
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A shear-thickening colloidal electrolyte for aqueous zinc-ion batteries with resistance on impact†
Haobo
Dong
ab,
Xueying
Hu
b and
Guanjie
He
 *a
*a
aElectrochemical Innovation Lab, Department of Chemical Engineering, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: g.he@ucl.ac.uk
bChristopher Ingold Laboratory, Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
First published on 12th September 2022
Abstract
A conventional aqueous electrolyte is a crucial component of zinc-ion batteries providing an ion conductive medium. However, the monofunction of a liquid electrolyte cannot bear any external load. With regard to applications in electric vehicles and stationary energy storage devices, complicated battery packing materials are required to improve the mechanical properties, resulting in reduced energy or power densities from the perspective of the entire device. In this work, an electrolyte suspension combining both fluid-like and solid-like performances was developed for rechargeable zinc-ion batteries. Cornstarch water suspension is utilized in the electrolyte design forming a shear-thickening electrolyte with impact resistance ability. The formed electrolyte becomes rigid at a high shear rate. In other words, under a sudden impact, a battery with this shear-thickening electrolyte could offer additional load bearing avoiding short-circuiting and improving safety. Although an additional functionality, namely impact resistance, was added to the electrolyte, the as-prepared electrolyte still performs with comparable electrochemical performances for which it exhibits a superior ionic conductivity of 3.9 × 10−3 S cm−1 and Zn2+ transference number. This electrolyte even suppresses side-effects on the zinc anode, exhibiting a lower voltage gap in the symmetric cell compared to the aqueous electrolyte. The integrated full cell also delivered a specific capacity of 255 mA h g−1 with commercial MnO2 as the cathode at a current density of 0.1 A g−1.
 Guanjie He | Dr Guanjie He is a Lecturer in Advanced Propulsion at the Department of Chemical Engineering, University College London (UCL). Prior to this, Guanjie held faculty positions at the University of Lincoln and Queen Mary University of London. He obtained his PhD degree from the Department of Chemistry, UCL. Guanjie is rapidly expanding his research activities through a number of collaborations in diverse areas from the core focus of aqueous energy storage and conversion materials and devices, such as Zn-ion batteries, fuel cells and electrocatalysts, to advanced characterisation and simulation. |
Introduction
With a clear timeline for the carbon emission peak and carbon neutrality, rechargeable batteries continue to attract wide attention from both academic and industrial researchers. Currently, lithium-ion batteries (LIBs) still dominate most of the market; however, their expense and the limited abundance restrict the progress of the final target and achieving net zero carbon. With advantages in price, ionic conductivity, easy fabrication and robustness in harsh environments, rechargeable aqueous multivalent ion batteries (Al3+, Mg2+, Zn2+, Ca2+)1 are considered as the next-generation alternatives for current energy storage strategies. Among these multivalent ion batteries, rechargeable aqueous Zn-ion batteries (AZIBs) attract the most attention due to the high theoretical volumetric capacity of zinc metal (5851 mA h cm−3)2–4 and high power density with a magnitude of 103–104 kW kg−1. Moreover, the cost of core components is only $25 for a single AZIB battery unit compared with $165 for LIBs,5,6 which is of interest in both grid/off-grid energy storage and electrical vehicles (EVs).For ZIBs, owing to inhomogeneous zinc electrodeposition at the anode, the formation of zinc dendrites is a risk resulting in short-circuiting. Side-effects such as hydrogen evolution reactions (HERs), accompanied by plating, also facilitate dendrite formation.7–9 This situation could be even worse if the energy storage device suffers from a sudden external impact, such as a car crash or damage caused by extreme weather. To avoid fire and explosion, extensive work related to battery structural design, such as the blade battery,10 has been pursued. However, additional materials contribute extra weight, which is detrimental to light-weight structures. Veith and co-workers11 have developed an impact resistance electrolyte for lithium-ion batteries based on fume silicas, although there is no improvement of electrolytes for aqueous batteries, especially for AZIBs. Hence, in this work, the electrolyte has been embedded as a load-bearing component protecting the battery in addition to offering electrochemical properties. A shear-thickening electrolyte (STE) based on cornstarch suspensions has been designed for AZIBs to protect the battery under a sudden external force while providing competitive performances. Under normal conditions, this STE is in the liquid phase exhibiting an excellent specific capacity (255 mA h g−1) and ionic conductivity (3.9 × 10−3 S cm−1), while under a large shear stress the electrolyte solidifies in a certain region, as shown in Fig. 1a, avoiding an internal short circuit under any sudden external force. Besides, the colloid electrolyte also delivers a stable zinc plating/striping ability, for which a lower overpotential of 3.1 mV has been attained compared to 6.3 mV of the aqueous electrolyte. This STE strategy could also be applied to other aqueous multivalent-ion batteries apart from AZIBs.
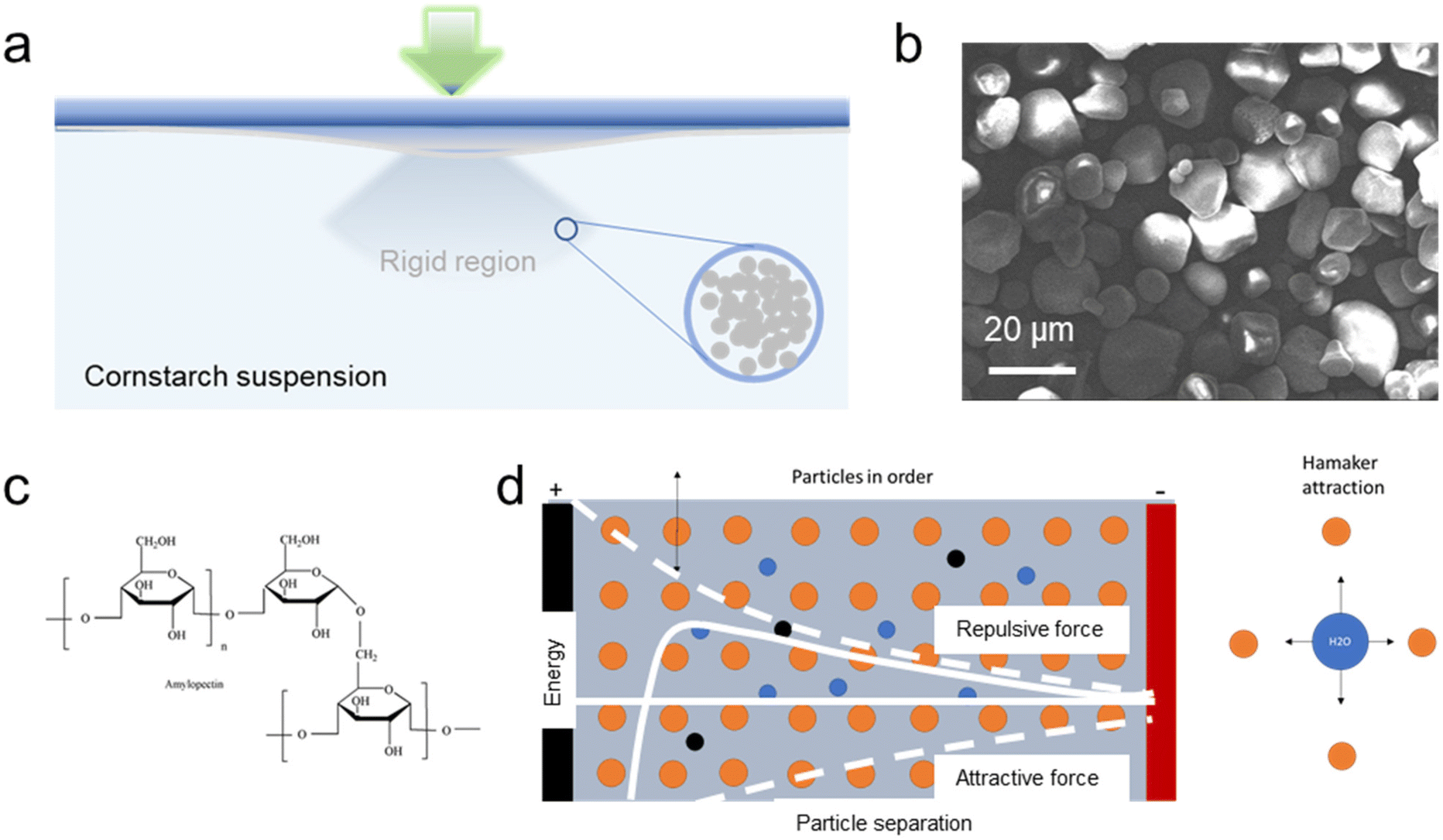 | ||
| Fig. 1 (a) Schematic diagram of the cornstarch shear-thickening electrolyte; (b) a SEM image of cornstarch particles; (c) cornstarch molecular structure; (d) a schematic diagram of Hamaker attraction in the suspension. | ||
Materials and methods
Cornstarch, a multi-branched polysaccharide of glucose, is a common type of biomass food additive and industrial adhesive, which can form a cornstarch–water suspension. As shown in an SEM image (Fig. 1b), cornstarch particles are of irregular shape but are relatively monodisperse. Amylose and amylopectin linked R-D-glucose residues are the main branch constituents in the structure (Fig. 1c). Non-Newtonian ability is exhibited once the weight ratio of cornstarch to water in the suspension is above 55%.12–14 The fine suspension displays peculiar phenomena, exhibiting solid-like and fluid-like behaviors at the same volume fraction.15 As reported, the typical viscosity–shear stress relation of a non-Newtonian cornstarch suspension is described as shear-thinning behaviours where viscosity decreases with an increase of the stress over a low stress range. However, after the shear rate exceeds a threshold magnitude, normally 2 s−1, there is a sudden increase in viscosity resulting in a shear-thickening behaviour.12,16 The suspension rheology can be described by the Hamaker theory combining the repulsion and attraction of two particles. A long-range repulsive force is dominant at long distances, while at a short distance, Hamaker attraction dominates, preventing the particles falling from the stable suspension. Hamaker attraction is inversely proportional to the square of the distance. As shown in Fig. 1d, when shearing a concentrated and stabilized solution at a relatively low shear rate, the repulsive particle–particle interactions keep the particles in an ordered, layered, and equilibrium structure. However, at shear rates elevated above the critical shear rate, the shear forces pushing the particles together overcome the repulsive particle–particle interactions, inducing the particles out of their equilibrium positions. This leads to a disordered structure, causing an increase in viscosity.17 Hydroclusters can be formed during the transition from an ordered to mobile state, for which the particles aggregate together momentarily and form an incompressible and jammed network.15,18 Because of the high concentration, Colin19 elucidated that for a concentrated suspension the rheological properties vary independently of the storage conditions or cell orientation. The equilibrium of the particle–particle Hamaker attractions prevents the collisions between cornstarch particles, alleviating amylopectin erosion for starch dissolution, which is beneficial for the battery electrolyte.The shear-thickening electrolyte suspension was prepared by forming a stable suspension with 55 wt% cornstarch in a 3 M ZnSO4 and 0.2 M MnSO4 aqueous electrolyte. Under a high-rate stirring process, the as-prepared electrolyte exhibits a strong resistance force; however, it performs like a liquid if the stirring rate is low. As shown by the FTIR spectrum of a cornstarch suspension in Fig. S1,† characteristic peaks at 1000 cm−1 and 1642 cm−1 are typical C–O stretching in glucose and bound water, respectively. The broad band at 3300 cm−1 corresponds to –OH in cornstarch, while the band at 2935 cm−1 is for CH stretching in the structure.20,21 The electrochemical performances of the cornstarch shear-thickening and aqueous electrolytes are compared in this work.
Results and discussion
The electrochemical performance of the batteries with the colloidal electrolytes has been compared with those with the 3 M ZnSO4 aqueous electrolyte. The Zn||Zn cyclic voltammetry (CV) test, illustrated in Fig. S2,† demonstrates a similar zinc stripping/plating window, where the plating/stripping voltages of the STE are located at ± 0.65 V close to electroplating in the aqueous electrolyte. Besides, the STE reduces the self-corrosion of the zinc anode. The corrosion test was performed by linear sweep voltammetry (LSV) at a scan rate of 1 mV s−1 with the reference electrode Ag/AgCl. As shown in the Tafel polarisation curve (Fig. 2a), the current density is confirmed to be reduced for the STE from −1.82 log(mA) to −2.59 log(mA). Meanwhile, the corrosion potential has also reduced to −0.97 V compared to −1.01 V for the aqueous electrolyte. The reduced current density and potential refer to low corrosion and polarisation at the anode.22,23 The zinc transference number was also calculated according to the equation shown below. The Zn2+ transference number (t+) was measured by potentiation polarisation (∇V = 5 mV) embedded with electrochemical impedance spectroscopy (EIS) before and after the polarisation. R0 and Rss are resistances for the initial and the steady-state impedance, respectively. I0 and Iss are the initial and the steady-state current after 1200 seconds. As displayed in Fig. 2b, the steady-state current of the STE is 1.8 μA, which is lower than the 6.3 μA of the aqueous electrolyte, indicating that the STE stabilises the electrode polarisation process. With regard to EIS spectra (Fig. 2c), the ionic conductivities of the STE are at a magnitude of 10−3 S cm−1 for bulk resistances, and are 3.9 × 10−3 S cm−1 and 3.3 × 10−3 S cm−1 for the initial and the steady-state, respectively. As shown in Fig. 2d, the cation transference number of the STE is 0.251, which is similar to that for aqueous electrolytes. Hence, the concentrated suspension also exhibits considerable Zn2+ transportation. The STE is able to expand the voltage potential range and reduce the electrode–electrolyte polarisation without affecting the cation transference number. Moreover, under a sudden external impact, the STE still exhibits a superior recovery in the transference number. As shown in Fig. S11c,† a quick current variation is attained under the impact, which has resulted from the phase variation of the colloid from fluid to rigid. The current firstly decreases because of the increase of the internal resistance of the rigid colloidal electrolyte; however, after the impact, the current quickly increases to its initial value and decreases during the relaxation process. The Zn2+ transference number determined for this impacted battery is 0.255, which is in the same range when compared to the colloid electrolyte without impact, namely 0.251. There is only an instantaneous influence of the Zn2+ transference number of the STE under external impact where the rigid state of the electrolyte exhibits a greater internal resistance, while the rigid colloid electrolyte recovers to the fluid state after the sudden impact.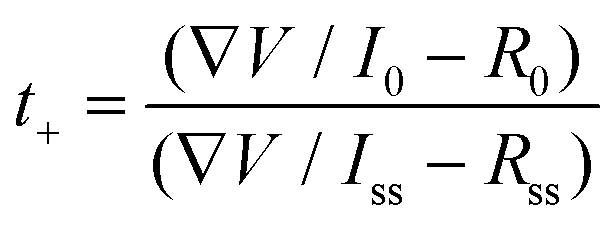 | (1) |
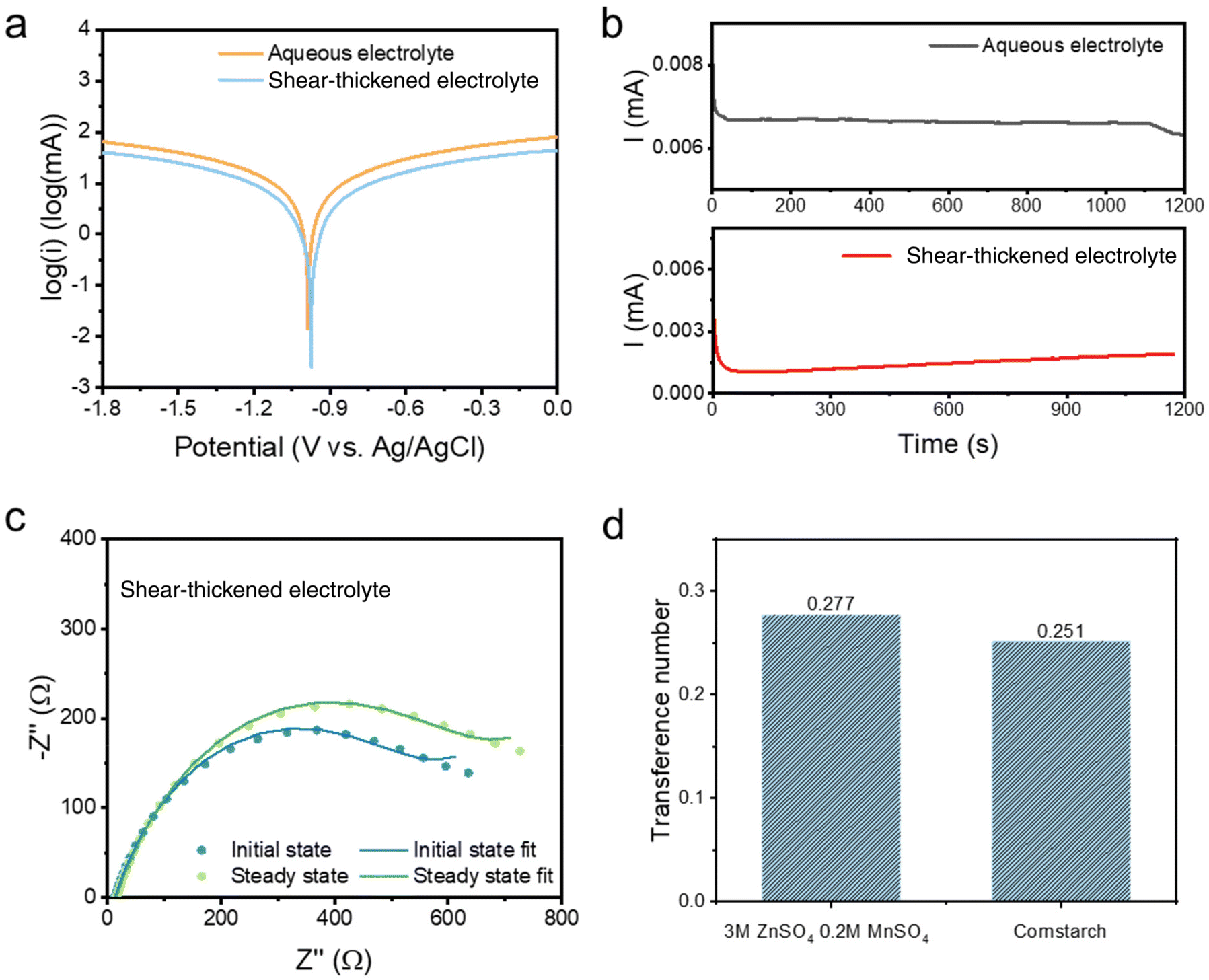 | ||
| Fig. 2 (a) Tafel polarisation profile in the symmetric cell. (b) Current profile of the STE and the aqueous electrolyte in the Zn||Zn symmetric cell under potentiostatic polarisation (∇V = 5 mV) with the reference electrode Ag/AgCl; (c) Nyquist plots before and after 1200 s of potentiostatic polarisation. (Initial and steady states represent the EIS profiles before and after chronoamperometry polarization, respectively.) (d) Zn2+ transference number of the STE and the aqueous electrolyte. | ||
The electrochemical performance evaluated in the symmetric cell also confirms the protection of the Zn anode in the STE. The zinc plating/stripping cycling performance of Zn|STE|Zn, as shown in Fig. 3a, exhibits a constant voltage profile over 200 hours at a low current density of 0.5 mA cm−2 and a low area capacity of 0.5 mA h cm−2, whereas the pristine aqueous electrolyte only maintains a stable profile for 100 hours followed by fluctuation short circuiting. As displayed in the enlarged plot, Fig. 3b, the voltage difference of the STE is about 0.095 V with a voltage hysteresis of 47 mV; however, as a comparison, the voltage difference of the pristine electrolyte is 0.135 V with a voltage hysteresis of 68 mV. Moreover, the flat plateaus are achieved with the STE after the relaxation period instead of a high slope profile, which also indicates the low polarisation within Zn|STE|Zn. A lower overpotential of 3.1 mV was achieved with the STE compared to 6.3 mV for the aqueous electrolyte (Fig. S4†). Hence, the concentrated suspension provides a uniformly distributed electrical field between electrodes and stabilises the zinc plating/stripping at the electrolyte–electrode interface. The concentrated cornstarch suspension acts as a donor and an acceptor not only restricting the movement of water molecules by strengthening hydrogen bonds, but also suppressing side effects by adsorbing anions. Combined with the low current in the Tafel polarisation test, the shear-thickening suspension delivers uniform plating and a thin passivation film by offering a uniform electric field for diffusion in the electrolyte and reducing the nucleation energy barrier at the interface.24 The low polarisation current and overpotential could also result in a dendrite-free Zn anode. The SEM images shown in Fig. S7† further elucidate the surface morphology of the Zn anode. After 50 cycles, a smooth anode surface was attained with the STE compared to a flower-like dendrite for the aqueous electrolyte.
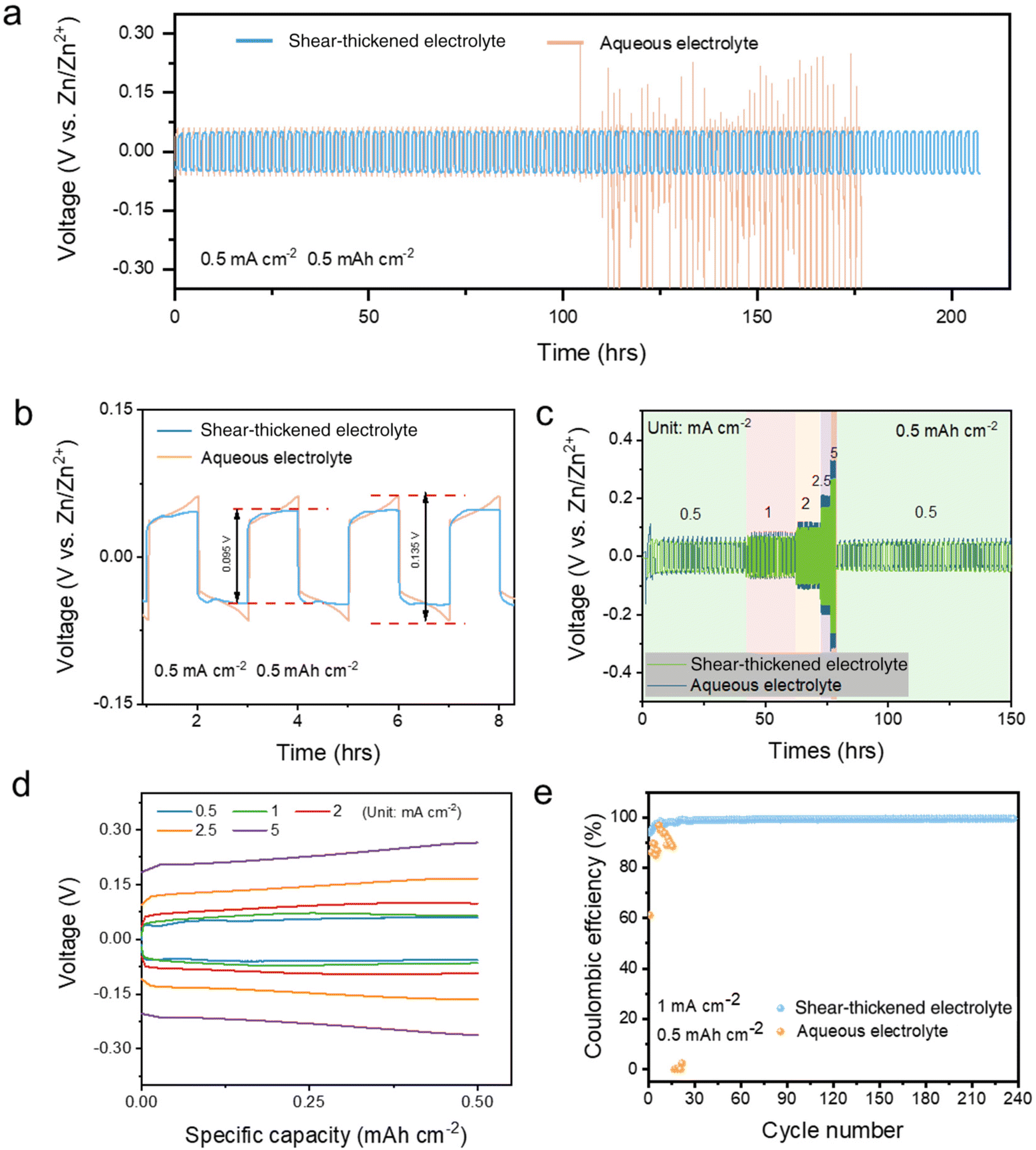 | ||
| Fig. 3 Electrochemical performances of the Zn|STE|Zn symmetric cell. (a) Zn plating and stripping with the STE and the aqueous electrolyte at a current density of 0.5 mA cm−2 and capacity of 0.5 mA h cm−2. (b) Enlarged diagram for the comparison of the STE and the aqueous electrolyte. (c) Zn plating and stripping rate test of the STE and the aqueous electrolyte at different current densities of 0.5, 1, 2, 2.5, and 5 mA cm−2 and capacity of 0.5 mA h cm−2. (d) Voltage–capacity profile of the Zn|STE|Zn symmetric cell. (e) Coulombic efficiency in Cu//Zn at 1 mA cm−2. | ||
The cornstarch suspension electrolyte also delivers a stable cycling performance at high current density. As shown in Fig. 3c, the plating/stripping rate performance was measured at different current densities of 0.5, 1, 2, 2.5 and 5 mA cm−2 within a constant area capacity of 0.5 mA h cm−2. Compared to the aqueous electrolyte, voltage differences obtained for the STE are less than those for the pristine electrolyte. The corresponding voltage gaps for each current density are ± 0.048, ± 0.065, ± 0.097, ± 0.166 and ± 0.262 mV, respectively (Fig. 3d). Owing to the uniform electric field distribution and low overpotential, Zn nucleation happens homogeneously resulting in flat voltage–capacity profiles. Coulombic efficiency (CE) was revealed in the Zn||Cu cell test under 1 mA cm−2. As shown in Fig. 3e, Zn|STE|Cu exhibited an initial CE of 95.3% and then it increased to 99.4% after 40 cycles, while the aqueous electrolyte could not maintain a stable plating reaction in Zn||Cu. Furthermore, as shown in Fig. S9a,† the STE exhibited a stable plating performance throughout a 240-hour test; however, the aqueous electrolyte only maintained a stable plating performance for less than 25 hours. A smaller potential difference of 105 mV was attained with the STE, when compared to 199 mV of the conventional aqueous electrolyte, which was accompanied by unstable plating. In utilising STE as the electrolyte for AZIBs, there is hardly any influence on the electrochemical plating/stripping mechanism, but on the contrary, it even suppresses the side effects on the Zn anode. In the STE, the electrostatic force of charged particles stabilising the electrolyte could be regarded as a long-range force. The presence of two or more particles in the suspension fundamentally alters Brownian motion and the entire dispersion is balanced by the equilibrium between stochastic and interparticle forces.15,18 OH− and H+ would be absorbed under the electrostatic field by the charged particles. The restriction of free water mobility by the concentrated long chains in cornstarch reduces the contact area of water at the electrolyte–electrode interface; hence, the electrochemically stable window is widened following the kinetic diffusion mechanism. In addition, a small contact area of water at the zinc anode surface results in a reduced corrosion situation.11 Hydrated Zn2+(H2O)6 could also be attracted by the long-range force reducing the transportation. However, the similar Zn2+ transference number attained for the STE indicates that the Zn2+ segmental motion could also be a transfer mechanism for the STE,25 where Zn2+ could hop from one active site on the cornstarch branch to a neighbouring site. Under the restriction of free water molecules, the desolvation of Zn2+(H2O)6 to Zn2+ could easily be initiated at the electrolyte–electrode interface.26
To further investigate the electrochemical properties of the STE, full cell tests were carried out by integrating commercial MnO2. The CV curves of Zn|STE|MnO2 were recorded at scan rates of 0.1, 0.2, 0.3, 0.4, 0.5, 1, 2 and 5 mV s−1. As shown in Fig. 4a, two redox peaks are observed. The reduction peaks at 1.36 and 1.23 V are relevant to H+ and Zn2+ intercalation, respectively. The oxidation peaks at 1.57 and 1.64 V correspond to Zn/Zn2+ and Mn2+/Mn3+ as reported. On increasing the scan rates, two pairs of redox peaks overlap to a single broad redox pair. The kinetic process was evaluated by calculating the contributions from diffusion and capacitive controlled processes. Theoretically, the peak current (i) and sweep rate (ν) can be correlated using the following formulae:27
| i = avb | (2) |
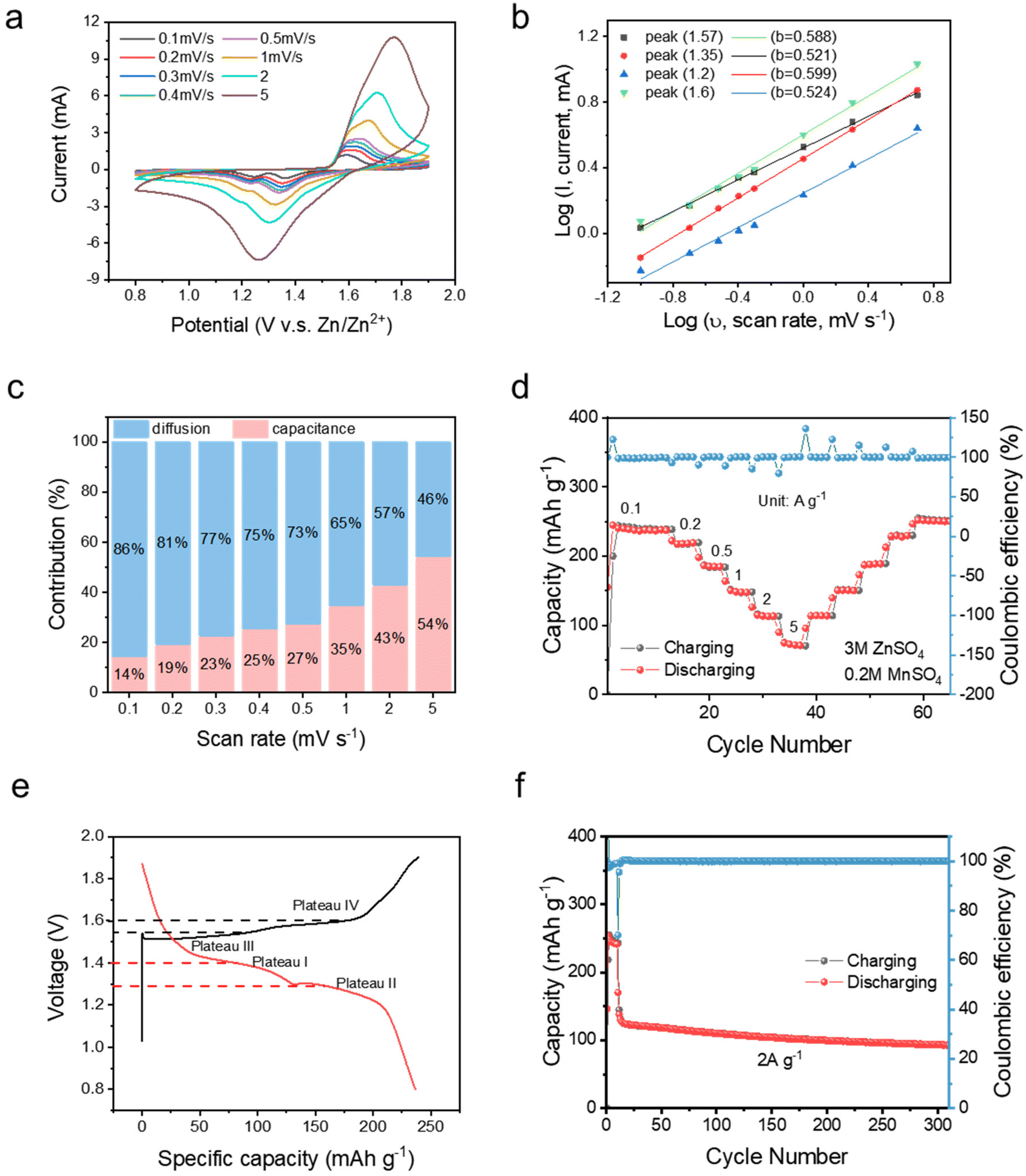 | ||
| Fig. 4 Electrochemical performances of the Zn|STE|commercial MnO2 full cell. (a) Cyclic voltammetry test at scan rates of 0.1, 0.2, 0.3, 0.4, 0.5, 1, 2, and 5 mV s−1. (b) Values of b calculated from the log–log plot of the current (i) and the scan rate (ν) of four peaks in CV curves. (c) Bar chart of diffusion and capacitive contributions integrated at different scan rates (ν). (d) Galvanic charging/discharging rate test at current densities of 0.1, 0.2, 0.5, 1, 2, and 5 A g−1. (e) Voltage–capacity profile with four plateaus. (f) Galvanostatic charge/discharge long-term cycling test at a current density of 2 A g−1. | ||
hence,
| log(i) = log(a) + b·log(v) | (3) |
By applying the logarithm, the equation could be deduced as eqn (3), for which a and b are the y-intercept and the slope of the log–log plot. If the coefficient b is close to 1, the electrochemical reaction is highly influenced by the capacitive behaviour, while if b is close to 0.5, the diffusion process is dominant. As revealed in Fig. 4b, the fitted b values are in the range of 0.52–0.59; hence, the kinetics of the ZIB with the STE is dominated by the diffusion controlled process. The quantitative analysis under each sweep rate is deduced from the integration of the following equation by assuming that the reaction is a combination of semi-infinite diffusion and capacitive processes:
| i = k1v + k2v1/2 | (4) |
The galvanostatic charge/discharge test was performed for the full cell with a 3 M ZnSO4 and 0.2 M MnSO4 electrolyte, as shown in Fig. 4d. At different current densities of 0.1, 0.2, 0.5, 1, 2, and 5 A g−1, the ZIB with the STE delivers specific capacities of 245, 220, 186, 150, 113, and 75 mA h g−1, respectively. The reasonable specific capacities exhibited in the ZIB with the STE further prove that there is no effect of electrochemical performance on the STE. On comparing the capacity attained at the upstream current densities, a superior reversibility was also achieved. ∼99% CE indicates negligible side effects when utilising the STE. As shown by the plateaus from voltage–capacity plots, two plateaus at potentials of 1.4 and 1.25 V during discharging are assigned to H+ and Zn2+ insertion, while plateaus at 1.55 and 1.6 V are attributed to the deintercalation of H+ and Zn2+. Under an external impact (see Fig. S12†), a voltage variation was observed at the charging/discharging plateaus. During the charging process, there is a sudden drop in the voltage to the open circuit potential (OCP) in the plateau, which means that the STE transforms into the rigid state thus blocking the Zn2+ transfer. However, the voltage profile returns to its initial profile once the impact is removed. This phenomenon is consistent with the chronoamperometry as shown in Fig. S11c.† The same voltage variation also happens during the discharging plateau. Therefore, under an external impact, the STE becomes rigid avoiding any short-circuiting, while after removal of the external impact, it recovers since the rigid state of STE transforms into a fluid state. In terms of the long-term cycling performance, 74.7% capacity retention was achieved after 300 cycles at a current density of 2 A g−1 with nearly 99% CE. The same excellent galvanostatic performance is also achieved for the STE with pristine 3 M ZnSO4. As shown in Fig. S5,† a capacity of 101 mA h g−1 obtained at a current density of 0.1 A g−1 is in the comparable range with respect to the value when using the aqueous electrolyte.
Conclusion
The shear-thickening suspension electrolyte exhibits a superior electrochemical performance while providing an impact load-carrying ability. The STE is a colloid electrolyte offering a competitive electrochemical performance and also an enhanced structural performance under a sudden impact, while for a normal aqueous electrolyte, only a mono-function is exhibited, which makes it difficult to modify the electric field at the electrolyte–electrode interface. There are no side effects using the cornstarch suspension as observed from the Zn2+ transference number and capacity achieved. In the meantime, it can also recover the Zn2+ transference ability after the sudden impact during the relaxation process. In addition, it can even protect the zinc anode by reducing the overpotential polarisation. The flat plateaus achieved using the symmetric cell further indicate that the application of the cornstarch water suspension not only offers an additional load-bearing property but can also suppress the side effect on the zinc anode, resulting in a low voltage difference of 95 mV compared to 135 mV for aqueous electrolytes at 0.5 mA cm−2 and 0.5 mA h cm−2. A similar zinc transference number achieved demonstrates the feasibility of the application of the STE as a functional electrolyte for rechargeable AZIBs. This work aims to provide an insight into constructing an impact-resistant battery based on an aqueous electrolyte, for which the designed impact-resistant aqueous battery could be applied as an energy storage system in body armour or armoured vehicle applications. In order to investigate its mechanical behaviour, a detailed mechanical testing of this impact resistant battery following the ASTM standard D256 should be conducted in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Engineering and Physical Sciences Research Council (EPSRC, EP/V027433/1).References
- Y. Liu, G. He, H. Jiang, I. P. Parkin, P. R. Shearing and D. J. L. Brett, Adv. Funct. Mater., 2021, 31, 2010445 CrossRef CAS.
- H. Dong, J. Li, S. Zhao, Y. Jiao, J. Chen, Y. Tan, D. J. L. Brett, G. He and I. P. Parkin, ACS Appl. Mater. Interfaces, 2021, 13, 745–754 CrossRef CAS PubMed.
- X. Li, Z. Chen, Y. Yang, S. Liang, B. Lu and J. Zhou, Inorg. Chem. Front., 2022, 2, 3986–3998 RSC.
- Z. Liu, L. Qin, B. Lu, X. Wu, S. Liang and J. Zhou, ChemSusChem, 2022, e202200348 CAS.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1(10), 16119–16126 CrossRef CAS.
- J. Frith, Lithium-Ion Batteries: The Incumbent Technology, 2019, https://ec.europa.eu/energy/sites/ener/files/documents/6.1_frith_energy_storage.pdf Search PubMed.
- V. Yufit, F. Tariq, D. S. Eastwood, M. Biton, B. Wu, P. D. Lee and N. P. Brandon, Joule, 2019, 3, 485–502 CrossRef.
- P.-C. Hsu, S.-K. Seol, T.-N. Lo, C.-J. Liu, C.-L. Wang, C.-S. Lin, Y. Hwu, C. H. Chen, L.-W. Chang, J. H. Je and G. Margaritondo, J. Electrochem. Soc., 2008, 155, D400 CrossRef CAS.
- Z. Liu, Y. Yang, S. Liang, B. Lu and J. Zhou, Small Struct., 2021, 2, 2100119 CrossRef CAS.
- X. G. Yang, T. Liu and C. Y. Wang, Nat. Energy, 2021, 6, 176–185 CrossRef CAS.
- G. M. Veith, B. L. Armstrong, H. Wang, S. Kalnaus, W. E. Tenhaeff and M. L. Patterson, ACS Energy Lett., 2017, 2, 2084–2088 CrossRef CAS.
- A. Fall, F. Bertrand, G. Ovarlez and D. Bonn, J. Rheol., 2012, 56, 575–591 CrossRef CAS.
- E. E. B. White, M. Chellamuthu and J. P. Rothstein, Rheol. Acta, 2010, 49, 119–129 CrossRef.
- P. Kilbride, M. V. Rull, A. Townsend, H. Wilson and J. Morris, Biorheology, 2019, 56, 39–50 CAS.
- A. S. Baumgarten and K. Kamrin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20828–20836 CrossRef CAS.
- S. E. Taylor, J. Dispersion Sci. Technol., 2013, 34, 887–897 CrossRef CAS.
- L. O. Gálvez, S. De Beer, D. Van Der Meer and A. Pons, Phys. Rev. E, 2017, 95, 1–6 Search PubMed.
- N. J. Wagner and J. F. Brady, Phys. Today, 2009, 62, 27–32 CrossRef CAS.
- C. Kusina, W. J. Smit, J. B. Boitte, O. Aubrun and A. Colin, Phys. Rev. E, 2021, 103, 1–7 CrossRef PubMed.
- J. F. Mendes, R. T. Paschoalin, V. B. Carmona, A. R. S. Neto, A. C. P. Marques, J. M. Marconcini, L. H. C. Mattoso, E. S. Medeiros and J. E. Oliveira, Carbohydr. Polym., 2016, 137, 452–458 CrossRef CAS PubMed.
- A. Willfahrt, E. Steiner, J. Hötzel and X. Crispin, Appl. Phys. A: Mater. Sci. Process., 2019, 125, 1–10 CrossRef.
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC.
- J. Gao, X. Xie, S. Liang, B. Lu and J. Zhou, Nano-Micro Lett., 2021, 13, 1–12 CrossRef CAS PubMed.
- L. Kang, M. Cui, F. Jiang, Y. Gao, H. Luo, J. Liu, W. Liang and C. Zhi, Adv. Energy Mater., 2018, 8, 1–8 Search PubMed.
- H. Dong, J. Li, J. Guo, F. Lai, F. Zhao, Y. Jiao, D. J. L. Brett, T. Liu, G. He and I. P. Parkin, Adv. Mater., 2021, 33, 2007548 CrossRef CAS PubMed.
- J. Li, X. Hou, R. Wang, X. He, T. P. Pollard, X. Ju, L. Du, E. Paillard, H. Frielinghaus, L. C. Barnsley, O. Borodin, K. Xu and M. Winter, Angew. Chem., Int. Ed., 2021, 60, 22812–22817 CrossRef PubMed.
- G. He, M. Qiao, W. Li, Y. Lu, T. Zhao, R. Zou, B. Li, J. A. Darr, J. Hu, M. M. Titirici and I. P. Parkin, Adv. Sci., 2017, 4, 1600214 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr04140e |
| This journal is © The Royal Society of Chemistry 2022 |