
Electrochemical regioselective C–H selenylation of 2H-indazole derivatives†
Shengsheng
Lin‡
,
Xiaomei
Cheng‡
,
Balati
Hasimujiang
,
Zhongnan
Xu
,
Fengtan
Li
and
Zhixiong
Ruan
 *
*
Key Laboratory of Molecular Target & Clinical Pharmacology and the State & NMPA Key Laboratory of Respiratory Disease, School of Pharmaceutical Sciences & the Fifth Affiliated Hospital, Guangzhou Medical University, Guangzhou, 511436, P.R. China. E-mail: zruan@gzhmu.edu.cn
First published on 27th November 2021
Abstract
Selenium-substituted heteroarenes are biologically active compounds and useful building blocks. In this study, we have developed a metal- and oxidant-free, environmentally friendly protocol for the regioselective selenylation of 2H-indazole derivatives by an electrochemical strategy. A number of selenylated 2H-indazoles with a wide range of functional groups have been synthesized in moderate to good yields under mild and environment-friendly reaction conditions.
Organoselenium compounds have been gaining much attention in pharmaceutical, biological, and materials applications as well as in organic synthesis in recent years because of their extensive use as therapeutically active agents such as antimicrobial, antioxidant, anti-inflammatory, antiviral, antibacterial, and anticancer agents etc.1 Moreover, several heterocyclic compounds such as pyrimidine, pyridine, quinoline, imidazole, ferrocene, indole and pyrazole in association with the selenium atom exhibit excellent pharmacological and biological activities.2 Among the numerous heterocyclic derivatives, selenium substituted imidazoles and 3-selenylindoles are particularly attractive due to their various biological and chemical activities.3 Several representative selenium-containing molecules are outlined in Fig. 1.
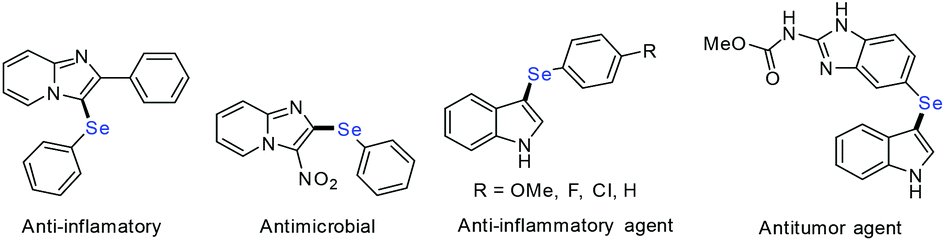 | ||
| Fig. 1 Selected examples of biologically important selenium-containing heterocycles. | ||
The application of selenium enriched compounds containing one or two heteroaromatic moieties has been observed to a greater extent in biological systems compared to their non-heteroaryl analogues.4 Thus, in recent years extensive research efforts have been devoted to the development of synthetic methodologies for C–Se bond formation in arenes and heteroarenes.5 In the past two decades transition metal catalyzed cross-coupling reactions have been the main powerful method for the synthesis of aryl/heteroaryl selenides.6 However, the use of expensive and in some cases toxic metal salts, ligands, harsh conditions, high temperature etc. has been a serious limitation of these protocols.7 In recent years, the preparation of novel organoselenium compounds using photoelectric technology for C–Se bond formation in a more ecological way has attracted much attention.8 In addition, it is known that indazole compounds are very important bioisosteres of the indole and benzimidazole moieties, which are commonly found in various marketed drugs and natural products along with useful synthons in organic synthesis.9 Considering its increasing importance much attempt has been made towards the synthesis and functionalization of 2H-indazoles in the last few years (Scheme 1a).10 Recently, Hajra and co-workers have developed a metal-free, environmentally friendly selenylation protocol of 2H-indazole in the presence of a catalytic amount of I2 under ambient conditions (Scheme 1b).11 However, these methods usually have some obvious drawbacks, such as the use of transition metals and strong oxidants, which make them unfriendly to the environment and lead to poor functional group compatibility. Therefore, due to the wide applications of indazoles, the development of more efficient and greener synthetic methods as well as novel entities of selenium-containing compounds is in high demand.
