
Molecular recognition-driven supramolecular nanoassembly of a hydrophobic uracil prodrug and hydrophilic cytarabine for precise combination treatment of solid and non-solid tumors†
Chang
Li
a,
Qiu
Wang
a,
Dan
Li
a,
Yubo
Liu
a,
Baichun
Hu
 b,
Yao
Feng
b,
Haotian
Zhang
c,
Zhonggui
He
a,
Cong
Luo
*a and
Jin
Sun
*a
b,
Yao
Feng
b,
Haotian
Zhang
c,
Zhonggui
He
a,
Cong
Luo
*a and
Jin
Sun
*a
aWuya College of Innovation, Shenyang Pharmaceutical University, Shenyang, 110016, P. R. China. E-mail: sunjin@syphu.edu.cn; luocong@syphu.edu.cn
bKey Laboratory of Structure-Based Drug Design and Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang, 110016, P. R. China
cSchool of Life Science and Biopharmaceutics, Shenyang Pharmaceutical University, Shenyang, 110016, P. R. China
First published on 20th January 2022
Abstract
Combination chemotherapy has shown distinct therapeutic advantages over monotherapy in clinical cancer treatment, especially for two chemotherapeutic drugs with different mechanisms of action. However, how to achieve efficient co-delivery of two or more drugs with different physicochemical and pharmacokinetic properties for synergistic therapy is still a huge challenge. In particular, it is even more difficult to efficiently co-deliver a hydrophilic drug and a hydrophobic drug into one nanosystem. Herein, inspired by the natural Watson–Crick base pair molecular recognition in nucleic acids, a reduction-sensitive uracil prodrug of doxorubicin (U-SS-DOX) is synthesized and performs supramolecular co-assembly with cytarabine (Ara-C). Interestingly, the hydrophilic Ara-C molecules could readily co-assemble with U-SS-DOX, and multiple hydrogen bonds are found in the nanoassembly with an ultra-high drug loading rate. Moreover, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) is used as a fluorescent probe to investigate the pharmacokinetics of U![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C NPs. It turns out that the DiR-labeled U:C NPs significantly prolong the systemic circulation and promote the tumor-specific accumulation of DiR when compared with DiR solution. Furthermore, the supramolecular nanoassembly demonstrates potent satisfactory therapeutic effects in treating both solid and non-solid tumors in vivo. This study provides a novel molecular co-assembly nanoplatform for efficient co-delivery of hydrophilic and hydrophobic drugs.
:C NPs. It turns out that the DiR-labeled U:C NPs significantly prolong the systemic circulation and promote the tumor-specific accumulation of DiR when compared with DiR solution. Furthermore, the supramolecular nanoassembly demonstrates potent satisfactory therapeutic effects in treating both solid and non-solid tumors in vivo. This study provides a novel molecular co-assembly nanoplatform for efficient co-delivery of hydrophilic and hydrophobic drugs.
New conceptsInspired by the natural Watson–Crick base pair molecular recognition in nucleic acids, we proposed a concept for nanodrug delivery that involves supramolecular nanoassembly driven by nucleic acid-like molecular recognition and prodrug self-assembly techniques. Intermolecular hydrogen bonds are formed between nucleoside analogues. Chemotherapeutic drugs are covalently linked to nucleoside analogues through tumor specific responsive sensitive bonds to be designed as nucleic acid functionalization prodrugs. Prodrugs with nucleic acid functionalization can form supramolecular nanoassemblies with nucleoside chemotherapeutic drugs driven by specific molecular interactions. Compared with traditional nanoparticles, these supramolecular nanoassemblies, on the one hand, retain the multiple drug delivery advantages of prodrug-based nanoassemblies, such as facile fabrication, high drug loading capacity, long systemic circulation, tumor-targeted accumulation, and tumor stimuli-responsive prodrug activation. On the other hand, the combined co-delivery of two chemotherapeutic drugs with different physicochemical properties (such as hydrophilicity and hydrophobicity) but with synergistic antitumor effects can be achieved through specific supramolecular assembly. Our findings provide new insights into approaches for the construction of nanosystems and the possibilities for the development of effective co-delivery strategies for hydrophilic and hydrophobic chemotherapeutic drugs with synergistic antitumor activity. |
1. Introduction
With the rapid development of nanotechnology, nanoparticle drug delivery systems have received widespread attention due to their distinct therapeutic advantages, especially in the field of cancer treatment.1 A variety of nanocarriers have been developed to improve the safety and efficacy of chemotherapy drugs, such as liposomes, vesicles, polymeric micelles, nanoemulsions and inorganic nanoparticles (NPs). However, these conventional nanosystems still have many limitations in clinical applications: (i) premature drug leakage in the blood; (ii) potential excipient-related toxicity due to the heavy use of carrier materials for maximum drug loading; and (iii) complex preparation methods with poor reproducibility.2 Therefore, there is an urgent need to develop new and efficient nanocarriers for anticancer drug delivery.As we all know, hydrophobic drugs can be more easily encapsulated into nanocarriers than hydrophilic drugs, due to the high risk of premature leakage of hydrophilic molecules in an aqueous environment from most nanocarriers. It is even more challenging to co-encapsulate a hydrophobic drug and a hydrophilic drug into one nanosystem, owing to the significant difference in terms of physicochemical properties. Notably, there is synergistic action between some hydrophobic drugs and hydrophilic drugs. For instance, Ara-C (a hydrophilic pyrimidine nucleoside analogue) could be metabolized by cellular deoxycytosine kinase to produce cytarabine triphosphate (Ara-CTP, the active form of Ara-C).3 Ara-CTP effectively inhibits the activity of DNA polymerase and stops DNA synthesis.4 Presently, induction chemotherapy with DNA damaging drugs, such as Ara-C and idarubicin, is the standard treatment of most acute myeloid leukemia (AML) patients in clinical therapy.5 Similar to idarubicin, doxorubicin (DOX) diffuses into the nucleus and inserts its anthracene ring into the DNA base pair, destroying the double helix structure of DNA and triggering a series of signaling events that eventually lead to cell apoptosis.6,7 The ability to incorporate Ara-C and DOX into the DNA of cancer cell lines and primary AML cells has been demonstrated in human trials.8 Previous studies have reported a clear relationship between DNA incorporation and cytotoxicity or patient response rate.9 Based on its antitumor mechanism and significant efficacy in clinical trials, it has been found that the combination of Ara-C and DOX produces synergistic cytotoxicity against multiple tumor cell lines. Nevertheless, so far, there is no practical strategy to realize the efficient co-loading and co-delivery of hydrophilic Ara-C and hydrophobic DOX in vivo.
Given that it is still challenging to efficiently co-deliver Ara-C and DOX using conventional nanocarriers, it is necessary to develop new drug delivery strategies based on the chemical characteristics of these two drugs. Notably, Ara-C as a pyrimidine nucleoside analogue possesses the capacity to form hydrogen bonds with nucleic acid analogues. A supramolecule usually refers to a complex formed by two or more molecules driven by specific intermolecular interactions.10 Prodrugs with tumor stimulus-responsive activation are inactive conjugates that can be activated in the tumor site to release parent drugs. A successful prodrug strategy can overcome a number of drug delivery obstacles, such as low water or lipid solubility, poor stability, lack of site specificity, inefficient cellular uptake and severe adverse effects.11,12 Based on this rationale, in order to develop an advanced nano-drug delivery strategy for co-delivery of Ara-C and DOX, we propose to construct a supramolecular nanoassembly driven by nucleic acid-like molecular recognition and prodrug self-assembly techniques. We hypothesize that this supramolecular nanoassembly, on the one hand, retains multiple drug delivery advantages of prodrug based nanoassembly, such as facile fabrication, high drug loading capacity, long systemic circulation, tumor-targeted accumulation, and tumor stimuli-responsive prodrug activation.13–16 On the other hand, the introduction of nucleic acid-like groups into prodrugs for molecular recognition with Ara-C through specific intermolecular hydrogen bonds further increases the sensitivity of the supramolecular nanoassembly in the micro-acidic tumor microenvironment.
To test our hypothesis, a reduction-sensitive uracil functionalized doxorubicin prodrug (U-SS-DOX) was synthesized by covalently linking DOX and uracil bases via a disulfide bond and subjected to supramolecular co-assembly with Ara-C (Fig. 1).
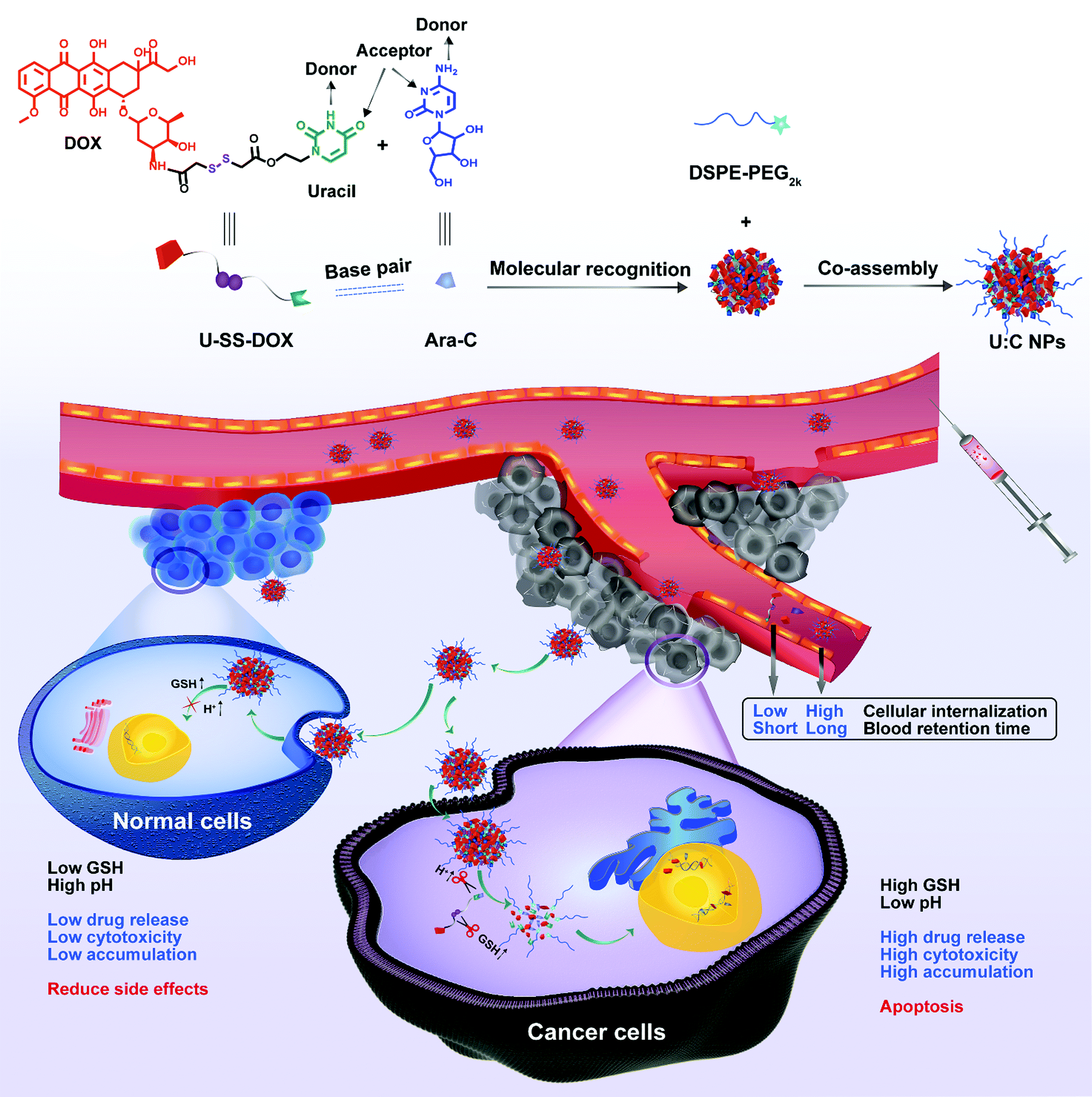 | ||
| Fig. 1 Schematic representation of the molecular recognition-driven supramolecular nanoassembly of U-SS-DOX and Ara-C, synchronous co-delivery and precise combination cancer treatment. The reduction sensitive supramolecular nanoassembly was based on U-SS-DOX and Ara-C specific molecular recognition, and DSPE-PEG2k was used to realize long blood circulation. After the introduction of the supramolecular nanoassembly into tumor cells, drug release can be triggered and controlled under conditions of low pH and excessive glutathione (GSH) production in tumor cells.18 | ||
Interestingly, hydrophilic Ara-C molecules could readily co-assemble with U-SS-DOX in water to form novel Ara-C and uracil functionalized doxorubicin prodrug supramolecular nanoparticles (U:C NPs) for high efficiency and precise combination therapy.17In vitro characterization and computer docking simulation further verified the feasibility of supramolecular recognition and co-assembly between U-SS-DOX and Ara-C. Moreover, the supramolecular nanoassembly significantly prolong the systemic circulation time, thus increasing tumor-specific accumulation. As expected, the supramolecular nanoassembly demonstrated potent therapeutic effects in treating both solid and non-solid tumors in vivo. This is the first attempt to elaborately formulate hydrophilic and hydrophobic drugs into one nanosystem based on the molecular recognition-driven supramolecular assembly technique.
2. Results and discussion
2.1 Design and synthesis of prodrugs (U-SS-DOX)
To test our hypothesis, a novel redox-sensitive DOX-nucleoside conjugate (U-SS-DOX) was synthesized by conjugating hydroxyl-containing uracil (U-OH) to DOX via a disulfide bond (Scheme S1, ESI†). The chemical structure of U-SS-DOX was characterized and confirmed by 1H NMR, 13C NMR and MS (Fig. S1–S3, ESI†). The spectroscopy of U-SS-DOX is shown in Fig. S4 (ESI†).2.2 Co-assembly of U-SS-DOX and Ara-C and optimum dose ratio screening
It is well known that hydrogen bonds, unlike van der Waals forces, are saturated and directional.19 The unique nature of hydrogen bonds significantly impacts the formation, particle size and polydispersity index (PDI) of U:C NPs. In particular, the molar ratios of U-SS-DOX and Ara-C might exert an influence on the supramolecular assembly process. We first investigated the self-assembly characteristics of U-SS-DOX and Ara-C. As shown in Fig. S5A (ESI†), U-SS-DOX alone could not self-assemble into stable and uniform NPs in water, with obvious precipitation observed after storage for 30 min. As expected, the hydrophilic Ara-C molecules also could not form NPs in water due to their excellent water solubility (Fig. S5B, ESI†). Interestingly, U-SS-DOX and Ara-C could co-assemble into NPs at molar ratios (U-SS-DOX/Ara-C) from 5:1 to 1:2 (Table S1, ESI†). Notably, the U-SS-DOX/Ara-C nanoassembly showed small and uniform nanostructures at molar ratios (U-SS-DOX/Ara-C) from 1:1 to 5:1 (Fig. S6, ESI†). In contrast, U-SS-DOX and Ara-C revealed poor co-assembly features at molar ratios from 1:2 to 1:5 (Table S1, ESI†). These results suggested the dominant role of the hydrophobic prodrug in the U:C NPs. The supramolecular recognition between U-SS-DOX and Ara-C drove the hydrophilic Ara-C molecules into the nanoassembly (Fig. S5C, ESI†).
In addition to assembly ratio optimization, it is even more important to figure out the optimal synergistic dose ratios of two drugs for efficient combination cancer therapy. The optimal synergistic dose ratios of DOX and Ara-C were screened by examining the synergistic cytotoxicity against two tumor cell lines. Notably, DOX and Ara-C showed synergistic cytotoxicity against MCF-7 cells at molar ratios from 1:1 to 5:1 (Fig. S7A, ESI†), while they only revealed synergistic effects in 4T1 cells at ratios of 2:1 and 3:1, respectively (Fig. S7B, ESI†). Moreover, DOX and Ara-C had more potent synergistic cytotoxicity against 4T1 cells at a ratio of 3:1, with a smaller CI value (Fig. S7B, ESI†). Additionally, the optimal assembly ratio of DOX and Ara-C was further verified by molecular docking simulation. As shown in Fig. S8 and Table S2 (ESI†), the number of intermolecular and intramolecular hydrogen bonds was higher at a molar ratio of 3:1 than at a molar ratio of 2:1. Taken together, the optimal molar ratio of U-SS-DOX and Ara-C in the supramolecular nanosystem was identified as 3:1 (U-SS-DOX/Ara-C) after a comprehensive consideration on the particle size, synergistic cytotoxicity and molecular docking simulation. The optimal formulation was utilized for subsequent investigations.
2.3 Fabrication and characterization of U![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :C NPs
:C NPs
According to the co-assembly and molar ratio optimization results, the U-SS-DOX/Ara-C supramolecular nanoassembly was fabricated at an optimal ratio of 3:1. The particle size and morphology of the U:C NPs were then characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM). As shown in Fig. 2A and B, the U:C NPs showed a spherical shape with an average diameter of approximately 117.2 nm. NPs of approximately 100 nm in size have been considered to facilitate tumor-specific accumulation through EPR effects.20 The loading efficiencies of DOX and Ara-C were about 44.98% and 10.17%, respectively.
