
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtease-activated indocyanine green nanoprobes for intraoperative NIR fluorescence imaging of primary tumors†
Benjamin
Lew
 a,
Mebin
George
a,
Steven
Blair
a,
Zhongmin
Zhu
a,
Zuodong
Liang
a,
Jamie
Ludwig
b,
Celeste Y.
Kim
a,
Kyekyoon (Kevin)
Kim
*acd,
Viktor
Gruev
*ade and
Hyungsoo
Choi
*a
a,
Mebin
George
a,
Steven
Blair
a,
Zhongmin
Zhu
a,
Zuodong
Liang
a,
Jamie
Ludwig
b,
Celeste Y.
Kim
a,
Kyekyoon (Kevin)
Kim
*acd,
Viktor
Gruev
*ade and
Hyungsoo
Choi
*a
aDepartment of Electrical and Computer Engineering, University of Illinois, Urbana, IL 61801, USA. E-mail: kevinkim@illinois.edu; vgruev@illinois.edu; hyungsoo@illinois.edu
bDivision of Animal Resources, University of Illinois, Urbana, IL 61801, USA
cDepartment of Bioengineering, University of Illinois, Urbana, IL 61801, USA
dBeckman Institute for Advanced Science and Technology, University of Illinois, Urbana, IL 61801, USA
eCarle Illinois College of Medicine, University of Illinois, Urbana, IL 61801, USA
First published on 1st July 2022
Abstract
Tumor-targeted fluorescent probes in the near-infrared spectrum can provide invaluable information about the location and extent of primary and metastatic tumors during intraoperative procedures to ensure no residual tumors are left in the patient's body. Even though the first fluorescence-guided surgery was performed more than 50 years ago, it is still not accepted as a standard of care in part due to the lack of efficient and non-toxic targeted probes approved by regulatory agencies around the world. Herein, we report protease-activated cationic gelatin nanoparticles encapsulating indocyanine green (ICG) for the detection of primary breast tumors in murine models with high tumor-to-background ratios. Upon intravenous administration, these nanoprobes remain optically silent due to the energy resonance transfer among the bound ICG molecules. As the nanoprobes extravasate and are exposed to the acidic tumor microenvironment, their positive surface charges increase, facilitating cellular uptake. The internalized nanoprobes are activated upon proteolytic degradation of gelatin to allow high contrast between the tumor and normal tissue. Since both gelatin and ICG are FDA-approved for intravenous administration, this activatable nanoprobe can lead to quick clinical adoption and improve the treatment of patients undergoing image-guided cancer surgery.
Introduction
Early detection and complete resection of localized and disseminated tumor cells are paramount for successful treatment and disease-free survival of patients with various types of solid cancers.1 The primary methods for screening patients with suspected cancerous lesions are physical exams and lab tests followed by biopsy and one or more imaging studies conducted with ultrasound, magnetic resonance, computerized tomography, positive emission tomography, and X-rays.2 However, during intraoperative procedures, physicians only rely on their eyesight, touch, pre-operative imaging results, and surgical experience to identify and remove all cancerous tissue.3–5 This approach toward cancer treatment has virtually remained identical to the ones adopted over 100 years ago. However, since cancerous tissue can easily blend with healthy tissue, especially at the tumor periphery, incomplete tumor resection is identified in 25% of patients with breast cancer, and 40% of patients with head and neck cancer, and similar statistics are observed in many other types of solid cancers.6Various imaging techniques, such as optical coherence tomography, photoacoustic imaging, label-free multispectral imaging, phase-contrast imaging, and fluorescence imaging are actively investigated and evaluated in clinical settings for identifying and differentiating cancerous from healthy tissue during intraoperative procedures.7–13 Among these imaging techniques, near-infrared (NIR) fluorescence imaging has shown great potential to facilitate cancer surgeries. Imaging in the NIR spectrum (700–1000 nm) has the inherent benefits of deep tissue imaging due to minimal light absorption by the primary constituents of tissue, i.e., water and hemoglobin, high tumor-to-background ratio (TBR) due to low tissue autofluorescence, and high patient and caregiver safety due to the use of non-radiation energy.
The clinical success of NIR fluorescence imaging greatly depends on the sensitivity and specificity of tumor-targeted fluorescent probes, which have been extensively investigated and evaluated in clinical settings over the last 20 years. An ideal molecular probe should have the following properties: excitation and emission in the NIR spectrum with large Stokes shift, high absorption coefficient and quantum yield, good water solubility to prevent probe aggregation, photo and chemical stability, and high affinity to quickly accumulate in tumors while minimizing accumulation in healthy tissue. A molecular probe that satisfies the above-cited criteria will yield a TBR within clinically relevant time.14 Furthermore, a probe must have minimal toxicity on healthy tissue to enable wide and rapid acceptance for in-human use.
Two general approaches have been adopted toward the realization of tumor-targeted probes: conjugation of NIR fluorophores with cancer-specific ligands, such as peptides, proteins, antibodies, affibodies, and others,14–18 or activatable probes which are triggered by the tumor microenvironment, such as enzymes, pH, glutathione, and reactive oxygen/nitrogen species.19–22 Both approaches have generated probes that have undergone clinical evaluation and demonstrated their potential during cancer surgeries.23 Despite initial promising results, regulatory approval for these probes has been very slow mainly due to toxicity concerns. Even though in both approaches, the individual fluorophores, quenching molecules, and cancer-specific ligands are typically approved by the regulatory organization, the final tumor-targeted probe is a new molecule and requires in-depth toxicity evaluation, taking a long and painstaking process.
To address these clinical and regulatory challenges, we have developed tumor-targeted nanoprobes by encapsulating indocyanine green (ICG) in protease-activated cationic gelatin nanoparticles (GNPs). Cationic gelatin (type A) is readily available and generally recognized as safe by the U.S. Food and Drug Administration, and can form polyionic complexes with anionic molecules, such as ICG.24–26 The ICG-encapsulating GNPs (ICG-GNPs) are optically silent due to the energy resonance transfer among the bound ICG molecules. Further, the positively charged surface of ICG-GNPs implies enhanced interactions with the cell membranes, facilitating their intratumoral accumulation. The nanoprobes are activated upon proteolytic degradation of gelatin to release ICG and emit fluorescence (Fig. 1), leading to high TBR at early times after administration. In this paper, we report the size-, time- and dose-dependent localization of ICG-GNPs in 4T1 tumor-bearing mice using both an in vivo imaging system (IVIS) and a novel bioinspired image sensor13 capable of intraoperative imaging in a clinical environment.
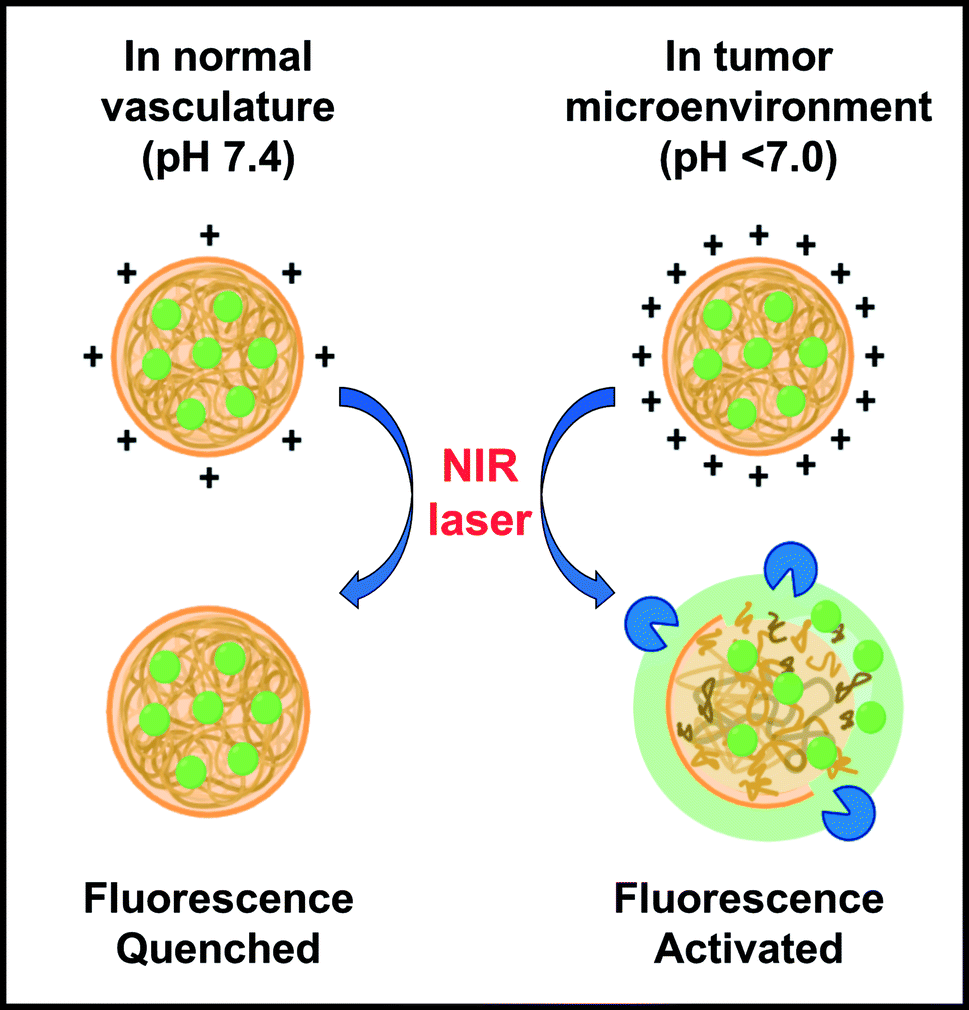 | ||
| Fig. 1 Schematic illustration of ICG nanoprobes (ICG-GNPs) administered intravenously, displaying increased surface charges in acidic tumor microenvironment and NIR fluorescence activation via upregulated tumor proteolytic activity. | ||
Experimental methods
Materials
Gelatin from porcine skin (type A, bloom 300), glutaraldehyde solution (grade II, 25% in H2O), Tween 20, and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich (Milwaukee, WI, USA). Indocyanine green (ICG) was purchased from Pfaltz & Bauer (Waterbury, CT, USA). Roswell Park Memorial Institute 1640 medium (RPMI-1640), fetal bovine serum (FBS), penicillin-streptomycin solution, and trypsin ethylenediaminetetraacetic acid (EDTA) solution (0.25%) were purchased from Fisher-Mediatech (Fisher Scientific, Waltham, MA, USA). D-Luciferin and 4T1-Red-FLuc (4T1) tumor cell line were purchased from PerkinElmer (Hopkinton, MA, USA). All chemicals were used without further purification.Preparation and characterization of ICG-GNPs
GNPs were synthesized using a modified two-step desolvation method as previously reported.27,28 Briefly, acetone was added dropwise to purified type A gelatin solution under vigorous stirring until the suspension reached the volume ratio of 2.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 3:1 to produce GNPs with an average diameter of 80 and 160 nm, respectively, which were crosslinked with 0.2% glutaraldehyde for 2 hours at 25 °C.
:1 and 3:1 to produce GNPs with an average diameter of 80 and 160 nm, respectively, which were crosslinked with 0.2% glutaraldehyde for 2 hours at 25 °C.
ICG-loaded GNPs of two different sizes, ICG-GNP-80 and ICG-GNP-160 were prepared by mixing GNPs and ICG with a 20:1 weight ratio in 1 ml of deionized water. The resulting suspension was incubated for 2 hours at 25 °C, followed by centrifugation to remove any free ICG. The morphology of the ICG-GNPs was observed by field emission scanning electron microscopy (FESEM, Hitachi S-4800, Japan). The hydrodynamic diameter and polydispersity index (PDI) of the ICG-GNPs were measured by dynamic light scattering using a Malvern Zetasizer (Zetasizer Nano S, Malvern, UK) (n = 3). The surface charge of ICG-GNPs was measured using a zeta-potential analyzer (Zetasizer Nano S, Malvern, UK) (n = 3). To assess the amount of encapsulated ICG in the GNPs, ICG-GNPs were digested in 1 ml of trypsin solution for 12 hours at 37 °C in an orbital shaker with continuous agitation (100 rpm). A 500 μl aliquot was collected by centrifugation (13000 rpm for 5 minutes) and the amount of ICG in each aliquot was determined by measuring the absorbance at 780 nm using a UV-Vis spectrophotometer (Genesys 10s UV-Vis Spectrophotometer, Thermo Scientific, Waltham, MA, USA) (n = 3). The same method was applied to prepare FITC-loaded GNPs (FITC-GNPs).
In vitro fluorescence activation of ICG-GNPs
Absorption spectra of ICG-free, ICG-GNP-80, and ICG-GNP-160 (equivalent to 10 μM ICG) solutions were obtained by the UV-Vis spectrophotometer. The mean fluorescence intensity (MFI) of ICG-free and ICG-GNP-80 at various ICG concentrations was recorded and quantified using an In Vivo Imaging System (IVIS, PerkinElmer, Waltham, MA, USA) (ex 740/em 800 nm).For in vitro release study, 1 mg of ICG-GNP-80 solution in 1 ml of PBS was prepared with or without 10 μl of trypsin solution to simulate the cancer microenviornment and incubated in an orbital shaker at 37 °C and 100 rpm.29–31 At different time intervals, samples were centrifuged to collect 500 μl aliquots from the supernatant. An equal amount of PBS with or without trypsin was added before placing the samples back in the incubator. The amount of ICG in each aliquot was determined by measuring the absorbance of ICG at 780 nm using the UV-Vis spectrophotometer (n = 3).
Analysis of in vitro cellular uptake of ICG-GNPs
4T1 cells were seeded in a 6-well plate (106 cells per well) 24 hours before the study and incubated in 1 ml of RPMI-1640 supplemented with FBS (10% v/v), penicillin (100 IU ml−1), and streptomycin (100 μg ml−1) with 5% CO2 at 37 °C. For flow cytometry analysis, 4T1 cells were incubated with ICG-free, ICG-GNP-80, or ICG-GNP-160 (equivalent to 1 μM ICG) for 1 hour at 37 °C. Subsequently, cells were washed three times with PBS and assessed by flow cytometry (BD LSRFortessa, New Jersey, USA) with an APC-Cy7 channel (Ex 647, Ex 785). A minimum of 10000 events was recorded (n = 3). Untreated cells were used as a negative control.
On a separate 6-well plate seeded with 4T1 cells, 1 mg FITC-GNPs with 80 nm diameters were seeded and incubated for 4 hours. After washing each well three times with PBS, the fluorescence images of the cells were captured using a fluorescence light microscope (Zeiss Observer Z1 inverted research-grade microscope, Oberkochen, Germany) (FITC Ex 488/Em 525 nm).
Animal tumor model
Female immunodeficient mice (J:NU, 2 months old, average body weight of 25 g) were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). All animal studies were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC, Protocol ID 20194) and the Division of Animal Resources at the University of Illinois. All mice were maintained in the animal care facility at the Beckman Institute for Advanced Science and Technology (University of Illinois at Urbana-Champaign, Urbana, IL, USA) under the required conditions with free access to water and food throughout the experiments. Breast cancer xenograft was established by subcutaneous administration of 100 μl of PBS with 4T1 cells (106 cells per injection) in the mammary fat pad of the second nipple on the left side of each mouse. Anesthesia was maintained by mask inhalation of 1.5–2.0% isoflurane throughout the procedure. Successful xenograft development was confirmed by in vivo bioluminescence imaging after treating each mouse with D-luciferin (150 mg kg−1).Quantification of fluorescence in blood plasma
Nine healthy mice were intravenously administered with ICG-free, ICG-GNP-80, or ICG-GNP-160 (n = 3 per group) with a dose equivalent to 2.0 mg kg−1 ICG. A few minutes after the administration, 100 μl of blood was drawn from each mouse from the lateral tail vein. The blood samples were centrifuged at 2500 rpm for 10 min to separate plasma from blood cells. The plasma was transferred to a 96-well plate and imaged using the IVIS. A plasma sample from the mouse without dye treatment was used as a negative control.In vivo NIR fluorescence imaging
Twelve tumor-bearing mice were randomly assigned for intravenous (IV) administration of ICG-free, ICG-GNP-80, or ICG-GNP-160 (n = 4 per group) in 100 μl of PBS with a dose equivalent to 2.0 mg kg−1 ICG and monitored for 24 hours. The animals were selected when the tumor area reached 10 mm2. At each time interval, the fluorescence signals from the primary tumor and muscle tissue (background) were recorded using the IVIS. A ratio between the mean fluorescence from the tumor and muscle was assessed. All mice were euthanized after imaging at 24 hours and all tumors, major organs, and muscle tissues were used for ex vivo imaging as described below. The fluorescence intensity from each organ or tissue was acquired using the IVIS.For the dose-dependent study, sixteen tumor-bearing mice were randomly assigned to four groups for IV administration of ICG-GNP-80 with concentrations of 0.25, 0.5, 1.0, and 2.0 mg kg−1 ICG (n = 4 per group). The IVIS was used to acquire the fluorescence intensity from the tumor and muscle tissue to assess the mean TBR at each time interval for 48 hours. Once the optimal dose was determined, a bioinspired image sensor was used to record visible and NIR images of a 4T1 tumor-bearing mouse at 6-, and 24 hour post-administration of ICG-GNP-80 excited by the 780 nm laser source (20 mW cm−2).