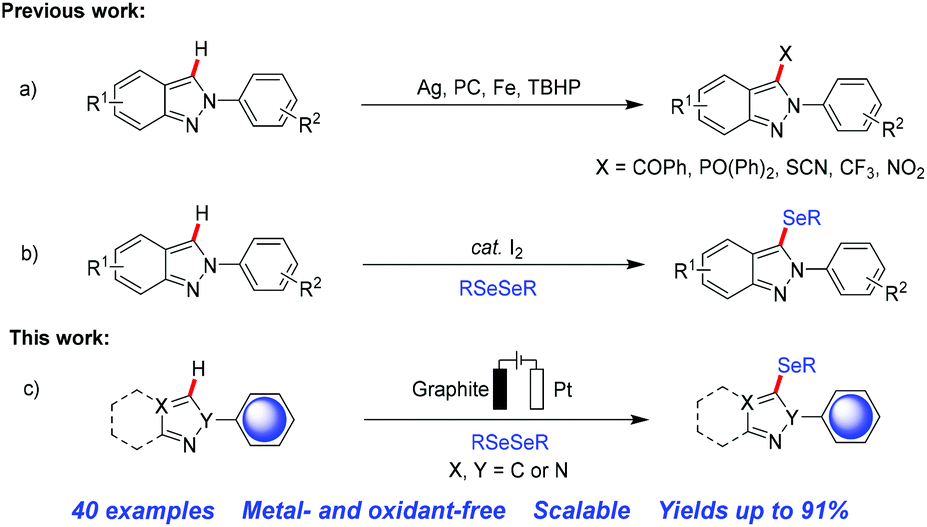 | ||
| Scheme 1 Regioselective C3–H functionalization of 2H-indazole. | ||
In recent years, the electrochemical functionalization of C–H bonds has become a powerful method in organic synthesis due to its convenience and environmental sustainability as well as controllability simply by altering the potential, current, or total charge.12 However, as far as we know, there is no previous report of an electrochemical regioselective selenylation method of 2H-indazole derivatives. Moreover, an environmentally friendly low-cost direct C–H selenylation procedure involving simple operation under mild conditions is highly desirable. In continuation of our interest in organic electrosynthesis of functionalized heteroarenes,13 we herein report an efficient, eco-friendly, scalable, transition metal- and oxidant-free approach for the direct C–H selenylation of 2H-indazole derivatives under aerobic electrolytic conditions (Scheme 1c).
Initially, 2-phenyl-2H-indazole 1a and diphenyldiselenide 2a were chosen as model substrates to explore various reaction conditions in an undivided cell equipped with a graphite anode and a platinum cathode under constant current electrolytic conditions (Table 1 and Table S1†). Optimal results were obtained when substrate 1a was directly electrolyzed at a constant current of 8 mA in an electrolyte solution of n-Bu4NPF6 in MeCN at 50 °C under atmospheric conditions without additional catalysts. Under these conditions, the desired selenylated product 3a was isolated in 85% yield, with 2a being recovered in 56% yield (Table 1, entry 1). The solvent and electrolyte were both found to be essential for the reaction to achieve the optimal yield (entries 2–6). Thus, other supporting electrolytes, such as n-Bu4NClO4, n-Bu4NBF4 and LiClO4, were explored, which displayed poor performance (entries 4–6). Furthermore, the choice of the electrode material proved to be critical because a dramatically reduced product yield was obtained when a Pt anode and a graphite or Ni cathode were used (entries 7–9). A further control experiment verified the necessity of the external electricity (entry 10).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) |
| a Standard conditions: Undivided cell, graphite as the anode (1.5 cm × 1 cm × 0.2 cm), Pt plate as the cathode (1 cm × 1 cm × 0.01 cm), 1a (0.2 mmol), 2a (0.3 mmol), n-Bu4NPF6 (0.5 mmol), and MeCN (5.0 mL), constant current = 8.0 mA, 5 h (7.5 F mol−1), under air, at 50 °C. b Yields of the isolated product. | ||
| 1 | None | 85 |
| 2 | MeOH as solvent | 0 |
| 3 | MeCN/DCE (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as solvent :1) as solvent |
30 |
| 4 | n-Bu4NClO4 instead of n-Bu4NPF6 | 29 |
| 5 | n-Bu4NBF4 instead of n-Bu4NPF6 | 22 |
| 6 | LiClO4 instead of n-Bu4NPF6 | 38 |
| 7 | Pt plate as anode | 31 |
| 8 | Graphite as cathode | 78 |
| 9 | Ni plate as cathode | 76 |
| 10 | No electricity | 0 |
With the optimized reaction conditions in hand, we explored the applicability of this protocol with a viable substrate scope by first testing the effect exerted by substituents on the arene moiety of 2H-indazoles (Scheme 2). The catalyst- and oxidant-free electrochemical selenylation exhibited excellent compatibility with a variety of electron-donating and electron-withdrawing groups at the ortho-, meta- or para-position on the 2-phenyl substituent of substrate 1a to afford the desired products (3b–3n). Thus, 2H-indazoles with electrophilic functional groups were fully tolerated, including sensitive fluoro, chloro, bromo and iodo substituents, which could serve as a handle for future late-stage modifications. It is noteworthy that the electrochemical selenylation of 2H-indazoles containing various multisubstituted benzenes at the 2-position smoothly produced the corresponding products in moderate yields under the standard reaction conditions (3o–3r). In addition, the electrolysis reaction was extended by exploring the effect of different substituents on the arene part of 2H-indazoles. A variety of halo and methoxy groups at different positions of the phenyl ring of 2H-indazoles were found to be well tolerated affording the desired products in moderate to excellent yields (3s–3z). It is particularly noteworthy that the 1-phenyl-1H-pyrazole and imidazo[1,2-a]pyridine substrates were well accepted by the robust metal-free electro-selenylation regime to generate the C4 site-selective selenylated product 3aa and 3-selenylated imidazopyridine derivative 3ab under the optimized conditions, respectively.
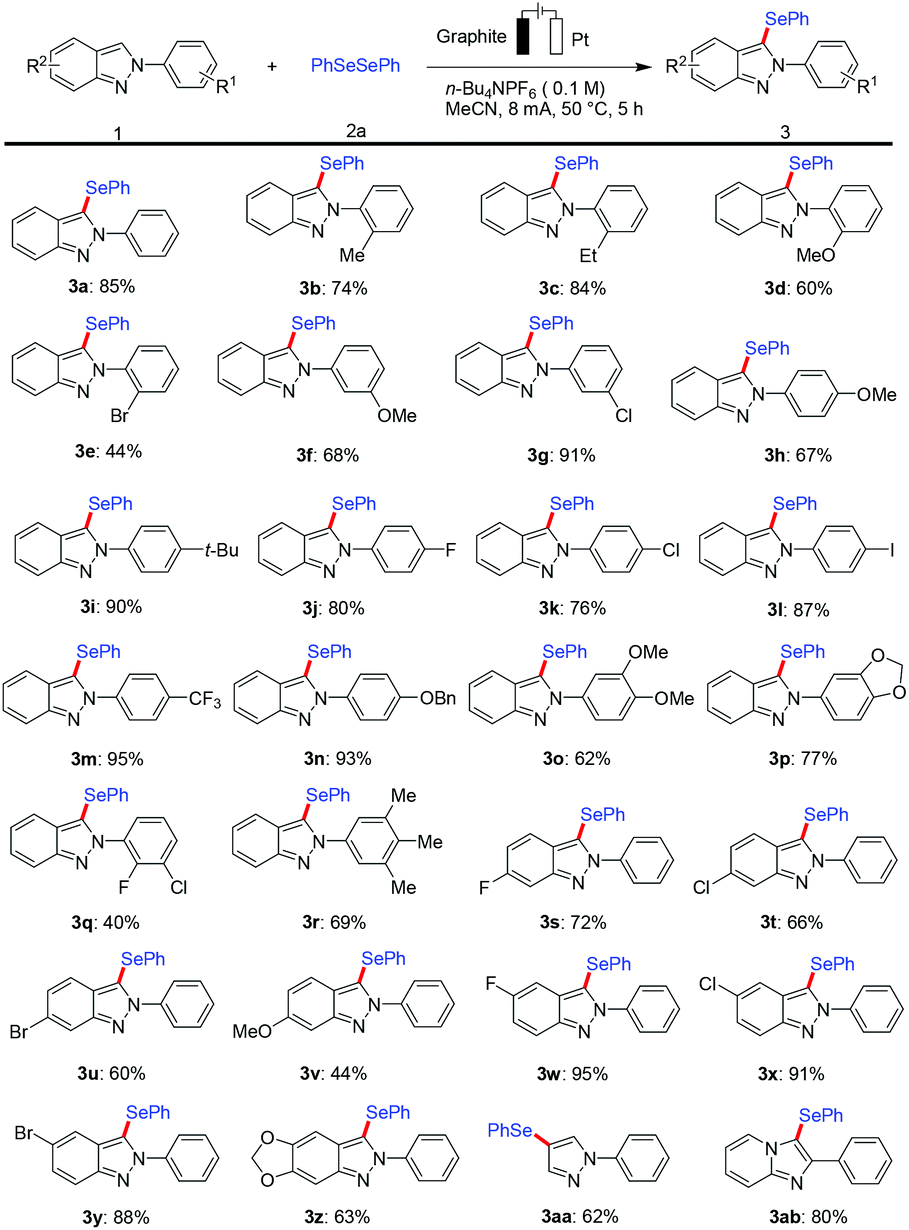 | ||
| Scheme 2 Substrate scope for the C–H selenylation of 2H-indazole with 2a. Reaction conditions: undivided cell, graphite as the anode (1.5 cm × 1 cm × 0.2 cm), Pt plate as the cathode (1 cm × 1 cm × 0.01 cm), 1 (0.2 mmol), 2a (0.3 mmol), n-Bu4NPF6 (0.5 mmol), and MeCN (5.0 mL), constant current = 8.0 mA, 5 h (7.5 F mol−1), under air, at 50 °C. Isolated yields. | ||
Furthermore, we evaluated the feasibility with various substituted diselenide coupling partners in the electrochemical C–H selenylation approach under the established reaction conditions (Scheme 3). A set of representative diaryl diselenides bearing electron-withdrawing groups such as fluoro and chloro groups at the meta-position, as well as a fluoro group at the para-position or electron-donating groups, such as methyl and methoxyl groups at the meta-position and an ethyl group at the ortho-position, were smoothly converted into the desired functionalized 2H-indazoles in good to moderate yields. Gratifyingly, the aliphatic diselenide derivatives proved to be suitable substrates to furnish the desired selenylated products in acceptable yields, albeit 4l was obtained in somewhat lower yield.
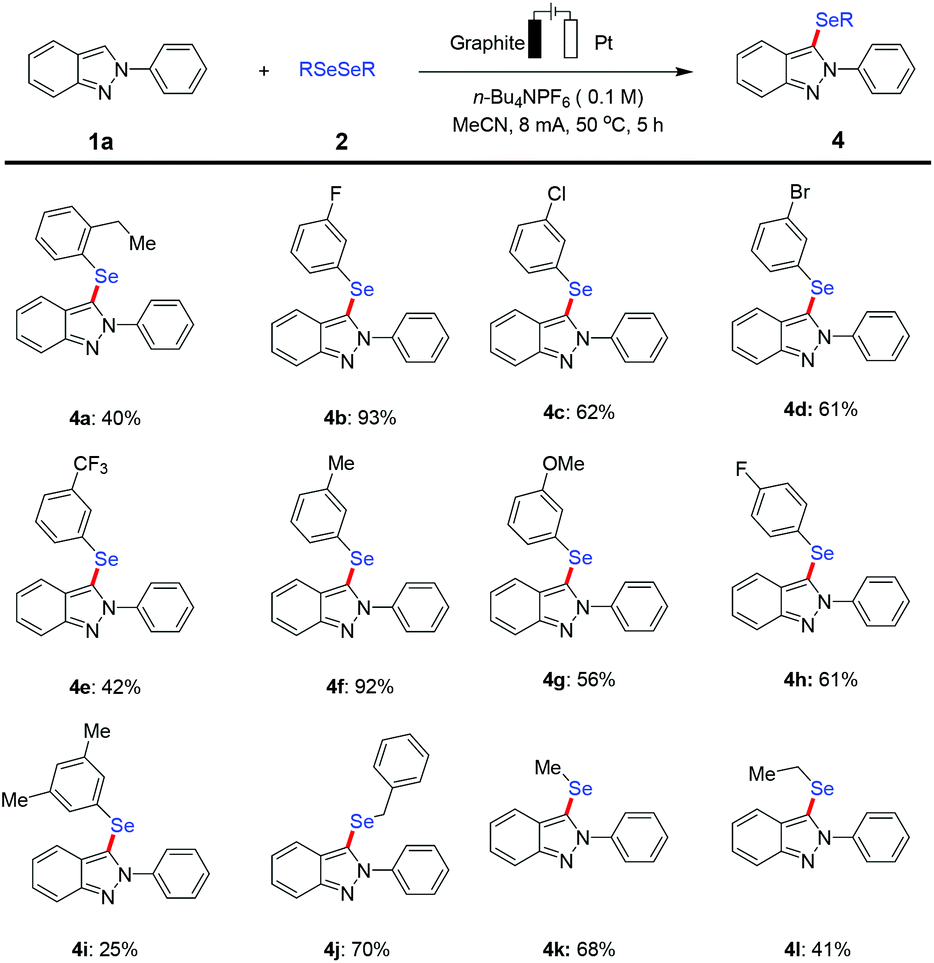 | ||
| Scheme 3 Electrochemical selenylation of 1a with variation of diorganyl selenides 2. Reaction conditions: Undivided cell, graphite as the anode (1.5 cm × 1 cm × 0.2 cm), Pt plate as the cathode (1 cm × 1 cm × 0.01 cm), 1a (0.2 mmol), 2 (0.3 mmol), n-Bu4NPF6 (0.5 mmol), and MeCN (5.0 mL), constant current = 8.0 mA, 5 h (7.5 F mol−1), under air, at 50 °C. Isolated yields. | ||
The outstanding potential of our electrochemical selenylation approach was further demonstrated by its easy scalability. For instance, the gram-scale synthesis of 3a was performed under slightly varied conditions with 76% isolated yield without appreciable loss in efficacy, in which larger electrodes and a higher constant current of 20 mA (3.0 F mol−1) were employed to ensure completion of the reaction in a short time (Scheme 4).
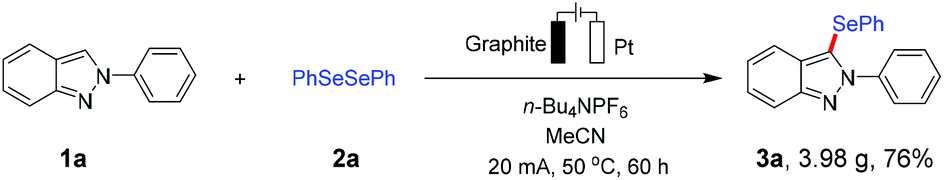 | ||
| Scheme 4 Gram-scale synthesis of 3a. Reaction conditions: Undivided cell, graphite as the anode (3 cm × 3 cm × 0.6 cm), Pt plate as the cathode (3 cm × 3 cm × 0.01 cm), 1a (15.0 mmol), 2a (22.5 mmol), n-Bu4NPF6 (15.0 mmol), and MeCN (150 mL), constant current = 20.0 mA, 60 h (3.0 F mol−1), under air, at 50 °C. | ||
Considering the remarkable performance of the electrochemical C–H selenylation, we have performed several control experiments to elucidate its mode of reaction as shown in Scheme 5. Intermolecular competition experiments between electronically discriminated substrates 1 revealed that electron-poor 2H-indazoles were inherently more reactive (Scheme 5a). This might be because the phenyl selenium radical is more electron-rich than the electron-deficient aromatic ring conjugated system.14 Therefore, the electron-deficient 2H-indazole is more capable of attracting phenyl selenium radicals. When the electrolysis was performed in the presence of the radical scavenger 2,6-di-tert-butyl-4-methylphenol (BHT) under the standard reaction conditions, the desired product 3a was obtained in an abruptly decreased yield (Scheme 5b), which implied that this process likely proceeded by a radical pathway. However, further investigation revealed that 1a afforded the desired product 3a in 76% yield in the presence of PhSeCl instead of diphenyldiselenide (2a), which could not exclude the ionic pathway (Scheme 5c). In addition, the substrate 1a also produced the desired selenylation product 3a in the presence of PhSeH with 60% yield under the electrochemical standard conditions (Scheme 5d), which proved that the reaction preferred a radical pathway.
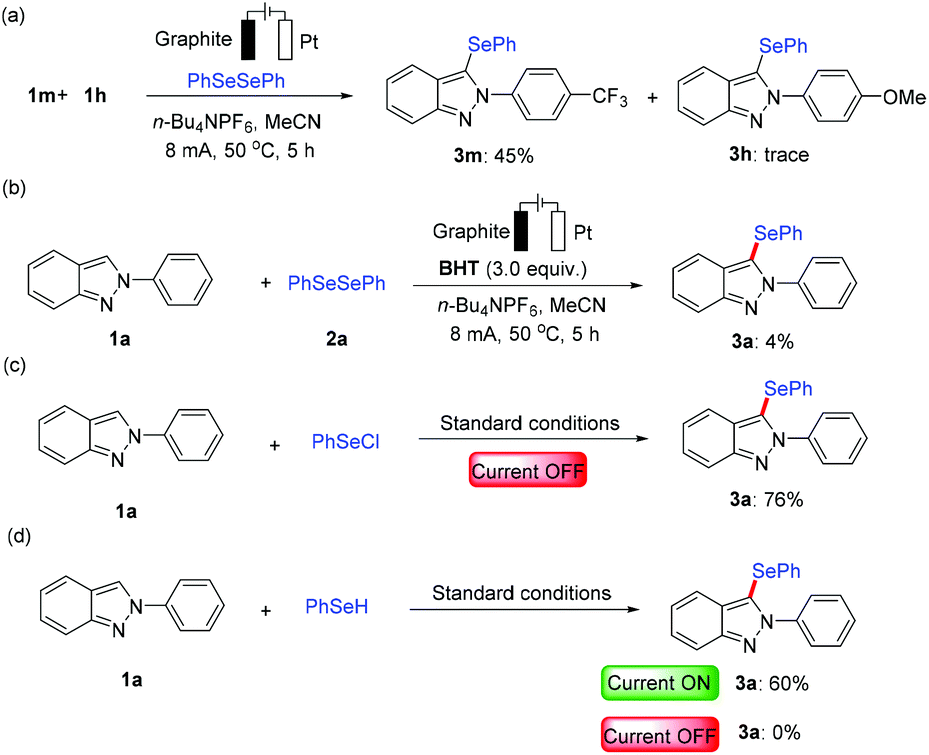 | ||
| Scheme 5 Control experiments. | ||
Furthermore, cyclic voltammetry (CV) experiments were also performed to gain key mechanistic insights into the electrochemical transformation (Fig. 2). It was found that diphenyl diselenide (2a) was oxidized preferentially at a low potential (1.459 V vs. Ag/AgCl), which was lower than the oxidation potential of 1a (1.565 V (vs. Ag/AgCl).