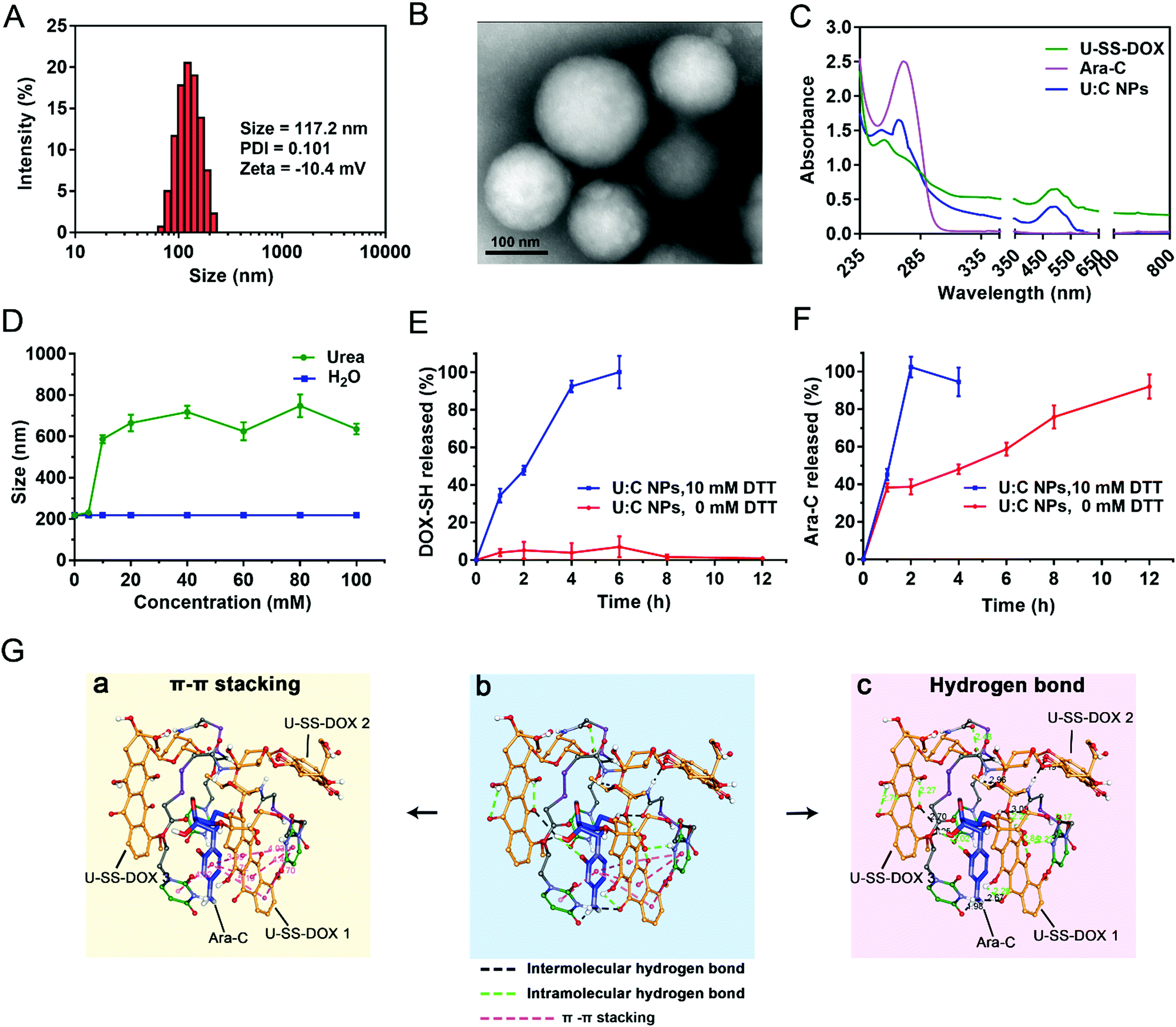 | ||
| Fig. 2 The preparation process and chemical characterization of co-assembled U:C NPs. (A) Particle size distribution profile of U:C NPs. (B) TEM image of U:C NPs. (C) UV-Vis spectra of U-SS-DOX, Ara-C and U:C NPs in aqueous solution. (D) The particle size changes of non-PEGylated U:C NPs after treatment with different concentrations of urea. Data are presented as mean ± SD (n = 3). (E) In vitro cumulative drug release profiles of DOX-SH from U:C NPs at pH 5.5. Data are presented as mean ± SD (n = 3) (F). In vitro cumulative drug release profiles of Ara-C from U:C NPs at pH 5.5. Data are presented as mean ± SD (n = 3). (G) Molecular simulation results of the noncovalent interactions and co-assembly mechanisms between U-SS-DOX and Ara-C. The color of the atoms in U-SS-DOX and Ara-C is the same as that of the structural formula in Fig. 1 The oxygen atoms of both drugs are red and nitrogen atoms are wathet blue. | ||
Afterward, the ultraviolet-visible (UV-vis) spectra of U:C NPs were scanned using a spectrophotometer. As shown in Fig. 2C, both Ara-C and U-SS-DOX had one absorption peak at wavelengths from 250 to 400 nm, while U:C NPs had two absorption peaks, demonstrating that U:C NPs were prepared successfully. Moreover, a clear blue shift was found in the UV-vis spectrum of the U:C NPs when compared with that of free Ara-C and U-SS-DOX, further indicating the existence of hydrogen bonding forces among Ara-C, U-SS-DOX and water molecules. Furthermore, as shown in Fig. 2D, the particle size of the non-PEGylated U:C NPs significantly increased when incubated with urea (a hydrogen bond breaker), suggesting the dominant role of hydrogen bonds in the formation process of U:C NPs.21,22
2.4
In vitro release from U:C NPs
Dithiothreitol (DTT), a prevailing GSH analogue, was utilized to investigate the reduction-response of the U:C NPs. Notably, DTT was utilized as a reducing reagent to simulate the redox stimuli of tumor cells, which could trigger the cleavage of the disulfide bond of U-SS-DOX. As a result, the cleavage of U-SS-DOX led to the disassembly of the U:C NPs and the rapid release of Ara-C. The in vitro release behavior of the U:C NPs in PBS (pH 5.5 or pH 6.5) with or without DTT at 37 °C was investigated. In the release medium added with DTT, a reductive condition resulted in degradation of U-SS-DOX and destruction of the assembly structure of the U:C NPs, so Ara-C would be released quickly. As shown in Fig. 2E, less than 10% of DOX-SH was released from the U:C NPs within 12 h in PBS (pH 5.5) without DTT, while almost 90% of DOX-SH was released within 4 h in the presence of DTT (10 mM). Moreover, more than 90% of Ara-C was completely released within 4 h in the presence of DTT (10 mM) (Fig. 2F). As shown in Fig. S9A (ESI†), DOX-SH was hardly released in the absence of DTT. In contrast, the release rate of Ara-C from the U:C NPs was much faster in the release medium at a lower pH value (pH 5.5) than that in the release medium of pH 6.5, even in the absence of DTT (Fig. S9B, ESI†). As shown in Fig. S9C (ESI†), only ∼20% of Ara-C could be released from the NPs within 12 h in the release medium (pH 7.4) without DTT. Additionally, the particle size of U:C NPs was scarcely changed under the condition of pH 7.4, while the particle size increased to ∼500 nm within 1 h under the condition of pH 6.5 (Fig. S9D, ESI†). Taken together, these results indicated that U:C NPs could remain stable in neutral media without DTT, and the drugs could be selectively released from the NPs in the presence of either acidic or redox stimuli. The good stability of U:C NPs at pH 7.4 would be beneficial for a long blood circulation time. U-SS-DOX and U:C NPs were co-incubated with 10 mM DTT, respectively, and the activation of DOX-SH from U-SS-DOX and U:C NPs was measured every 0.5 h. The results showed that DOX-SH was completely released from U-SS-DOX and U:C NPs after 4 h, and there was no significant difference in the release rate (Fig. S10, ESI†).
We then conducted an exploratory experiment on the mechanism of drug release. U-SS-DOX was co-incubated with excess DTT for 24 h. As shown in Fig. S11A (ESI†), the release mechanism of U-SS-DOX was as follows: under the attack of GSH, the hydrolysis product was the doxorubicin thiol intermediate (DOX-SH). After all the disulfide bonds in U-SS-DOX were broken, the product was scanned by mass spectrometry. The results of the mass spectrum showed that the released product was sulfhydryl-modified doxorubicin. The structural formula, molecular formula and m/z of DOX-SH are shown in Fig. S11B (ESI†).
