
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDark peptide discs for the investigation of membrane proteins in supported lipid bilayers: the case of synaptobrevin 2 (VAMP2)†
Alessandra
Luchini‡
 *a,
Frederik Grønbæk
Tidemand
b,
Nicolai Tidemand
Johansen
b,
Federica
Sebastiani
c,
Giacomo
Corucci
de,
Giovanna
Fragneto
de,
Marité
Cárdenas
c and
Lise
Arleth
f
*a,
Frederik Grønbæk
Tidemand
b,
Nicolai Tidemand
Johansen
b,
Federica
Sebastiani
c,
Giacomo
Corucci
de,
Giovanna
Fragneto
de,
Marité
Cárdenas
c and
Lise
Arleth
f
aEuropean Spallation Source – ERIC, Partikelgatan, Lund, 224 84, Sweden
bDepartment of Plant and Environmental Sciences, University of Copenhagen, Thorvaldsensvej 40, 1871 Frederiksberg C, Denmark
cBiofilms Research Center for Biointerfaces and Department of Biomedical Science, Faculty of Health and Society, Malmö University, Per Albin Hanssons Väg 35, 21432 Malmö, Sweden
dInstitut Laue-Langevin, 71 Avenue des Martyrs, BP 156, 38042 Grenoble, France
eUniversité Grenoble Alpes, Ecole Doctorale de Physique, 110 Rue de la Chimie, 38400 Saint-Martin-d'Hères, France
fNiels Bohr Institute, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark
First published on 12th September 2022
Abstract
Supported lipid bilayers (SLBs) are commonly used as model systems mimicking biological membranes. Recently, we reported a new method to produce SLBs with incorporated membrane proteins, which is based on the application of peptide discs [Luchini et al., Analytical Chemistry, 2020, 92, 1081–1088]. Peptide discs are small discoidal particles composed of a lipid core and an outer belt of self-assembled 18A peptides. SLBs including membrane proteins can be formed by depositing the peptide discs on a solid support and subsequently removing the peptide by buffer rinsing. Here, we introduce a new variant of the 18A peptide, named dark peptide (d18A). d18A exhibits UV absorption at 214 nm, whereas the absorption at 280 nm is negligible. This improves sample preparation as it enables a direct quantification of the membrane protein concentration in the peptide discs by measuring UV absorption at 280 nm. We describe the application of the peptide discs prepared with d18A (dark peptide discs) to produce SLBs with a membrane protein, synaptobrevin 2 (VAMP2). The collected data showed the successful formation of SLBs with high surface coverage and incorporation of VAMP2 in a single orientation with the extramembrane domain exposed towards the bulk solvent. Compared to 18A, we found that d18A was more efficiently removed from the SLB. Our data confirmed the structural organisation of VAMP2 as including both α-helical and β-sheet secondary structure. We further verified the orientation of VAMP2 in the SLBs by characterising the binding of VAMP2 with α-synuclein. These results point at the produced SLBs as relevant membrane models for biophysical studies as well as nanostructured biomaterials.
1 Introduction
Biological membranes include a large variety of chemical species, mostly proteins and lipids. Because of their high level of compositional complexity, the investigation of biological membranes with biophysical or physico-chemical methods is very challenging. Therefore, most of the research in this field is based on the application of simpler model systems, mainly including lipids.1–3 In particular, lipid bilayers, produced in solution in form of vesicles or deposited in the proximity of a solid substrate, are among the most used model systems to mimic some of the fundamental features of biological membranes.4,5Recently, our team introduced a method for the preparation and characterisation of supported lipid bilayers (SLBs) including membrane proteins.6,7 The method is based on the application of peptide discs, discoidal lipid particles, which are stabilised in solution by an outer belt of self-assembled 18A peptide molecules.8,9 Initially, peptide discs are deposited on the hydrophilic surface of a solid support, and by rinsing with fresh buffer solution, the 18A peptide is removed, leaving a SLB. We showed that the peptide discs can be used to produce SLBs with variable lipid composition7 and also for incorporating membrane proteins6 on different kinds of solid surfaces. The incorporation of membrane proteins in SLBs is achieved by first producing peptide discs loaded with the membrane protein of interest.8 Because of the small size of a peptide disc, i.e. approx. 10 nm diameter, they can approach the support surface with different orientations. Typically, the orientation enabling energetically favourable interactions between the lipid headgroups and the support surface is selected. Therefore, in case of a membrane protein with an asymmetric structure, i.e. one large extramembrane domain, the orientation of the membrane protein with the extramembrane domain exposed towards the bulk solvent is spontaneously selected. Subsequent removal of the peptide leads to the formation of the SLB with oriented membrane proteins.
Here we introduce a new variant of the 18A peptide, named dark peptide (d18A), and demonstrate its application to producing peptide discs with improved performances for the formation of SLBs and SLBs with incorporated membrane proteins. The d18A sequence differs from 18A by only two residues: a tryptophan is replaced by a glutamine and a tyrosine is replaced by a leucine. These replacements were designed to minimize the UV absorption of the d18A peptide molecules at 280 nm, leaving the absorption at this wavelength available for monitoring the concentration of the membrane protein loaded in the peptide discs. Because of the lack of absorption at 280 nm, we name the peptide discs prepared with the d18A peptide “dark peptide discs”. The formation of SLBs by d18A peptide discs as well as the structure of the SLBs were characterised by different surface sensitive techniques; quartz crystal microbalance with dissipation monitoring (QCM-D), neutron reflectometry (NR), and attenuated total reflectance – Fourier transform infrared spectroscopy (ATR-FTIR). The collected data confirmed the successful formation of SLBs by the dark peptide discs and suggested an improved peptide removal compared to 18A peptide discs.
The dark peptide discs were also used to produce SLBs with a membrane protein incorporated. More specifically, we investigated synaptobrevin 2, also known as VAMP2. VAMP2 is a small membrane protein (approx. 13 kDa) that is highly abundant in the brain and more specifically in the synaptic vesicles.10 Together with syntaxin-1 and SNAP-25, it forms the protein complex known as SNARE, which allows the synaptic vesicle and the neuron plasma membrane to fuse.11,12 This event is crucial for neuronal communication.13,14 The structure of VAMP2 has been studied in different membrane model systems.15–18 In particular in a recent study investigating only the transmembrane domain (TMD) of VAMP2, it was shown that the TMD stabilised in a lipid multilayer could form larger β-sheet structures.18 Here, we used the dark peptide discs to produce SLBs with VAMP2. We monitored the formation of the SLBs with VAMP2 by QCM-D and characterised the orientation of VAMP2 within the SLBs by NR measurements. Finally, we validated the formation of β-sheet structures in the case of the full-length VAMP2 by ATR-FTIR.
In order to further confirm the orientation of VAMP2 in the SLB with the extramembrane domain exposed towards the bulk solvent, we tested the binding of α-synuclein (α-syn) to the SLBs with VAMP2. α-syn is a small soluble protein highly abundant in the brain and well-known for its role in neurodegenerative diseases. α-syn has been reported to bind synaptic vesicles and more specifically the extramembrane domain of VAMP2.19,20 Our results show that α-syn can bind to SLBs containing VAMP2. This interaction is favoured by the presence of VAMP2 and negatively charged lipids in the SLB. Interesting, as for pure lipid membranes, α-syn binding to VAMP2 appeared to strongly depend on the ionic strength of the buffer used for the experiments.
Altogether, we show the preparation of SLBs with oriented VAMP2 molecules and their application for the structural investigation of VAMP2 and its binding to α-syn. We introduce a new variant of the 18A peptide, i.e. d18A, which allows the general production of SLBs with oriented membrane proteins to be used for biophysical experiments as well as for producing functional biomaterials for drug testing and discovery.
2 Experimental section
2.1 Chemicals
1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) (≥99% purity), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS) (≥99% purity), were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and used without further purification. The d18A peptide (DQLKAFLDKVAEKLKEAF, acetylated and amidated at the N-terminal and C-terminal respectively, ≥95% purity) was purchased from GenScript and used without further purification. Heavy water (D2O 99.9% purity), chloroform (≥99.5% purity), ethanol (98% purity), acetone (98% purity), methanol (99.8% purity), were purchased from Sigma-Aldrich.2.2 Protein production
Codon-optimised genes were synthesised and cloned into a pET28 vector by Twist Bioscience. Proteins were produced in E. coli and purified by affinity chromatography. Protein sequences and the details of the production are reported in the ESI.† The sequences are also shown in SI: briefly, VAMP2 was added a C-terminal His-tag and α-synuclein was added an N-terminal His-tag and a cleavable tobacco etch virus (TEV) site. The plasmids were transformed into heat competent E. coli BL21star(DE3) (ThermoFisher) and plated on selective (kanamycin) medium. Starter cultures were prepared by inoculating a single colony into 10 ml TB medium and grown over night at 37 °C. The next day, a 1 L culture was inoculated to an OD(600 nm) of 0.05 and grown at 37 °C until the OD reached 0.6–0.8. Further details about the α-synuclein and VAMP2 production are reported in ESI.†2.3 Dark peptide disc preparation and SLB formation
The dark peptide discs were prepared according to the same protocol previously reported for the 18A peptide discs.6,7 To produce the dark peptide discs loaded with VAMP2, the film containing 1 mg d18A and 1 mg lipids was dissolved with 0.5 ml of purified VAMP2 (20 μM) in 20 mM Tris, 100 mM NaCl, 10 mM CaCl2, 1.1 mM 12:0 lysoPC, pH = 7.5. The rest of the preparation protocol was identical to the one used for the empty dark peptide discs. The collected fractions were pooled (2 ml in total), the absorbance at 280 nm was measured using a Nanodrop 1000 and the sample was stored at −20 °C until use.SLBs were prepared in situ in solid/liquid cells designed for the QCM-D, NR or ATR-FTIR experiments. The dark peptide disc solution (with or without VAMP2) was injected in the solid/liquid cell containing a freshly cleaned solid support (SiO2-coated sensor for QCM-D, monocrystalline silicon for NR and ATR-FTIR). After 2–10 min, 20 ml of fresh buffer solution was flushed through the solid/liquid cell to remove the d18A peptide and form the SLB.
2.4 Sample characterisation
Quartz Crystal Microbalance with Dissipation monitoring (QCM-D) was performed with a Q-Sense E4 instrument (Q-Sense, Biolin Scientific AB, Sweden), using SiO2-coated 5 MHz quartz sensors. Crystals and O-rings were placed in Hellmanex 2% for 10 min, extensively flushed with ethanol and ultrapure water, and then dried under a nitrogen flow. Immediately before use, the crystals were treated with a UV ozone cleaner for 20 min. Before data acquisition, the fundamental frequency and six overtones (3rd, 5th, 7th, 9th, 11th and 13th) were recorded and the system was equilibrated in ultrapure water at 25 °C, until stable baselines were obtained. After further equilibration in buffer (20 mM Tris, 100 mM NaCl, 10 mM CaCl2, pH = 7.4), the samples were introduced in the flow cell at 0.1 ml min−1. During the experiments, real-time shifts in the resonance frequencies (ΔFn) with respect to the calibration value were measured, with n indicating the overtone number. Simultaneously, the energy dissipation factor (ΔD) was recorded for all the overtones. The measured ΔFn is inversely proportional to the adsorbed mass on the sensor.Neutron Reflectometry (NR) experiments were performed on the SURF reflectometer at ISIS neutron source, Chilton, (UK) and on the FIGARO reflectometer at the Institut Laue Langevin (ILL), Grenoble (France). SURF is a horizontal time of flight reflectometer and three incoming angles (θ), typically of 0.35°, 0.65° and 1.5°, were used to cover the q-range 0.01 Å−1 < q < 0.2 Å−1, where 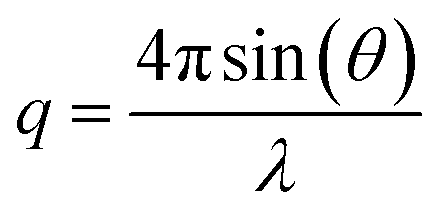 . FIGARO is also a horizontal time of flight reflectometer and two incoming angles, namely 0.8° and 3.2°, were used to collect data in the q-range 0.003 Å−1 < q < 0.25 Å−1. The reflected intensity measured both on SURF and FIGARO was converted to an absolute reflectivity scale (R(q)) by normalisation to the direct beam measured at the same slit settings. Slits were chosen to vary with the incident angle in such a way to provide a constant illumination of the sample (30 × 60 mm on SURF and 35 × 65 mm on FIGARO). In the case of the data collected on FIGARO, the background was measured on the left and right side of the reflectivity signal and subtracted from the measured reflected intensity. At both reflectometers, the neutron beam is focused on a solid/liquid cell containing the solid support and the sample. Monocrystalline silicon supports (80 mm × 50 mm × 15 mm) were cleaned with sequential sonication (∼20 min) in chloroform, ethanol, acetone followed by either 30 min treatment with UV-ozone lamp or 3 min with Plasma Cleaner. The freshly cleaned silicon supports were assembled in the solid/liquid cells. 2 ml dark peptide disc (with or without VAMP2) solution were introduced in the solid/liquid cells through manual injection with a syringe. 20 ml of buffer were injected with a HPLC pump at 1 ml min−1 flow rate. The temperature of the solid/liquid cells was kept at 25 °C during data collection with a thermalised water bath. In order to achieve different contrasts between the samples and the solvents, NR measurements were performed with three buffers (20 mM Tris, 100 mM NaCl, 10 mM CaCl2, pH = 7.4) prepared with 100% D2O (named D2O-buffer, ρ = 6.35 × 10−6 Å−2), 38% D2O 62%H2O (named SMW-buffer, ρ = 2.07 × 10−6 Å−2) and 100% H2O (named H2O-buffer, ρ = −0.56 × 10−6 Å−2). Data collected in the different contrasts were simultaneously analysed with the same structural model. NR data were analysed as detailed in ESI.†
. FIGARO is also a horizontal time of flight reflectometer and two incoming angles, namely 0.8° and 3.2°, were used to collect data in the q-range 0.003 Å−1 < q < 0.25 Å−1. The reflected intensity measured both on SURF and FIGARO was converted to an absolute reflectivity scale (R(q)) by normalisation to the direct beam measured at the same slit settings. Slits were chosen to vary with the incident angle in such a way to provide a constant illumination of the sample (30 × 60 mm on SURF and 35 × 65 mm on FIGARO). In the case of the data collected on FIGARO, the background was measured on the left and right side of the reflectivity signal and subtracted from the measured reflected intensity. At both reflectometers, the neutron beam is focused on a solid/liquid cell containing the solid support and the sample. Monocrystalline silicon supports (80 mm × 50 mm × 15 mm) were cleaned with sequential sonication (∼20 min) in chloroform, ethanol, acetone followed by either 30 min treatment with UV-ozone lamp or 3 min with Plasma Cleaner. The freshly cleaned silicon supports were assembled in the solid/liquid cells. 2 ml dark peptide disc (with or without VAMP2) solution were introduced in the solid/liquid cells through manual injection with a syringe. 20 ml of buffer were injected with a HPLC pump at 1 ml min−1 flow rate. The temperature of the solid/liquid cells was kept at 25 °C during data collection with a thermalised water bath. In order to achieve different contrasts between the samples and the solvents, NR measurements were performed with three buffers (20 mM Tris, 100 mM NaCl, 10 mM CaCl2, pH = 7.4) prepared with 100% D2O (named D2O-buffer, ρ = 6.35 × 10−6 Å−2), 38% D2O 62%H2O (named SMW-buffer, ρ = 2.07 × 10−6 Å−2) and 100% H2O (named H2O-buffer, ρ = −0.56 × 10−6 Å−2). Data collected in the different contrasts were simultaneously analysed with the same structural model. NR data were analysed as detailed in ESI.†
Attenuated total reflectance-FTIR (ATR-FTIR) Measurements were performed on a Nicolet Nexus 670/870 (Thermo Fisher Scientific) with a mercury-cadmium-telluride detector. A solid/liquid cell from Specac attenuated total reflectance accessory was mounted on the instrument. The trapezoidal monocrystalline silicon support (50 mm × 10 mm × 10 nm) was cleaned with sonication (∼20 min) in ethanol, and subsequently water, followed by ∼30 min treatment with UV-ozone lamp. The freshly cleaned support was sealed with D2O in the solid/liquid cell and purged overnight, together with the instrument, from water vapour and CO2 with a air purge system. The infrared beam is focused on the short side of the silicon support (10 mm) at a fixed angle such that the radiation entering the support is totally reflected multiple times at the solid/liquid interface. The evanescent wave associated with the reflected beam has a penetration depth of ∼1 μm.21 The adsorbance (IAbs) is reported as a function of the radiation wavenumber (cm−1). ATR-FTIR spectra were collected at 25 °C in the wavenumber range 3100–1600 cm−1. To enhance the absorption signal of the hydrogenous lipids and d18A peptide, both the peptide disc solution and the buffer (20 mM Tris-DCl, 100 mM NaCl, 10 mM CaCl2, pH = 7.4 or 0.5 mM Tris-DCl, pH = 7.4) were prepared with D2O. The samples were manually injected with a syringe in the solid/liquid cell, while a syringe pump was used to rinse the cell with fresh buffer solution at 1 ml min−1 flow rate. Each of the time-resolved spectrum is the average of 16 consecutively collected spectra (9.68 seconds total acquisition time). Spectra collected after injection of α-syn are the average of 128 consecutively spectra. A single Gaussian function or the sum of two Gaussian functions was used to estimate the position of the absorbance bands in the collected spectra.
3 Results and discussion
3.1 Dark peptide discs as precursor of pure lipid SLBs
As reference sample, we prepared POPC loaded peptide discs using our new d18A (DQLKAFLDKVAEKLKEA). Compared to 18A (DWLKAFYDKVAEKLKEAF), a Trp residue is replaced by a more polar amino acid, Gln, while a Tyr residue is replaced by a more hydrophobic residue, Leu. These changes were made based on a helical wheel representation of 18A (Fig. 1a), which suggests that the Trp is located at the interface between the hydrophobic and polar regions of the peptide, whereas the Tyr is located in the middle of the hydrophobic region. As mentioned, these changes were made in order to eliminate UV absorption at 280 nm of the peptide. We note that d18A still contains two Phe residues, which absorb UV at 280 nm. However, the extinction coefficient of Phe (200 M−1 cm−1) is often negligible, especially, when membrane proteins containing one or more Trp (5500 M−1 cm−1) residues are incorporated in the peptide discs.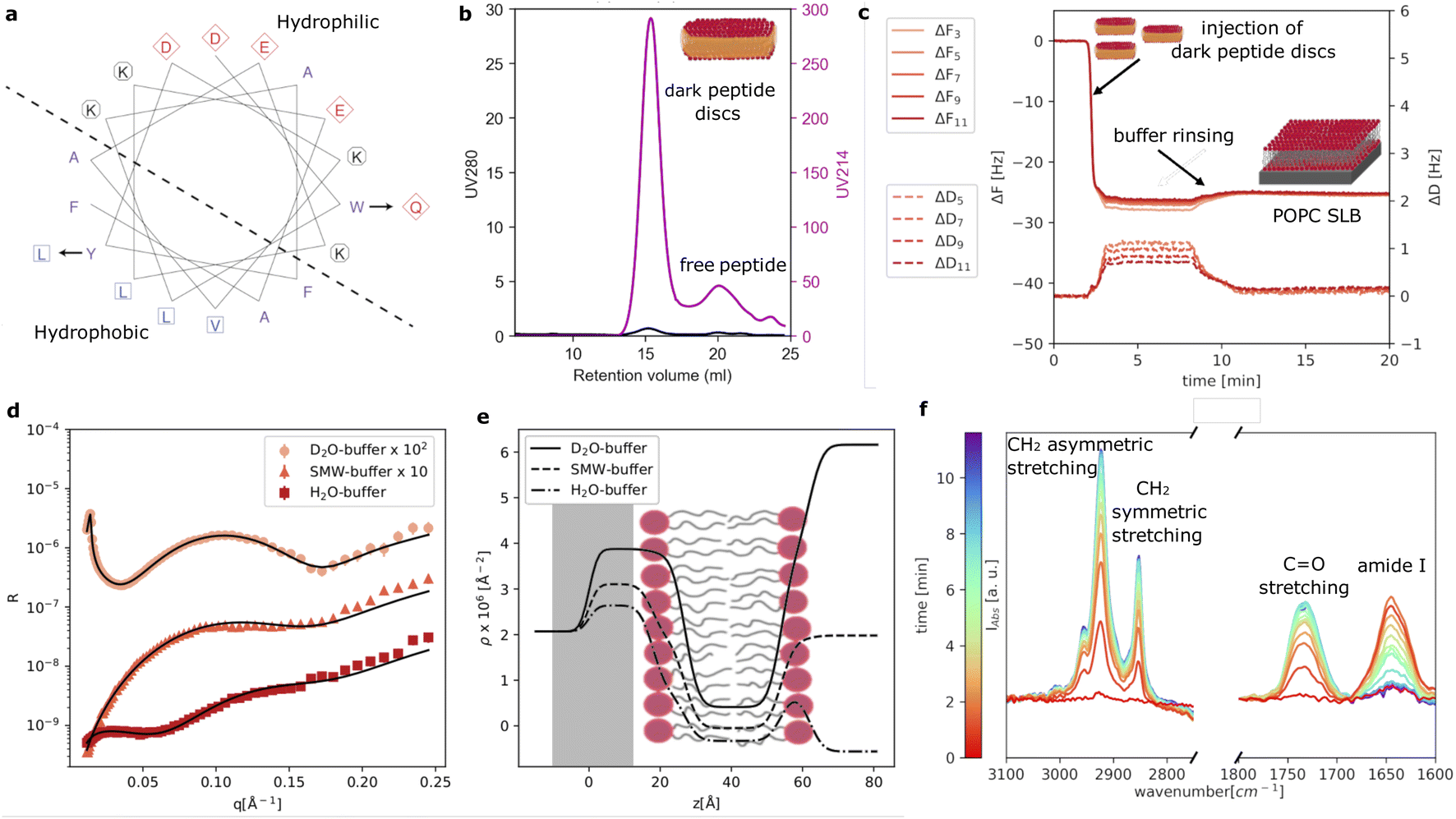 | ||
| Fig. 1 Helical wheel representation (calculated with emboss pep wheel available at https://emboss.bioinformatics.nlcgi-binembosspepwheel?_pref_hide_optional1̄) of 18A showing the replacement of residues designed to obtain d18A (a). Size exclusion chromatogram showing UV absorption at 214 and 280 nm and collected during purification of the dark peptide discs (b). QCM-D data showing the formation of the SLB by deposition of the dark peptide discs and peptide removal (c). Experimental NR data together with the fitting curves (d). Data and fits are offset for clarity, as reported in the legend. Scattering length density profiles calculated from NR data analysis (e). Time resolved ATR-FTIR spectra showing the formation of the SLB by the dark peptide discs (f). | ||
Fig. 1b shows the SEC purification of the dark peptide discs formed upon the spontaneous self-assembly of the d18A peptide with the POPC lipids in an aqueous solution. Notably, the absorption at 280 nm was low, as expected, whereas the absorption at 214 nm, used for monitoring peptide bonds, was significant. This shows the successful elimination of UV280 nm absorption as desired. The chromatogram exhibits a well-defined main peak containing the dark peptide discs, followed by a minor peak, likely containing free peptide not associated with lipids. A 2 ml fraction was collected from the main peak and used for further experiments. We note that contrary to the use of 18A, we obtain these two peaks for all reconstitutions using d18A and lipids, indicating that the two mutations slightly alter the lipid affinity of the peptide as expected given the replacement of the lipophilic Trp by the more polar Gln. However, this does not constitute a major drawback for d18A since the majority of the peptides participate in the peptide discs with the fractions clearly resolved from each other.
QCM-D data were collected to evaluate the formation of SLBs using the dark peptide discs (Fig. 1c). The fundamental frequency and higher harmonics of the quartz sensor were measured at the beginning of the experiment and corresponded to a frequency shift (ΔF) of zero. Upon injection of the dark peptide discs, ΔF rapidly decreased and a parallel increase of the dissipation (ΔD) was observed. This suggests that the dark peptide discs are adsorbed on the sensor surface. After approx 7 min, the sensor was rinsed with buffer solution, which resulted in a small increase of ΔF and a decrease in ΔD. This observation indicates that the d18A peptide molecules are removed from the sensor surface during the rinsing step, which could lead to a rearrangement of the initially adsorbed lipid bilayer patches and the formation of a SLB. The latter is further confirmed by the stable ΔF value of −25 Hz after rinsing, which is consistent to a POPC SLB formed by, for example, vesicle fusion.22
Fig. 1d shows the experimental NR data that was collected to characterise the structure of the SLB. The model used to analyse the data is based on the representation of the sample structure as a stack of layers with different composition. More specifically 3 layers were used to describe the SLB: (i) the inner lipid headgroup layer; (ii) the lipid acyl chains; (iii) the outer lipid headgroup. The inner and outer lipid headgroup layers were constrained to have the same structural parameters (further details can be found in ESI†). As also reported by the scattering length density profile (Fig. 1e), NR confirmed the formation of a POPC bilayer with structural parameters in agreement with previously reported results for deposition with 18A and by vesicle fusion.6,7 From the solvent volume fraction among the lipid acyl chain, a surface coverage of 90% was estimated, which is also consistent with previous depositions using 18A (Table 1) and generally indicating the formation of good quality SLB for structural characterisation.
| Parameters | POPC | POPC/VAMP2 | |||
|---|---|---|---|---|---|
| Headgroups | Acyl chains | Headgroups | Acyl chains + VAMP2 TMD | VAMP2 EMD | |
| t [Å] | 9 ± 1 | 27 ± 3 | 7 ± 3 | 29 ± 2 | 30 ± 5 |
| ρ × 10−6 [Å−2] | 1.87 ± 0.03 | − 0.30 ± 0.01 | 1.9 ± 0.1 | − 0.25 ± 0.02 | 3.06 (D2O buffer) |
| 2.28 (SMW buffer) | |||||
| 1.81 (H2O buffer) | |||||
| Φ | 0.45 ± 0.04 | 0.1 ± 0.2 | 0.41 ± 0.05 | 0.03 ± 0.02 | 0.94 ± 0.03 |
The formation of the POPC bilayer was also monitored by time-resolved ATR-FTIR spectra (Fig. 1f). Spectra were collected during the injection of the dark peptide discs and a following wash step with buffer solution. A few minutes after starting data collection, the spectra included five bands in the wavenumber-region 3100–2750 cm−1:23,24 (i) the choline CH3 asymmetric stretching (∼3007 cm−1), (ii) the acyl chain CH3 asymmetric (∼2956), (iii) the CH2 asymmetric (2925 cm−1), (iv) the acyl chain CH3 symmetric ∼2872 cm−1 and (v) the CH2 symmetric stretching (2855 cm−1). In the wavenumber-range 1800–1600 cm−1, two bands were observed: the POPC C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1733 cm−1 and the amide I band at 1644 cm−1 associated with CO stretching in the d18A peptide backbone.24 The position of the amide I band suggests a mixture of random coil and α-helical conformations,25 in agreement with previous results collected for peptide discs produced with the 18A peptide.7 The amide I band rapidly increased in intensity during the deposition of the dark peptide discs and decreased to the baseline value during buffer rinsing. This suggests a rapid removal of the d18A peptide from the lipids upon rinsing with buffer. The disappearance of the amide I band further suggests that the d18A peptide was more effectively removed compared to the 18A peptide, for which a low signal remained after buffer rinsing that indicates the presence of residual 18A molecules in the SLB.7 This is most likely favoured by the replacement in the d18A sequence of the tryptophan with a more polar residue, i.e. glutamine.
O stretching at 1733 cm−1 and the amide I band at 1644 cm−1 associated with CO stretching in the d18A peptide backbone.24 The position of the amide I band suggests a mixture of random coil and α-helical conformations,25 in agreement with previous results collected for peptide discs produced with the 18A peptide.7 The amide I band rapidly increased in intensity during the deposition of the dark peptide discs and decreased to the baseline value during buffer rinsing. This suggests a rapid removal of the d18A peptide from the lipids upon rinsing with buffer. The disappearance of the amide I band further suggests that the d18A peptide was more effectively removed compared to the 18A peptide, for which a low signal remained after buffer rinsing that indicates the presence of residual 18A molecules in the SLB.7 This is most likely favoured by the replacement in the d18A sequence of the tryptophan with a more polar residue, i.e. glutamine.
QCM-D, NR and ATR-FTIR data were also collected for the dark peptide discs prepared with a mixture of POPC and POPS (70:30 mol mol−1) (ESI,†Fig. 1). The obtained results were similar to those for the pure POPC, which confirms the compatibility of the dark peptide discs with the production of SLBs containing anionic phospholipids.
Altogether, Fig. 1 demonstrates that the sequence modification introduced in the d18A peptide compared to 18A did not affect the formation of SLB, as confirmed by QCM-D and NR. However, the ATR-FTIR data showed that compared to the results previously reported for 18A, d18A can be more efficiently removed from the lipids during the formation of the SLB.
3.2 SLBs with oriented VAMP2
To further validate the application of the dark peptide discs to form complex biological samples, we reconstituted a small membrane protein, VAMP2, with POPC lipids and the d18A peptide. VAMP2 consists of a single transmembrane helix (transmembrane domain, TMD) along with an extramembrane domain (EMD) of around 90 residues. After reconstitution, the VAMP2 loaded dark peptide discs were purified by SEC, which yielded a single, almost symmetric peak (Fig. 2a). This time, the absorption at 280 nm was significantly higher compared to the unloaded peptide discs (Fig. 1b) owing to the presence of VAMP2. This highlights the straight-forward application of the dark peptide to visualize successful reconstitution of membrane proteins, and furthermore, it allows for an accurate determination of membrane protein concentration from UV absorption measurements. Notably, the chromatographic elution of the VAMP2 loaded peptide discs only showed a single main peak, and no free peptide. This is likely due to the presence of VAMP2 and/or the detergent 12:0 lysoPC used for purifying VAMP2, which are both capable of interacting with lipids and peptides. On the SEC, 12:0 lysoPC is diluted well below its CMC, ensuring that little to none of this detergent is carried over in the peptide disc fraction. A 2 ml sample was collected from the main peak, and the concentration of VAMP2 was determined to be 4 μM from a UV280 nm measurement. This sample was used for further experiments. | ||
| Fig. 2 Size exclusion chromatogram of the purification of the dark peptide discs loaded with VAMP2 (a). The magenta curve refers to the UV absorption at 214 nm, whereas the black curve refers to the UV absorption at 280 nm. QCM-D data showing the formation of the SLB with incorporated VAMP2 molecules (b). NR experimental data together with the fitting curves (c). Data and fits are offset for clarity. Scattering length density profiles calculated from NR data analysis (d). | ||
As also observed for QCM-D data on pure POPC, the ΔF rapidly decreased and the ΔD increased upon injection of the dark peptide discs with VAMP2 (Fig. 2b). Such signal variation indicates the adsorption of the dark peptide discs with VAMP2 on the sensor surface. Upon buffer rinsing, the ΔF slightly increased due to the removal of the d18A peptide and subsequent rearrangement of the adsorbed lipid and VAMP2 molecules. The final ΔF of −32 Hz is considerably smaller than the −25 Hz observed for pure POPC. This observation suggests the formation of a layer at the sensor surface heavier than a pure lipid bilayer, which is expected for a SLB with incorporated membrane proteins. The dissipation is higher than 1 ppm which, together with some spreading of overtones, suggests a more heterogeneous adsorbed layer due to the VAMP2 protrusion outside of the lipid bilayer. NR data was collected on the SLB with VAMP2 incorporated (Fig. 2c). The model used to analyse the data was similar to the one used for the pure lipid SLB with the addition of an outer layer representing the extramembrane domain of VAMP2. More specifically, we used 4 layers: (i) lipid headgroup layer partially incorporating VAMP2; (ii) lipid acyl chains including VAMP2 transmembrane domain (TMD); (iii) outer lipid headgroups partially incorporating VAMP2 and (iv) VAMP2 extramembrane domain (EMD). The VAMP2 volume fraction in layer (iii) and (iv) were constrained to reflect the same number of protein molecules (further details are reported in ESI†). The parameters obtained from optimisation of the model to the experimental data are reported in Table 1. The overall surface coverage of the SLB with VAMP2 resulted to be 97%. Unfortunately, the volume fraction of the VAMP2 extramembrane domain was too low (<10% v/v) for a reliable determination of the location of this protein domain with respect to the membrane surface. Nevertheless, the presence of VAMP2 molecules in the bilayer was detected by comparing the scattering length density value associated to the acyl chain region in the samples prepared with and without VAMP2 (Table 1). The value obtained for the POPC/VAMP2 SLB is consistent with a protein/lipid ratio of about 170 lipids per VAMP2 molecule. NR also provided indirect information on the overall orientation of the VAMP2 molecules. Indeed, if all or some of the VAMP2 molecules were oriented with the extramembrane domain towards the support surface, a low-density layer of protein would be expected between the bilayer and the support surface. By estimating a thickness of at least 10 Å, which would correspond to the extramembrane domain of VAMP2 partially penetrating the lipid headgroups, the presence of this protein layer between the support surface and the lipid bilayer would produce a considerably different data trend than the one observed (see ESI†). Therefore, even if NR cannot directly detect the extramembrane domain of VAMP2, the collected data can still suggests that the VAMP2 molecules incorporated in the bilayer do not expose such domain towards the solid substrate, and that this latter is instead exposed towards the bulk solvent as depicted in Fig. 2d.
To further investigate VAMP2 in the POPC bilayer, ATR-FTIR data was collected during the formation of the SLB (Fig. 3a). Similar results were collected in case of the POPC/POPS mixture (Fig. 3b and ESI†). Also in case of VAMP2-loaded dark peptide discs, the absorption was a fast process, and already after a few tens of seconds, the characteristic signals of the lipids and the proteins clearly appeared in the spectra. Compared to the data reported in Fig. 1e, the amide I band is much more intense, which strongly indicates the presence of VAMP2 among the deposited lipids and d18A peptide. The amide I band intensity decreased during buffer rinsing, but importantly, instead of going back the baseline value, it remained the most intense signal in the spectra. Such observation suggests that the buffer rinsing process probably removed the d18A molecules from the support surface, while leaving VAMP2 in the bilayer. The position of the bands previously ascribed to the lipid molecules is comparable to Fig. 1e.