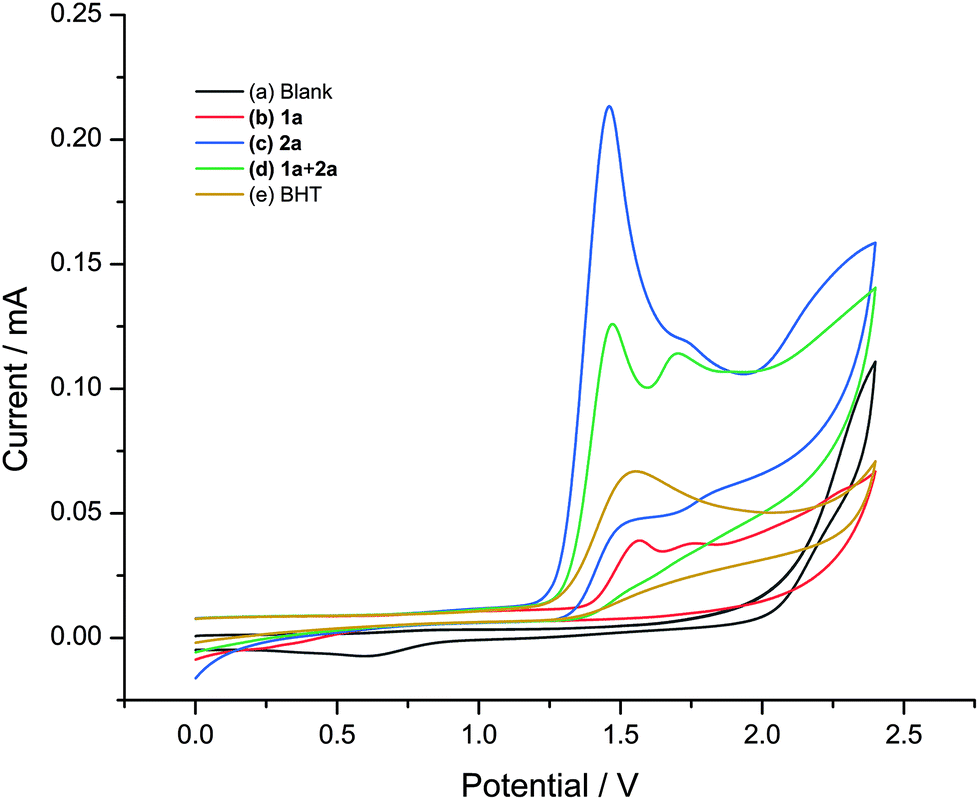 | ||
| Fig. 2 Cyclic voltammograms. Conditions: A glassy carbon working electrode, a Ag/AgCl (3 M KCl) reference electrode, and a platinum wire counter electrode, n-Bu4NPF6 (0.1 M in MeCN), 100 mV s−1 scan rate with (a) background; (b) 1a (1 mM); (c) 2a (1 mM); and (d) 1a (1 mM) + 2a (1 mM). | ||
Thus, based on our mechanistic studies and previousl reports,15 a plausible mechanistic scenario was proposed for the electrochemical oxidative C–H selenylation of 2H-indazoles as shown in Scheme 6. The reaction was initiated by the formation of the selenium radical A and selenium cation Bvia direct anodic oxidation of 2a. Thereafter, the radical addition of A to 1a generates the radical intermediate C, followed by deprotonation, affording the desired product 3a. Alternatively, the ionic pathway cannot be omitted, that is, the selenium cation B can also add to 1a to provide the cationic intermediate D, followed by deprotonation, achieving the selenylated product 3a. At the cathode, protons are concurrently reduced to release molecular hydrogen.
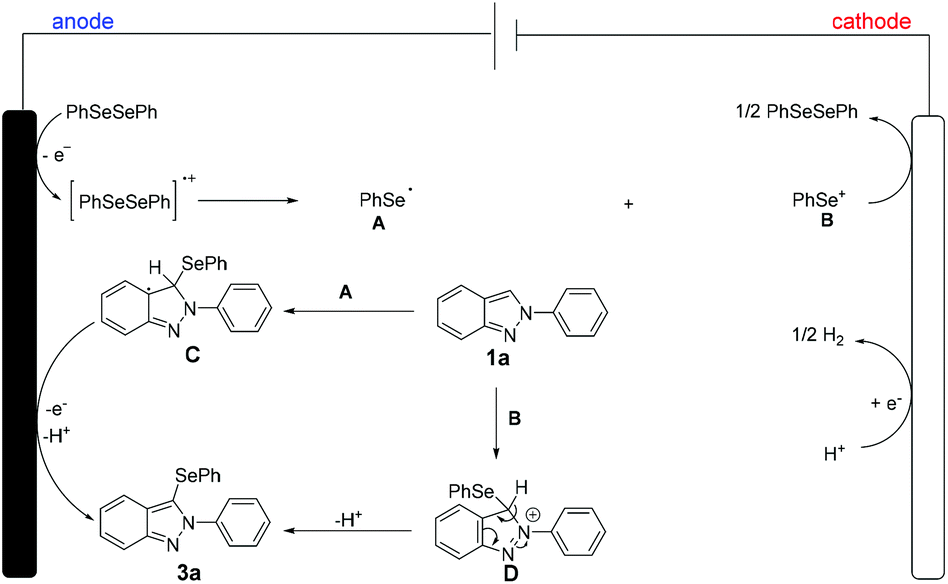 | ||
| Scheme 6 A plausible mechanistic pathway. | ||
Conclusions
In summary, we have reported the first electrochemical C–H selenylation of 2H-indazoles with various diselenides under metal- and external oxidant-free aerobic conditions. The electrochemical selenylation strategy is highlighted by its ample scope and high functional group tolerance. Importantly, this protocol was easily conducted on a gram-scale owing to its simple operation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21901052), the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2019), the Guangdong Basic and Applied Basic Research Foundation (2020A1515010722), Guangzhou Science and Technology Project (202102020979) and the High-level University Construction Fund of Guangdong Province (06-410-2107233 and 06-410-2107227).Notes and references
- (a) D. A. Bellnier, D. N. Young, M. R. Detty, S. H. Camacho and A. R. Oseroff, Photochem. Photobiol., 1999, 70, 630 CrossRef CAS; (b) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed; (c) D. M. Freudendahl, S. Santoro, S. A. Shahzad, C. Santi and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 8409 CrossRef CAS; (d) S. T. Manjare, Y. Kim and D. G. Churchill, Acc. Chem. Res., 2014, 47, 2985 CrossRef CAS; (e) M. Chhowalla, Z. Liu and H. Zhang, Chem. Soc. Rev., 2015, 44, 2584 RSC; (f) K. Selvakumar and H. B. Singh, Chem. Sci., 2018, 9, 7027 RSC; (g) M. Michalczuk, M. Batorska, U. Sikorska, D. Bień, J. Urban, K. Capecka and P. Konieczka, Anim. Sci. J., 2021, 92, e13662 CrossRef PubMed; (h) Y. Yu, Y. Jin, J. Zhou, H. Ruan, H. Zhao, S. Lu, Y. Zhang, D. Li, X. Ji and B. H. Ruan, ACS Chem. Biol., 2017, 12, 3003 CrossRef CAS PubMed; (i) X. Jing, C. Chen, X. Deng, X. Zhang, D. Wei and L. Yu, Appl. Organomet. Chem., 2018, 32, e4332 CrossRef; (j) H. Cao, R. Qian and L. Yu, Catal. Sci. Technol., 2020, 10, 3113 RSC.
- (a) Q. Guan, C. Han, D. Zuo, M. Zhai, Z. Li, Q. Zhang, Y. Zhai, X. Jiang, K. Bao, Y. Wu and W. Zhang, Eur. J. Med. Chem., 2014, 87, 306 CrossRef CAS PubMed; (b) Z. Wen, J. Xu, Z. Wang, H. Qi, Q. Xu, Z. Bai, Q. Zhang, K. Bao, Y. Wu and W. Zhang, Eur. J. Med. Chem., 2015, 90, 184 CrossRef CAS PubMed.
- (a) K. A. Leonard, J. P. Hall, M. I. Nelen, S. R. Davies, S. O. Gollnick, S. Camacho, A. R. Oseroff, S. L. Gibson, R. Hilf and M. R. Detty, J. Med. Chem., 2000, 43, 4488 CrossRef CAS; (b) Y. Jia, J. Zhang, J. Feng, F. Xu, H. Pan and W. Xu, Chem. Biol. Drug Des., 2014, 83, 306 CrossRef CAS.
- C. W. Nogueira, G. Zeni and J. B. Rocha, Chem. Rev., 2004, 104, 6255 CrossRef CAS.
- (a) P. Gandeepan, J. Koeller and L. Ackermann, ACS Catal., 2017, 7, 1030 CrossRef CAS; (b) D. Kundu, RSC Adv., 2021, 11, 6682 RSC; (c) W. Ma, N. Kaplaneris, X. Fang, L. Gu, R. Mei and L. Ackermann, Org. Chem. Front., 2020, 7, 1022 RSC; (d) P. Ni, J. Tan, W. Zhao, H. Huang and G.-J. Deng, Adv. Synth. Catal., 2019, 361, 5351 CrossRef CAS; (e) Y. Wu, X. Tian, H. Zhang, R. Chen and T. Cao, Chin. J. Org. Chem., 2020, 40, 1384 CrossRef CAS; (f) L.-H. Lu, Z. Wang, W. Xia, P. Cheng, B. Zhang, Z. Cao and W.-M. He, Chin. Chem. Lett., 2019, 30, 1237 CrossRef CAS.
- I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS.