2.5 Molecular docking simulation
Molecular docking simulation was utilized to disclose the noncovalent interactions and co-assembly mechanisms between U-SS-DOX and Ara-C. A unit of supramolecular assembly consisted of three U-SS-DOX molecules and one Ara-C molecule. As shown in Fig. 2G(b), hydrogen bonding and π–π stacking interaction were found to drive the assembly of U-SS-DOX and Ara-C molecules. The three-dimensional molecule cluster was formed by three U-SS-DOX molecules (1, 2 and 3) and one Ara-C molecule. As shown in Fig. 2G(a) and Table S2 (ESI†), there are 6 π–π stacking interactions present mainly between the pyrimidine rings of Ara-C and the quinone rings and pyrimidine rings of two U-SS-DOX molecules (1 and 2), with bond lengths ranging from 3.72 to 5.7 Å. Moreover, the carbonyl group and pyrimidine rings on another U-SS-DOX provided hydrogen bond receptors, and formed intermolecular hydrogen bonds with the donors of Ara-C (Fig. 2G(c)). As shown in Table S2 (ESI†), the number of intermolecular and intramolecular hydrogen bonds in each supramolecular assembly unit was 6 and 9, respectively, far exceeding the π–π stacking interactions. In addition, the bond lengths of all the hydrogen bonds are less than 3.3 Å, indicating that there were strong hydrogen bonding forces. These results confirmed the vital role of hydrogen bonds in the supramolecular nanoassembly.2.6 Cellular internalization and intracellular drug release
Flow cytometry, confocal laser scanning microscopy (CLSM) and a multimode reader were used to explore the cellular internalization of U:C NPs in 4T1 cells. As shown in Fig. S12 (ESI†), the fluorescence intensity of DOX in U:C NPs was lower than that of free DOX at an equivalent concentration, due to the intermolecular aggregation-caused quenching effect. The flow cytometry results showed that, after incubation with 4T1 cells at 1 h and 2 h, the fluorescence intensities of the U-SS-DOX and U:C NPs were weaker than that of DOX (Fig. 3A). At 6 h, there was no significant difference between the fluorescence intensities of the U:C NPs and DOX, probably due to the release of the parent drug and recovery of the fluorescence under the action of intracellular GSH and low pH. As shown in Fig. 3B, the GLSM results were the same as flow cytometry, both showing that the uptake rate of U-SS-DOX was slow, and weak fluorescence could be detected in the cells after 6 h. Due to the aggregation quenching effect of U:C NPs, the lower fluorescence intensity of the U:C NPs than DOX did not indicate that the uptake efficiency of the U:C NPs was lower than that of DOX. To solve this problem, the 4T1 cells were incubated with the free DOX, U-SS-DOX, U:C NPs or PBS (control) for 2 h, respectively. Then the free medium was discarded, the cells were destroyed and the intracellular drugs were extracted with methanol. Since methanol can destroy U:C NPs, the uptake of intracellular U:C NPs could be determined by measuring the fluorescence intensity of intracellular prodrugs using a multimode reader. As shown in Fig. S13 (ESI†), there was no significant difference in the fluorescence intensity between the U:C NPs group and DOX group, and both of them were stronger than the U-SS-DOX group. This indicated that the uptake of U:C NPs has reached a similar level to DOX at 2 h.
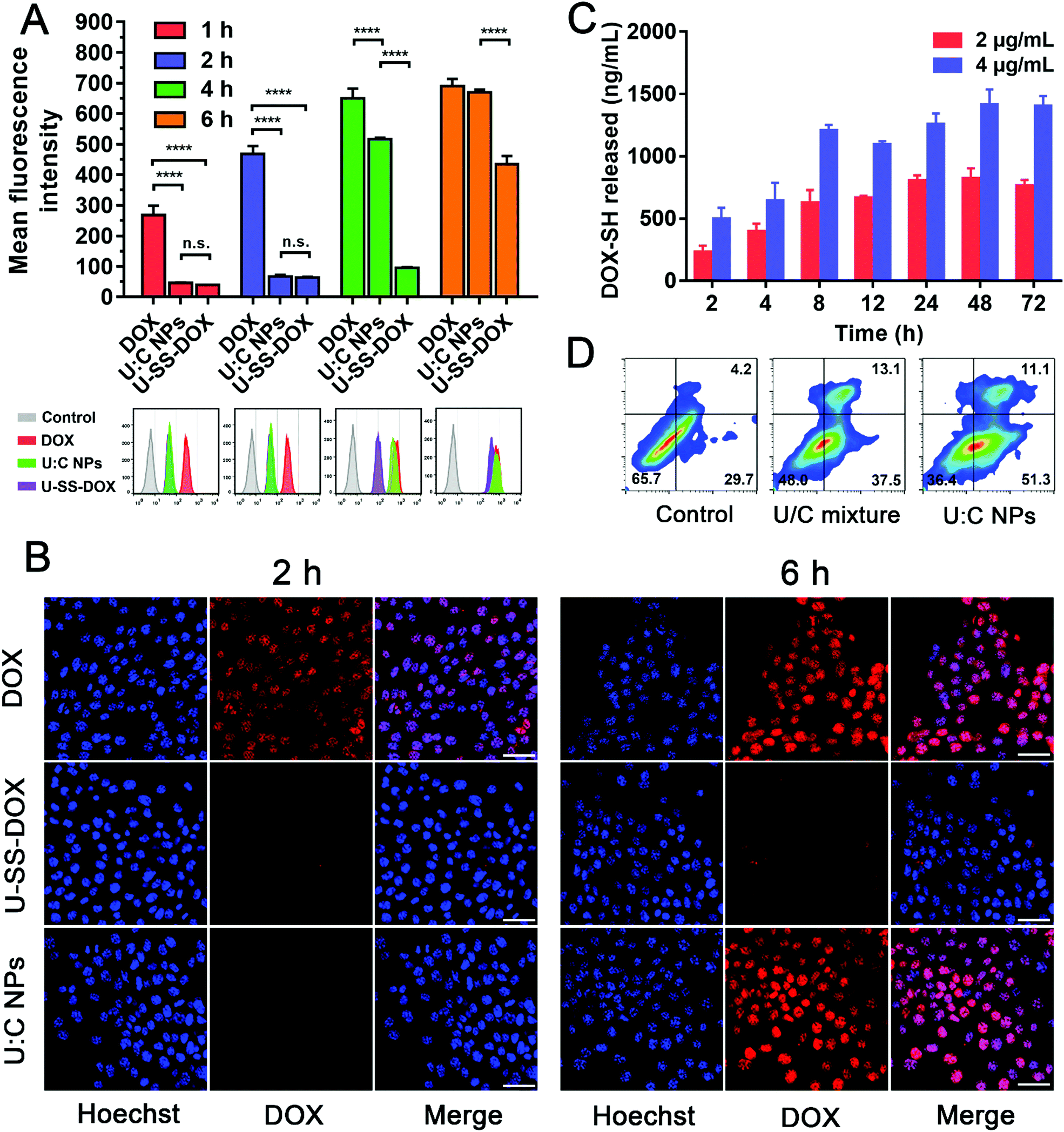 | ||
| Fig. 3
In vitro cellular uptake, intracellular drug release, and cell apoptosis of U:C NPs. (A) Flow cytometry results of cellular uptake in 4T1 cells after incubation with free DOX, U-SS-DOX or U:C NPs for 1, 2, 4 and 6 h. (B) CLSM images of 4T1 cells incubated with the free DOX, U-SS-DOX or U:C NPs for 2 h and 6 h (the scale bar represents 20 μm). (C) The intracellular release of the free DOX-SH level from U:C NPs at different incubation times with 4T1 cells: 2, 4, 8, 12, 24, 48 and 72 h. (D) Flow cytometry analysis for apoptosis of 4T1 cells induced by the U/C mixture and U:C NPs, at the same drug concentration for 24 h. Lower left (Q4): living cells; lower right (Q3): early apoptotic cells; upper right (Q2): late apoptotic cells; upper left (Q1): necrotic cells. Inserted numbers in the profiles indicate the percentage of cells present in this area. The data are presented as mean ± SD, n = 3. Error bars represent ± S.D, ns, not significant, *P < 0.1, **P < 0.01, ***P < 0.001, ****P < 0.0001. | ||
We further investigated the intracellular drug release behavior of DOX-SH from U:C NPs in 4T1 cells. As shown in Fig. 3C, the release of DOX-SH in 4T1 cells after incubation with U:C NPs (2 and 4 μg mL−1) showed a time-dependent manner within 24 h. After 24 h, the release quantity tended to be stable (∼40.58%) in the 2 μg mL−1 group. This phenomenon might be attributed to a comprehensive result of uptake, efflux and cytotoxicity. However, after 12 h, the total release quantity of DOX in the 4 μg mL−1 group was lower than that in the 2 μg mL−1 group (Table S3, ESI†). This may be due to the fact that the toxicity of a high concentration of U:C NPs increased with the incubation time. As shown in Fig. 4C and D, after incubation with 4 μg mL−1 U:C NPs for 48 and 72 h, the cell survival rate was less than 20%.
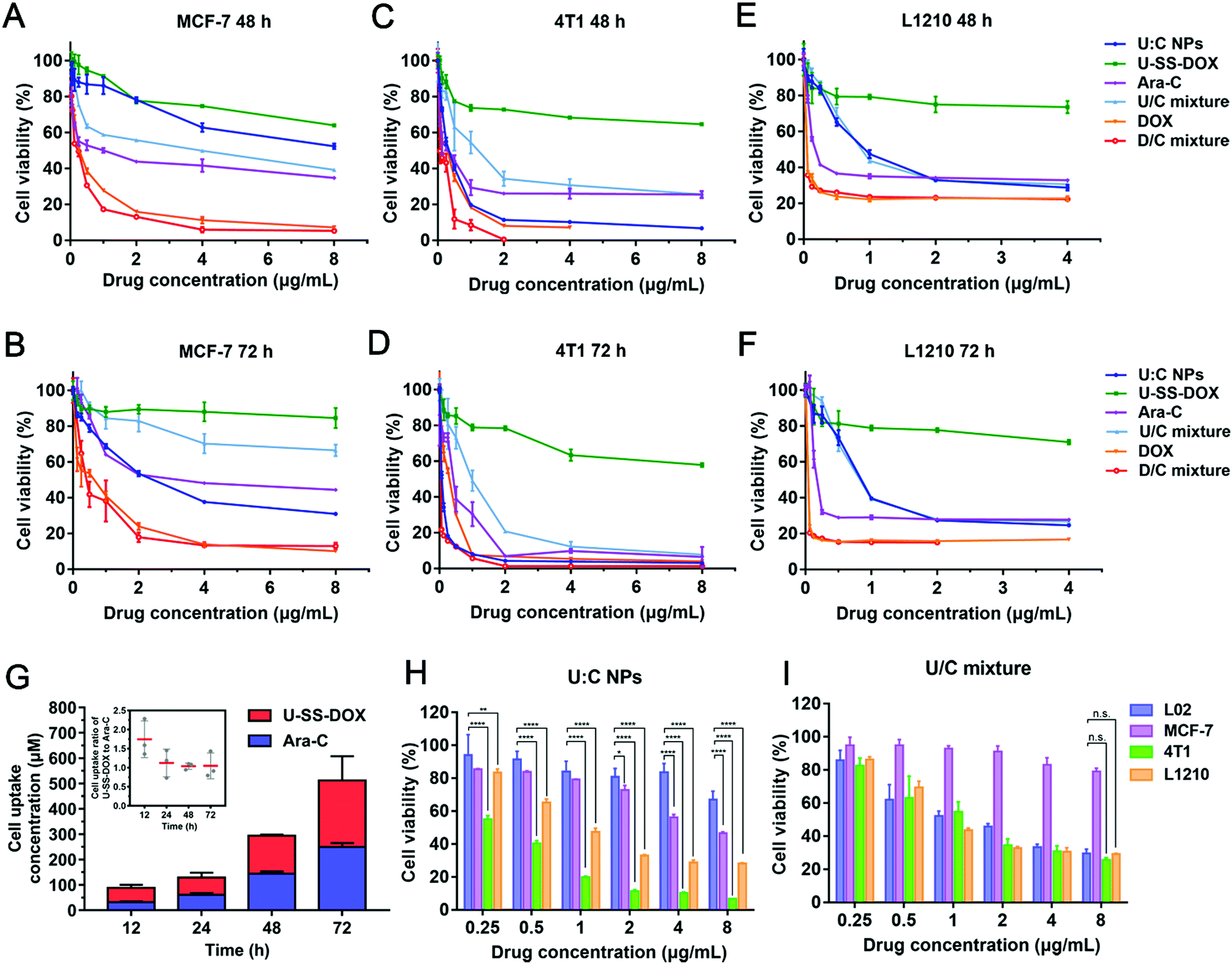 | ||
| Fig. 4 Cytotoxicity of U:C NPs, U-SS-DOX, Ara-C, U/C mixture, DOX, and D/C mixture against (A and B) MCF-7 cells, (C and D) 4T1 cell and (E and F) L1210 cells after 48 and 72 h treatment. (G) Cell uptake of U-SS-DOX and Ara-c at 12, 24, 48 and 72 h, respectively. The comparison of cell viability between (H) U:C NPs and (I) U/C mixture against four types of cells. *P < 0.1, **P < 0.01, ***P < 0.001, ****P < 0.0001 as compared with incubation with L02 cells at the same concentration point. All the data are presented as mean ± SD (n = 3). | ||
It has been reported that DOX and Ara-C induce apoptosis of tumor cells.7,23 To investigate the nanoassembly-induced apoptosis, 4T1 cells were treated with U:C NPs and a U/C mixture, respectively. After incubation for 24 h, FITC-annexin V/propidium iodide staining of the 4T1 cells revealed a higher induction of apoptosis in U:C NP-treated cells when compared with that of the U/C mixture-treated cells under the same conditions (Fig. 3D). Specifically, the cells receiving U:C NPs resulted in 62.4% cell apoptosis. More precisely, 55.3% of the cells were in the early stage of apoptosis and 11.1% were in the late stage of apoptosis, while the control group and U/C mixture-treated group only resulted in 33.9% and 50.6% apoptosis, respectively. Notably, most of the cells treated with U:C NPs were at an early stage of apoptosis. This may be due to the time required for the cellular uptake and disassembly of the supramolecular nanoassembly in tumor cells. Moreover, DOX release from the U-SS-DOX prodrug was a time-consuming process.
2.7 In vitro cytotoxicity
Given the effective intracellular uptake of the supramolecular nanoassembly, we further examined the antitumor efficacy of U:C NPs against 4T1, MCF-7, L1210 and L02 cells at equivalent DOX concentrations using the MTT assay. Cytotoxicity significantly depends on cellular uptake and drug release. After cellular internalization of the U:C NPs, DOX could be released at a high concentration of GSH in tumor cells.24 Therefore, the cytotoxicity of the U:C NPs was stronger than that of the free U-SS-DOX to three kinds of cells. Notably, the free DOX and the D/C mixture showed stronger cytotoxicity against MCF-7 cells than other groups (Fig. 4A and B). Meanwhile, the U:C NPs at high concentrations showed similar cytotoxicity with the free DOX and the D/C mixture in 4T1 cells (Fig. 4C and D). This phenomenon was not seen in the other two cell types, which could be attributed to the higher GSH levels in 4T1 cells than in MCF-7 and L1210 cells (Fig. 4E and F and Fig. S14, ESI†). To investigate why the cytotoxicity of U:C NPs was higher than that of the U/C mixture in 4T1 cell lines with a higher GSH content, we measured the uptake of U-SS-DOX and Ara-C at 12, 24, 48 and 72 h, respectively. As shown in Fig. 4G, the uptake ratio of U-SS-DOX and Ara-C was about 2:1 at 12 h, whereas the uptake ratio was close to 1:1 after 24 h to 72 h. It could be seen from the optimal synergistic dose ratio results that there was no synergistic cytotoxicity when the ratio of the two was 1:1 (Fig. S7 (B), ESI†). However, the U:C NPs are absorbed intactly by cells according to the optimal synergistic ratio of the two drugs, so the cytotoxicity of the U:C NPs was stronger than that of the U:C mixture. We then investigated whether U:C NPs can selectively kill cancer cells instead of normal cells (L02 cells). According to the results, the U/C mixture showed much higher cytotoxicity against L02 cells than U:C NPs in 48 h, indicating the non-selectivity of the U/C mixture to normal cells (Fig. 4H and I).
2.8 Ex vivo and in vivo biodistribution
The retention behaviors of DiR-labeled U:C NPs and DiR sol in the blood were evaluated in Sprague-Dawley rats. The particle size distribution profile of DiR-loaded U:C NPs and the TEM image of DiR-loaded U:C NPs are shown in Fig. S15 (ESI†). As shown in Fig. 5A and B, the average fluorescence intensity in the blood of DiR sol decreased sharply within 1 h. In contrast, the fluorescence intensity in the blood of DiR-loaded U:C NPs remained strong within 24 h. These results indicated that the supramolecular nanoassembly significantly extended the systemic circulation of drugs, which had the potential to promote tumor-specific accumulation. To further assess the biodistribution of the supramolecular nanoassembly, the fluorescence intensities of DiR-labeled U:C NPs and DiR sol in the major organs and tumors were examined in 4T1 tumor-bearing mice. As displayed in Fig. 5C and D, DiR sol showed the strongest fluorescence intensity in the lungs at 12 h after intravenous injection, followed by the liver, spleen, tumor, kidneys and heart. The accumulation of DiR sol in the lungs significantly decreased at 24 h, while bright signals were still found in the liver and kidneys. In contrast, DiR-labeled U:C NPs were predominately accumulated in the liver and tumor at 12 h, and the fluorescence signals in the tumor presented an upward trend from 12 h to 24 h.
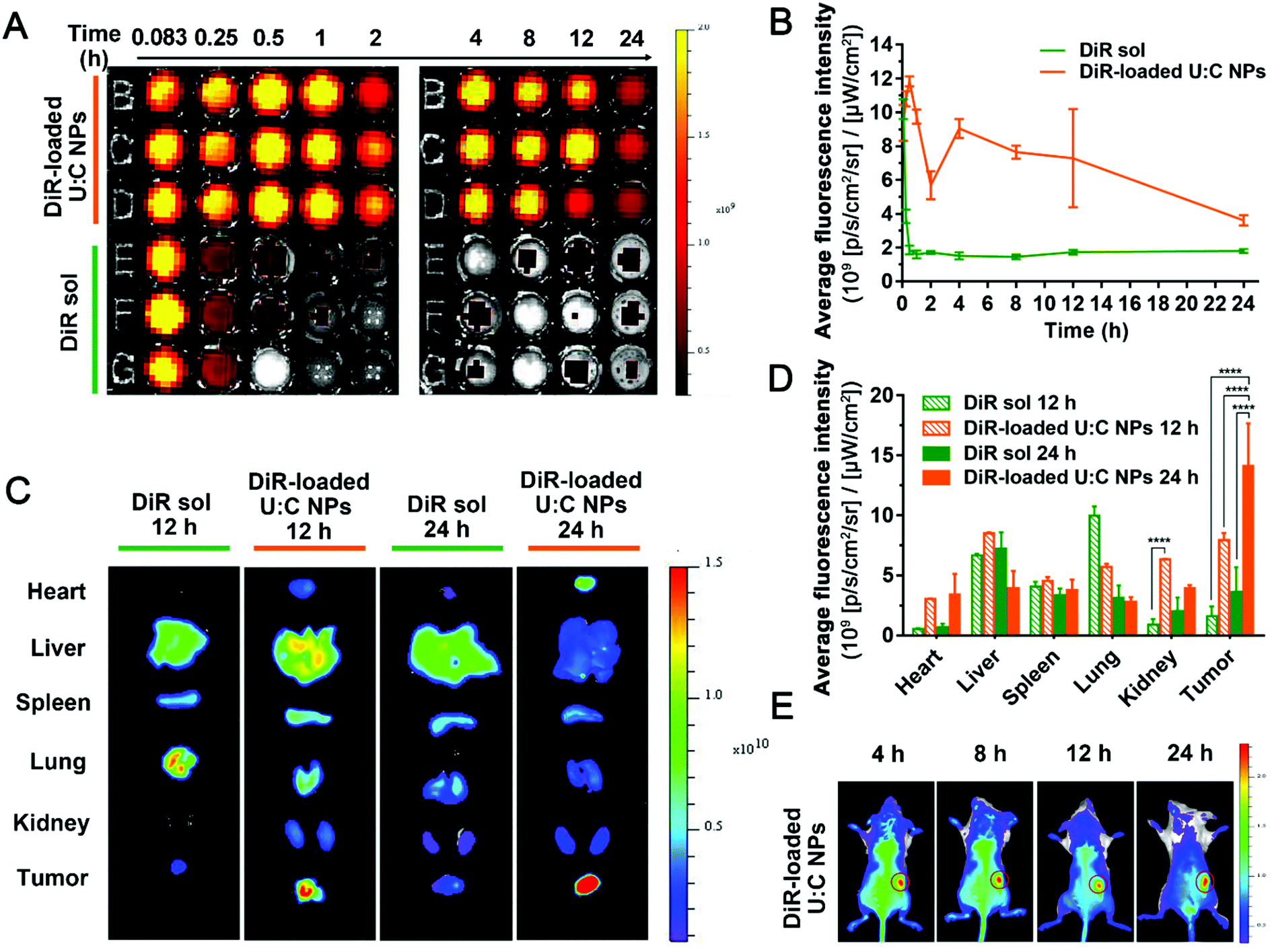 | ||
| Fig. 5
In vivo distribution and IVIS imaging of DiR-labeled U:C NPs (n = 3). (A) Fluorescence imaging of blood samples at different points in time. (B) Quantified graph of the average fluorescence intensity–time of DiR sol and DiR-loaded U:C NPs (1 mg kg−1 equivalent DiR, n = 3). (C) Ex vivo fluorescence imaging of major organs and tumors at 12 and 24 h. (D) Quantitation of the average fluorescence intensity of organ and tumor accumulation at 12 and 24 h; error bars represent ± S.D *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (E) In vivo IVIS whole body imaging of time dependent of 4T1 tumor-bearing mice after the intravenous injection of DiR-loaded U:C NPs at 4, 8, 12 and 24 h. The tumor is circled in red. | ||
In addition to the ex vivo biodistribution, real-time animal imaging in tumor-bearing mice was further carried out to vividly observe the in vivo biodistribution of DiR sol and DiR-labeled U:C NPs. As shown in Fig. S16 (ESI†), the signal was strong in the lungs and liver of DiR sol-treated mice at 4 h and 8 h after intravenous injection. In contrast, DiR-labeled U:C NPs were rapidly distributed throughout the whole animal body at 4 h post intravenous injection, and a much stronger fluorescence intensity was observed in the tumor tissues than that of DiR sol under the same conditions. More importantly, the fluorescence signals in the tumors of the mice receiving DiR-labeled U:C NPs revealed a gradually increasing trend from 8 h to 24 h, accompanying the decrease of the fluorescence intensity in other major organs (Fig. 5E). These results indicated that supramolecular nanoassembly could not only significantly prolong the circulation time of the drugs in the blood, but also greatly facilitate the accumulation in tumors via the EPR effect.25
2.9
In vivo anticancer efficacy of U:C NPs against 4T1 breast tumors
A 4T1 breast tumor-bearing xenograft mouse model was established as a solid tumor animal model to evaluate the in vivo antitumor activity of the supramolecular nanoassembly (Fig. 6A). As shown in Fig. 6B–D, compared with the saline group, Ara-C, U-SS-DOX, U/C mixture, DOX and D/C mixture showed a moderate antitumor effect with delayed tumor progression. Notably, both the U/C mixture and D/C mixture showed almost no advantage over the monotherapy groups (DOX, Ara-C and U-SS-DOX), suggesting the poor synergy effect of drug mixture solutions (Fig. 6B). In contrast, the mice treated with U:C NPs exhibited a significant tumor suppression. The superior therapeutic effect of the U:C NPs could be ascribed to their long blood retention, high accumulation in tumors via the EPR effect, and efficient cellular uptake.26,27 Furthermore, the strong antitumor effect of the U:C NPs could also be attributed to the rapid and differential release of active DOX molecules in tumor cells and the synergistic effect of two drugs with different antitumor mechanisms. Furthermore, the biosafety and biocompatibility of these formulations were evaluated. As shown in Fig. 6E and Fig. S17 (ESI†), no significant changes in the body weight and hematological parameters were observed in all the mice. The H&E staining results also showed no obvious histological variations in the major organs of the mice (Fig. S18, ESI†). These results indicated that the U:C NPs had good biosafety and biocompatibility in vivo.
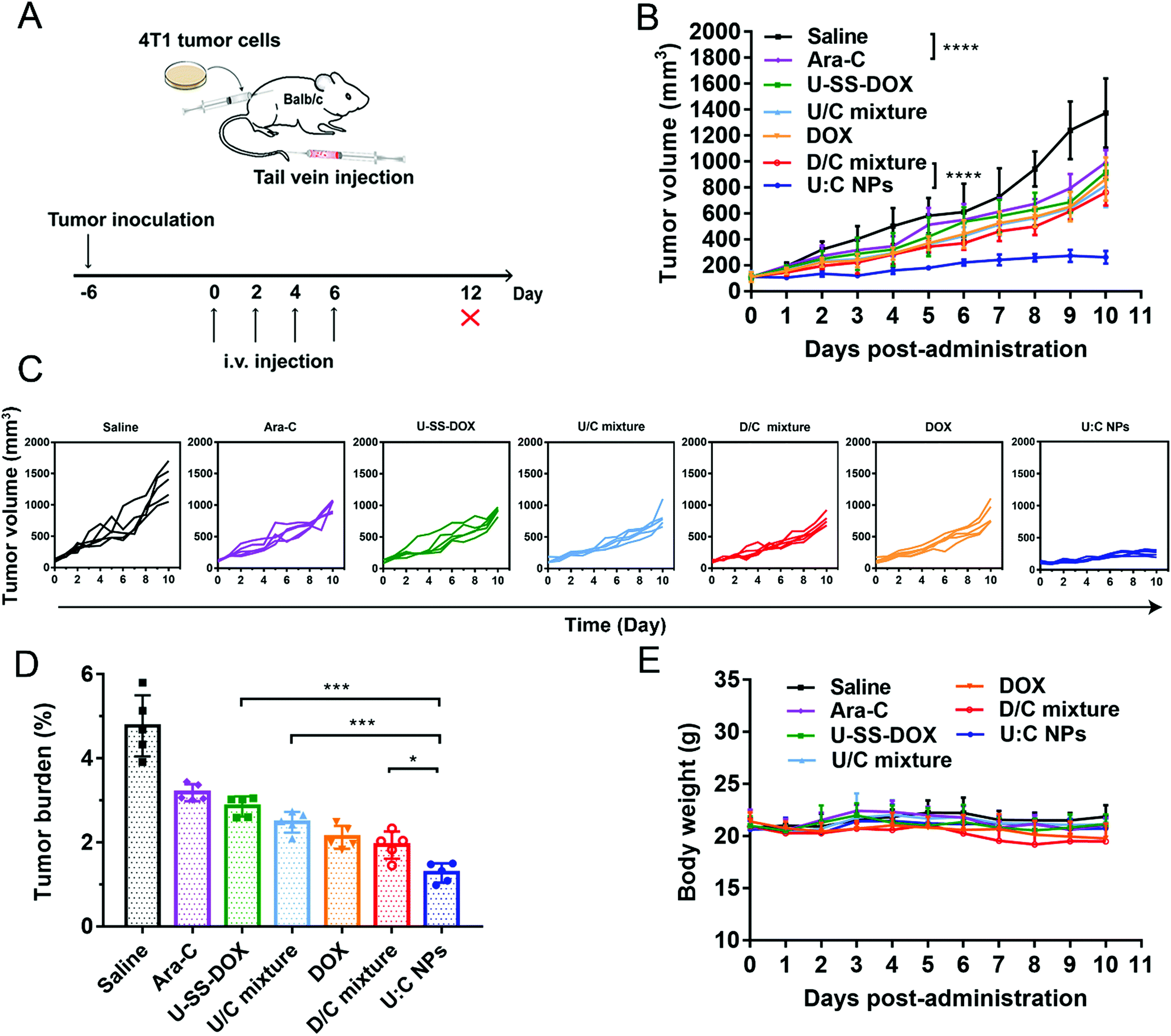 | ||
| Fig. 6 In vivo antitumor efficacy of the supramolecular nanoassembly against 4T1 tumors (n = 5). (A) Schematic diagram of treatment of Balb/c mice subcutaneously inoculated with 4T1 cells by the supramolecular nanoassembly. (B) Tumor growth profiles treated with different formulations. (C) Individual tumor growth kinetics in different groups. (D) Tumor burden after the last treatment. (E) Weight changes of mice in different groups. The data are presented as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. | ||
2.10
In vivo anticancer efficacy of U:C NPs against leukemia
In addition to solid tumors, chemotherapy plays a more important role in the clinical treatment of non-solid tumors, such as leukemia.28 We further investigated the anticancer efficacy of U:C NPs against leukemia. The DBA/2 mice inoculated with L1210 cells were utilized to study the anticancer efficacy of the supramolecular nanoassembly against non-solid tumors (Fig. 7A). As shown in Fig. 7B, all mice in the other groups died within 22 days except for the U:C NP group. It was encouraging to see that both the U:C NPs-L (low dose U:C NPs) group and U:C NPs-H (high dose U:C NPs) group prolonged the survival cycle of the mice owing to high co-delivery efficacy.29 In the survival experiment, the DOX, U:C NPs-L and U:C NPs-H groups had a significant difference with the saline group. As shown in Fig. 7C, the weight of mice in the D/C mixture group was decreased sharply after the beginning of drug administration. Death occurred on day 9 and the weight measurement was stopped when the number of mice was less than 3 on day 13th. The weight of mice in the DOX group also decreased with the increase of dosing times. It was suggested that the DOX and D/C mixture had certain toxicity. The body weight of other groups did not change. Compared with the U:C NPs-L group, the survival time of the U:C NPs-H group was longer and the weight change was more stable. Blood routine examination was used to assess the antitumor effect of U:C NPs on L1210 cells. As shown in Fig. 7D, several representative indicators of the U:C NPs-H group and the control group were within the normal range, indicating that the hematological examination results of leukemia mice were basically returned to normal after the treatment of U:C NPs-H.
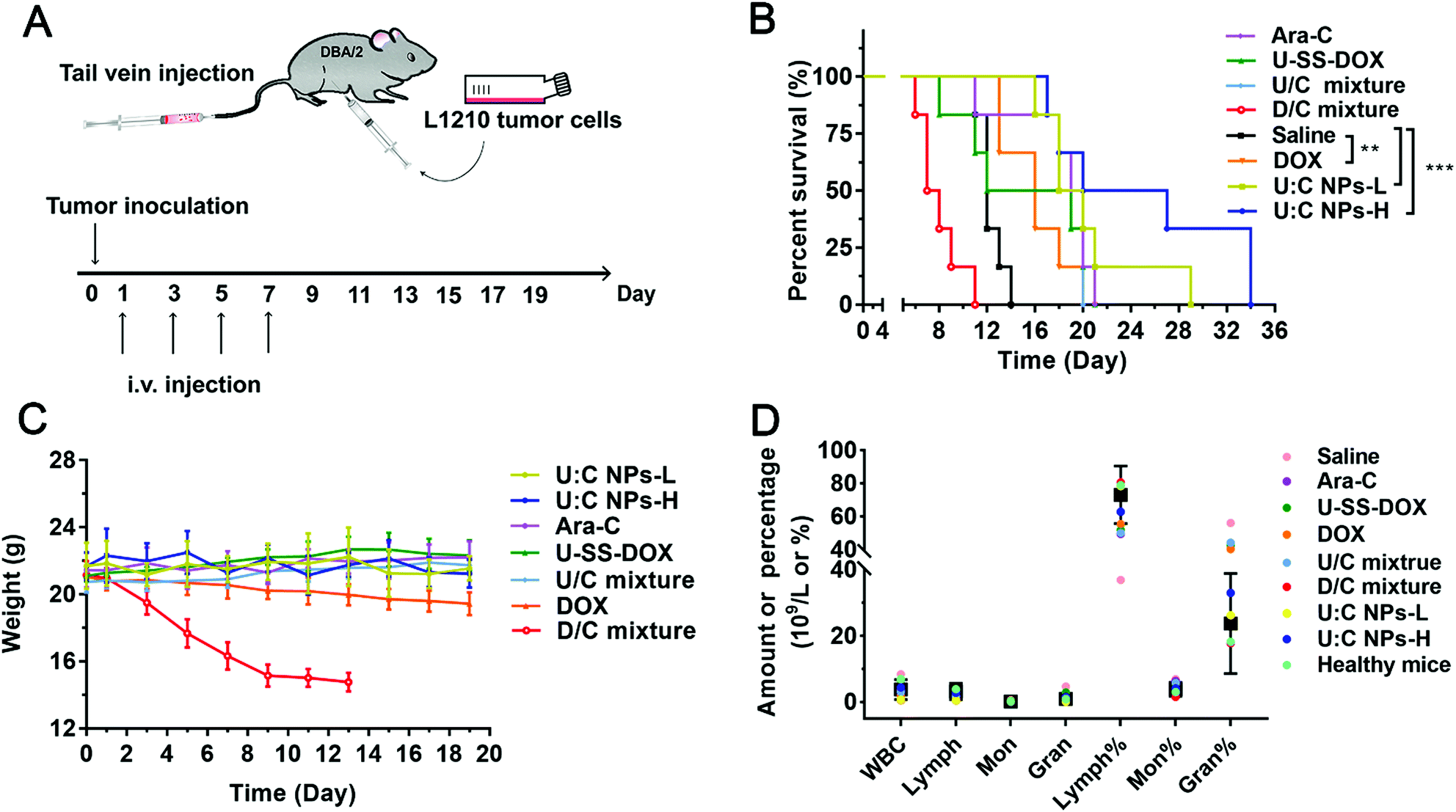 | ||
| Fig. 7 In vivo anticancer efficacy of the supramolecular nanoassembly in non-solid tumors (n = 6). (A) Schematic diagram of the supramolecular nanoassembly for the treatment of DBA/2 mice intraperitoneally inoculated with L1210 cells. (B) Survival rate of mice after different treatment groups as indicated. The data are presented as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (C) Weight changes of healthy mice in different groups. (D) The parameters of blood routine in different groups. The black squares and error lines are the normal ranges. | ||
3. Conclusions
Here, we successfully developed a reduction-sensitive supramolecular nanoassembly based on the specific molecular recognition of U-SS-DOX and Ara-C. Both the computer docking simulation and nanoassembly characterization results verified the successful fabrication of U:C NPs. This novel supramolecular nanoassembly demonstrated multiple drug delivery advantages, including impressively high drug loading, long blood circulation, favorable intra-tumor accumulation, and selective drug release within tumor cells. As expected, the ingenious combination of one hydrophilic and one hydrophobic anticancer drug in a supramolecular nanoassembly exerted synergistic antitumor activities against both solid and non-solid tumors. This unique supramolecular assembly strategy provides a new fabrication approach for the development of co-delivery nanomedicines.
Author contributions
Chang Li: conceptualization, visualization, investigation, software, validation, data curation, and writing – original draft. Qiu Wang: formal analysis, software, data curation, and writing – original draft. Dan Li: conceptualization, software, investigation, and data curation. Yubo Liu: software, investigation, and validation. Baichun Hu: investigation and data curation. Yao Feng: investigation and data curation. Haotian Zhang: supervision. Zhonggui He: funding acquisition. Cong Luo: supervision, writing – review and editing. Jin Sun: supervision and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Excellent Youth Science Foundation of Liaoning Province (No. 2020-YQ-06), the National Natural Science Foundation of China (no. 81773656), the Liaoning Revitalization Talents Program (no. XLYC1808017), and the Shenyang Youth Science and Technology Innovation Talents Program (no. RC190454).References
- H. Zhao, H. Feng, D. Liu, J. Liu, N. Ji, F. Chen, X. Luo, Y. Zhou, H. Dan, X. Zeng, J. Li, C. Sun, J. Meng, X. Ju, M. Zhou, H. Yang, L. Li, X. Liang, L. Chu, L. Jiang, Y. He and Q. Chen, ACS Nano, 2015, 9, 9638–9651 CrossRef CAS PubMed.
- C. Luo, J. Sun, B. Sun and Z. He, Trends Pharmacol. Sci., 2014, 35, 556–566 CrossRef CAS PubMed.
- P. Pourquier, Y. Takebayashi, Y. Urasaki, C. Gioffre, G. Kohlhagen and Y. Pommier, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1885–1890 CrossRef CAS PubMed.
- X. Wang, Z. Chen, A. K. Mishra, A. Silva, W. Ren, Z. Pan and J. H. Wang, Haematologica, 2018, 103, 466–476 CrossRef CAS PubMed.
- A. H. Wei and I. S. Tiong, Blood, 2017, 130, 2469–2474 CrossRef CAS PubMed.
- C. Perez-Arnaiz, N. Busto, J. M. Leal and B. Garcia, J. Phys. Chem. B, 2014, 118, 1288–1295 CrossRef CAS PubMed.
- S. Ashrafizaveh, M. Ashrafizadeh, A. Zarrabi, K. Husmandi, A. Zabolian, M. Shahinozzaman, A. R. Aref, M. R. Hamblin, N. Nabavi, F. Crea, Y. Wang and K. S. Ahn, Cancer Lett., 2021, 508, 104–114 CrossRef CAS PubMed.
- T. M. Scharadin, M. A. Malfatti, K. Haack, K. W. Turteltaub, C. X. Pan, P. T. Henderson and B. A. Jonas, Chem. Res. Toxicol., 2018, 31, 1042–1051 Search PubMed.
- A. Raza, S. Gezer, J. Anderson, J. Lykins, J. Bennett, G. Browman, J. Goldberg, R. Larson, R. Vogler and H. D. Preisler, Exp. Hematol., 1992, 20, 1194–1200 CAS.
- D. Wang, B. Liu, Y. Ma, C. Wu, Q. Mou, H. Deng, R. Wang, D. Yan, C. Zhang and X. Zhu, J. Am. Chem. Soc., 2017, 139, 14021–14024 CrossRef CAS PubMed.
- L. Bildstein, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2011, 63, 3–23 CrossRef CAS PubMed.
- B. Sun, C. Luo, X. Zhang, M. Guo, M. Sun, H. Yu, Q. Chen, W. Yang, M. Wang, S. Zuo, P. Chen, Q. Kan, H. Zhang, Y. Wang, Z. He and J. Sun, Nat. Commun., 2019, 10, 3211 CrossRef PubMed.
- L. Shi, Y. Wang, Q. Wang, Z. Jiang, L. Ren, Y. Yan, Z. Liu, J. Wan, L. Huang, B. Cen, W. Han and H. Wang, J. Controlled Release, 2020, 324, 289–302 CrossRef CAS PubMed.
- S. K. Misra, Z. Wu, F. Ostadhossein, M. Ye, K. Boateng, K. Schulten, E. Tajkhorshid and D. Pan, ACS Appl. Mater. Interfaces, 2019, 11, 18074–18089 CrossRef CAS PubMed.
- S. Zhang, Z. Wang, Z. Kong, Y. Wang, X. Zhang, B. Sun, H. Zhang, Q. Kan, Z. He, C. Luo and J. Sun, Theranostics, 2021, 11, 6019–6032 CrossRef CAS PubMed.
- S. Zhang, Y. Wang, Z. Kong, X. Zhang, B. Sun, H. Yu, Q. Chen, C. Luo, J. Sun and Z. He, Acta Pharm. Sin. B, 2021, 11, 3636–3647 CrossRef CAS PubMed.
- C. Ghosh, A. Nandi and S. Basu, Nanoscale, 2019, 11, 3326–3335 RSC.
- X. An, A. Zhu, H. Luo, H. Ke, H. Chen and Y. Zhao, ACS Nano, 2016, 10, 5947–5958 CrossRef CAS PubMed.
- L. J. Karas, C. H. Wu, R. Das and J. I. Wu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10 Search PubMed.
- J. Wang, X. Sun, W. Mao, W. Sun, J. Tang, M. Sui, Y. Shen and Z. Gu, Adv. Mater., 2013, 25, 3670–3676 CrossRef CAS PubMed.
- H. R. Jia, Y. X. Zhu, K. F. Xu and F. G. Wu, Adv. Healthcare Mater., 2018, 7, e1800380 CrossRef PubMed.
- Y. X. Zhu, H. R. Jia, G. Y. Pan, N. W. Ulrich, Z. Chen and F. G. Wu, J. Am. Chem. Soc., 2018, 140, 4062–4070 CrossRef CAS PubMed.
- F. Guo, C. Sigua, P. Bali, P. George, W. Fiskus, A. Scuto, S. Annavarapu, A. Mouttaki, G. Sondarva, S. Wei, J. Wu, J. Djeu and K. Bhalla, Blood, 2005, 105, 1246–1255 CrossRef CAS PubMed.
- L. H. Fu, Y. Wan, C. Qi, J. He, C. Li, C. Yang, H. Xu, J. Lin and P. Huang, Adv. Mater., 2021, 33, e2006892 CrossRef PubMed.
- H. Deng, L. Lin, S. Wang, G. Yu, Z. Zhou, Y. Liu, G. Niu, J. Song and X. Chen, Adv. Mater., 2019, 31, e1903443 CrossRef PubMed.
- Y. Huang, Y. He, Z. Huang, Y. Jiang, W. Chu, X. Sun, L. Huang and C. Zhao, Nanoscale, 2017, 9, 10002–10019 RSC.
- J. Wang, W. Mao, L. L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu and Y. Shen, ACS Nano, 2015, 9, 7195–7206 CrossRef CAS PubMed.
- I. Truebenbach, S. Kern, D. M. Loy, M. Hohn, J. Gorges, U. Kazmaier and E. Wagner, Mol. Pharm., 2019, 16, 2405–2417 CrossRef CAS PubMed.
- Y. Abebe Alemayehu, B. Tewabe Gebeyehu and C. C. Cheng, Biomacromolecules, 2019, 20, 4535–4545 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nh00590a |
| This journal is © The Royal Society of Chemistry 2022 |