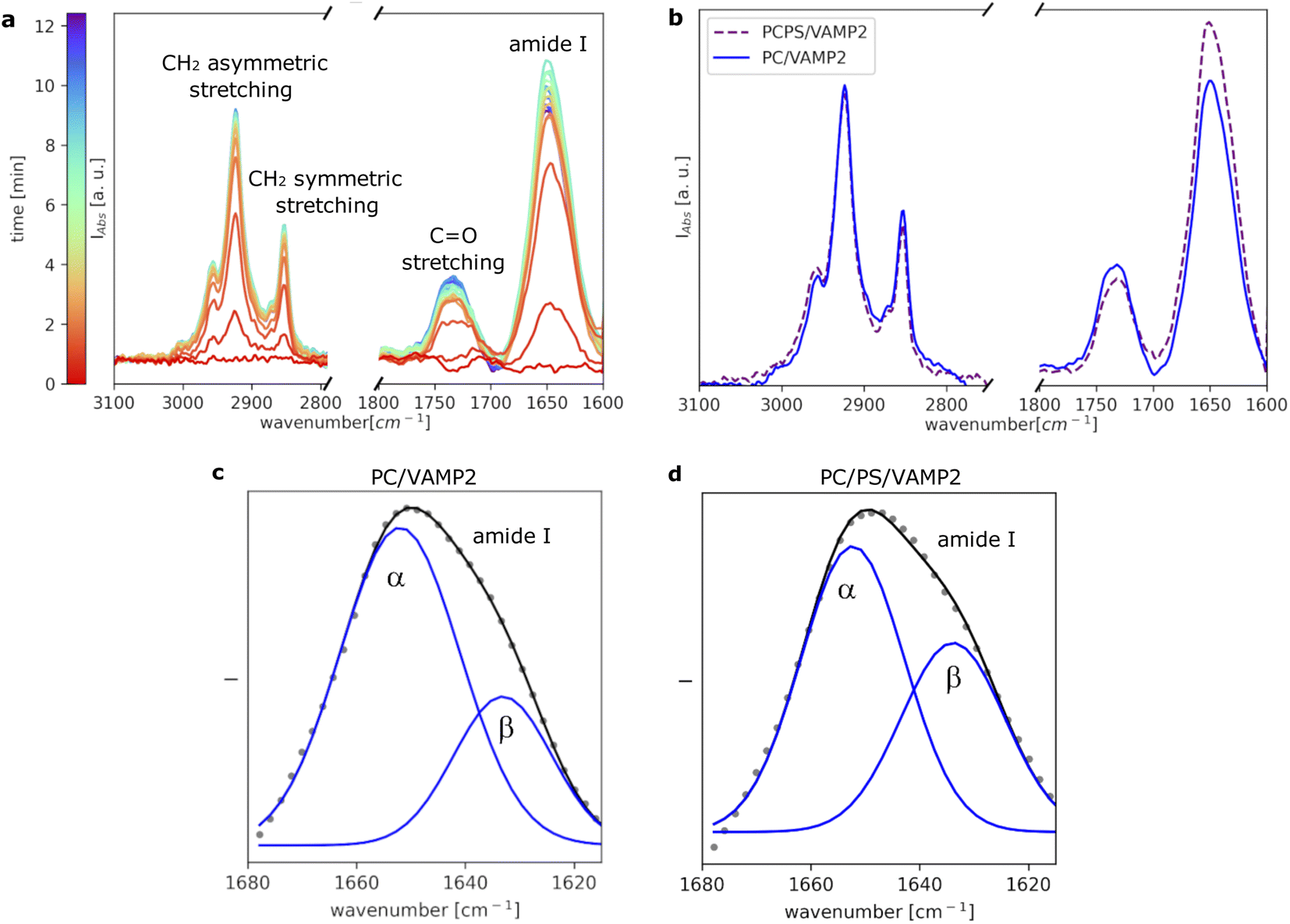 | ||
| Fig. 3 Time-resolved ATR-FTIR data collected during the formation of the SLB with VAMP2 (a). Comparison between the final spectra collected for VAMP2 in POPC and POPC/POPS (b). Analysis of the amide I band for VAMP2 in POPC (c) and VAMP2 in POPC/POPS (d). | ||
A closer look at the amide I band shows an apparent asymmetric shape of this peak (Fig. 3c and d). The amide I band was analysed as described in the Materials and Method section and previously reported.26 As a result, the band was deconvoluted into two bands centered at 1652 cm−1 and 1632 cm−1 (Fig. 3c), which suggests the presence of both α-helical and β-sheet secondary structures.24 Previous studies on the TMD of proteins belonging to the VAMP family have suggested the presence of these two secondary structure motifs.18,27,28 The TMD of VAMP2 is expected to be a single-pass α-helix, as indicated by the structural model of VAMP2 stabilised in micelles.16 However, lateral diffusion in the SLB can bring VAMP2 molecules close together and favour the formation of the β-sheet structure involving TMDs belonging to different VAMP2 molecules. We hypothesise that such lateral diffusion could take place in the SLB therefore leading to the formation of the β-sheet structure indicated by the collected ATR-FTIR data. Such a phenomenon would not be observed in micelles, where only one VAMP2 is incorporated in each particle, but it is of high biological relevance given the relatively high concentration of VAMP2 in the synaptic vesicles.10 The amount of β-sheet structure versus α-helix has also been reported to depend on the protein-lipid ratio and the presence of PS lipids.18 In agreement with these results, we observed that the contribution of the β-sheet structure is larger in the case of VAMP2 in the POPC/POPS (45% ± 3%) compared to pure POPC (30% ± 2%).
3.3 Interaction with α-synuclein
Controlled membrane protein orientation is one of the major advantages of peptide disc mediated SLB formation.6 For VAMP2, the bulky extramembrane domain contains the SNARE motif, which is available for interaction with proteins in the bulk solvent. In order to further confirm this orientation of VAMP2 in our SLBs, we tested the binding of α-synuclein (α-syn) to the SLBs with VAMP2. Indeed, α-synuclein has been reported to bind the extramembrane domain of VAMP2,19,20 and therefore promote the fusion between synaptic vesicles and the neuron membrane together with the SNARE complex.20,29 α-synuclein has also been found to bind negatively charged membranes, e.g. phosphatidylserine lipids, but to a less extent to neutral membranes, such as those composed of phosphatydilcholine lipids.30,31A preliminary characterisation was carried out with QCM-D. The collected data showed that in the presence of 100 mM NaCl, no α-syn binding to any of the investigated bilayers (both pure lipids and with VAMP2) could be detected (SI). Such observation agrees with previous studies on α-syn binding to lipid membranes, where buffer solutions without additional salt were typically used for biophysical and physico-chemical experiments.31–34 Thus, unlike our other experiments reported here, we used a very dilute buffer without salt (i.e. 0.5 mM Tris, pH = 7.5) to study α-syn binding. QCM-D data were collected also in the case of this buffer without additional salt (ESI†). As a result the frequency shift change upon injection of α-syn was very small and the characteristic signal of the SLB with VAMP2 was restored after rinsing with fresh buffer solution. On the other hand, as discussed below, ATR-FTIR resulted to be more sensitive to α-syn binding to the SLBs, and therefore represented a more suited experimental method to investigate the interaction between α-syn and VAMP2.
ATR-FTIR data were collected to evaluate the binding of α-syn to both pure SLBs (i.e. POPC and POPC/POPS) and SLBs with VAMP2 inserted (i.e. PC/VAMP2 and PCPS/VAMP2). Data were collected before and after injection of the α-syn solution (10 μM) and after buffer rinsing. We observed that rinsing with the buffer removed some of the initially adsorbed α-syn molecules while leaving a detectable fraction still attached to the bilayer (ESI†).
Fig. 4a shows a comparison of the amide I bands collected for all the investigated samples after subtraction of the signal before the addition of α-syn. For the POPC SLB, we observed only a weak amide I band, whereas for the POPC/POPS SLB, the amide I band substantially increased, suggesting a higher affinity of α-syn for the negatively charged membrane, as expected. Incorporation of VAMP2 in the POPC SLB also promoted the binding of α-syn, as indicated again by the increased intensity of the amide I band compared to pure POPC. This observation suggests that the VAMP2 extramembrane domain was exposed on the bilayer surface and available for interacting with α-syn. The largest adsorption of α-syn was observed in case of the POPC/POPS SLB with VAMP2, which is likely the result of synergy between the negatively charged lipids, i.e. PS, and VAMP2 in the bilayer.
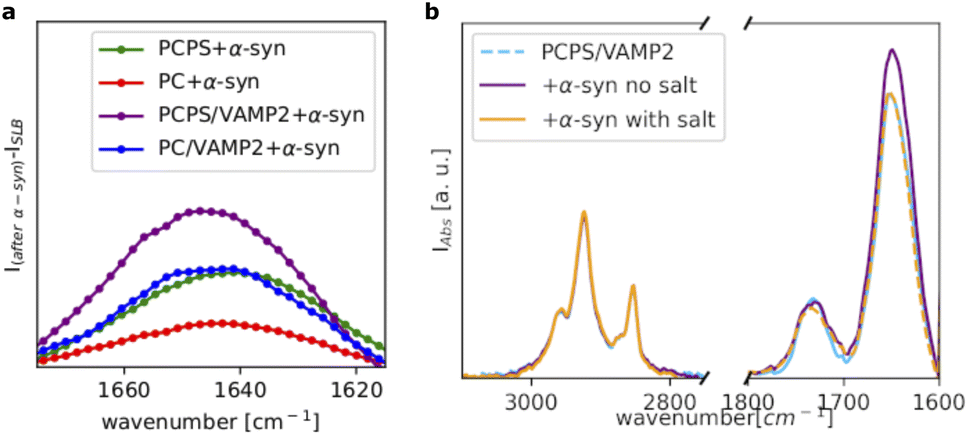 | ||
| Fig. 4 Amide I band corresponding to the α-syn contribution to the ATR-FTIR spectra collected for the different samples, as reported in the legend (a). Comparison of the ATR-FTIR spectra collected for POPC/POPS/VAMP2 before and after the addition of α-syn and after rinsing with the buffer with salt, i.e. 20 mM Tris, 100 mM NaCl, pH = 7.5 (b). The curve corresponding to the POPC/POPS/VAMP2 (light blue) and the curve for the POPC/POPS/VAMP2 after the addition of α-syn and upon rising with the buffer with salt (orange) are difficult to distinguish as they exhibit a very similar profile. | ||
Interestingly, we observed that even in the case of the SLB with VAMP2, the binding of α-syn was extremely sensitive the ionic strength of the buffer. Fig. 4b shows the spectra collected for the POPC/POPS SLB with VAMP2 before and after the addition of α-syn and after rinsing with the buffer containing salt (i.e. 20 mM Tris, 100 mM NaCl, pH = 7.5). Upon injection of this buffer, we observed a decrease in the amide I band intensity, which ultimately became almost identical to the one in the spectrum collected before the injection of α-syn. Therefore, rinsing with the buffer with salt, promoted the complete release of α-syn from the bilayers. This further supports the choice of the buffer with low salt concentration for the characterisation of the α-syn membrane binding.
4 Conclusions
Incorporation of membrane proteins with controlled orientation in supported lipid bilayers (SLBs) is an important aspect for the production of biologically relevant membrane model systems. Here, we showed the application of a modified 18A peptide, d18A, for the production of peptide discs, named dark peptide discs, with improved optical properties and more efficient peptide dissociation from the lipids.The d18A sequence differs from 18A as the Trp and Tyr residues are replaced by Gln and Leu, respectively, in order to minimise the light absorption at 280 nm. From time-resolved ATR-FTIR spectra, we discovered that removal of the d18A peptide during SLB formation was more efficient than for the 18A peptide. While not intentional, this is an additional advantage of having replaced an aromatic residue with a more polar one, which is expected to improve the solubility of the d18A peptide in water. In addition, we demonstrated by QCM-D and NR that the dark peptide discs can be used as precursors of SLBs with different phospholipid composition and also with a membrane protein incorporated, i.e. VAMP2.
Despite the small size of VAMP2 (13 kDa), we were successful in incorporating this membrane protein with a single orientation in the SLB composed of either POPC or POPC/POPS. These SLBs with VAMP2 were used to investigate the structural organisation of VAMP2. We found the VAMP2 molecules in the SLBs to exhibit both α-helical and β-sheet secondary structure. Similar structural motifs have previously been suggested from data collected on only the transmembrane domain of VAMP2.18,27,28 Here, we confirm these results in a single bilayer and with full-length VAMP2.
By measuring the binding of α-syn to pure lipid SLBs and SLBs with VAMP2, we further confirmed the orientation of VAMP2 in the SLBs with its extramembrane domain exposed towards the bulk solvent. Furthermore, our results support previous studies that found the C-terminal domain of α-syn can bind the extramembrane domain of VAMP2.19,29 Indeed, a larger α-syn adsorption was detected on the SLBs with VAMP2 compared to the pure lipid SLBs.
Altogether, the dark peptide disc is a promising new tool for generating complex biological samples. While we have shown its application for generating SLBs compatible with a range of surface sensitive techniques, it is likely well-suited for a wide range of other biophysical techniques as well. In particular, the removal of the aromatic residues Trp and Tyr allows for fluorescence-based techniques to study membrane proteins containing those residues.
Author contributions
AL: conceptualisation, formal analysis, investigation, resources, writing–original draft; FGT: conceptualisation, resources, investigation, writing–review & editing; NTJ: resources, writing–review & editing; FS: investigation, writing–review & editing; GC: investigation, writing–review & editing; GF: investigation, writing–review & editing; MC: writing–review & editing; LA: funding acquisition, writing–review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the grant from the Lundbeck foundation “BRAINSTRUC” project and the Villum Foundation (grant 00035955). The authors also thank the ISIS neutron source (10.5286/ISIS.E.RB1910248) and the Institut Laue-Langevin (ILL) (10.5291/ILL-DATA.8-02-929, 10.5291/ILL-DATA.EASY-619) for the allocation of beamtime and provision of support facilities for sample preparation (PSCM labs at the ILL). Authors are grateful to Dr Mario Campana and Dr Samantha Micciulla for technical support during the NR experiments at ISIS and ILL, respectively. Marité Cárdenas thanks the Swedish Research Council for financial support.Notes and references
- W. F. Zeno, K. J. Day, V. D. Gordon and J. C. Stachowiak, Annu. Rev. Biophys., 2020, 49, 19–39 CrossRef CAS.
- Y.-H. M. Chan and S. G. Boxer, Curr. Opin. Chem. Biol., 2007, 11, 581–587 CrossRef CAS PubMed.
- G. Fragneto, R. Delhom, L. Joly and E. Scoppola, Curr. Opin. Colloid Interface Sci., 2018, 38, 108–121 CrossRef CAS.
- V. Kiessling, S.-T. Yang and L. K. Tamm, Lipid Domains, Academic Press, 2015, vol. 75, pp. 1–23 Search PubMed.
- A. Luchini and G. Vitiello, Biomimetics, 2021, 6(1), 3 CrossRef CAS PubMed.
- A. Luchini, F. G. Tidemand, N. T. Johansen, M. Campana, J. Sotres, M. Ploug, M. Cárdenas and L. Arleth, Anal. Chem., 2020, 92, 1081–1088 CrossRef CAS PubMed.
- A. Luchini, F. Sebastiani, F. G. Tidemand, K. C. Batchu, M. Campana, G. Fragneto, M. Cárdenas and L. Arleth, J. Colloid Interface Sci., 2021, 585, 376–385 CrossRef CAS.
- S. R. Midtgaard, M. C. Pedersen, J. J. K. Kirkensgaard, K. K. Sørensen, K. Mortensen, K. J. Jensen and L. Arleth, Soft Matter, 2014, 10, 738–752 RSC.
- A. N. Larsen, K. K. Sørensen, N. T. Johansen, A. Martel, J. J. K. Kirkensgaard, K. J. Jensen, L. Arleth and S. R. Midtgaard, Soft Matter, 2016, 12, 5937–5949 RSC.
- S. Hussain and S. Davanger, PLoS One, 2015, 10(10), e0140868 CrossRef PubMed.
- C. Wang, J. Tu, S. Zhang, B. Cai, Z. Liu, S. Hou, Q. Zhong, X. Hu, W. Liu, G. Li, Z. Liu, L. He, J. Diao, Z.-J. Zhu, D. Li and C. Liu, Nat. Commun., 2020, 11, 1531 CrossRef CAS PubMed.
- T.-Y. Yoon and M. Munson, Curr. Biol., 2018, 28, PR397–R401 CrossRef PubMed.
- C. C. Winkle and S. L. Gupton, International Review of Cell and Molecular Biology, Academic Press, 2016, vol. 322, pp. 247–280 Search PubMed.
- Y. Yang, J. Y. Shin, J.-M. Oh, C. H. Jung, Y. Hwang, S. Kim, J.-S. Kim, K.-J. Yoon, J.-Y. Ryu, J. Shin, J. S. Hwang, T.-Y. Yoon, Y.-K. Shin and D.-H. Kweon, Proc. Natl. Acad. Sci., 2010, 107, 22145–22150 CrossRef CAS PubMed.
- N.-A. Lakomek, H. Yavuz, R. Jahn and Á. Pérez-Lara, Proc. Natl. Acad. Sci., 2019, 116, 8699–8708 CrossRef CAS PubMed.
- J. F. Ellena, B. Liang, M. Wiktor, A. Stein, D. S. Cafiso, R. Jahn and L. K. Tamm, Proc. Natl. Acad. Sci., 2009, 106, 20306–20311 CrossRef CAS PubMed.
- B. Liang, D. Dawidowski, J. F. Ellena, L. K. Tamm and D. S. Cafiso, Biochemistry, 2014, 53, 1485–1494 CrossRef CAS PubMed.
- Z. Fezoua-Boubegtiten, B. Hastoy, P. Scotti, A. Milochau, K. Bathany, B. Desbat, S. Castano, R. Oda and J. Lang, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 670–676 CrossRef CAS PubMed.
- J. Lautenschläger, A. D. Stephens, G. Fusco, F. Ströhl, N. Curry, M. Zacharopoulou, C. H. Michel, R. Laine, M. F. Nadezhda Nespovitaya, W. Z. Dorothea Pinotsi, P. Fraser, A. Tandon, P. S. George-Hyslop, E. Rees, J. J. Phillips, A. D. Simone, C. F. Kaminski and G. S. K. Schierle, Nat. Commun., 2018, 9, 712 CrossRef PubMed.
- J. Sun, L. Wang, H. Bao, S. Premi, U. Das, E. R. Chapman and S. Roy, Proc. Natl. Acad. Sci., 2019, 116, 11113–11115 CrossRef CAS PubMed.
- M. Y. Arteta, D. Berti, C. Montis, R. A. Campbell, C. Eriksson, L. A. Clifton, M. W. A. Skoda, O. Soltwedel, A. Koutsioubas, P. Baglioni and T. Nylander, Soft Matter, 2015, 11, 1973–1990 RSC.
- A. Åkesson, T. Lind, N. Ehrlich, D. Stamou, H. Wacklin and M. Cárdenas, Soft Matter, 2012, 8, 5658–5665 RSC.
- S. A. Tatulian, Biochemistry, 2003, 42, 11898–11907 CrossRef CAS PubMed.
- L. K. Tamm and S. A. Tatulian, Q. Rev. Biophys., 1997, 30, 365–429 CrossRef CAS.
- Y. Shai, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2306–2313 CrossRef CAS.
- S. E. Glassford, B. Byrne and S. G. Kazarian, Biochim. Biophys. Acta, Proteins Proteomics, 2013, 1834, 2849–2858 CrossRef CAS.
- W. Yassine, N. Taib, S. Federman, A. Milochau, S. Castano, W. Sbi, C. Manigand, M. Laguerre, B. Desbat, R. Oda and J. Lang, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1722–1730 CrossRef CAS PubMed.
- B. Hastoy, P. A. Scotti, A. Milochau, Z. Fezoua-Boubegtiten, J. Rodas, R. Megret, B. Desbat, M. Laguerre, S. Castano, D. Perrais, P. Rorsman, R. Oda and J. Lang, Sci. Rep., 2017, 7, 2835 CrossRef PubMed.
- J. Burré, M. Sharma, T. Tsetsenis, V. Buchman, M. R. Etherton and T. C. Südhof, Science, 2010, 329, 1663–1667 CrossRef PubMed.
- C. Galvagnion, J. Parkinson's Dis., 2017, 7, 433–450 CAS.
- C. Galvagnion, J. W. P. Brown, M. M. Ouberai, P. Flagmeier, M. Vendruscolo, A. K. Buell, E. Sparr and C. M. Dobson, Proc. Natl. Acad. Sci., 2016, 113, 7065–7070 CrossRef CAS PubMed.
- E. Hellstrand, M. Grey, M.-L. Ainalem, J. Ankner, V. T. Forsyth, G. Fragneto, M. Haertlein, M.-T. Dauvergne, H. Nilsson, P. Brundin, S. Linse, T. Nylander and E. Sparr, ACS Chem. Neurosci., 2013, 4, 1339–1351 CrossRef CAS PubMed.
- Z. Lv, M. Hashemi, S. Banerjee, K. Zagorski, J.-C. Rochet and Y. L. Lyubchenko, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 802–812 CrossRef CAS PubMed.
- W. K. Man, A. De Simone, J. D. Barritt, M. Vendruscolo, C. M. Dobson and G. Fusco, Front. Neurosci., 2020, 14 DOI:10.3389/fnins.2020.00018.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00384h |
| ‡ Current affiliation: Department of Physics and Geology, University of Perugia, I-06123 Perugia, Italy, alessandra.luchini@unipg.it. |
| This journal is © The Royal Society of Chemistry 2022 |