Tumor resection and ex vivo analysis
Four tumor-bearing mice administered with ICG-GNP-80 at a dose equivalent to 1 mg kg−1 ICG were euthanized at 24 hours to harvest the tumors, which were immediately embedded in optimal cutting temperature (OCT) compound and sectioned with a cryostat (CM3050S, Leica, Germany) at −20 °C into a series of 100 μm-thick specimens. The distribution of ICG molecules within each specimen was determined by measuring the fluorescence using a NIR fluorescence scanner (Odyssey CLx imager, Li-Cor, Lincoln, NE, USA). Subsequently, each specimen was stained with hematoxylin and eosin (H&E) and imaged with an optical slide scanning system (Nanozoomer slide scanning system, Hamamatsu, Japan) to confirm the cellular morphology of malignant tissues.For ImageJ analysis, a method described in Mahalingam et al. was used.32 The whole body image was acquired using the IVIS in grayscale, which was processed by ImageJ software to obtain a surface plot. A region of interest was marked with a black square for quantitative analyses.
Statistical analysis
Data are expressed as mean values ± SD. For statistical analysis, one-way ANOVA with Tukey post hoc test was performed using SPSS Statistics 28. Differences between samples were considered significant at p < 0.05.Results and discussion
Synthesis and characterization of ICG-GNPs
Cationic GNPs with diameters of 80 and 160 nm were prepared to investigate the effect of particle size on the efficacy of ICG delivery to primary tumors. The DLS analysis of ICG-GNP-80 and ICG-GNP-160 exhibited mean hydrodynamic diameters of 83.06 ± 4.22 nm and 167.63 ± 5.27 nm, respectively, with small PDI values (<0.2) indicating their monodispersity (Fig. 2a). The SEM images show that both ICG-GNPs have a well-defined spherical shape and smooth surface with no signs of aggregation of the particles. The zeta-potentials of GNP-80 and GNP-160 at pH 7 were 25.30 ± 4.42 and 26.00 ± 3.86 mV, respectively, showing no difference in surface charge density between the two. Furthermore, the net positive charges of the GNPs enabled the gelatin matrix to stably incorporate the anionic ICG molecules with a high loading efficiency (>90%). After ICG encapsulation, the zeta-potentials of ICG-GNP-80 and ICG-GNP-160 at pH 7 were 23.60 ± 3.13 and 23.40 ± 2.89 mV, respectively, to establish particle stability to resist aggregation in a dispersion. Fig. 2b shows increases in the zeta-potential of ICG-GNPs as the pH decreased from 7 to 4, implying that the positive surface charges of the nanoprobes would further increase as they extravasate into the acidic tumor microenvironment. No statistically significant difference between the zeta-potentials of ICG-GNP-80 and ICG-GNP-160 was observed at each pH level.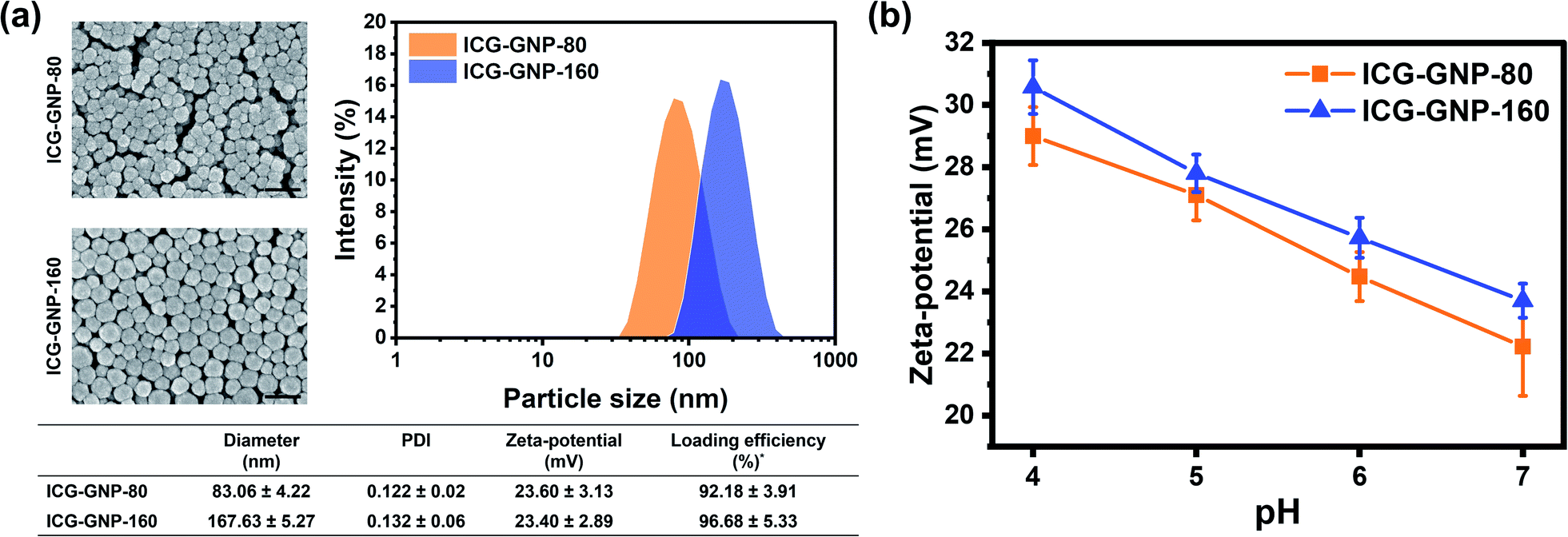 | ||
| Fig. 2 Physical properties of ICG-GNPs. (a) Particle morphology, size, PDI, zeta-potential, and loading efficiency of ICG-GNPs (scale bar = 200 nm). (b) A plot of zeta-potential of ICG-GNPs versus pH. *Loading efficiency (w/w%) = [amount of ICG loaded/amount of ICG used] × 100. | ||
In vitro fluorescence activation of ICG-GNPs
Tumor-responsive fluorescence activation of NIR nanoprobes could lead to improved contrast between tumors and normal tissues during intraoperative procedures.33,34 In this context, we first evaluated the fluorescence activation of ICG-GNPs in vitro. Fig. 3a shows the absorption spectra of 10 μM ICG-free and ICG-GNPs (equivalent to 10 μM ICG) solutions. The primary and secondary absorption peaks were observed in all cases at 780 nm and 710 nm, respectively. However, the absorbance of ICG-GNPs was about 1.43-folds lower than that of ICG-free at the primary peak, which could be attributed to the scattering of the incident light beam by GNPs.35 No significant difference was observed between ICG-GNP-80 and ICG-GNP-160.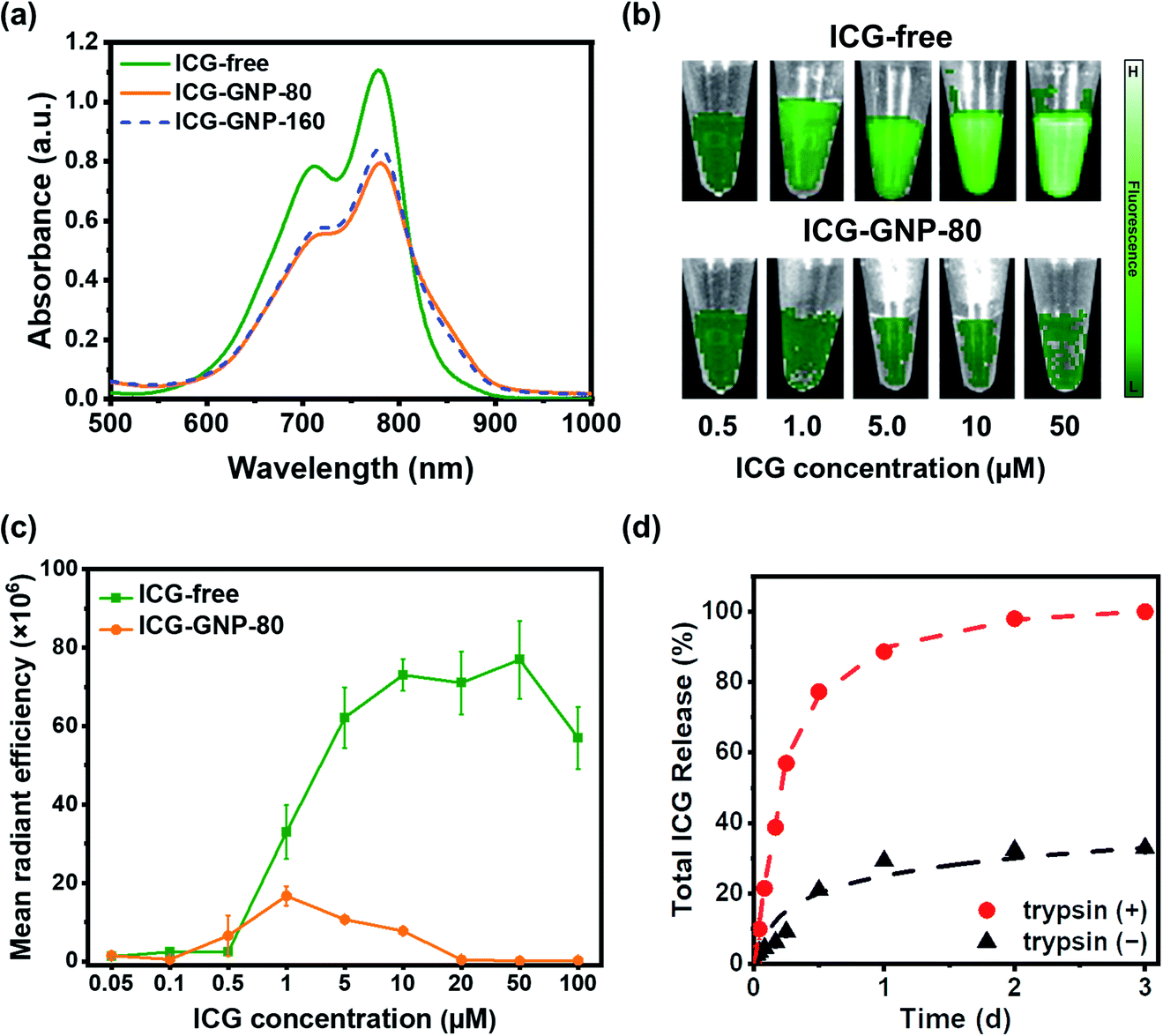 | ||
| Fig. 3 Self-quenching and proteolytic activation of ICG-GNPs in vitro. (a) Absorbance spectra of ICG-free and ICG-GNP solutions (equivalent to 10 μM ICG). (b) Representative fluorescence images, and (c) MFI of ICG-free and ICG-GNP-80 solutions at varying concentrations of ICG, expressed as mean radiant efficiency ([p s cm2 sr]−1/[μW cm−2]). (d) Cumulative ICG release from ICG-GNP-80 in the absence (−) and presence (+) of trypsin at each time point. | ||
To investigate the emission behavior of ICG-GNPs, the MFI of ICG-free and ICG-GNP-80 solutions with different ICG concentrations was evaluated using the IVIS. As depicted in Fig. 3b, the fluorescence of ICG-free was minimal below 0.5 μM and increased significantly with concentrations up to 10 μM. The MFI was similar between 10 and 50 μM and decreased afterward due to concentration-dependent quenching (Fig. 3c). Interestingly, however, only minimal fluorescence was measured from the ICG-GNP-80 solutions with concentrations of 0.5–20 μM, and no notable fluorescence was detected above 20 μM demonstrating the self-quenching of ICG fluorescence when it is in a bound state within GNPs.
The release behavior of ICG from the GNPs was measured using ICG-GNP-80 in cell culture media in the absence or presence of trypsin simulating the protease-upregulated intratumor environment. Fig. 3d depicts the release kinetics of ICG-GNP-80, indicating >60% total release of ICG within 6 hours and 100% in 2 days in the presence of trypsin, whereas only ∼30% was released in a day and no further release afterward in the absence of trypsin. The unbound ICG was released mainly by diffusion without the protease-triggered degradation, and the ICG molecules complexed to the gelatin matrix remained intact through electrostatic attraction and ICG-protein binding, conferring the site-specific release of ICG in the intratumor environment expressing high proteolytic activity.36–38 We have previously reported the degradation of GNPs by upregulated proteases to locally release hydrophilic as well as hydrophobic therapeutics for the treatment of central nervous system disorders and cancers.27,28,39,40 These findings show that ICG-GNPs could provide a significant advantage in improving the tumor-specificity of ICG over ICG-free.
Cellular internalization of ICG-GNPs
The size and surface charge of nanoprobes play an important role in determining the kinetics of their internalization into tumor cells.41,42 Herein, we evaluated in vitro cellular internalization of ICG-free, ICG-GNP-80 and ICG-GNP-160 using flow cytometry. Fig. 4a shows the histograms of the cells treated with these probes for 1 hour. All ICG-treated cells exhibit various levels of fluorescence intensity to differentiate the fluorescence-positive population (blue histogram) from the untreated population (red histogram). Note that the histograms of the cells treated with ICG-GNPs were shifted further to the right compared to those treated with ICG-free. As a result, about 60.4%, 90%, and 78.5% of total cells were fluorescence-positive for ICG-free, ICG-GNP-80 and ICG-GNP-160, respectively (Fig. 4b). This is in line with the previous studies on facilitated internalization of cationic nanoparticles into tumor cells.43,44 ICG-GNP-80 displayed a higher population of fluorescence-positive cells than ICG-GNP-160, demonstrating the beneficial effect of smaller GNP size on cellular internalization.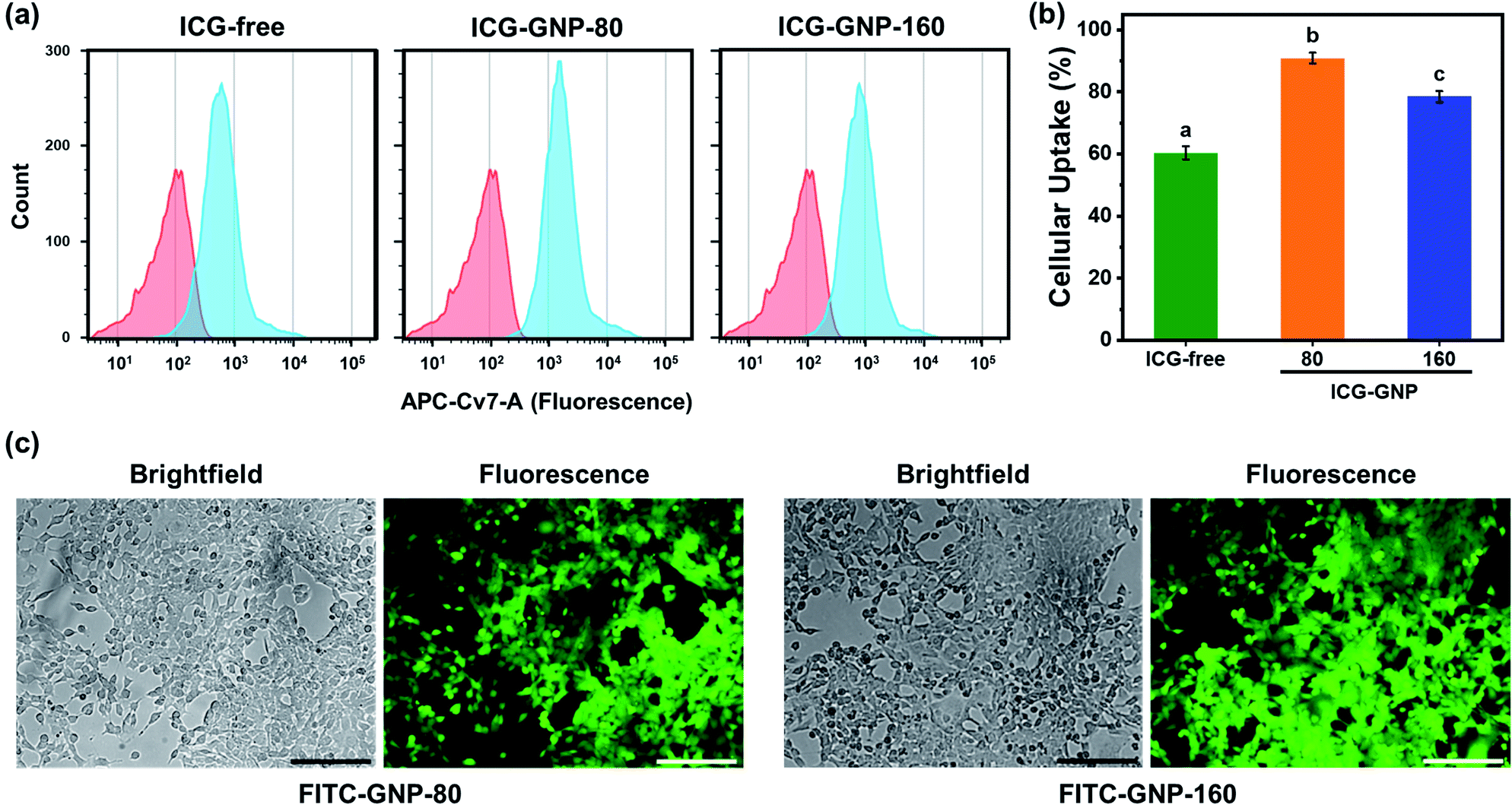 | ||
| Fig. 4 In vitro cellular internalization of ICG-GNPs. (A) Representative flow cytometry histograms tracing the distribution of fluorescence from 4T1 cells after incubating with ICG-free and ICG-GNPs (equivalent to 1 μM ICG) for 1 hour (blue). Untreated cells were used as negative control (red). (B) Quantitative analysis of cellular uptake of ICG-free and ICG-GNPs by 4T1 cells over 1 hour. Different letters indicate a significant difference between groups (p <0.05). (C) Representative brightfield and fluorescence images of 4T1 cells after 1 hour incubation with FITC-GNP-80. Scale bar = 100 μm. | ||
To visualize the internalization of GNPs in 4T1 cells, FITC-loaded GNPs with diameters of 80 (FITC-GNP-80) and 160 nm (FITC-GNP-160) were incubated with 4T1 cells and observed under a fluorescence microscope. Fig. 4c shows FITC-loaded GNPs of both sizes internalized in 4T1 cells.
In vivo tumor imaging and biodistribution study
Next, we performed a series of in vivo studies with our nanoprobes to highlight the beneficial role of GNP-mediated delivery of ICG. In the first study, we compared the MFI of ICG-free, ICG-GNP-80 and ICG-GNP-160 in the blood plasma of healthy mice immediately after systemic administration. Fig. 5a shows that the MFI of ICG-free was about 5-fold higher compared to that of ICG-GNPs, indicating that the fluorescence from the ICG intact in GNPs was quenched in the absence of proteolytic activity. No statistically significant difference was found between ICG-GNP-80 and ICG-GNP-160 as expected. This is consistent with the in vitro release behavior of the nanoprobes, discussed in the previous section. In the absence of proteolytic activity, only a negligible amount of ICG is expected to be released from the nanoprobes during the initial period (<30 min) through diffusion, which is not sufficient to produce any meaningful fluorescence (Fig. 3c and d).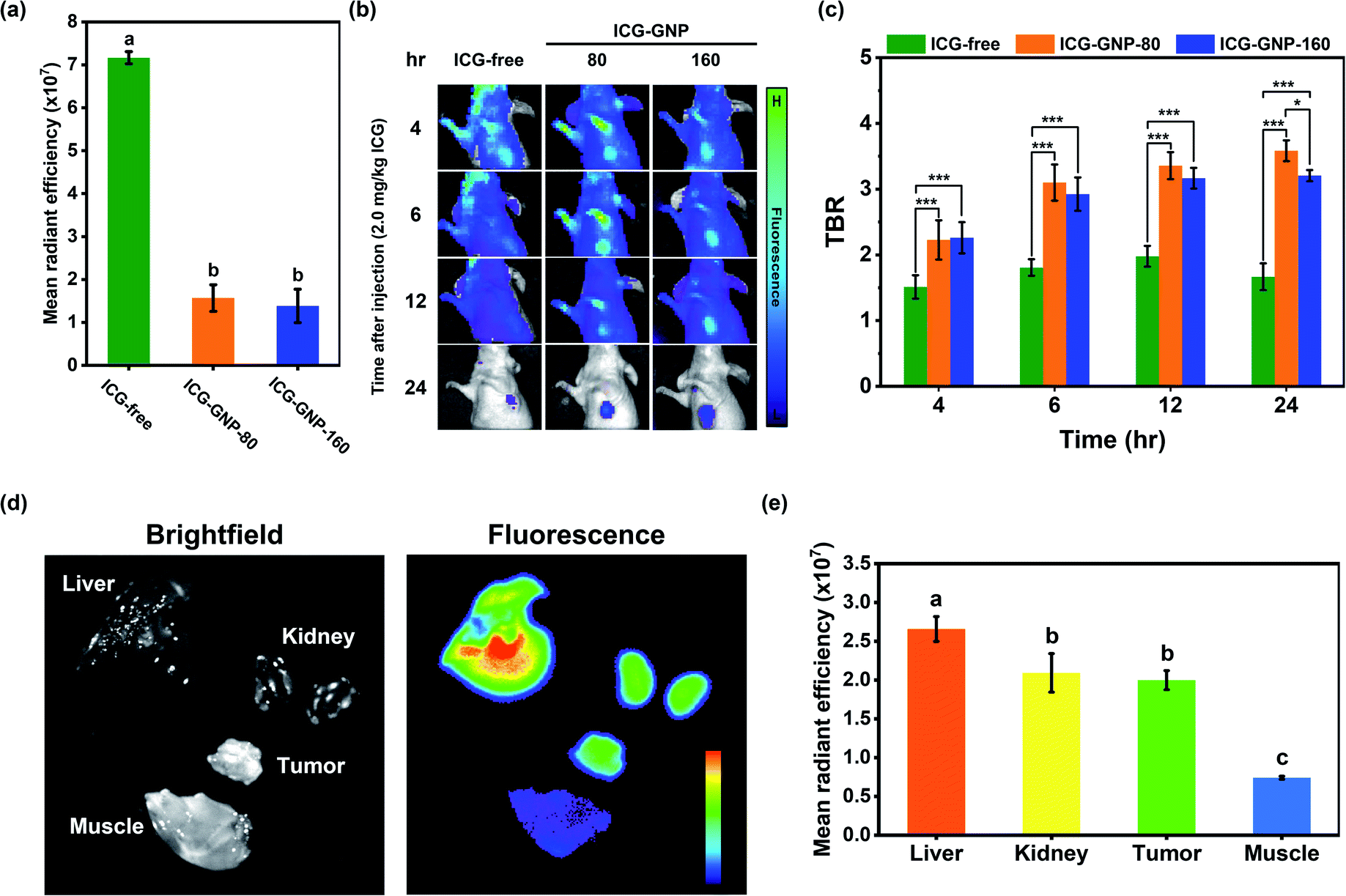 | ||
| Fig. 5 In vivo imaging and biodistribution of ICG-GNPs. (a) Quantitative analysis of MFI in the mouse blood plasma collected immediately after administering ICG-free and ICG-GNPs. (b) Representative fluorescence images and (c) quantitative analysis of mean TBR of tumor-bearing mice over 24 hours after administering ICG-free and ICG-GNPs. (d) Representative fluorescence image and (e) quantitative analysis of MFI in kidneys, liver, tumor, and muscle tissue harvested at 24 hours after administering ICG-GNP-80. The dose used for all probes was equivalent to 2.0 mg kg−1 ICG. Different letters indicate a significant difference between groups. *p < 0.05, ***p < 0.005. MFI is expressed as mean radiant efficiency. | ||
Thereupon, we prepared three sets of 4T1 tumor-bearing mice, administered with ICG-free, ICG-GNP-80, or ICG-GNP-160 (equivalent to 2 mg kg−1 ICG), and imaged over 24 hours using the IVIS. Fig. 5b shows the changes in MFI in the tumor and background tissues of each mouse as a function of time. The mice treated with ICG-free exhibited a slightly higher level of fluorescence in the tumor than in the background at 4 hour post-administration. However, the fluorescence decreased in both the background and tumor at 6- and 12 hour post-administration, and only minimal fluorescence remained at 24 hour. On the contrary, mice administered with ICG-GNPs exhibited improved contrast between the tumor and background over time, resulting in strong fluorescence in the tumor with minimal background fluorescence at 24 hour post-administration. Fig. 5c depicts the contrast between the tumor and the background analyzed quantitatively for the three mice groups at each time point. A TBR above 2.0–2.5 is considered adequate for the clinical intraoperative imaging.45–47 Mice treated with ICG-free maintained a mean TBR of 1.74 ± 0.20 over 24 hours indicating the insufficient tumor contrast for intraoperative NIR imaging at the given dose. Starting from 4 hour post-administration, both groups of ICG-GNPs exhibited significantly higher tumor contrasts than those with ICG-free throughout the monitoring period. The mean TBR with ICG-GNP-80 reached 3.58 ± 0.16 at 24 hour, which was significantly higher than that with ICG-GNP-160 (3.21 ± 0.09), demonstrating preferential intratumor ICG accumulation mediated by the GNPs in a size-dependent manner. Previous studies have shown that larger nanoparticles (>100 nm) tend to accumulate around the tumor vasculature, whereas smaller nanoparticles (<100 nm) diffuse into the tumor matrix.48,49 The location of the primary tumor in each mouse was confirmed by the bioluminescence imaging with the IVIS (Fig. S1†). These results indicate that primary tumors could be visualized effectively by ICG-GNPs using a much lower dose (2.0 mg kg−1) than the doses reported as optimal for ICG-free (5–10 mg kg−1) in clinical practices.45,50 Since ICG-GNP-80 exhibited a higher TBR than ICG-GNP-160, we used the former for all further studies.
To assess the biodistribution of ICG-GNP-80, the fluorescence in the tumor, liver, kidneys, and muscle tissues, harvested at 24 hour post-administration, was measured (Fig. 5d). The highest MFI was detected in the liver followed by the tumor and kidneys, whereas no notable fluorescence was observed in the muscle.51–53 As shown in Fig. 5e, the MFI measured from the tumor was significantly higher than that from the muscle, authenticating the selective internalization of ICG-GNPs by tumor cells. Whole-section images of the resected tumor and healthy muscle tissues were obtained and evaluated using H&E staining to show the clear distinction between the malignant and normal tissues at a cellular level (Fig. S2†).
Optimization of ICG-GNPs for intraoperative NIR fluorescence imaging
Fig. 6a depicts the changes in the tumor and background fluorescence in mice administered with ICG-GNP-80 at doses equivalent to 0.25, 0.5, 1.0, and 2.0 mg kg−1 ICG over 48 hours. All doses contributed to strong background fluorescence at 4 hour post-administration, which progressively decreased in a dose-dependent manner. This resulted in prominent differences between the tumor and surrounding tissues, allowing tumor identification as early as 6- and 12 hour post-administration with doses equivalent to 0.25 and 0.5 mg kg−1 ICG, respectively. The peak TBRs for 0.25 and 0.5 mg kg−1 were 2.32 ± 0.19 and 2.50 ± 0.28, respectively, which decreased with time. For 1.0 and 2.0 mg kg−1, the TBR increased gradually over time and reached 4.00 ± 0.21 and 3.63 ± 0.21 at 24- and 48 hour, respectively (Fig. 6b). These results showed that ICG-GNPs at a dose equivalent to 1.0 mg kg−1 ICG achieved the highest peak TBR at 24 hour to effectively visualize the contrast between the primary tumor and surrounding tissues with minimized background fluorescence.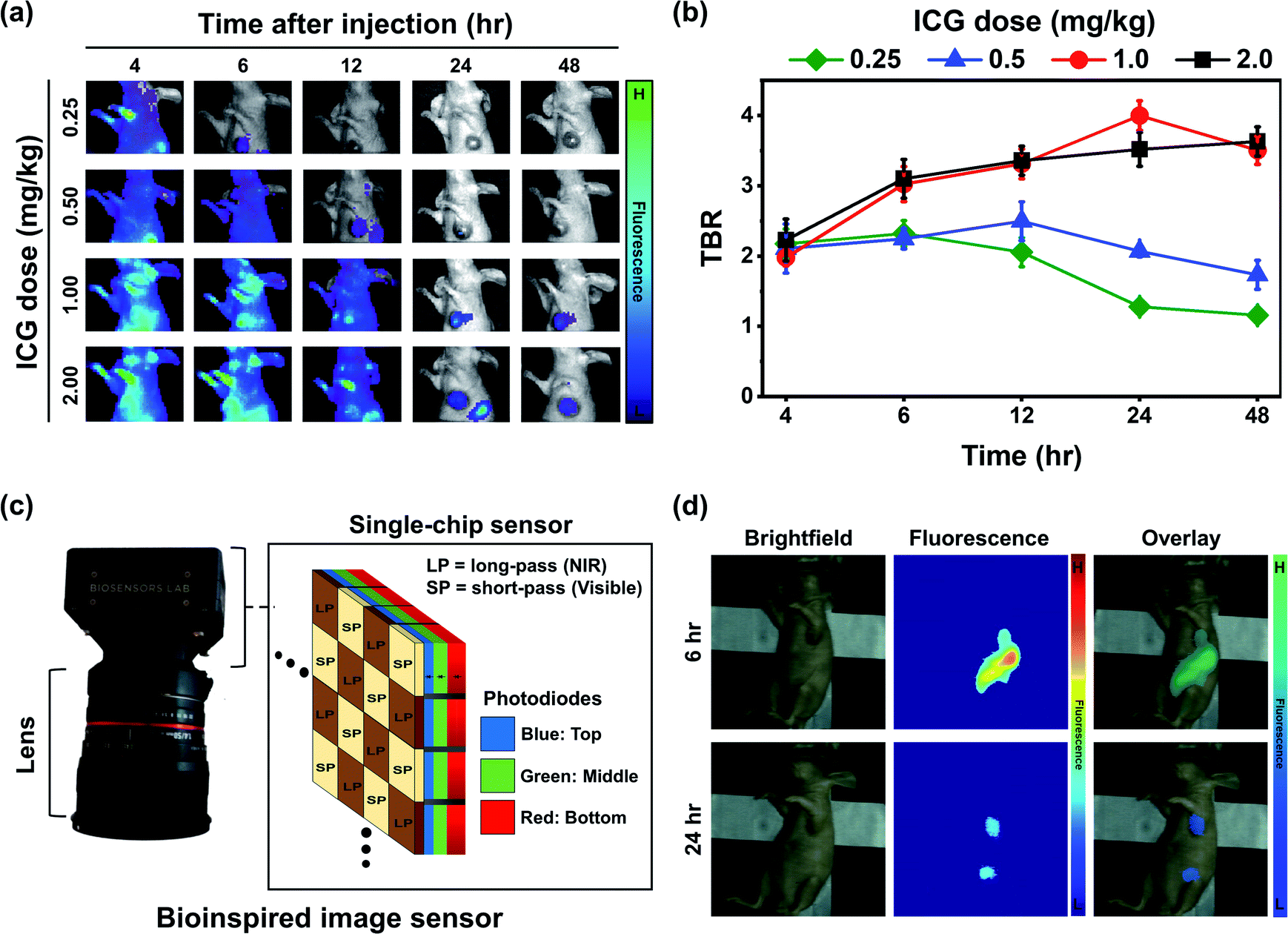 | ||
| Fig. 6 Optimization of ICG-GNP-80 dose for intraoperative NIR fluorescence imaging. (a) Representative fluorescence images and (b) mean TBR of tumor-bearing mice administered with ICG-GNP-80 solutions of different concentrations during a 48 hour period, acquired and analyzed using the IVIS. (c) Schematic illustration of a bioinspired image sensor and (d) representative visible, fluorescence, and overlay images of tumor-bearing mice recorded using the sensor at 6- and 24 hour post-administration of ICG-GNP-80 (equivalent to 1.0 mg kg−1 ICG). | ||
Based on this, we injected ICG-GNP-80 into several tumor-bearing mice at a dose equivalent to 1.0 mg kg−1 ICG and imaged them using a novel bioinspired image sensor. This versatile single-chip sensor mimics the visual system of a mantis shrimp that features three vertically stacked photodiodes (blue, green, and red channels) each having a distinct spectral response (Fig. 6c). By incorporating pixelated spectral filters, the sensor could capture both visible and NIR images simultaneously. Previously, the sensor was employed to enable sentinel lymph node mapping to guide the surgeons during the surgical resection of patients with breast cancer after the intratumoral injection of ICG and methylene blue (MB).13 In this in vivo study, a 780 nm laser with 20 mW cm−2 optical power was used to excite the ICG-GNPs in mice. At 6 hour post-administration, high fluorescence signals from both the liver and tumor were recorded while only minimal fluorescence was observed from the surrounding healthy tissues (Fig. 6d). After 24 hours, the fluorescence signal from the tumor decreased while the TBR increased in a similar fashion observed in the in vivo images acquired using the IVIS system. These findings indicate the possibility of imaging tumors with ICG-GNPs using an intraoperative type of imaging instrument at any given time within 24 hours to facilitate the needs of surgeons and patients.
Ex vivo imaging and analysis of resected tumor
For curative cancer surgery, the removal of the entire tumor with negative margins is crucial. In this context, we analyzed the distribution of fluorescence in the tumor and surrounding tissues in vivo as well as ex vivo. The surface plot in Fig. 7a depicts the distribution of in vivo fluorescence within a region containing a primary tumor and its surrounding tissues of a mouse 24 hours after the administration of ICG-GNP-80 (equivalent to 1.0 mg kg−1 ICG). The signal depth between the center and the edge of the surface plot represents a well-defined boundary between the tumor and normal tissues, enabling accurate detection of tumor margins using the NIR fluorescence imaging. Subsequently, the mouse was euthanized to harvest and section the primary tumor for ex vivo imaging. The fluorescence images of the harvested tumor and corresponding tissue specimens in Fig. 7b display the distribution of ICG within the tumor. The harvested tumor exhibited the highest fluorescence intensity near the center when measured with the IVIS. Given the higher fluorescence detected on the specimen from the center compared to the edge of the tumor, we could estimate that the tumor vasculature was leading the ICG-GNPs to the core of the primary tumor until it reached the necrotic regions with avascularity. Necrosis emerges typically at the core of the mammary tumor after 14 days of growth suggesting that the cellular internalization of ICG-GNPs could also depend on the stage of tumor development and corresponding angiogenesis.54 Notably, the high fluorescence signal was also observed at the periphery of the specimens, enabling the facile identification of the boundary between the tumor and normal tissues.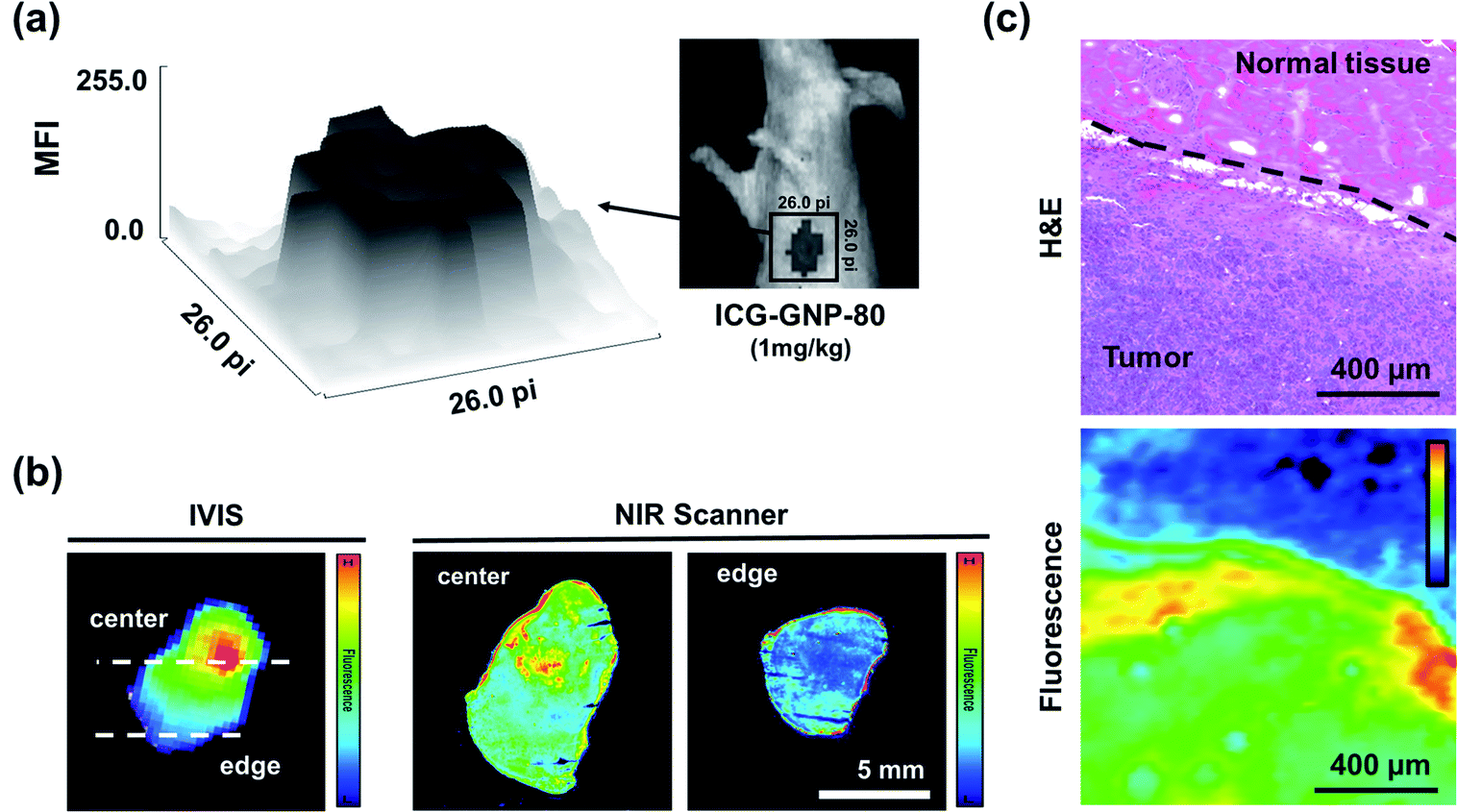 | ||
| Fig. 7 Ex vivo tumor contrast analysis. (a) Fluorescence image of a 4T1 tumor-bearing mouse at 24 hours after administering ICG-GNP-80 (equivalent to 1 mg kg−1 ICG) with a surface plot showing the distribution of fluorescence within the region of interest (marked with a 26 × 26 pixel square). (b) Representative fluorescence image of a resected tumor and the corresponding tissue specimens taken by the IVIS and NIR scanner, respectively. (c) Histological and fluorescence images of the tissue specimen showing the contrast between the tumor and normal tissue. The dotted line shows the border between the tumor and normal tissue. | ||
To further visualize the tumor margins, one of the tissue specimens containing both the tumor and normal tissue was histologically analyzed and compared with the corresponding fluorescence image (Fig. 7c). The H&E-stained tissue specimen shows two distinct regions, malignant and normal, distinguishable by their cellular morphology and distribution of nuclei stained in purple. The corresponding fluorescence image of the same area of the tissue exhibits significantly higher fluorescence intensity in the malignant region than in the normal region, matching the H&E image. The clear contrast between the malignant and normal regions of the specimen suggests that accurate assessment of tumor margins would be facilitated by ICG-GNPs.
Conclusions
In summary, we designed and fabricated proteases-activated ICG-GNPs for imaging tumors in murine models of breast cancer. ICG molecules formed stable polyionic complexes with GNPs leading to optically silent nanoprobes in the absence of proteases. In the in vitro studies, ICG-GNPs exhibited higher cellular uptake compared to free-ICG in 4T1 mammary tumor cells and released ICG in response to protease-triggered degradation of GNPs intratumorally. Additionally, the pharmacokinetics of ICG-GNPs in vivo was assessed by administering ICG-GNPs intravenously into breast tumor-bearing mice and monitored over 48 hours. The results show improved intratumoral accumulation of ICG via GNP-mediated delivery confirmed by the significantly increased mean TBR of the mice treated with ICG-GNPs compared to those with free-ICG. Especially, we found ICG-GNP-80 at a dose equivalent to 1.0 mg kg−1 ICG to be optimal for clinically relevant intraoperative NIR fluorescence imaging to exhibit a TBR of ∼4.0 within 24 hours, confirmed by imaging with a bioinspired image sensor. The ex vivo imaging indicated the accumulation of ICG-GNPs at the core and periphery of the tumor, thus enabling the visualization of the boundary between malignant and normal tissues. Taken together, all the in vitro and in vivo results manifested the beneficial role of GNPs to improve intratumoral ICG delivery for the visualization of primary tumors via intraoperative NIR fluorescence imaging.Conflicts of interest
All authors declare that they have no known financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Kim Research Fund [Grant No. UIUC-933008-633134], the Congressionally Directed Medical Research Programs [Grant No. W81XWH-19-1-0299], and the National Science Foundation [Grant No. 2030421]. We thank Prof. Zeynep Madak-Erdogan for use of the Odyssey CLx imager.References
- S. B. Mondal, S. Gao, N. Zhu, R. Liang, V. Gruev and S. Achilefu, in Advances in Cancer Research, ed. M. G. Pomper and P. B. Fisher, Academic Press, 2014, vol. 124, pp. 171–211 Search PubMed.
- J. V. Frangioni, J. Clin. Oncol., 2008, 26, 4012–4021 CrossRef PubMed.
- M. Koller, S.-Q. Qiu, M. D. Linssen, L. Jansen, W. Kelder, J. de Vries, I. Kruithof, G.-J. Zhang, D. J. Robinson, W. B. Nagengast, A. Jorritsma-Smit, B. van der Vegt and G. M. van Dam, Nat. Commun., 2018, 9, 3739 CrossRef PubMed.
- P. S. Low, S. Singhal and M. Srinivasarao, Curr. Opin. Chem. Biol., 2018, 45, 64–72 CrossRef CAS PubMed.
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. H. van de Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10, 507–518 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, CA A Cancer J. Clin., 2020, 70, 7–30 CrossRef PubMed.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 1–22 CrossRef.
- V. Ntziachristos, J. Ripoll, L. V. Wang and R. Weissleder, Nat. Biotechnol., 2005, 23, 313–320 CrossRef CAS PubMed.
- B. M. Greenberg and E. Frohman, Ther. Adv. Neurol. Disord., 2010, 3, 153–160 CrossRef PubMed.
- J. Shapey, Y. Xie, E. Nabavi, R. Bradford, S. R. Saeed, S. Ourselin and T. Vercauteren, J. Biophot., 2019, 12, e201800455 Search PubMed.
- A. Momose, Phys. Med., 2020, 79, 93–102 CrossRef PubMed.
- C. W. Barth and S. L. Gibbs, Proc. SPIE-Int. Soc. Opt. Eng., 2020, 11222, 112220J Search PubMed.
- S. Blair, M. Garcia, T. Davis, Z. Zhu, Z. Liang, C. Konopka, K. Kauffman, R. Colanceski, I. Ferati, B. Kondov, S. Stojanoski, M. B. Todorovska, N. T. Dimitrovska, N. Jakupi, D. Miladinova, G. Petrusevska, G. Kondov, W. L. Dobrucki, S. Nie and V. Gruev, Sci. Transl. Med., 2021, 13, eaaw7067 CrossRef CAS PubMed.
- R. R. Zhang, A. B. Schroeder, J. J. Grudzinski, E. L. Rosenthal, J. M. Warram, A. N. Pinchuk, K. W. Eliceiri, J. S. Kuo and J. P. Weichert, Nat. Rev. Clin. Oncol., 2017, 14, 347–364 CrossRef CAS PubMed.
- N. Zhao, Y. Qin, H. Liu and Z. Cheng, Anticancer Agents Med. Chem., 2018, 18, 74–86 CrossRef CAS PubMed.
- J.-J. Carine, C. Mylene, M. Sylvie, J. Charlotte, B.-L. Corinne, O. L'Houcine and E.-B. Assou, J. Biomed. Opt., 2014, 19, 1–7 Search PubMed.
- A. G. T. Terwisscha van Scheltinga, G. M. van Dam, W. B. Nagengast, V. Ntziachristos, H. Hollema, J. L. Herek, C. P. Schröder, J. G. W. Kosterink, M. N. Lub-de Hoog and E. G. E. de Vries, J. Nucl. Med., 2011, 52, 1778 CrossRef CAS PubMed.
- S. Qi, Z. Miao, H. Liu, Y. Xu, Y. Feng and Z. Cheng, Bioconjugate Chem., 2012, 23, 1149–1156 CrossRef CAS PubMed.
- J. J. Yim, S. Harmsen, K. Flisikowski, T. Flisikowska, H. Namkoong, M. Garland, N. S. van den Berg, J. G. Vilches-Moure, A. Schnieke and D. Saur, Proc. Natl. Acad. Sci. U.S.A., 2021, 118 Search PubMed.
- S. Siriwibool, N. Kaekratoke, K. Chansaenpak, K. Siwawannapong, P. Panajapo, K. Sagarik, P. Noisa, R.-Y. Lai and A. Kamkaew, Sci. Rep., 2020, 10, 1283 CrossRef CAS PubMed.
- L. Tang, F. Yu, B. Tang, Z. Yang, W. Fan, M. Zhang, Z. Wang, O. Jacobson, Z. Zhou, L. Li, Y. Liu, D. O. Kiesewetter, W. Tang, L. He, Y. Ma, G. Niu, X. Zhang and X. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 27558–27567 CrossRef CAS PubMed.
- J. Weber, L. Bollepalli, A. M. Belenguer, M. D. Antonio, N. De Mitri, J. Joseph, S. Balasubramanian, C. A. Hunter and S. E. Bohndiek, Cancer Res., 2019, 79, 5407–5417 CrossRef CAS PubMed.
- F. Azari, G. Kennedy, E. Bernstein, C. G. Hadjipanayis, A. L. Vahrmeijer, B. L. Smith, E. L. Rosenthal, B. D. Sumer, J. Tian and E. R. Henderson, J. Biomed. Opt., 2021, 26, 050901 Search PubMed.
- J. Zhang, J. Song, X. Liang, Y. Yin, T. Zuo, D. Chen and Q. Shen, Nanomedicine, 2019, 14, 447–464 CrossRef CAS PubMed.
- Y. I. Jeong, B. Cha, H. L. Lee, Y. H. Song, Y. H. Jung, T. W. Kwak, C. Choi, G. W. Jeong, J. W. Nah and D. H. Kang, Int. J. Pharm., 2017, 532, 194–203 CrossRef CAS PubMed.
- C. Xu, P. Wang, J. Zhang, H. Tian, K. Park and X. Chen, Small, 2015, 11, 4321–4333 CrossRef CAS PubMed.
- E. Joachim, I.-D. Kim, Y. Jin, K. Kim, J.-K. Lee and H. Choi, Drug Deliv. Transl. Res., 2014, 4, 395–399 CrossRef CAS PubMed.
- I. D. Kim, E. Sawicki, H. K. Lee, E. H. Lee, H. J. Park, P. L. Han, K. K. Kim, H. Choi and J. K. Lee, Nanomed. Nanotechnol. Biol. Med., 2016, 12, 1219–1229 CrossRef CAS PubMed.
- C. Tramontano, B. Miranda, G. Chianese, L. De Stefano, C. Forestiere, M. Pirozzi and I. Rea, Int. J. Mol. Sci., 2021, 22, 10755 CrossRef CAS PubMed.
- K. Soreide, E. Janssen, H. Körner and J. Baak, J. Pathol., 2006, 209, 147–156 CrossRef CAS PubMed.
- X. Li, M. Bottini, L. Zhang, S. Zhang, J. Chen, T. Zhang, L. Liu, N. Rosato, X. Ma, X. Shi, Y. Wu, W. Guo and X.-J. Liang, ACS Nano, 2019, 13, 176–186 CrossRef CAS PubMed.
- S. M. Mahalingam, S. A. Kularatne, C. H. Myers, P. Gagare, M. Norshi, X. Liu, S. Singhal and P. S. Low, J. Med. Chem., 2018, 61, 9637–9646 CrossRef CAS PubMed.
- E. Walker, Y. Liu, I. Kim, M. Biro, S. R. Iyer, H. Ezaldein, J. Scott, M. Merati, R. Mistur, B. Zhou, B. Straight, J. J. Yim, M. Bogyo, M. Mann, D. L. Wilson, J. P. Basilion and D. L. Popkin, Cancer Res., 2020, 80, 2045–2055 CrossRef CAS PubMed.
- L. Wang and C. Li, J. Mater. Chem., 2011, 21, 15862–15871 RSC.
- T. K. Hill, A. Abdulahad, S. S. Kelkar, F. C. Marini, T. E. Long, J. M. Provenzale and A. M. Mohs, Bioconjugate Chem., 2015, 26, 294–303 CrossRef CAS PubMed.
- S. Zucker, J. M. Wieman, R. M. Lysik, D. Wilkie, N. S. Ramamurthy, L. M. Golub and B. Lane, Cancer Res., 1987, 47, 1608–1614 CAS.
- A. K. Verma, K. Sachin, A. Saxena and H. B. Bohidar, Curr. Pharmaceut. Biotechnol., 2005, 6, 121–130 CrossRef CAS PubMed.
- Y. B. Choy, F. Cheng, H. Choi and K. Kim, Macromol. Biosci., 2008, 8, 758–765 CrossRef CAS PubMed.
- E. Joachim, R. Barakat, B. Lew, K. K. Kim, C. Ko and H. Choi, Nanomed. Nanotechnol. Biol. Med., 2020, 29, 102246 CrossRef CAS PubMed.
- G. Kulsharova, M. Lee, F. Cheng, M. Haque, H. Choi, K. Kim, W. O'Brien and L. Liu, IEEE Trans. NanoBioscience, 2013, 12 Search PubMed.
- M. Zhu, G. Nie, H. Meng, T. Xia, A. Nel and Y. Zhao, Acc. Chem. Res., 2013, 46, 622–631 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- J. Lin, H. Zhang, Z. Chen and Y. Zheng, ACS Nano, 2010, 4, 5421–5429 CrossRef CAS PubMed.
- P. R. Leroueil, S. A. Berry, K. Duthie, G. Han, V. M. Rotello, D. Q. McNerny, J. R. Baker Jr, B. G. Orr and M. M. Holl, Nano Lett., 2008, 8, 420–424 CrossRef CAS PubMed.
- J. X. Jiang, J. J. Keating, E. M. Jesus, R. P. Judy, B. Madajewski, O. Venegas, O. T. Okusanya and S. Singhal, Am. J. Nucl. Med. Mol. Imaging, 2015, 5, 390–400 Search PubMed.
- J. D. Predina, A. D. Newton, J. Keating, A. Dunbar, C. Connolly, M. Baldassari, J. Mizelle, L. Xia, C. Deshpande, J. Kucharczuk, P. S. Low and S. Singhal, Ann. Thorac. Surg., 2018, 105, 901–908 CrossRef PubMed.
- A. D. Newton, J. D. Predina, C. J. Corbett, L. G. Frenzel-Sulyok, L. Xia, E. J. Petersson, A. Tsourkas, S. Nie, E. J. Delikatny and S. Singhal, J. Am. Coll. Surg., 2019, 228, 188–197 CrossRef PubMed.
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. W. Chan, Nano Lett., 2009, 9, 1909–1915 CrossRef CAS PubMed.
- W. Yu, R. Liu, Y. Zhou and H. Gao, ACS Cent. Sci., 2020, 6, 100–116 CrossRef CAS PubMed.
- S. S. Cho, R. Salinas and J. Y. K. Lee, Front. Surg., 2019, 6, 11 CrossRef PubMed.
- V. Saxena, M. Sadoqi and J. Shao, Int. J. Pharm., 2006, 308, 200–204 CrossRef CAS PubMed.
- E. I. Altinoğlu, T. J. Russin, J. M. Kaiser, B. M. Barth, P. C. Eklund, M. Kester and J. H. Adair, ACS Nano, 2008, 2, 2075–2084 CrossRef PubMed.
- J. Xu, F. Gattacceca and M. Amiji, Mol. Pharm., 2013, 10, 2031–2044 CrossRef CAS PubMed.
- J. C. Forster, W. M. Harriss-Phillips, M. J. Douglass and E. Bezak, Hypoxia, 2017, 5, 21–32 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00276k |
| This journal is © The Royal Society of Chemistry 2022 |