- (a) G. Perin, E. J. Lenardão, R. G. Jacob and R. B. Panatieri, Chem. Rev., 2009, 109, 1277 CrossRef CAS; (b) D. Prasad Ch, S. J. Balkrishna, A. Kumar, B. S. Bhakuni, K. Shrimali, S. Biswas and S. Kumar, J. Org. Chem., 2013, 78, 1434 CrossRef; (c) J. Rafique and S. Saba, Chem. – Eur. J., 2016, 22, 11854 CrossRef CAS; (d) S. Shu, Z. Fan, Q. Yao and A. Zhang, J. Org. Chem., 2016, 81, 5263 CrossRef CAS.
- (a) J. Li, X. Liu, J. Deng, Y. Huang, Z. Pan, Y. Yu and H. Cao, Chem. Commun., 2020, 56, 735 RSC; (b) D. A. Sonawane and M. Koketsu, Curr. Org. Chem., 2019, 23, 3206 CrossRef; (c) P.-O. Schwartz, S. Förtsch, A. Vogt, E. Mena-Osteritz and P. Bäuerle, Beilstein J. Org. Chem., 2019, 15, 1379 CrossRef CAS PubMed; (d) M. Sacramento, G. P. Costa, A. M. Barcellos, G. Perin, E. J. Lenardão and D. Alves, Chem. Rec., 2021, 21, 2855 CrossRef CAS PubMed; (e) X. Wang, Y. Zhang, K. Sun, J. Meng and B. Zhang, Chin. J. Org. Chem., 2021 DOI:10.6023/cjoc202109046.
- Y. Jia, J. Zhang, J. Feng, F. Xu, H. Pan and W. Xu, Chem. Biol. Drug Des., 2014, 83, 306 CrossRef CAS PubMed.
- (a) G. Bogonda, H. Y. Kim and K. Oh, Org. Lett., 2018, 20, 2711 CrossRef CAS PubMed; (b) A. Murugan, K. R. Gorantla, B. S. Mallik and D. S. Sharada, Org. Biomol. Chem., 2018, 16, 5113 RSC; (c) M. Singsardar, A. Dey, R. Sarkar and A. Hajra, J. Org. Chem., 2018, 83, 12694 CrossRef CAS; (d) A. Dey and A. Hajra, Adv. Synth. Catal., 2019, 361, 842 CrossRef CAS; (e) Y. L. Liu, Y. L. Pan, G. J. Li, H. F. Xu and J. Z. Chen, Org. Biomol. Chem., 2019, 17, 8749 RSC; (f) A. Murugan, V. N. Babu, A. Polu, N. Sabarinathan, M. Bakthadoss and D. S. Sharada, J. Org. Chem., 2019, 84, 7796 CrossRef CAS.
- A. Dey and A. Hajra, J. Org. Chem., 2019, 84, 14904 CrossRef CAS.
- (a) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605 RSC; (b) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786 RSC; (c) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086 CrossRef CAS; (d) D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573 CrossRef CAS PubMed; (e) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef CAS; (f) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef CAS; (g) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339 CrossRef CAS; (h) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300 CrossRef CAS PubMed; (i) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309 CrossRef CAS; (j) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415 CrossRef CAS PubMed; (k) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484 CrossRef CAS.
- (a) Z. Ruan, Z. Huang, Z. Xu, S. Zeng, P. Feng and P.-H. Sun, Sci. China: Chem., 2021, 64, 800 CrossRef CAS; (b) Y. Li, Z. Huang, G. Mo, W. Jiang, C. Zheng, P. Feng and Z. Ruan, Chin. J. Chem., 2021, 39, 942 CrossRef CAS; (c) X. Cheng, B. Hasimujiang, Z. Xu, H. Cai, G. Chen, G. Mo and Z. Ruan, J. Org. Chem., 2021, 86, 16045 CrossRef CAS; (d) Z. Xu, Y. Li, G. Mo, Y. Zheng, S. Zeng, P.-H. Sun and Z. Ruan, Org. Lett., 2020, 22, 4016 CrossRef CAS; (e) Z. Xu, Z. Huang, Y. Li, R. Kuniyil, C. Zhang, L. Ackermann and Z. Ruan, Green Chem., 2020, 22, 1099 RSC.
- (a) J. M. Tedder, Angew. Chem., Int. Ed. Engl., 1982, 21, 401 CrossRef; (b) B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25 RSC; (c) G. Lei, M. Xu, R. Chang, I. Funes-Ardoiz and J. Ye, J. Am. Chem. Soc., 2021, 143, 11251 CrossRef CAS.
- (a) X.-Y. Wang, Y.-F. Zhong, Z.-Y. Mo, S.-H. Wu, Y.-L. Xu, H.-T. Tang and Y.-M. Pan, Adv. Synth. Catal., 2021, 363, 208 CrossRef CAS; (b) A. Dey and A. Hajra, Adv. Synth. Catal., 2019, 361, 842 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob02108g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |