
UV-absorbing benzamide-based dendrimer precursors: synthesis, theoretical calculation, and spectroscopic characterization†
Chidera C.
Nnadiekwe
a,
Ahmed
Nada
a,
Ismail
Abdulazeez
b,
Mohammad R.
Imam
c,
Muhammad Ramzan Saeed Ashraf
Janjua
 a and
Abdulaziz A.
Al-Saadi
*ad
a and
Abdulaziz A.
Al-Saadi
*ad
aChemistry Department, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia
bInterdisciplinary Research Center for Membranes & Water Security, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia
cDepartment of Mathematics and Natural Sciences, American University of Iraq, Sulaimani, Kurdistan Region 46001, Iraq
dInterdisciplinary Research Center for Refining & Advanced Chemicals, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia. E-mail: asaadi@kfupm.edu.sa
First published on 17th November 2021
Abstract
The ability to tune the structural properties with respect to size, polarity, and terminal functionalities makes dendrimers a highly attractive class of compounds with potential biological and catalytic applications. In the present study, Janus (J) and Twin (T) benzamide-based branched structures were synthesized from a primary precursor of methyl gallate and then further characterized using spectroscopic and theoretical techniques. The optimized structures of these dendrimer precursors at the B3LYP/6-311G(d) level revealed that the aliphatic chains in the most stable forms are oriented in the least strained arrangement with the (–benzene–O–CH2−) linkage adopting the phenol-like configuration. UV–Vis spectra showed a consistent absorption at nearly 290 nm, suggesting that both molecules absorb within the ultraviolet region and are colorless. Prominent bands, which include the –OH vibrational stretch, the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– stretch, the –CO stretch, the –C–O in-plane stretch, and the –NH in-plane bending, were assigned and correlated with the structural and electronic aspects of the molecules.
C– stretch, the –CO stretch, the –C–O in-plane stretch, and the –NH in-plane bending, were assigned and correlated with the structural and electronic aspects of the molecules.
1. Introduction
Dendrimers are characterized1,2 with their monodispersed macromolecules through a regular and well-defined three-dimensional architecture. They have attracted the attention of a wide scope of disciplines3–6 due to their distinctive structural aspects and synthetic accessibility of a variety of sizes, making them multipurpose building blocks for several applications in catalysis,7 drug delivery,8 organic light-emitting diodes (OLEDs)9 and sensors.10 Recently, there has been high interest in potentially utilizing dendrimers in nanoscience.11Twin-tapered dendrimers consist of two identical dendritic building blocks (i.e., dendrons) attached to each other. They can self-assemble into supramolecular columns generated by an orthogonal H-bonded stack of supramolecular columns.12 These supramolecular columns self-organize into periodically ordered hexagonal assemblies or co-assemble into more complex hexagonal superlattices.13 Janus dendrimers are structurally similar to Twin dendrimers but contain two different structural motifs attached via covalent bonds. Linking two chemically distinct dendritic building blocks forms a Janus dendrimer, which breaks the roughly spherical symmetry that characterizes most dendrimers. The rational design of Janus dendrimers can incorporate fluorophobic, polar and non-polar, hydrophilic, and hydrophobic elements in the same molecule that can function as a powerful structure-directing amphiphile.14 The self-assembly of Janus dendrimers can incorporate a large diversity of supramolecular architectural manners, such as columnar, globular, and bilayer structures.15 These supramolecular Janus dendrimers can exhibit thermotropic liquid crystalline (LC) properties in their self-assembled state.13
Understanding the potential of Twin- and Janus-based dendrimeric systems depends on our ability to identify their structural and electronic properties. Vibrational and UV–Vis spectroscopies are informative tools to explore and understand the structural functionalities of dendrimers.16 In this study, we report the synthesis and spectroscopic assessment of two novel higher-generation dendrimer precursors, namely 3,4,5-tris(11-hydroxyundecyloxy)-N-(3,4,5-tris(octyloxy)phenyl)benzamide (Janus) and 3,4,5-tris(octyloxy)-N-(3,4,5-tris(octyloxy)phenyl)benzamide (Twin), characterized with octyloxy and hydroxyundecyloxy terminal groups (Fig. 1a and b). These triply-substituted cores show potential for the synthesis of new, higher-generation, biologically active and non-toxic benzamide-based dendrimers.17 With the aid of spectroscopic techniques, the elucidation of the structural properties, conformational stability, and chemical reactivity of the two synthesized dendrimers can be achieved.
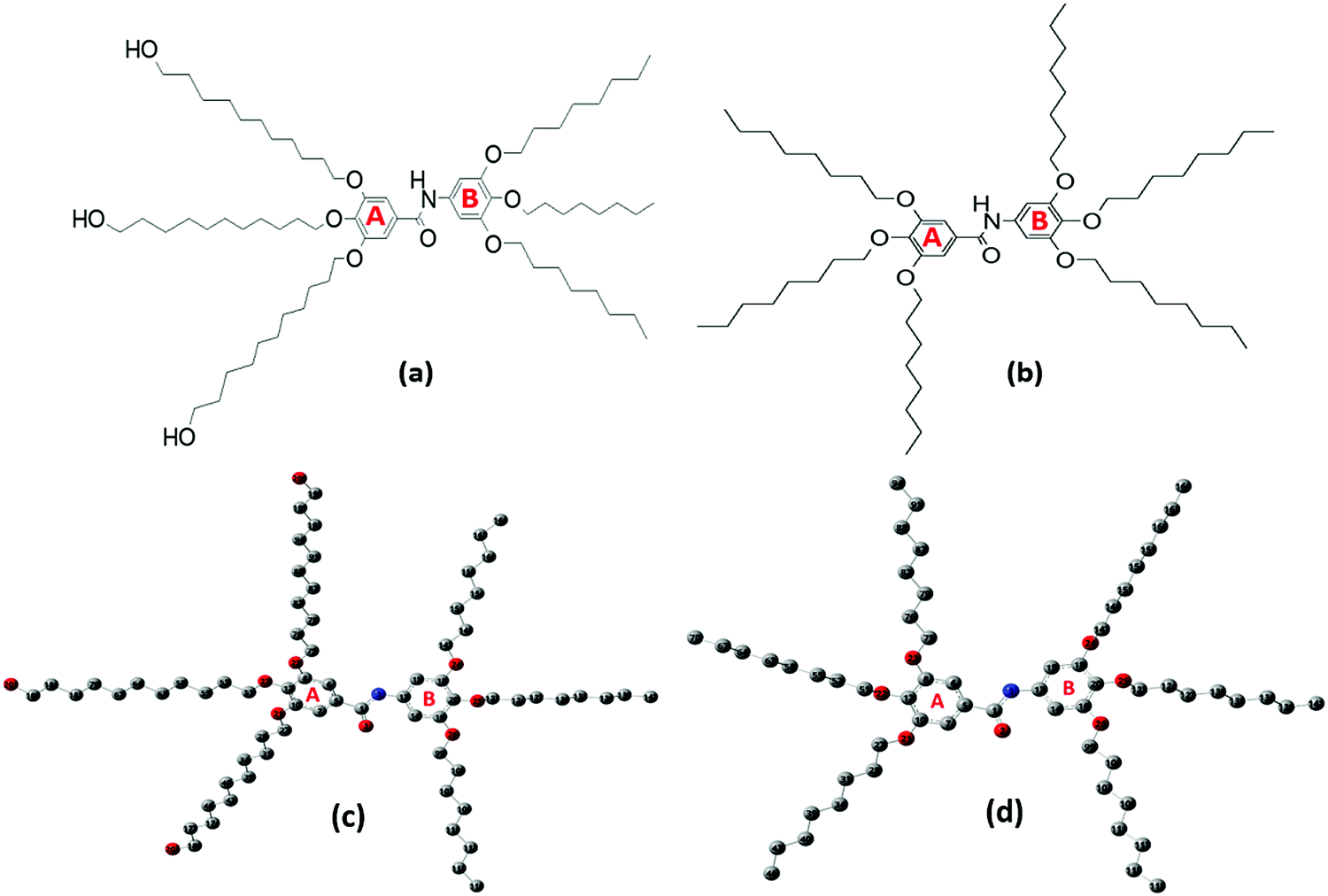 | ||
| Fig. 1 Structures of (a) 3,4,5-tris(11-hydroxyundecyloxy)-N-(3,4,5-tris(octyloxy)phenyl)benzamide (Janus) and (b) 3,4,5-tris(octyloxy)-N-(3,4,5-tris(octyloxy)phenyl)benzamide (Twin). Optimized geometry and numbering of atoms of the (c) Janus 1 (J1) and (d) Twin (T1) benzamide configurations. Hydrogen atoms are omitted for the sake of clarity. | ||
2. Experimental
All the chemicals used in the synthesis processes are of standard grades and the synthetic techniques were adopted as reported in the literature18–23 with specific major modifications. The structures of the two benzamide systems obtained (Fig. 1a and b) were confirmed via their nuclear magnetic resonance spectra recorded using an Advance 400, Bruker NMR 400 MHz spectrometer from National Scientific Company Limited (NSC). The 13C-NMR and 1H-NMR spectra of the synthesized benzamides are shown in the supplementary data (see Fig. S1 and S2, ESI†). The infrared, Raman and UV–vis spectra of the final Twin and Janus dendrimer precursors were collected and analysed. Vibrational assignments of the key fundamental modes were also performed based on the reported group frequencies and by comparison with the computed wavenumbers. The detailed synthesis and spectroscopic verifications of intermediate products are described as follows:Methyl 3,4,5-tris(octyloxy)benzoate (2)
To a thoroughly degassed suspension of anhydrous K2CO3 (16.6 g, 0.12 mol) in DMF (100 mL), methyl gallate (1) (3.70 g, 0.02 mol) was added. The reaction mixture was heated to 80 °C and 1-bromooctane (15.45 g, 0.08 mol) was added under a N2 atmosphere. After stirring for 8 h at 80 °C, the reaction mixture was poured into ice-cooled water and extracted with CH2Cl2. The CH2Cl2 extract was washed with H2O and brine, then dried over MgSO4 and filtered. The solvent was removed using a rotatory evaporator and passed through a short column of basic Al2O3 using CH2Cl2 as the eluent to give 9.8 g (93%) as a yellow oil.TLC (EtOAc/hexane = 1/9): Rf = 0.56; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 0.88 (t, 9H, CH3, J = 6.56 Hz), 1.31 (overlapped m, 24H, CH3(CH2)4), 1.47 (m, 6H, CH2(CH2)2O), 1.79 (m, 6H, CH2CH2O), 3.88 (s, 3H, CO2CH3), 4.01 (m, 6H, CH2O), and 7.26 (s, 2H, ArH); 13C NMR (125 MHz, CDCl3, 25 °C, TMS): δ = 14.11, 22.73, 26.12–30.18, 31.91, 52.07, 69.11, 73.42, 107.71, 124.59, 142.34, 152.81, and 166.92.
Methyl 3,4,5-tris(11-hydroxyundecyloxy)benzoate (3)
To a thoroughly degassed suspension of anhydrous K2CO3 (5.5 g, 0.04 mol) in DMF (50 mL), methyl gallate (1) (1.22 g, 0.0066 mol) was added. The reaction mixture was heated to 80 °C and 11-bromo-1-undecanol (5.0 g, 0.02 mol) was added under a N2 atmosphere. After stirring for 8 hours at 80 °C, the reaction mixture was poured into ice-cooled water and the precipitate was collected by filtration and vacuum dried. The crude product was passed through a short column of basic Al2O3 using CH2Cl2 as the eluent. Recrystallization from acetone yielded 4.2 g (91%).TLC (EtOAc/hexane = 5/5): Rf = 0.26; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 1.31 (overlapped m, 36H, (CH2)6CH2CH2OH), 1.47 (m, 6H, CH2(CH2)2O), 1.56 (m, 6H, CH2CH2OH), 1.77 (m, 6H, CH2CH2O), 2.16 (s, 3H, OH), 3.64 (t, 6H, CH2OH, J = 6.56 Hz), 3.89 (s, 3H, CO2CH3), 4.01 (m, 6H, CH2O), and 7.25 (s, 2H, ArH); 13C NMR (125 MHz, CDCl3, 25 °C, TMS): δ = 25.68, 25.70, 25.97, 26.00, 29.22, 29.29, 29.37, 29.41, 29.46, 29.48, 29.52, 29.54, 29.56, 29.60, 29.62, 30.24, 32.67, 52.08, 62.97, 62.99, 69.10, 73.44, 107.96, 124.68, 142.35, 152.85, and 167.03.
3,4,5-Tris(octyloxy)benzoic acid (4)
Compound 2 (2.5 g, 0.0048 mol), EtOH (30 mL, 95%), and KOH (1.61 g, 0.029 mol) were added to a 50 mL round-bottom flask containing a Teflon-coated magnetic stirring bar and heated to reflux for 2 h under constant stirring. After the reaction was completed, the flask was cooled to room temperature and acidified with dil. HCl (1 M) to pH 1. The reaction mixture was poured into ice-cooled water, then filtered, and vacuum dried to yield 2.4 g (98%).TLC (EtOAc/hexane = 1/9): Rf = 0.05; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 0.89 (t, 9H, CH3, J = 6.87 Hz), 1.31 (overlapped m, 24H, CH3(CH2)4), 1.47 (m, 6H, CH2(CH2)2O), 1.79 (m, 6H, CH2CH2O), 4.03 (m, 6H, CH2O), and 7.29 (s, 2H, ArH); 13C NMR (125 MHz, CDCl3, 25 °C, TMS): δ = 14.11, 22.73, 26.12–30.18, 31.91, 69.21, 73.62, 108.51, 123.69, 143.14, 152.91, and 172.22.
3,4,5-Tris(11-hydroxyundecyloxy)benzoic acid (5)
Compound 3 (2.0 g, 0.0029 mol), EtOH (20 mL, 95%), and KOH (0.97 g, 0.017 mol) were added to a 50 mL round-bottom flask containing a Teflon-coated magnetic stirring bar and heated to reflux for 2 h under constant stirring. After the reaction was completed, it was cooled to room temperature and acidified with dil. HCl (1 M) to pH 1. The reaction mixture was poured into ice-cooled water, then filtered, and vacuum dried to yield 1.8 g (92%). The synthetic route for 1–5 is shown in Scheme 1.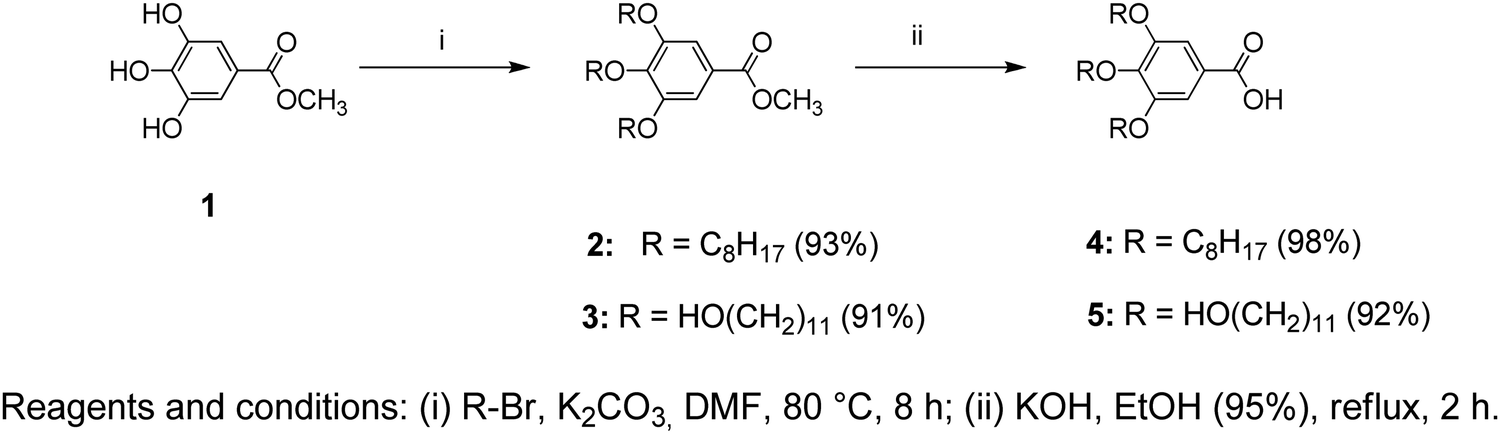 | ||
| Scheme 1 Synthesis pathway leading to benzoate and benzoic acid derivatives (1–5). | ||
TLC (EtOAc/hexane = 5/5): Rf = 0.05; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 1.30 (overlapped m, 36H, (CH2)6CH2CH2OH), 1.48 (m, 6H, CH2(CH2)2O), 1.57 (m, 6H, CH2CH2OH), 1.78 (m, 6H, CH2CH2O), 3.09 (s, 3H, OH), 3.65 (t, 3H, CH2OH, J = 6.56 Hz), 4.04 (m, 6H, CH2O), and 7.31 (s, 2H, ArH); 13C NMR (125 MHz, CDCl3, 25 °C, TMS): δ = 25.68, 25.70, 25.97, 26.00, 29.22, 29.29, 29.37, 29.41, 29.46, 29.48, 29.52, 29.54, 29.56, 29.60, 29.62, 30.24, 32.67, 62.97, 62.99, 69.10, 73.44, 108.96, 123.88, 143.35, 152.95, and 173.23.
1,2,3-Tris(octyloxy)benzene (7)
To a thoroughly degassed suspension of anhydrous K2CO3 (24.9 g, 0.18 mol) in DMF (100 mL), pyrogallol (6) (3.8 g, 0.03 mol) was added. The reaction mixture was heated to 80 °C and 1-bromooctane (23.2 g, 0.12 mol) was added under a N2 atmosphere. After stirring for 8 h at 80 °C, the reaction mixture was poured into ice-cooled water and extracted with CH2Cl2. The CH2Cl2 extract was washed with H2O and brine, then dried over MgSO4 and filtered. The solvent was removed using a rotatory evaporator and passed through a short column of basic Al2O3 using CH2Cl2 as the eluent to yield 13.2 g (95%) as a colorless oil.TLC (EtOAc/hexane = 1/9): Rf = 0.80; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 0.91 (t, 9H, CH3, J = 6.71 Hz), 1.33 (overlapped m, 24H, CH3(CH2)4), 1.49 (m, 6H, CH2(CH2)2O), 1.80 (m, 6H, CH2CH2O), 3.98 (m, 6H, CH2O), 6.56 (d, 2H, ArH, 4,6 positions, J = 8.24 Hz), and 6.92 (t, 1H, ArH, 5 position, J = 8.39 Hz); 13C NMR (125 MHz, CDCl3, 25 °C, TMS): δ = 14.31, 22.93, 26.12–30.18, 32.21, 69.41, 73.62, 107.21, 123.29, 138.54, and 153.71.
5-Nitro-1,2,3-tris(octyloxy)benzene (8)
To a stirred suspension of SiO2·HNO3 (25.2 g, 0.08 mol, 20% HNO3 w/w) in CH2Cl2 (130 mL) at room temperature, compound 7 (7.4 g, 0.016 mol dissolved in 20 mL of CH2Cl2) was added rapidly. The resulting red solution was stirred for 15 min, filtered, and washed several times with CH2Cl2. The solution was concentrated using a rotary evaporator and precipitated in cold MeOH (100 mL). The yellow precipitate was collected by filtration and air-dried. Recrystallization from acetone yielded 5.7 g (70%).TLC (EtOAc/hexane = 1/9): Rf = 0.76; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 0.89 (t, 9H, CH3, J = 7.63 Hz), 1.31 (overlapped m, 24H, CH3(CH2)4), 1.49 (m, 6H, CH2(CH2)2O), 1.75 (m, 2H, CH2CH2O, 4 position), 1.84 (m, 4H, CH2CH2O, 3,5 positions), 4.05 (m, 6H, CH2O), and 7.47 (s, 2H, ArH); 13C NMR (125 MHz, CDCl3, 25 °C, TMS): δ = 14.31, 22.93, 26.12–30.18, 32.11, 69.71, 73.95, 102.71, 143.39, 144.14, and 152.91.
3,4,5-Tris(octyloxy)aniline (9)
A mixture of compound 8 (5.0 g, 0.01 mol), NH2NH2·H2O (1.50 g, 0.03 mol), and graphite (4.0 g) in EtOH (50 mL) was refluxed under a N2 atmosphere for 24 hours. The cooled mixture was diluted with CH2Cl2 (60 mL). Graphite was filtered and washed several times with CH2Cl2. The colorless solution was concentrated using a rotary evaporator and the resulting white solid was dissolved in CH2Cl2. After precipitation in MeOH, the obtained solid was collected by filtration and washed with cold MeOH. After drying under vacuum, 4.0 g (86%) of white powder was obtained (Scheme 2).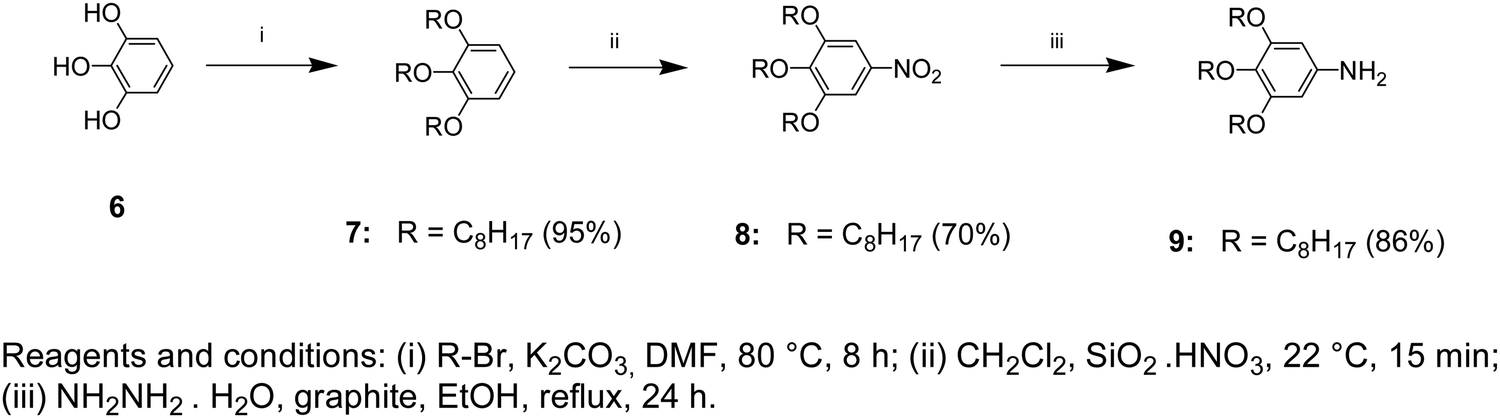 | ||
| Scheme 2 Synthesis pathway leading to benzene, nitrobenzene, and aniline derivatives (6–9). | ||
TLC (EtOAc/hexane = 1/9): Rf = 0.13; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 0.89 (t, 9H, CH3, J = 7.32 Hz), 1.30 (overlapped m, 24H, CH3(CH2)4), 1.45 (m, 6H, CH2(CH2)2O), 1.76 (m, 6H, CH2CH2O), 3.47 (br s, 2H, NH2), 3.85 (t, 2H, CH2O, 4 position, J = 6.56 Hz), 3.91 (t, 4H, CH2O, 3,5 positions, J = 6.41 Hz), and 5.92 (s, 2H, ArH); 13C NMR (125 MHz, CDCl3, 25 °C, TMS): δ = 14.51, 23.13, 26.12–30.18, 32.41, 69.51, 73.99, 95.01, 131.61, 142.68, and 154.11.
3,4,5-Tris(octyloxy)-N-(3,4,5-tris(octyloxy)phenyl)benzamide (10)
In a 50 mL two-neck flask, compound 4 (1.27 g, 0.0025 mol) was dissolved in dry THF (20 mL). After adding CDMT (0.48 g, 0.0028 mol) and NMM (0.38 g, 0.0038 mol), the mixture was stirred for 1 hour at room temperature under a N2 atmosphere. Then compound 9 (1.20 g, 0.0025 mol) was added and the reaction mixture was further stirred for 6 hours at room temperature. The mixture was diluted with CH2Cl2 (10 mL) and precipitated in methanol (100 mL) and the product was collected by filtration and dried. Purification by column chromatography (EtOAc/Hexane = 1/9) followed by precipitation in methanol yielded 1.32 g (54%).TLC (EtOAc/hexane = 1/9): Rf = 0.40; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 0.89 (t, 18H, CH3, J = 6.87 Hz), 1.31 (overlapped m, 48H, CH3(CH2)4), 1.48 (m, 12H, CH2(CH2)2O), 1.79 (m, 12H, CH2CH2O), 3.94 (t, 2H, CH2O para to NH, J = 5.80 Hz), 4.01 (overlapped t, 10H, CH2O), 6.91 (s, 2H, ArH, ortho to NH), 7.03 (s, 2H, ArH, ortho to CO), and 7.64 (s, 1H, NH); 13C NMR (125 MHz, CDCl3, 25 °C, TMS): δ = 14.02, 22.60, 26.01, 29.22, 29.31, 29.46, 29.51, 30.25, 31.77, 31.84, 69.14, 69.47, 73.51, 73.54, 99.15, 105.77, 129.95, 133.57, 135.06, 141.58, 153.30, and 165.55.
3,4,5-Tris(11-hydroxyundecyloxy)-N-(3,4,5-tris(octyloxy)phenyl)benzamide (11)
In a 50 mL two-neck flask, compound 5 (1.36 g, 0.002 mol) was dissolved in dry THF (20 mL). After adding CDMT (0.39 g, 0.0022 mol) and NMM (0.31 g, 0.003 mol), the mixture was stirred for 1 hour at room temperature under a N2 atmosphere. Then compound 9 (0.96 g, 0.002 mol) was added and the reaction mixture was further stirred for 6 hours at room temperature. The mixture was diluted with CH2Cl2 (10 mL) and precipitated in methanol (100 mL) and the product was collected by filtration and dried. Purification by column chromatography (EtOAc/Hexane = 7/3) followed by precipitation in methanol yielded 1.4 g (61%). The route of the synthesis is presented in Scheme 3.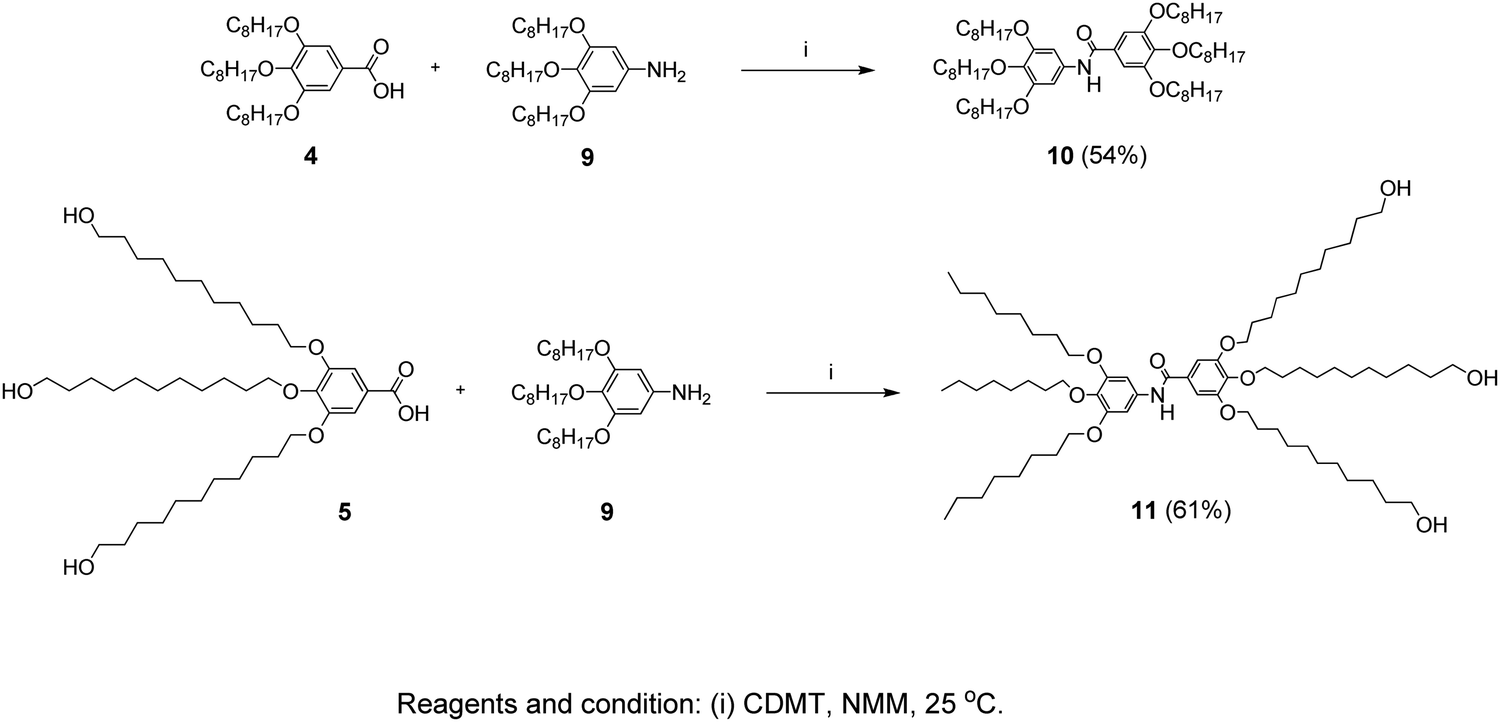 | ||
| Scheme 3 Synthesis pathway leading to the inner-shell (10) and the branched-structure (11) benzamide molecules. | ||
TLC (EtOAc/hexane = 7/3): Rf = 0.56; 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 0.88 (t, 9H, CH3, J = 6.40 Hz), 1.30 (overlapped m, 60H, CH3(CH2)4, (CH2)6CH2CH2OH), 1.47 (m, 15H, CH2(CH2)2O, OH), 1.56 (m, 6H, CH2CH2OH), 1.79 (overlapped m, 12H, CH2CH2O), 3.64 (t, 6H, CH2OH, J = 6.60 Hz), 3.93 (t, 2H, CH2O para to NH, J = 6.10 Hz), 4.01 (overlapped t, 10H, CH2O), 6.91 (s, 2H, ArH, ortho to NH), 7.04 (s, 2H, ArH, ortho to CO), and 7.68 (s, 1H, NH); 13C NMR (125 MHz, CDCl3, 25 °C, TMS): δ = 14.02, 22.60, 25.68, 25.99, 26.02, 26.06, 29.23, 29.31, 29.36, 29.41, 29.45, 29.51, 29.59, 30.24, 31.78, 31.85, 32.73, 63.00, 69.14, 69.43, 73.51, 99.19, 105.79, 129.98, 133.59, 135.05, 141.52, 153.26, and 165.60.
3. Computation
A comprehensive computational scan was conducted to detect the possible stable forms of each Janus and Twin benzamide structure. This was performed using density functional theory (DFT) at the hybrid Becke 3-Parameter (Exchange), Lee, Yang, and Parr functional (B3LYP) and the 6-311G(d) basis set. The chosen basis set provides a reliable estimation of the solvation energies when utilizing an implicit solvent model (compared with higher basis sets) and generates data consistent with experiments at a considerable cost.24,25 Full optimization of the structures was performed to the minima on the potential energy surface without thrusting symmetry constraints. Five stable configurations were successfully optimized for each molecule (Table 1). The optimized structures of the most stable forms are depicted in Fig. 1(c and d), where the hydrogen atoms are excluded for the sake of clarity. The key structural parameters of the most stable conformations are listed in Tables S1–S3 (ESI†). The descriptors of the quantum chemical reactivity, namely the HOMO–LUMO energy gap (ΔE), the energy of the highest occupied molecular orbital (EHOMO), the energy of the lowest unoccupied molecular orbital (ELUMO), the general hardness (η), ionization energy (IE), electron affinity (EA), electrophilicity (ω), electronegativity (χ), and the chemical potential (μ) of the molecules, were deduced using the DFT Koopman's theorem as reported by Pearson.26 The pictorial representations of the HOMO and LUMO frontier molecular orbitals are depicted in Fig S3 (ESI†). All of the calculations performed herein were executed using the Gaussian 09 software,27 while the extraction of data and analysis was achieved using the GaussView 6.0 graphical user interface.28 The equations used to compute the quantum chemical descriptors were adopted as reported by Furer et al.16 and Parr et al.29 The vibrational spectra of the most stable J1 and T1 forms were calculated and used as a basis to compare with the experimental ones. Scaling factors of 0.985 and 0.961 were used for wavenumbers below 2000 cm−1 and above 2000 cm−1, respectively.| Conformation | Key torsional anglesa (deg.) | Dipole moment (Debye) | Relative energy (kcal mol−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ϕ 1 | ϕ 2 | ϕ 3 | ϕ 4 | ϕ 5 | ϕ 6 | ϕ 7 | |||
| a ϕ1 = C(6)–C(8)–O(23)–C(75), ϕ2 = C(8)–C(12)–O(22)–C(51), ϕ3 = C(7)–C(10)–O(21)–C(27), ϕ4 = C(15)–C(18)–O(24)–C(147), ϕ5 = C(18)–C(20)–O(25)–C(123), ϕ6 = C(14)–C(16)–O(26)–C(99), and ϕ7 = O(2)–C(1)–N(3)–H(4). | |||||||||
| Janus benzamide | |||||||||
| J1 | 5.4 | −95.6 | −2.9 | 3.0 | −92.3 | −2.1 | 170.5 | 1.561 | 0.000 |
| J2 | −1.2 | 97.1 | 2.6 | 3.3 | −94.4 | −3.9 | 170.4 | 4.517 | 0.070 |
| J3 | −104.7 | 86.3 | 109.7 | −111.8 | 101.4 | 3.1 | 172.9 | 3.956 | 1.712 |
| J4 | −110.1 | 97.7 | 105.6 | −106.9 | 95.6 | 2.8 | 171.8 | 5.128 | 2.964 |
| J5 | −110.8 | 97.1 | 104.8 | −111.7 | 101.1 | 2.5 | 172.1 | 3.529 | 1.394 |
| Twin benzamide | |||||||||
| T1 | 4.5 | −68.6 | 143.0 | 111.2 | −100.6 | −2.1 | 170.1 | 4.241 | 0.000 |
| T2 | 4.0 | −96.5 | −1.7 | 2.4 | −94.4 | −1.7 | 170.4 | 2.877 | 0.134 |
| T3 | −1.9 | 97.1 | 3.8 | 3.4 | −94.7 | −3.5 | 170.4 | 3.929 | 1.777 |
| T4 | −108.1 | 86.5 | 112.3 | −109.9 | 97.0 | 1.0 | 172.2 | 3.579 | 3.029 |
| T5 | −111.9 | 95.2 | 106.3 | −105.2 | 92.3 | 2.8 | 171.5 | 5.492 | 1.556 |
4. Results and discussion
The possible stable conformations of the benzamides were identified using the B3LYP/6-311G(d) level of calculation (Table 1 and Fig. S4, ESI†) with the optimized most stable forms being presented in Fig. 1c and d. J1 (for Janus benzamide) and T1 (for Twin benzamide), which exercise the least hindrance, were predicted to be the most stable configurations. Furthermore, the conformers J1, J2, T1, T2, and T3 have their aliphatic chains (described using ϕ1⋯ϕ6) and the amide moiety (described using ϕ7) adopting the least structural strain due to their co-planar orientation with respect to the benzyl groups. This suggests that the major factor in determining the stability of the investigated precursor systems is the pattern of the –benzene–O–CH2− linkage, which assumes the phenol-like configuration. Hence, the subsequent discussions will focus on the most stable J1 and T1 configurations as representative structures of the Janus and Twin benzamide molecular systems.The donor–acceptor mechanism of electron pairs governs the reactivity of the molecules.24,30–32 This is generally connected to the energetics of the frontier molecular orbitals. According to de Souza et al.33 and Wang et al.,34 the energy gap (ΔE) reflects the readiness and ease with which electron pairs are promoted. The smaller the ΔE magnitude, the more readily and easily the electron pairs are promoted from their HOMO to the LUMO for improved reactivity and selectivity performance. Table 2 lists the global reactivity descriptors for the different configurations of the Janus and Twin structures. Both benzamides were predicted to exhibit comparable reactivity, which can be attributed to their similar structural patterns and energetics. The molecular orbital diagram (Fig. 2) shows that in both molecules the HOMOs are centered at ring “B”, while ring “A” is more associated with the LUMOs, suggesting a viable π → π* type of transition. Thus, the feasibility of the electronic migration should be more significant across the B rings. The higher electron affinity of J1 against T1, on the other hand, indicates the readiness of its LUMO to accept electron pairs when interacted. Due to the comparable magnitude of ΔE for both J1 and T1, however, both molecules have an almost equivalent electron-pair-transfer nature, indicating that the most stable configurations of these molecules are comparatively reactive.35,36 Their comparability can also be confirmed from the measured UV–Vis spectra shown in Fig. 4 and 5. The analysis of the frontier orbital energies confirms the potential of these benzamides to self-assemble in a π–π intramolecular columnar stacking manner when appropriate conditions are maintained, similar to previously reported dendrimeric systems.37–39
| Configuration | E HOMO | E LUMO | χ | η | IE | EA | ω | μ |
|---|---|---|---|---|---|---|---|---|
| Janus benzamide | ||||||||
| J1 | −5.425 | −1.126 | 3.276 | 2.150 | 5.425 | 1.126 | 2.496 | −3.275 |
| J2 | −5.426 | −1.124 | 3.275 | 2.151 | 5.426 | 1.124 | 2.493 | −3.274 |
| J3 | −5.568 | −1.250 | 3.409 | 2.159 | 5.568 | 1.250 | 2.691 | −3.408 |
| J4 | −5.588 | −1.209 | 3.399 | 2.189 | 5.588 | 1.209 | 2.638 | −3.398 |
| J5 | −5.561 | −1.227 | 3.394 | 2.167 | 5.561 | 1.227 | 2.658 | −3.394 |
| Twin benzamide | ||||||||
| T1 | −5.536 | −1.060 | 3.298 | 2.238 | 5.536 | 1.060 | 2.430 | −3.298 |
| T2 | −5.418 | −1.118 | 3.268 | 2.150 | 5.418 | 1.118 | 2.483 | −3.267 |
| T3 | −5.419 | −1.115 | 3.267 | 2.152 | 5.419 | 1.115 | 2.479 | −3.266 |
| T4 | −5.571 | −1.242 | 3.406 | 2.165 | 5.571 | 1.242 | 2.680 | −3.406 |
| T5 | −5.594 | −1.200 | 3.397 | 2.197 | 5.594 | 1.200 | 2.626 | −3.396 |
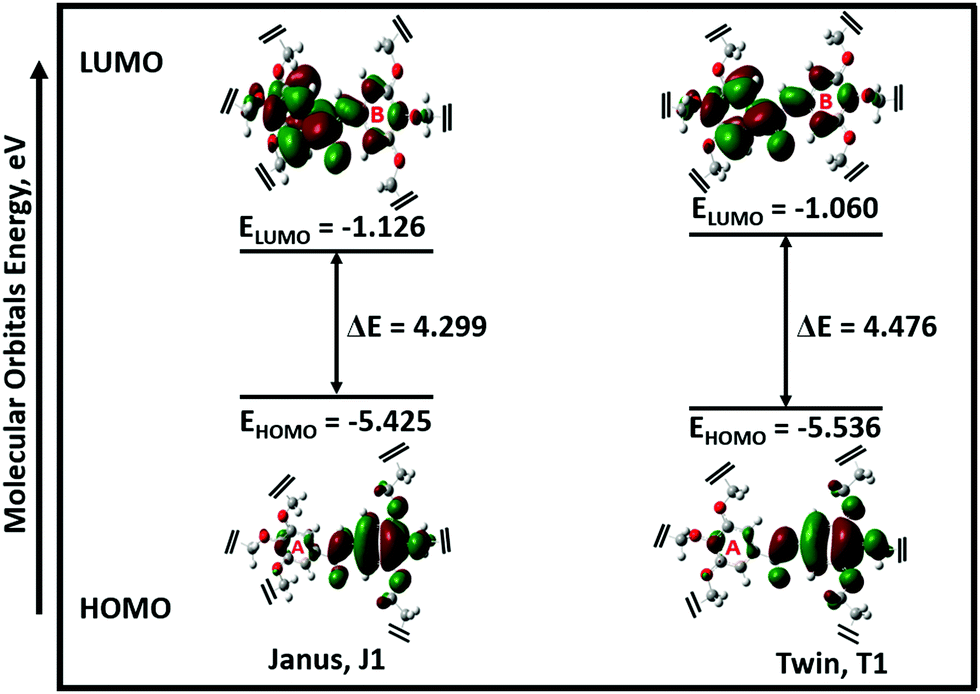 | ||
| Fig. 2 Frontier molecular orbitals (MOs) of the most stable configurations of the Janus (J1) and Twin (T1) structures. For clarity, 3,4,5-tris(11-hydroxyundecyloxy) and 3,4,5-tris(octyloxy) groups were detached from the MO profiles (refer to Fig. S3, ESI† for the full structures). | ||
The electronegativity (χ) represents the electronic attraction strength of a molecule. It is important as it indicates the tendency of a molecule to interact with appropriate species. It can be noticed from Table 2 that the electronegativity of J1 is slightly lower than that of T1, indicating that the Twin molecule is better positioned to attract electron pairs. The absence of the –OH groups in the T1 dendrimer results in a more positive nature of the inner shell of the molecule compared with the J1 analogue (Fig. 3). This could also explain the lower hardness (being softer), dipole moment, ionization energy, and chemical potential of J1 compared with T1. Dipole moments imply polarizability and chemical reactivity,40 and a comparison with the dipole moment of water41 concludes that both the Janus and Twin molecules are ionic, polarizable, and chemically reactive.
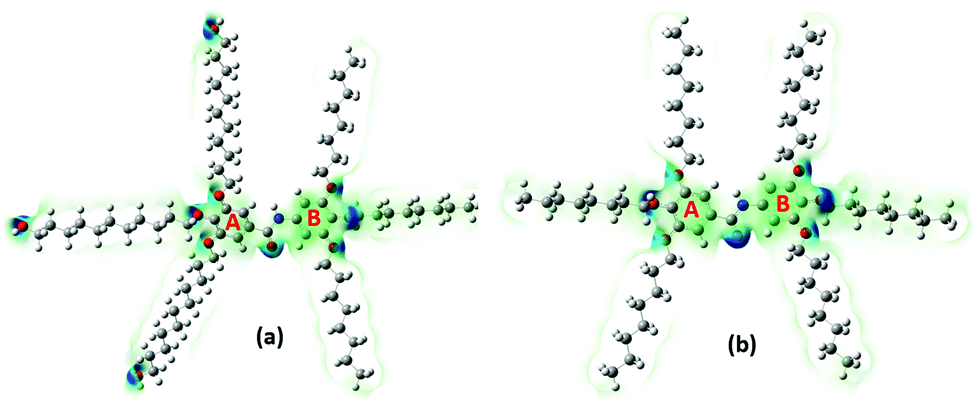 | ||
| Fig. 3 Total electron density maps of the two most stable configurations of the (a) Janus (J1) and (b) Twin (T1) benzamide structures. Different charge densities are shown via the varying color intensities. | ||
The computed surface electrostatic charge distributions depicted in Fig. 3 shed more light on the chemical reactivity. To better analyze the density maps, regions with a low potential are characterized with a relative electron abundance. On the other hand, regions with more transparent colors, such as the aliphatic chains, are of high potential and specify electron deficiency. The calculated charge distributions suggest that the Janus and Twin molecules would interact effectively through either the benzyl rings or the hydroxyl terminal moieties. One important observation from the calculated physical properties is the conformational dependency of the dipole moments on the benzene–O–CH2 ether linkage orientations. The two most stable forms of each branched benzamide system were predicted to have a moderate to substantial difference in dipole moment values. In particular, the dipole moment of J2 was calculated to be three times that of J1, while the T1 dipole moment is less than double the value for T2 (Table 1). The Janus molecule's dipole moment is more sensitive due to the presence of the terminal hydroxyl moieties. The electrical properties should be adjustable when an appropriate conformationally controlled synthesis is employed.
The absorption spectra of the molecules (Fig. 4) show intense peaks at λmax ≈ 292 nm, which is considered to be in the ultraviolet region and can be designated to the electronic π → π* transition42,43 that is primarily associated with the benzyl rings. The absorption band assigned to the n → π* amide carbonyl and CO group transitions44,45 should appear at a lower energy. However, this absorption could not be traced, probably due to band overlapping. Experimental spectral values were found to be consistent with the theoretical ones, in particular with the observed maximum wavelength. The electronic spectral characteristics indicate that both of the colorless benzamide compounds strongly absorb at the UV–Vis region. The shoulder peaks appearing from 235 nm to 250 nm are attributed to the chloroform solvent.42 Moreover, the experimental and theoretical (J1 and T1) UV–Vis data were treated using the Tauc plot extrapolation method,46,47 as presented in Fig. 5 and Fig. S5 (ESI†). The energy band gaps obtained from the experimental UV–Vis spectra were in excellent agreement with the theoretical UV–Vis values of the Janus (J1) and Twin (T1) molecules. Given that the EHOMO–ELUMO gap can be closely related to the energy band gap,48 the consistent experimental and theoretical band gaps deduced from the UV–Vis spectra showed a reliable agreement with the computed ΔE (Fig. 2). The Janus benzamide, thus, depicts a very slightly smaller energy band.
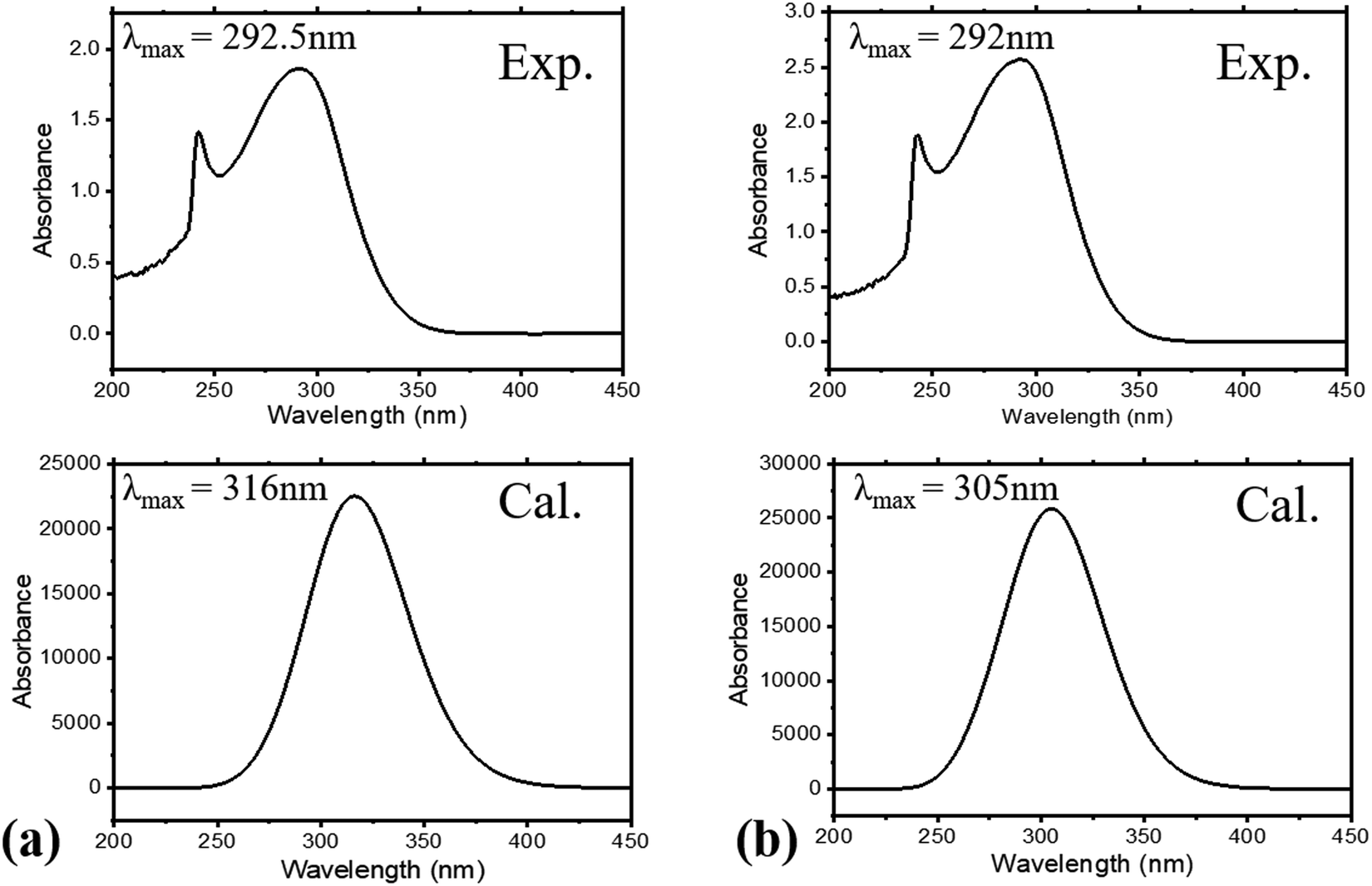 | ||
| Fig. 4 Experimental and calculated UV–Vis spectra for (a) Janus benzamide (J1) and (b) Twin benzamide (T1) systems. | ||
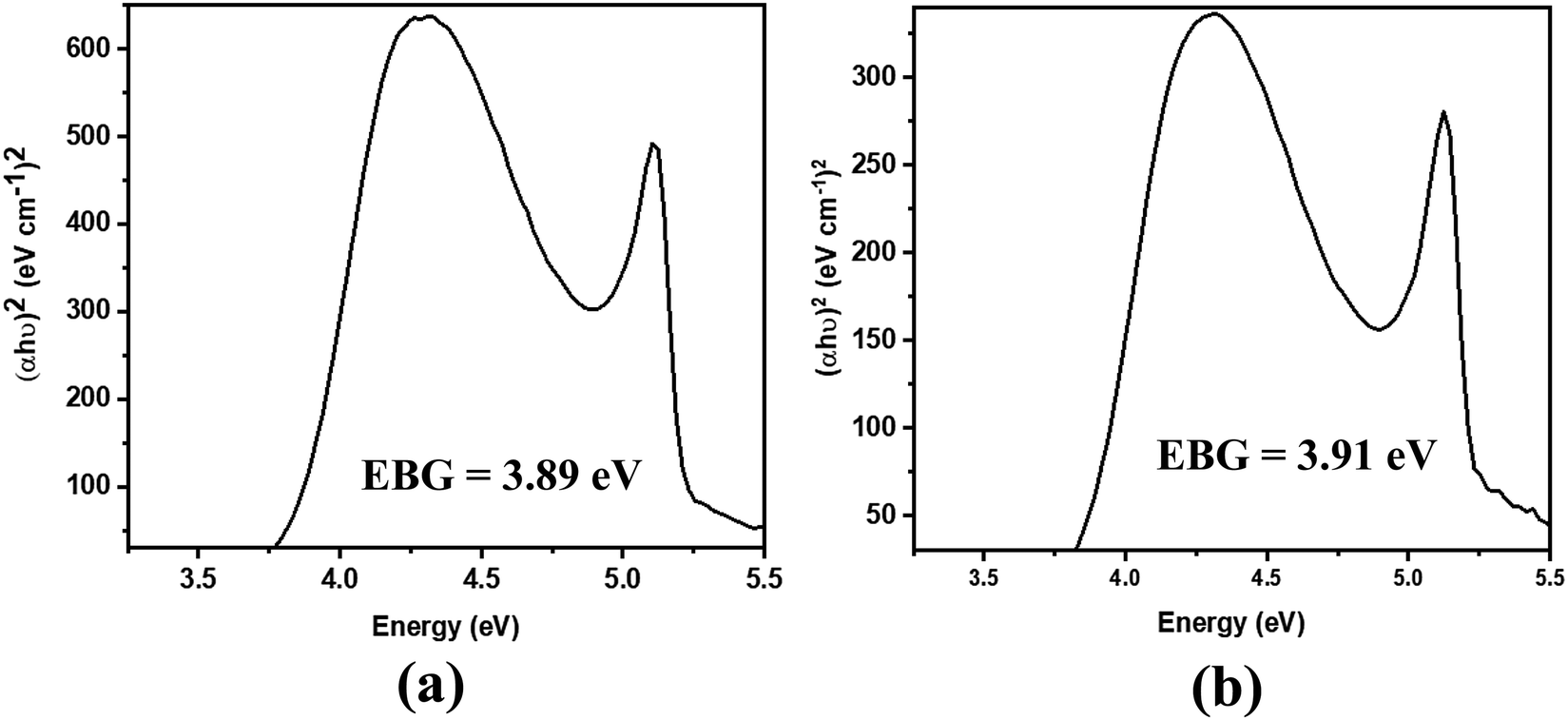 | ||
| Fig. 5 Derived energy band gaps (EBGs) of the (a) Janus benzamide (J1) and (b) Twin benzamide (T1) systems using the Tauc method. | ||
The calculated atomic polar tensor (APT) charges49 listed in Table 3 can be useful in identifying sites agile for nucleophilic and electrophilic interactions. The O(2) and N(3) atoms of the amide group in both the Janus and Twin benzamides are considered nucleophilic and would facilitate electrophilic types of attack. For ring “A” where the unoccupied molecular orbitals are positioned, the atoms C(5), C(6), and C(7) are more nucleophilic in nature, whereas their C(8), C(10), and C(12) counterparts are considered electrophilic and hence are better electron acceptors during a self-assembly charge-transfer process. Such behavior can be attributed to the π-electron resonance across the benzene rings and the inductive effect caused by the aliphatic chain substituents bonded to the electrophilic side of the ring.50,51 All of the hydroxyl oxygens are nucleophilic and hence are capable sites for the electrophilic attack of substrates like acids. Although the APT charge values are consistently comparable for both molecules, the absence of the nucleophilic oxygen atoms [O(201), O(203), and O(205)] from the Twin structure suggests that the Janus structure would more likely be nucleophilic, as was concluded from their total electron density maps (Fig. 3). The terminal hydroxyl oxygen atoms in the most stable J1 form are characterized with an sp3 environment reflected by the calculated angles (C–O–H = 108°) compared with the inside ether-type oxygen atoms (C–O–C = 115°–119° for J1 and 117°–121° for T1), which mimic the phenol-like sp2 hybridization (Table S2, ESI†).
| Janus benzamide (J1) | Twin benzamide (T1) | ||||||
|---|---|---|---|---|---|---|---|
| Atom | Charge | Atom | Charge | Atom | Charge | Atom | Charge |
| C1 | 1.43731 | C16 | 0.78907 | C1 | 1.45337 | C16 | 0.73773 |
| O2 | −0.82486 | C18 | 0.77343 | O2 | −0.83092 | C18 | 0.70361 |
| N3 | −1.06227 | C20 | 0.25580 | N3 | −1.08025 | C20 | 0.28217 |
| H4 | 0.18988 | O21 | −1.08337 | H4 | 0.19284 | O21 | −1.07465 |
| C5 | −0.12633 | O22 | −1.04750 | C5 | −0.14829 | O22 | −1.03386 |
| C6 | −0.27018 | O23 | −1.02031 | C6 | −0.26543 | O23 | −1.00661 |
| C7 | −0.24880 | O24 | −1.03640 | C7 | −0.21012 | O24 | −0.99641 |
| C8 | 0.64936 | O25 | −1.00740 | C8 | 0.61329 | O25 | −1.01128 |
| C10 | 0.72039 | O26 | −1.03995 | C10 | 0.69781 | O26 | −1.03276 |
| C12 | 0.38690 | O201 | −0.62781 | C12 | 0.39327 | ||
| C13 | 0.72934 | O203 | −0.62774 | C13 | 0.71949 | ||
| C14 | −0.41835 | O205 | −0.62819 | C14 | −0.37551 | ||
| C15 | −0.40137 | C15 | −0.36975 | ||||
The recorded infrared and Raman spectra of the Janus and Twin benzamides are presented in Fig. 6 and 7 and are compared with the calculated spectra of the J1 and T1 conformers. The assignments of the key vibrational modes are listed in Table 4. The infrared spectrum of the Janus molecule is characterized by two broad bands located at 3355 cm−1 (assigned to the terminal OH stretching) and 3223 cm−1 (attributed to the NH stretching of the core amide). According to Fig. 6, these two bands appear broad, indicating strong intermolecular hydrogen bonding. Hydrogen bonding is a crucial determinant of the structure and features of the dendrimers. The corresponding calculated wavenumbers for the isolated J1 were shown to be considerably higher at 3642 and 3489 cm−1, respectively, due to the absence of such forces. In the case of the Twin analogue and due to the absence of hydroxyl groups, only one broad band in the infrared spectrum at 3231 cm−1 (DFT at 3486 cm−1) could be observed and is attributed to NH stretching. The slight difference in the wavenumbers the between Twin and Janus molecules (of the order of 8 cm−1) could support the more pronounced intramolecular N–H⋯N type of bonding in the case of the Twin molecule. Generally, these observed infrared bands confirm that both benzamides are capable of undergoing hydrogen bonding with appropriate species.16 It is established that there is a relationship between the strength of the hydrogen bond and the shift of NH to low frequency. The in-plane NH bending bands were observed at 1598 cm−1 (infrared) and 1597 cm−1 (Raman) for the Janus molecules and at 1596 cm−1 (both infrared and Raman) for the Twin molecules (Table 4). The corresponding calculated wavenumbers are 1619 cm−1 and 1644 cm−1 for J1 and T1, respectively. The frequency difference, Δν (calc. − expt.), turns out to be 22 cm−1 in the case of the Janus molecule and 48 cm−1 in the case of the Twin molecule, confirming the stronger intermolecular interaction associated with the amide moiety. Similarly, the O–H bending IR absorption appears nearly at 1388 cm−1 (DFT at 1416 cm−1) in the case of the Janus benzamide.
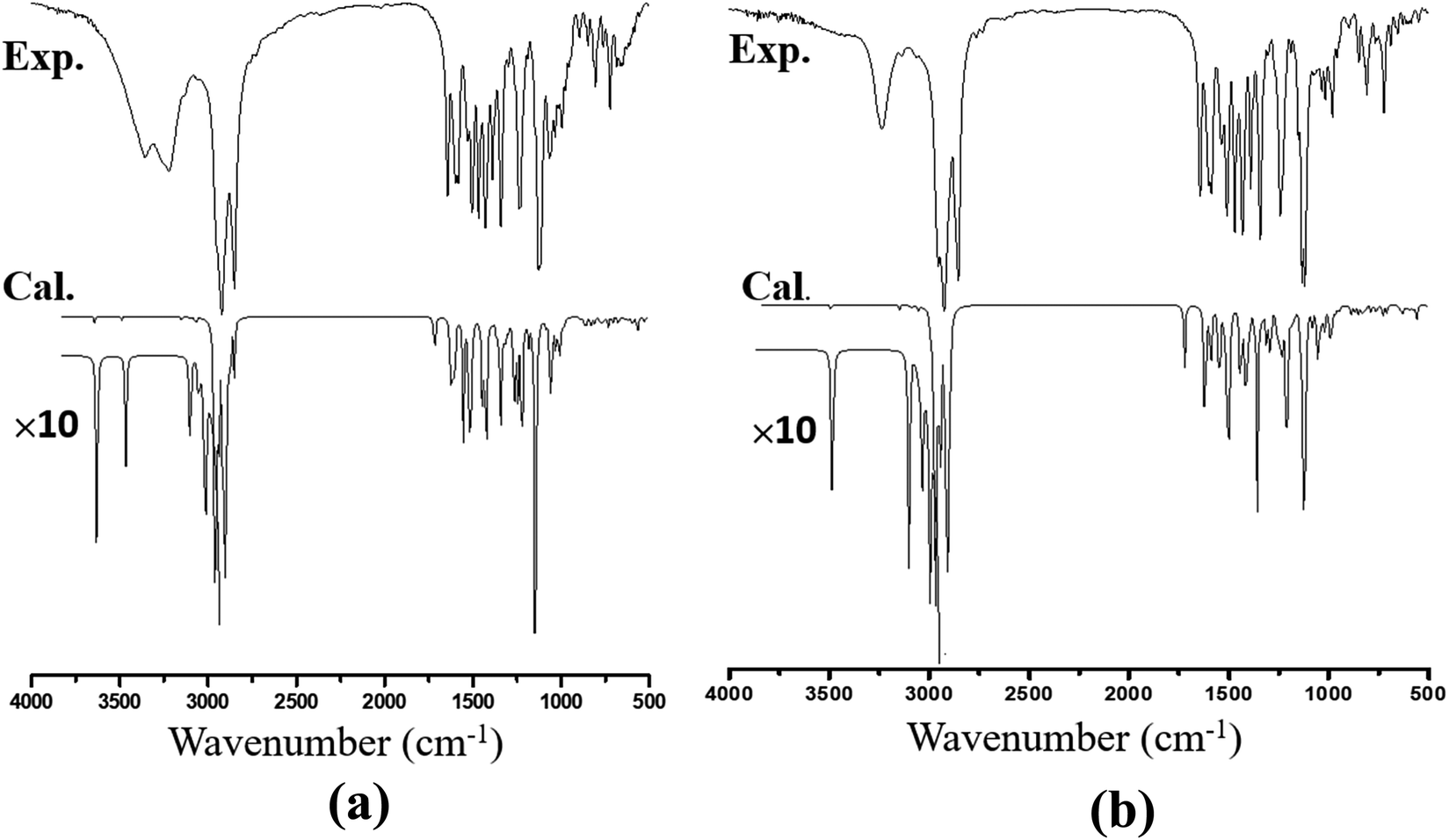 | ||
| Fig. 6 Calculated and experimental FT-IR spectra of (a) Janus benzamide and (b) Twin benzamide molecules. “×10” represents a ten-fold magnification of the theoretical intensities in the regions of interest. | ||
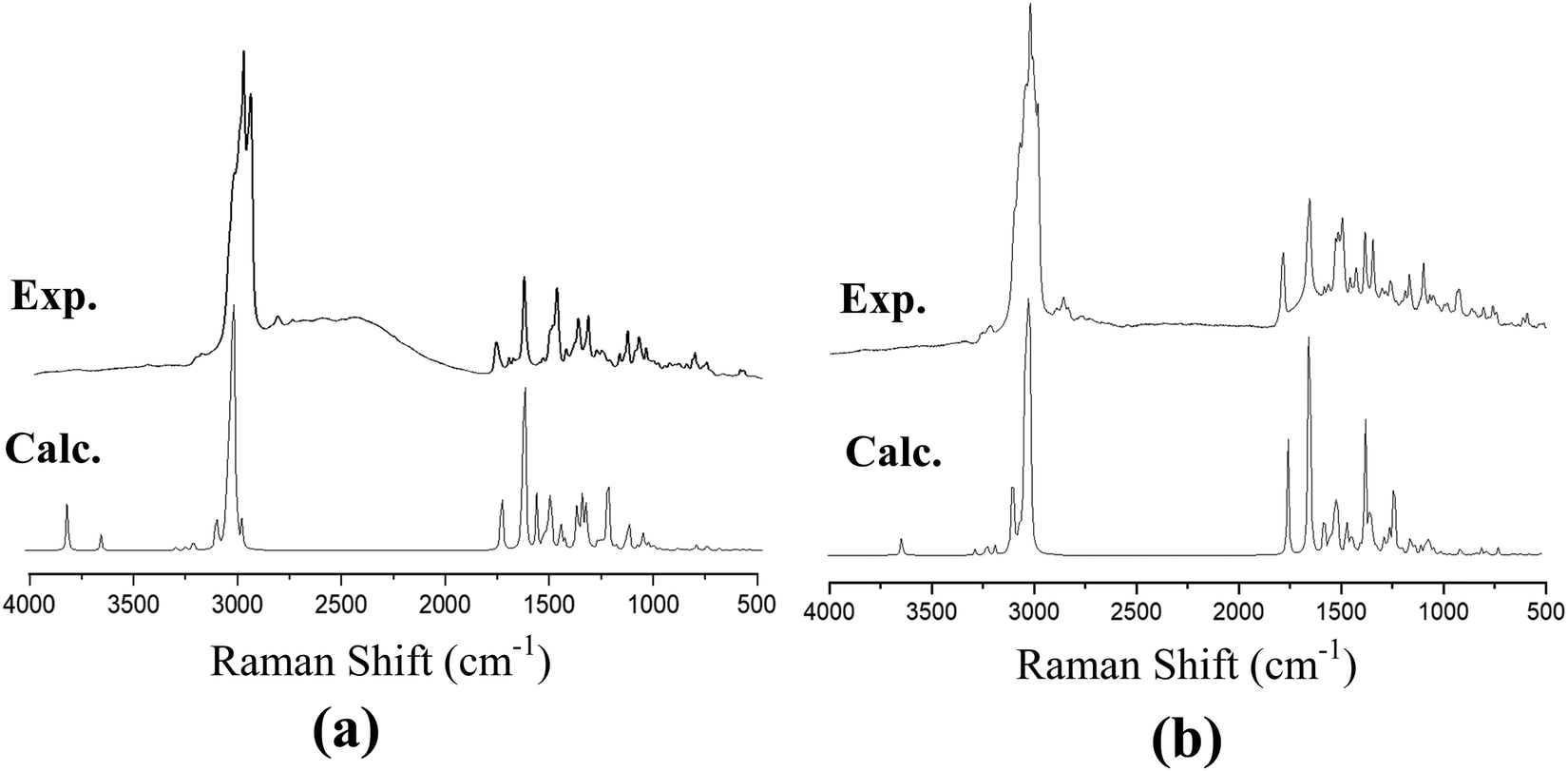 | ||
| Fig. 7 Calculated and experimental Raman spectra of (a) Janus benzamide, and (b) Twin benzamide molecules. | ||
| Janus dendrimer | Twin dendrimer | Assignment | ||||
|---|---|---|---|---|---|---|
| Exp. | Calc. | Exp. | Calc. | |||
| IR | Raman | IR | Raman | |||
| 3355(s) | 3351(w) | 3642 | — | — | — | OH stretching |
| 3223(s) | 3226(w) | 3489 | 3231(s) | 3239(w) | 3486 | NH stretching |
| 3139(w) | 3136(m) | 3148 | 3134(w) | 3130(m) | 3141 | CH stretching |
| 2922(vs) | 2918(vs) | 2966 | 2951(vs) | 2956(s) | 2987 | CH2 stretching |
| 2850(vs) | 2855(vs) | 2847 | 2852(vs) | 2845(vs) | 2879 | CH2 stretching, CH3 stretching |
| 1642(s) | 1637(w) | 1708 | 1640(s) | 1644(m) | 1714 | CO stretching |
| 1598(s) | 1597(s) | 1619 | 1596(s) | 1596(s) | 1644 | NH bending |
| 1583(s) | 1588(s) | 1610 | 1584(s) | 1590(m) | 1607 | Ring stretching |
| 1504(s) | 1509(w) | 1594 | 1507(s) | 1503(s) | 1584 | Ring stretching |
| 1429(s) | 1422(w) | 1451 | 1428(s) | 1420(w) | 1495 | CH2 bending |
| 1388(s) | 1386(w) | 1416 | — | — | — | OH bending |
| 1341(s) | 1347(s) | 1406 | 1341(s) | 1344(s) | 1405 | CH3 bending |
| 1299(w) | 1301(s) | 1356 | 1300(w) | 1300(s) | 1372 | Ring (A) breath |
| 1237(s) | 1233(w) | 1323 | 1240(s) | 1242(w) | 1364 | Ring (B) breath |
| 1121(s) | 1120(s) | 1191 | 1120(s) | 1125(s) | 1181 | CH in-plane bending |
| 1128(s) | 1133(m) | 1180 | 1131(s) | 1130(s) | 1180 | CN stretching |
| 1066(s) | 1069(m) | 1134 | 1117(s) | 1120(m) | 1128 | CO stretching |
| 807(s) | 810(s) | 875 | 807(s) | 810(s) | 876 | CH out-of-plane bending |
| 760(m) | 762(w) | 780 | 763(m) | 764(w) | 780 | CH2 bending |
| 480(m) | 486(w) | 495 | 476(m) | 470(w) | 493 | CH3 bending |
The stretching of the C–H bonds associated with the aromatic rings is straightforwardly assigned to the intense Raman peaks in the spectral area just above 3000 cm−1, as can be seen in Fig. 7, which is in excellent agreement with the DFT results (Table 4). The corresponding in-phase and out-of-phase C–H bending vibrations (normally mixed with other vibrations) can be best assigned, respectively, to 1120 cm−1 (DFT at 1191 cm−1) and 810 cm−1 (DFT at 875 cm−1) for the Janus molecule and 1125 cm−1 (DFT at 1181 cm−1) and 810 cm−1 (DFT at 876 cm−1) for the Twin molecule in the Raman spectra. The infrared bands observed in the region below 3000 cm−1 (Fig. 6) are assigned to the aliphatic chain CH2 and CH3 stretching modes. The CO stretching vibration of the amide group is assigned to the infrared peak centered at 1642 cm−1 (1637 cm−1 in the Raman spectra) as can be seen in the Janus spectra. This peak was calculated at 1708 cm−1. For the Twin counterpart, the carbonyl group absorbs strongly in the infrared spectrum at 1640 cm−1 (1644 cm−1 in Raman and 1714 cm−1 from DFT).
5. Conclusion
Two novel Janus and Twin generations of benzamide-based dendrimer precursors were successfully synthesized, and their structural, electronic, and vibrational properties were reported. Spectroscopic and theoretical methods were complementarily employed to develop a fundamental understanding of their energetics and reactivity. Of the five conformations theoretically determined, the J1, J2, T1 and T2 conformers were more structurally stable due to their least strained configurations. They are characterized with comparable EHOMO and ELUMO values exhibiting similar electronic features. Analysis of the frontier orbital energies suggested that these benzamides have the potential to undergo self-assembly with a π–π intramolecular stacking process. Furthermore, atomic charge analysis revealed that these benzamide structures possess potential electrophilic and nucleophilic sites within their structural backbone. The associated electron transfer is likely to be facilitated by the benzene rings in the core shell in an alternating A–B–A manner, as revealed from analysis of the frontier orbitals. Vibrational infrared and Raman spectra yielded consistent wavenumbers that are also consistent with the corresponding fundamental modes, confirming the structural properties. The structural properties, functional units, branching points, molecular weight, polarizability, and electronic features of these molecules indicate that they show potential for biomedical, catalytic, and pharmaceutical applications, especially in targeted drug delivery. Furthermore, they could serve as precursor materials for the synthesis of early generation benzamide-based dendrimers.Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to time limitations and will be made available by the corresponding author upon request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to acknowledge King Fahd University of Petroleum and Minerals (KFUPM) for supporting this work.References
- D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci. Part A Polym. Chem., 2002, 40, 2719–2728 CrossRef CAS.
- D. Astruc and F. Chardac, Chem. Rev., 2001, 101, 2991–3024 CrossRef CAS.
- Z. Xu, M. Kahr, K. L. Walker, C. L. Wilkins and J. S. Moore, J. Am. Chem. Soc., 2002, 116, 4537–4550 CrossRef.
- J.-P. Majoral and A.-M. Caminade, in Dendrimers, Springer Berlin Heidelberg, Berlin, Heidelberg, 1998, pp. 79–124 Search PubMed.
- A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665–1688 CrossRef CAS PubMed.
- S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74–91 CrossRef CAS.
- J. P. W. Eggert and U. Lüning, European J. Org. Chem., 2007, 2007, 6046–6052 CrossRef.
- M. Sajid, M. K. Nazal, I. Ihsanullah, N. Baig and A. M. Osman, Sep. Purif. Technol., 2018, 191, 400–423 CrossRef CAS.
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- M. Albrecht, M. Schlupp, J. Bargon and G. van Koten, Chem. Commun., 2001, 1874–1875 RSC.
- V. M. Rotello, J. Am. Chem. Soc., 2007, 129, 14524 CrossRef CAS.
- D.-Y. Li, S.-W. Li, Y.-L. Xie, X. Hua, Y.-T. Long, A. Wang and P.-N. Liu, Nat. Commun., 2019, 10, 2414 CrossRef PubMed.
- V. Percec, C.-H. Ahn, T. K. Bera, G. Ungar and D. J. P. Yeardley, Chem. – Eur. J., 1999, 5, 1070–1083 CrossRef CAS.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009LP–1014 CrossRef.
- V. Percec, M. R. Imam, T. K. Bera, V. S. K. Balagurusamy, M. Peterca and P. A. Heiney, Angew. Chem., Int. Ed., 2005, 44, 4739–4745 CrossRef CAS.
- V. L. Furer, A. E. Vandyukov, V. Tripathi, J. P. Majoral, A. M. Caminade and V. I. Kovalenko, Spectrochim. Acta, Part A, 2018, 194, 211–221 CrossRef CAS.
- A.-M. Caminade, Chem. Commun., 2017, 53, 9830–9838 RSC.
- A.-M. Caminade, R. Laurent, B. Delavaux-Nicot and J.-P. Majoral, New J. Chem., 2012, 36, 217–226 RSC.
- J. Nithyanandhan and N. Jayaraman, J. Org. Chem., 2002, 67, 6282–6285 CrossRef CAS.
- Y. Ito, I. Washio and M. Ueda, Macromolecules, 2008, 41, 2778–2784 CrossRef CAS.
- L. Zhou, Y. Shan, H. Hu, B. Yu and H. Cong, Curr. Org. Chem., 2018, 22, 600–612 CrossRef CAS.
- Y. Ito, T. Higashihara and M. Ueda, Macromolecules, 2012, 45, 4175–4183 CrossRef CAS.
- Ö. İnce, E. Akyol, E. Sulu, T. Şanal and B. Hazer, J. Polym. Res., 2015, 23, 5 CrossRef.
- I. Abdulazeez, M. Khaled and A. A. Al-Saadi, J. Mol. Struct., 2019, 1196, 348–355 CrossRef CAS.
- J. H. Shibata, J. Chem. Educ., 2012, 89, 1489–1490 CrossRef CAS.
- R. G. Pearson, Inorg. Chem., 2002, 27, 734–740 CrossRef.
- D. J. F. M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada and M. Ehar, 2016.
- J. Dennington, R. Keith and T. Millam, 2009.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 2002, 105, 7512–7516 CrossRef.
- M. Umar, C. C. Nnadiekwe, I. Abdulazeez, K. Alhooshani and A. A. Al-Saadi, Arab. J. Sci. Eng., 2021 DOI:10.1007/s13369-021-05648-x.
- C. C. Nnadiekwe, I. Abdulazeez, M. Haroon, Q. Peng, A. Jalilov and A. Al-Saadi, Front. Chem., 2021, 9, 274 Search PubMed.
- C. C. Nnadiekwe, I. Abdulazeez, A. Matin, M. M. Khaled, M. Khan, D. Anand and I. Ahmad, ACS ES&T Water, 2021, 1(5), 1136–1144, DOI:10.1021/acsestwater.0c00168.
- T. N. V. de Souza, S. M. L. de Carvalho, M. G. A. Vieira, M. G. C. da Silva, D. do and S. B. Brasil, Appl. Surf. Sci., 2018, 448, 662–670 CrossRef.
- H. Wang, X. Wang, H. Wang, L. Wang and A. Liu, J. Mol. Model., 2007, 13, 147–153 CrossRef CAS PubMed.
- T. Tada, D. Nozaki, M. Kondo and K. Yoshizawa, J. Phys. Chem. B, 2003, 107, 14204–14210 CrossRef CAS.
- J. Giarolla, K. F. M. Pasqualoto and E. I. Ferreira, Mol. Diversity, 2013, 17, 711–720 CrossRef CAS PubMed.
- V. Percec, M. Glodde, T. K. Bera, Y. Miura, I. Shiyanovskaya, K. D. Singer, V. S. K. Balagurusamy, P. A. Heiney, I. Schnell, A. Rapp, H.-W. Spiess, S. D. Hudson and H. Duan, Nature, 2002, 419, 384–387 CrossRef CAS PubMed.
- D. Adam, P. Schuhmacher, J. Simmerer, L. Häussling, K. Siemensmeyer, K. H. Etzbachi, H. Ringsdorf and D. Haarer, Nature, 1994, 371, 141–143 CrossRef CAS.
- S. P. Brown, I. Schnell, J. Diedrich Brand, K. Müllen and H. Wolfgang Spiess, J. Am. Chem. Soc., 1999, 121, 6712–6718 CrossRef CAS.
- B. Haddad, S. A. Brandán, M. A. Assenine, A. Paolone, D. Villemin and S. Bresson, J. Mol. Struct., 2020, 1212 DOI:10.1016/j.molstruc.2020.128104.
- J. K. Gregory, D. C. Clary, K. Liu, M. G. Brown and R. J. Saykally, Science, 1997, 275, 814LP–817 CrossRef.
- H. Kunkely and A. Vogler, Inorg. Chem. Commun., 2001, 4, 692–694 CrossRef CAS.
- B. Liang, L. Wang, Y. H. Xu, H. H. Shi and Y. Cao, Adv. Funct. Mater., 2007, 17, 3580–3589 CrossRef CAS.
- M. Gruzdev, U. Chervonova, T. Frolova and A. Kolker, Liq. Cryst., 2017, 44, 322–331 CAS.
- V. Romanova, V. Begishev, V. Karmanov, A. Kondyurin and M. F. Maitz, J. Raman Spectrosc., 2002, 33, 769–777 CrossRef CAS.
- J. Tauc and A. Abraham, Phys. Lett. A, 1968, 27, 98–99 CrossRef CAS.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- J. Roshan De Lile, S. Gu Kang, Y.-A. Son and S. Geol Lee, ACS Omega, 2020, 5, 15052–15062 CrossRef.
- M. M. C. Ferreira, J. Mol. Struct., 1993, 294, 75–78 CrossRef CAS.
- P. Rani, A. Husain, A. Shukla, N. Singla, A. K. Srivastava, G. Kumar, K. K. Bhasin and G. Kumar, CrystEngComm, 2021, 23, 1859–1869 RSC.
- J. Park, J. Sung and D. Kim, J. Phys. Chem. Lett., 2021, 12, 2226–2231 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nj04366h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |