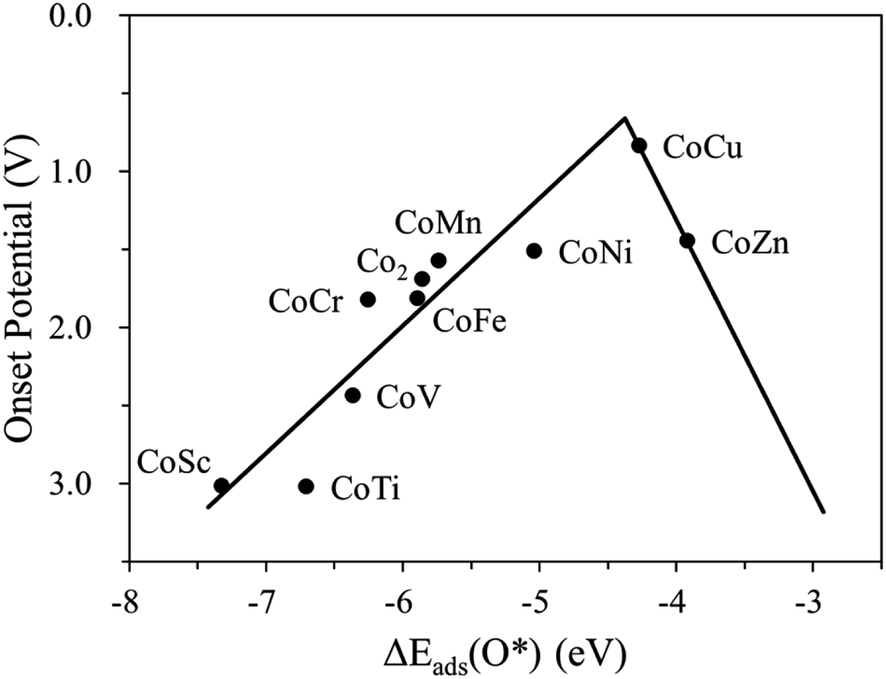
Fig. 4 and S4† display the variation of the onset potential as a function of O*, OH*, OOH* adsorption energy for the homo Co2/N4G and hetero CoM/N4G catalysts. We discovered that a key descriptor representing the electrocatalytic activity for the OER was the O* binding energy [note the clear volcanic correlation between Uonset and ΔEads(O*)]. Here, the volcano plot [Uonsetversus ΔEads(O*)] demonstrated that the OER activity could be increased when the ΔEads(O*) = −5.86 eV of a homo Co2/N4G catalyst rose to the peak position of about −4.37 eV, whereas the activity decreased when the ΔEads(O*) further decreased over the peak position. Therefore, indicating that there was still a probability to develop new CoM/N4G catalysts surpassing the OER performance of the CoCu/N4G catalyst.
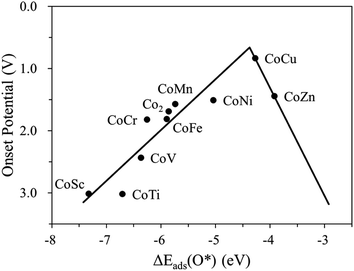 |
| | Fig. 4 Volcano curve indicating the activity (onset potential) for the CoM/N4G catalysts (M = 3d block metals) depending on the oxygen adsorption energy, ΔEads(O*). | |
In addition to the N4G support material, we investigated the OER activity when the N4G support (which had a di-vacancy of two carbon atoms surrounded by four nitrogen atoms) (Fig. S5†) was replaced by the N5G support (which had two mono-vacancies, and five nitrogen atoms covering the edge site of two mono-vacancies) (Fig. S5†) to examine the support vacancy arrangement effect on the activity of hetero CoCu double-atom catalyst. We found out that the most active CoCu double atoms in the N5G support stably existed when Co and Cu atoms were in each mono-vacancy site, and Co and Cu atoms were bound horizontally to each other (Fig. S5†). The result of the OER activity prediction showed that the hetero CoCu/N5G catalyst had a higher onset potential (Uonset = 1.25 V) than CoCu/N4G (Uonset = 0.83 V). The decreased OER activity of CoCu/N5G came from the increase in O* binding strength as well as the reduction of the OOH* affinity. Note the adsorption energies for CoCu/N5G [ΔEads(O*) = −4.45 eV, ΔEads(OH*) = −3.14 eV, and ΔEads(OOH*) = −1.81 eV]. Thus, the endothermicity of the free energy change at the PLS (O* + OH− → OOH* + e−) increased (see Fig. S6†) and in turn the OER activity was reduced. In addition, our DFT calculations showed the higher stability (formation energy) of di-vacancy by 6.12 eV than for the two mono-vacancy systems. Note that we considered the N4G + N → N5G + C reaction for calculation of the formation energy difference.
Origin of the enhanced OER activity in the hetero double-atoms catalysts
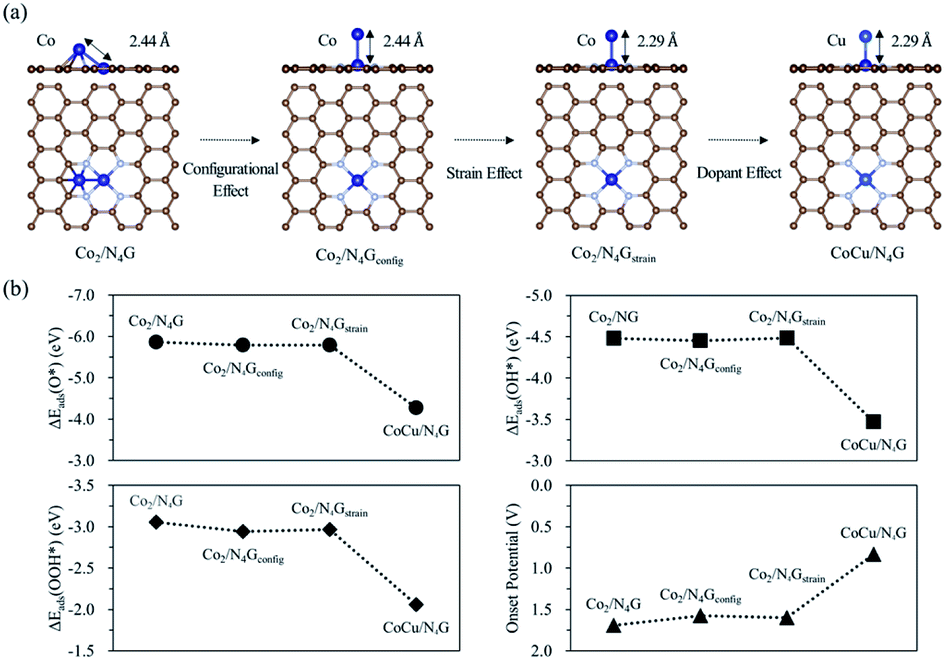
To better understand the role of the Cu atom in the hetero CoCu/N4G catalyst in boosting OER activity, we attempted to decouple the strain, dopant, and configurational effects in a CoCu/N4G catalyst. Note that the OER activity of the hetero CoCu/N4G catalyst compared to the homo Co2/N4G case could be changed by the three effects.58–60 The first factor (the so-called strain effect) was related to the atomic size mismatch between the Co and Cu atom in the hetero CoCu/N4G catalyst, which could affect the d-band structure and in turn the surface reactivity. The second one (the so-called dopant effect) was related to the modification in the d-band states of both Cu and Co atoms induced by the mixing of valence electrons between Cu and Co atoms.61,62 The last one (the so-called configurational effect) was related to the modification of the catalytic activity caused by the ensemble structure change of the CoCu dimer by the strong interaction between the hetero CoCu double atom and the nitrogen-doped graphene. As shown in Fig. 5(a), when the homo Co2/N4G catalyst turned into the hetero CoCu/N4G system, the Co2/N4G catalyst underwent the above three effects in series. The first change of the inclined hexagon Co double atom (Co2/N4G) into the vertical Co double atom (as indicated by the Co2/N4Gconfig model) represented the contribution of the configurational effect to the total OER activity. Here, the bond distance between Co atoms in the Co2/N4Gconfig model (2.44 Å) was the same as the Co2/N4G case. Our DFT calculations predicted that the affinity of the reaction intermediates on the Co2/N4Gconfig model was slightly decreased by 0.03∼0.12 eV compared to the Co2/N4G case. Note ΔEads(O*) = −5.86 eV, ΔEads(OH*) = −4.48 eV, ΔEads(OOH*) = −3.06 eV for the Co2/N4G catalyst, while ΔEads(O*) = −5.79 eV, ΔEads(OH*) = −4.45 eV, ΔEads(OOH*) = −2.94 eV for the Co2/N4Gconfig catalyst [See Fig. 5(b)]. As a result, the onset potential of the Co2/N4Gconfig model was slightly decreased by 0.11 V, suggesting that the configurational effect tended to raise the OER activity.
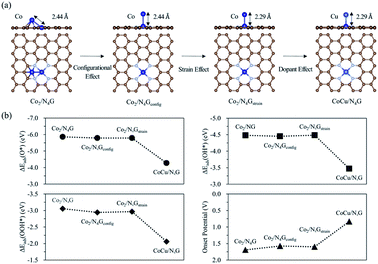 |
| | Fig. 5 (a) Decoupled models of the configurational (Co2/N4Gconfig), strain (Co2/N4Gstrain) and dopant effects. (b) Adsorption energy and onset potential variation of each decoupled model. | |
The second change of the Co2/N4Gconfig model to the compressively strained vertical Co double atom (referred to the Co2/N4Gstrain model) indicated the relative contribution of the strain effect to the total OER catalysis. Here, the Co–Co bond of the Co2/N4Gstrain model was 2.29 Å (whose bond length was the same as the Co–Cu bond of the stable CoCu/N4G catalyst), which was contracted by 6.15% compared to the Co2/N4Gconfig model. We saw almost no change of the binding strength for all reaction intermediates [ΔEads(O*) = −5.79 eV, ΔEads(OH*) = −4.48 eV, ΔEads(OOH*) = −2.97 eV] for the Co2/N4Gstrain model compared to the Co2/N4Gconfig case. Here, although there was a relatively large compressive strain effect on the Co2 double atom, the adsorption energies were little changed. This may come from the adsorption states of the intermediates where only the top Co atom in the vertical Co double atom actively interacted with the intermediates, such that the strain effect may hardly affect the affinity for the intermediates (see Fig. S7†). Accordingly, the OER activity of the Co2/N4Gstrain model (the onset potential = 1.60 V) was almost the same as for the Co2/N4Gconfig model (the onset potential = 1.58 V), implying that the strain effect was not important in determining the OER catalysis. Finally, a top Co atom of Co2/N4Gstrain model was substituted for a Cu atom as it maintained the bond distance of 2.29 Å (CoCu/N4G model), which could help understanding the Cu dopant effect on the total OER activity. Our DFT calculations showed the great reduction in the affinity of the reaction intermediates by the dopant effect. For example, the ΔEads(O*) of CoCu/N4G was reduced by 1.52 eV in comparison with the Co2/N4Gstrain model. Similarly, the CoCu/N4G catalyst had strikingly higher OH* (−3.47 eV) and OOH* (−2.06 eV) adsorption energies than the Co2/N4Gstrain case [ΔEads(OH*) = −4.48 eV, and ΔEads(OOH*) = −2.97 eV]. This large reduction of adsorption strength gave rise to a considerable decrease in the free energy change of the potential limiting step. Thus, the OER activity was significantly enhanced with the onset potential of 0.83 V. These results demonstrated that the dopant effect was critical for boosting the OER activity, while the configurational and strain effects played a minor role.
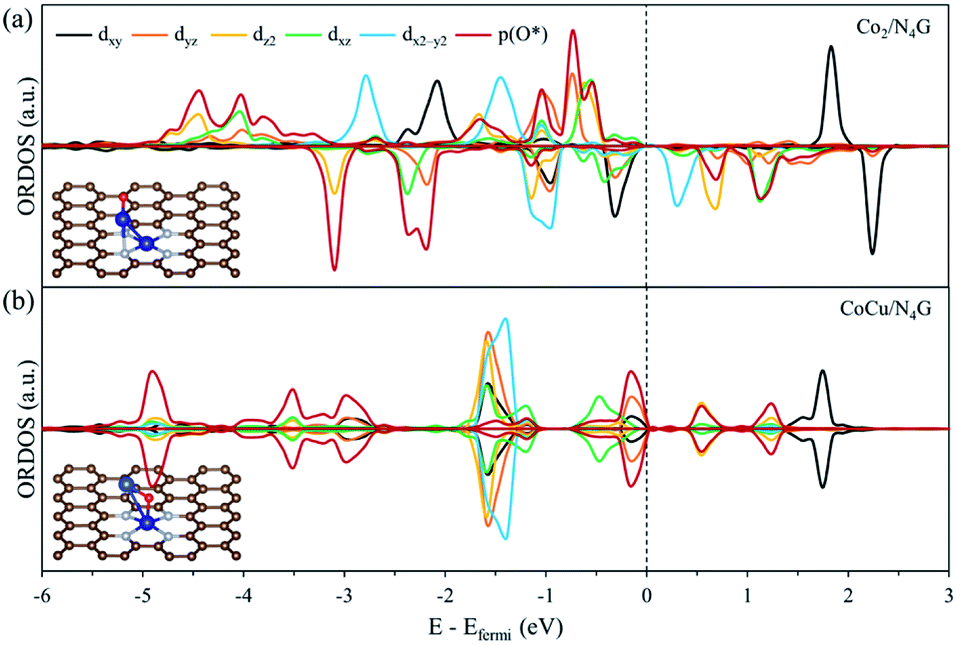
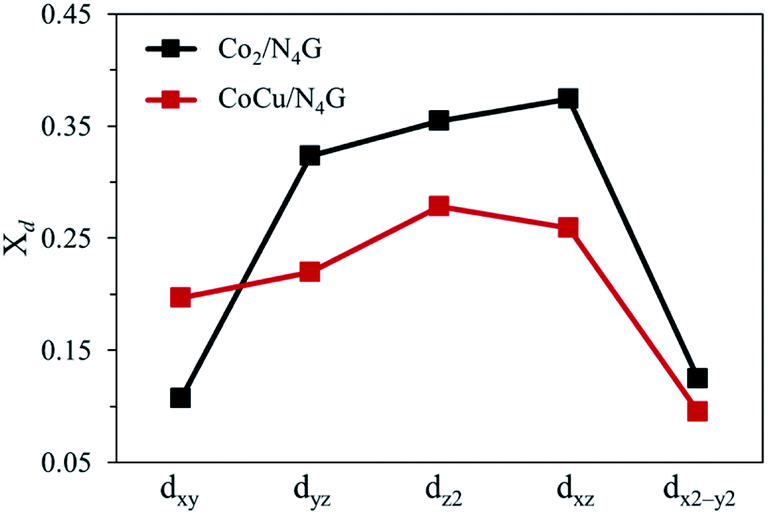
Next, to precisely comprehend the fundamental properties relevant to the OER catalysis, we predicted the d-orbital resolved density of states (ORDOS) (dz2, dxz, dyz, dxy, and dx2−y2) of the Co and Cu atoms and the p density of states (DOS) of the O atom for the O-adsorbed homo Co2/N4G and hetero CoCu/N4G catalysts (see Fig. 6). For validity of this analysis, the precedent work about the hetero double-atom catalysts for electrochemical NH3 production has been already reported and its validity proven.37 Here, the oxygen adsorption structure is key to the understanding of the descriptor for representing the OER activity as mentioned in previous section. For the homo Co2/N4G catalyst, we identified a strong overlap between the dz2, dxz, dyz, dxy, and dx2−y2 orbitals of the Co dimers and the p orbital of the O atom in the energy ranges from −4.80 to −3.60, from −3.40 to −2.90, from −2.50 to −2.05, and from −1.10 eV to −0.50 eV. To understand the degree of orbital overlap quantitatively, the overlap ratio (Xd), which denotes the fraction of p–d orbital overlap in the total p orbital, was estimated as shown in Fig. 7. We found that the dz2, dxz, and dyz orbitals, which feature an orientation toward z-direction, were closely involved in the bonding to the oxygen compared to the dx2−y2 and dxy for the homo Co2/N4G catalyst. Note that the order of overlap was dxz (Xd = 0.37) > dz2 (Xd = 0.35) > dyz (Xd = 0.32) ≫ dx2−y2 (Xd = 0.12) > dxy (Xd = 0.11). On the other hand, for the hetero CoCu/N4G case, the pattern of orbital overlap was greatly different from the homo Co2/N4G catalyst. Note the overlap energy ranged from −5.10 to −4.80, from −3.70 to −2.75, from −1.60 to −1.10, and from −0.25 eV to −0.0 eV for the hetero CoCu/N4G catalyst in Fig. 6(b). This led to a change in the key orbitals in determining the oxygen adsorption process. That is, in addition to the dz2 (Xd = 0.28), dxz (Xd = 0.26), and dyz (Xd = 0.22) orbitals, the dxy (Xd = 0.20) orbital was actively involved in oxygen adsorption. Here, the activation of dxy in the hetero CoCu/N4G catalyst was associated with the adsorption of an oxygen atom at the bridge site of the vertical CoCu double atom. Note that the p orbital of oxygen could strongly interact with the dxy (Xd = 0.20) orbital of the Cu atom, as shown in Fig. 7. In addition to the activation of the dxy orbital, another noticeable change was observed in the CoCu/N4G catalyst. Even though an additional orbital of dxy was involved in the adsorption process, the overlap ratio of most of the key orbitals (dz2, dxz and dyz) decreased, which means the interaction between CoCu atoms and O atom was reduced in comparison with Co2. As a result, the decrease in the overlap ratio of dz2, dxz and dyz orbitals made the oxygen adsorption weak (see Table 2).
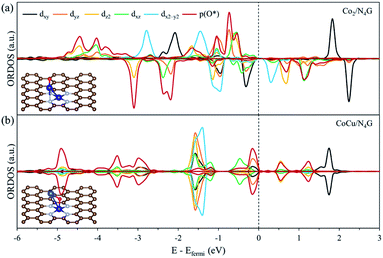 |
| | Fig. 6 The d-orbital resolved density of state (ORDOS) analysis of the oxygen adsorbed (a) Co2/N4G and (b) CoCu/N4G. | |
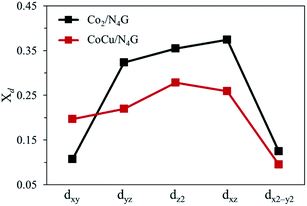 |
| | Fig. 7 Overlap ratio (Xd) of d-orbitals to p-orbitals. The black dots correspond to the overlap ratio of Co2/N4G, and the red dots are equivalent to the overlap ratio of CoCu/N4G. | |
Computational screening for the hetero CoM (M = 4d, 5d block metals) double-atoms
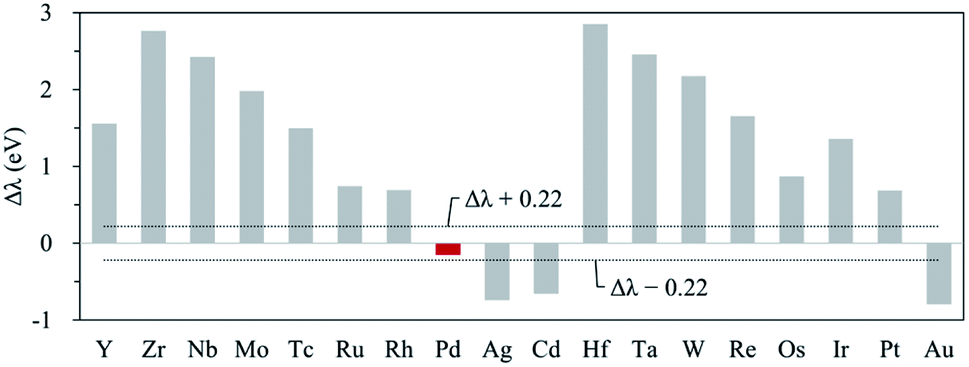
Next, to find another hetero double-atom catalyst, we searched the possible dopants that could enhance the OER activity of the hetero CoM (M = 4d and 5d block metals) double-atom catalysts supported on the defective N-doped graphene. For such a purpose, as discussed in the 3d dopant case, we first determined the thermodynamically stable structure of the hetero CoM (M = 4d and 5d block metals)/N4G catalyst (based on the formation energy) and then calculated the descriptor [which is ΔEads(O*) and denoted as λ] on the surface of the stable CoM/N4G catalysts. Fig. 8 depicts the relative descriptor value (indicated by Δλ), which is defined as the deviation from the peak position (λ = −4.37 eV) in the volcano plot of Fig. 4. We saw a large variation of Δλ as the dopant type changes. Here, we selected a promising CoPd/N4G candidate whose λ was placed near the peak position (−0.22 eV < Δλ < 0.22 eV). Fig. 9 shows the molecular configuration of the reaction intermediates and the free energy diagram for the OER on the proposed CoPd/N4G catalyst. Our DFT calculations predicted a reduced onset potential by 0.80 V (Uonset = 0.89 V) compared to the homo Co2/N4G catalyst, suggesting the improved OER activity.
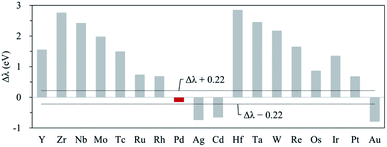 |
| | Fig. 8 Relative descriptor value (Δλ) of CoM/N4G (M = 4d, 5d block metals). The black dotted line denotes the range for selecting candidates (−0.22 eV < Δλ < 0.22 eV), and left column means the candidate M chosen within the range. | |
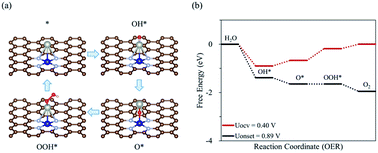 |
| | Fig. 9 (a) Schematic diagram of OER on the CoPd/N4G catalyst with the most favorable adsorption configuration of each intermediate, (b) the free energy diagram for the OER on the CoPd/N4G catalyst at OCV and the onset potential. | |
Conclusions
Using spin-polarized density functional theory (DFT) calculations, we investigated the electrochemical water oxidation on Co-based homo and hetero double-atom catalysts under alkaline conditions. The following is the summary findings from our study.
(1) The ensemble structures of the Co-based homo and double atom catalysts supported on nitrogen-doped graphene (CoM/N4G) were examined based on their nine possible configurations (C1–C9) and determined by the formation energy. We confirmed that the most stable structures of the CoM/N4G catalysts converged into three geometries: C1 (vertical Co double-atom shape) (CoCr, CoMn, CoFe, CoCu, CoZn); C2 (vertical M double-atom shape) (CoTi, CoV); and C4 (inclined hexagon M double-atom shape) (CoSc, CoNi).
(2) The OER activity of CoM/N4G (M = 3d block metals) was predicted based on the reaction mechanism under alkaline conditions. We discovered hetero CoCu, CoMn, CoNi, and CoZn double-atom catalysts had advanced activity compared to the homo Co2 double-atom catalyst. In particular, the hetero CoCu/N4G double-atom catalyst showed a strikingly lower onset potential by 0.86 V (Uonset = 0.83 V) than the Co2/N4G (Uonset = 1.69 V) case, which was related to the PLS shift from the OOH* → O2(g) step [Co2/N4G] to the O* → OOH* step and in turn the decrease in the endothermicity in PLS owing to the large reduction in the affinity of the reaction intermediates.
(3) The origin of the boosted activity of the hetero CoCu/N4G double-atom catalyst was described by the decoupled strain (bond length change of the double atom), dopant (substitute Co atom for Cu atom), and configurational (ensemble structure alteration) effects. Decoupling of the effects revealed that the dopant Cu atom, where the d-band states of both the Cu and Co atoms were modified by valence electrons mixing, had a great impact on improving the OER performance by decreasing the adsorption strength of O*, OH*, and OOH*, whereas the strain and configurational effect had only a slight influence on the activity or barely affected the variation of the onset potential. The weakened adsorption strength was also explained by the d-orbital resolved density of states (ORDOS) (dz2, dxz, dyz, dxy, and dx2−y2) of the double-metal atom and p density of states (DOS) of the adsorbed O atom, and the degree of overlap was quantitatively estimated by the p–d orbital overlap ratio (Xd). We found that the overlap ratio of the key orbitals for the homo Co2/N4G catalyst, which was characterized by the z-direction involved orbitals (dz2, dxz, and dyz), exhibited a higher overlap ratio [dz2 (Xd = 0.35), dxz (Xd = 0.37) and dyz (Xd = 0.32)] than the CoCu/N4G case. On the other hand, a distinctive change in the overlap ratio for the hetero CoCu/N4G catalyst, where the key orbitals were the dz2 (Xd = 0.28), dxz (Xd = 0.26), dyz (Xd = 0.22), and dxy (Xd = 0.20), was verified. As a result, a drop in the binding strength of the O atom arose in the hetero CoCu/N4G catalyst.
(4) Finally, we explored other dopants to improve the OER activity of the hetero CoM (M = 4d and 5d block metals) double-atom catalysts using the descriptor [ΔEads(O*) or λ] determined by volcano curve. The promising CoPd/N4G candidate was chosen by the deviation (Δλ) range from −0.22 eV to 0.22 eV on the basis of the peak position (λ = −4.37 eV). The onset potential of the selected CoPd/N4G catalyst presented a reduced onset potential by 0.80 V as compared to the homo Co2/N4G catalyst, verifying the suitability of the descriptor. It implied that there is still a chance to discover another efficient hetero double-atom catalyst having the descriptor value near the peak position.
In conclusion, our theoretical study offers useful insights into developing a strategy for high-efficiency OER catalysts using a variety of double-atom composites for the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2021M3I3A1084813, NRF-2020R1A2C1099711). This work was also financially supported by Korea Institute of Science and Technology (KIST) and Inha University Research Grant.
References
- J. Chi and H. Yu, Chin. J. Catal., 2018, 39, 390–394 CrossRef CAS.
- J. Brauns and T. Turek, Processes, 2020, 8, 248 CrossRef CAS.
- F. Barbir, Sol. Energy, 2005, 78, 661–669 CrossRef CAS.
- S. S. Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef.
- K. Zhu, T. Wu, M. Li, R. Lu, X. Zhu and W. Yang, J. Mater. Chem. A, 2017, 5, 19836–19845 RSC.
- H. Xu, H. Shang, C. Wang, L. Jin, C. Chen, C. Wang and Y. Du, Appl. Catal., B, 2020, 265, 118605 CrossRef CAS.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao and S. Wei, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. r. Selve, A. Bergmann, H. N. Nong and R. Schlögl, J. Am. Chem. Soc., 2015, 137, 13031–13040 CrossRef CAS PubMed.
- L. Zhang, Q. Fan, K. Li, S. Zhang and X. Ma, Sustainable Energy Fuels, 2020, 4, 5417–5432 RSC.
- J. Luo, L. Wang, D. Mott, P. N. Njoki, N. Kariuki, C.-J. Zhong and T. He, J. Mater. Chem., 2006, 16, 1665–1673 RSC.
- K. Bergamaski, A. L. Pinheiro, E. Teixeira-Neto and F. C. Nart, J. Phys. Chem. B, 2006, 110, 19271–19279 CrossRef CAS.
- R. A. Hameed and S. S. Medany, J. Colloid Interface Sci., 2018, 513, 536–548 CrossRef.
- J. N. Stuecker, J. E. Miller, R. E. Ferrizz, J. E. Mudd and J. Cesarano, Ind. Eng. Chem. Res., 2004, 43, 51–55 CrossRef CAS.
- R. H. Tammam, A. M. Fekry and M. M. Saleh, Korean J. Chem. Eng., 2019, 36, 1932–1939 CrossRef CAS.
- H. Sim, J. Lee, T. Yu and B. Lim, Korean J. Chem. Eng., 2018, 35, 257–262 CrossRef CAS.
- K. Min, S. Kim, E. Lee, G. Yoo, H. C. Ham, S. E. Shim, D. Lim and S.-H. Baeck, J. Mater. Chem. A, 2021, 9, 17344–17352 RSC.
- K. Min, M. Hwang, S. E. Shim, D. Lim and S.-H. Baeck, Chem. Eng. J., 2021, 424, 130400 CrossRef CAS.
- H. J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- C. Zhu, Q. Shi, S. Feng, D. Du and Y. Lin, ACS Energy Lett., 2018, 3, 1713–1721 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- Y. Peng, B. Lu and S. Chen, Adv. Mater., 2018, 30, 1801995 CrossRef.
- N. Cheng, L. Zhang, K. Doyle-Davis and X. Sun, Electrochem. Energy Rev., 2019, 2, 539–573 CrossRef.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- A. Morozan, V. Goellner, Y. Nedellec, J. Hannauer and F. Jaouen, J. Electrochem. Soc., 2015, 162, H719 CrossRef CAS.
- M. D. Hossain, Z. Liu, M. Zhuang, X. Yan, G. L. Xu, C. A. Gadre, A. Tyagi, I. H. Abidi, C. J. Sun and H. Wong, Adv. Energy Mater., 2019, 9, 1803689 CrossRef.
- L. Osmieri, R. Escudero-Cid, A. H. M. Videla, P. Ocón and S. Specchia, Appl. Catal., B, 2017, 201, 253–265 CrossRef CAS.
- M. M. Hossen, K. Artyushkova, P. Atanassov and A. Serov, J. Power Sources, 2018, 375, 214–221 CrossRef CAS.
- S. Dilpazir, H. He, Z. Li, M. Wang, P. Lu, R. Liu, Z. Xie, D. Gao and G. Zhang, ACS Appl. Energy Mater., 2018, 1, 3283–3291 CrossRef CAS.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1803498 CrossRef.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao and S. Wei, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- Y. Li, H. Su, S. H. Chan and Q. Sun, ACS Catal., 2015, 5, 6658–6664 CrossRef CAS.
- S.-h. Kim, H. C. Song, S. J. Yoo, J. Han, K.-Y. Lee and H. C. Ham, J. Mater. Chem. A, 2022, 10, 6216–6230 RSC.
- X.-Q. Han, Z.-L. Lang, F.-Y. Zhang, H.-L. Xu and Z.-M. Su, Appl. Surf. Sci., 2022, 579, 152214 CrossRef CAS.
- X. Lv, W. Wei, B. Huang, Y. Dai and T. Frauenheim, Nano Lett., 2021, 21, 1871–1878 CrossRef CAS PubMed.
- H. Li, Z. Zhao, Q. Cai, L. Yin and J. Zhao, J. Mater. Chem. A, 2020, 8, 4533–4543 RSC.
- W. Yang, H. Huang, X. Ding, Z. Ding, C. Wu, I. D. Gates and Z. Gao, Electrochim. Acta, 2020, 335, 135667 CrossRef CAS.
- H. Yan, Y. Lin, H. Wu, W. Zhang, Z. Sun, H. Cheng, W. Liu, C. Wang, J. Li and X. Huang, Nat. Commun., 2017, 8, 1–11 CrossRef.
- P.-F. Yuan, H. Liu, Q. Sun and Y. Jia, Comput. Mater. Sci., 2018, 151, 189–195 CrossRef CAS.
- Q. Luo, W. Zhang, C.-F. Fu and J. Yang, Int. J. Hydrogen Energy, 2018, 43, 6997–7006 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS PubMed.
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- H. Niu, X. Wang, C. Shao, Z. Zhang and Y. Guo, ACS Sustainable Chem. Eng., 2020, 8, 13749–13758 CrossRef CAS.
- X. Zhai, L. Li, X. Liu, Y. Li, J. Yang, D. Yang, J. Zhang, H. Yan and G. Ge, Nanoscale, 2020, 12, 10035–10043 RSC.
- Z. Xu, R. Song, M. Wang, X. Zhang, G. Liu and G. Qiao, Phys. Chem. Chem. Phys., 2020, 22, 26223–26230 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- Q. Liang, G. Brocks and A. Bieberle-Hütter, J. Phys.: Energy, 2021, 3, 026001 CAS.
- M. Plevová, J. Hnát and K. Bouzek, J. Power Sources, 2021, 230072 CrossRef.
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819 CrossRef.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund and H. Ogasawara, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- A. Khorshidi, J. Violet, J. Hashemi and A. A. Peterson, Nat. Catal., 2018, 1, 263–268 CrossRef.
- T. Bligaard and J. K. Nørskov, Electrochim. Acta, 2007, 52, 5512–5516 CrossRef CAS.
- H. Kim, H. Park, H. Bang and S.-K. Kim, Korean J. Chem.
Eng., 2020, 37, 1275–1294 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab