
Yb(OTf)3 catalyzed [1,3]-rearrangement of 3-alkenyl oxindoles†
Chaofei
Wu‡
,
Junlin
Wan‡
,
Chao
Song
,
Lingchen
He
,
Hongxin
Liu
 ,
Xinhua
Li
,
Juan
Li
,
Xin-Gen
Hu
*,
Hong-Ping
Xiao
and
Jun
Jiang
*
,
Xinhua
Li
,
Juan
Li
,
Xin-Gen
Hu
*,
Hong-Ping
Xiao
and
Jun
Jiang
*
College of Chemistry and Materials Science, Wenzhou University, Wenzhou 325035, PR China. E-mail: junjiang@wzu.edu.cn; hxgwzu@126.com
First published on 26th November 2021
Abstract
A Yb(OTf)3 catalyzed [1,3]-rearrangement of 3-alkenyl oxindoles was achieved, affording a variety of multifunctional 3-ylideneoxindoles with good yields and Z/E selectivities (64%–89% yield, 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22–>99:1 Z/E). Importantly, an operationally simple, one-pot sequential catalytic synthesis of 3-ylideneoxindoles was also developed. Additionally, a cross [1,3]-rearrangement experiment and nonracemic transformation were also carried out, which indicated a concerted rearrangement mechanism of this methodology.
:22–>99:1 Z/E). Importantly, an operationally simple, one-pot sequential catalytic synthesis of 3-ylideneoxindoles was also developed. Additionally, a cross [1,3]-rearrangement experiment and nonracemic transformation were also carried out, which indicated a concerted rearrangement mechanism of this methodology.
The development of atom-economical transformations with high efficiency for the construction of multifunctional molecules is a very challenging but attractive topic both in organic synthesis and medicinal chemistry.1 Compared with step-by-step approaches, rearrangement reactions especially carbon-transfer ones afforded a direct way to obtain multifunctional compounds in many cases because of their great capability of reconstruction of bond(s) and molecular skeletons, thus always enabling the fast formation of multifunctional architectures from relatively simple starting materials. Taking advantage of these properties, catalytic rearrangement reactions such as the Claisen2 and Cope3 rearrangements received continuous attention in the past several decades, and fruitful results have been obtained by many research groups.4 Driven by these achievements and our continuous interest in the synthesis of multifunctional heterocycles,5 we hoped to explore the possibility of designing an efficient rearrangement transformation on the basis of our previous research; in 2012, we developed a catalytic stereoselective conjugate addition reaction of oxindoles to electron-deficient alkynes,5a providing a variety of 3-alkenyl oxindoles with good yields and stereoselectivities. With these adducts in hand, a 1,3 carbon–carbon rearrangement was envisioned due to the migration-possibility of the carbon–carbon π bond from a maleate moiety to the 3-position of oxindoles, which may simultaneously initiate the reconstruction of sigma bonds and transform 3-alkenyl oxindoles A into 3-ylideneoxindoles B,8 a more stable structure with a larger conjugation system (Fig. 1). This hypothesis brought us to a careful examination of the literature. Surprisingly, in contrast to [3,3]- and [2,3]-sigmatropic rearrangements, [1,3]-alkyl migration transformations6 were much less developed despite the thermal [1,3]-alkyl shift of vinyl ethers being firstly discovered in 1896 by Claisen.7 Besides, the possible rearrangement product B is an important type of nitrogen heterocyclic motif which is commonly found in various natural products and pharmaceuticals (Fig. 1).9 Hence, exploring such a novel catalytic [1,3]-alkyl shift methodology could be an attractive target. Herein, we report our preliminary exploration on this hypothesis, in which a Yb(OTf)3 catalyzed [1,3]-rearrangement of 3-alkenyl oxindoles was achieved, affording a variety of multifunctional 3-ylideneoxindoles with good yields and Z/E selectivities under mild conditions. Additionally, a one-pot version combining a catalytic addition with a [1,3]-rearrangement process starting from diethyl but-2-ynedioates and (indol-3-yl)methyl substituted oxindoles was also successfully achieved.
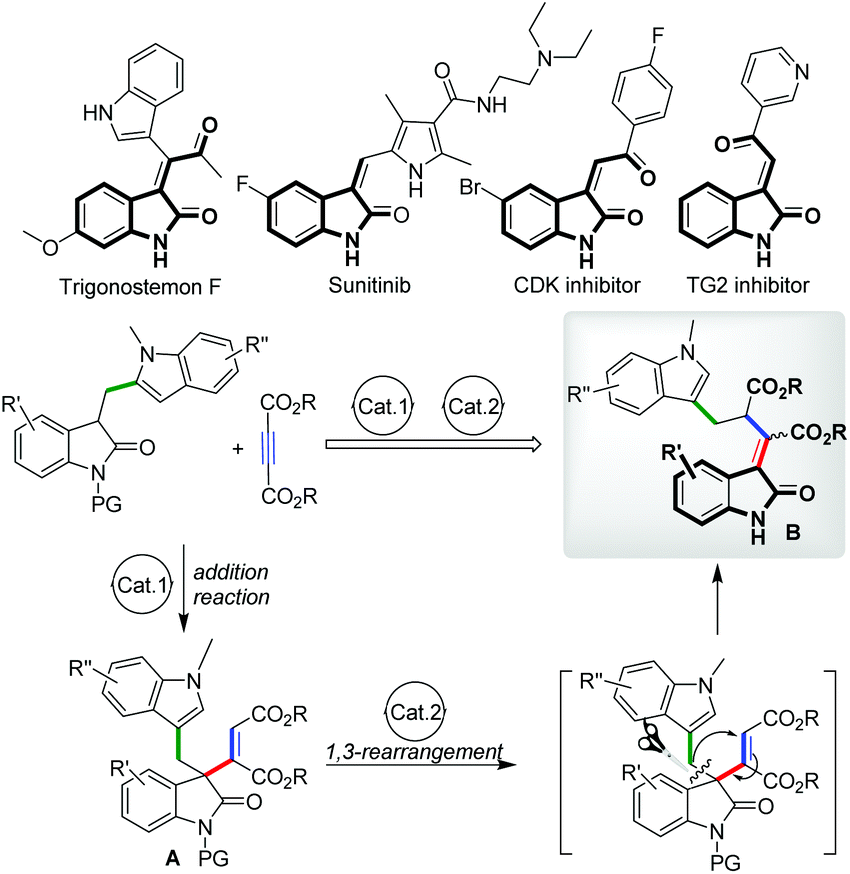 | ||
| Fig. 1 Lewis acid catalyzed 1,3-rearrangement of 3-alkenyl oxindoles. | ||
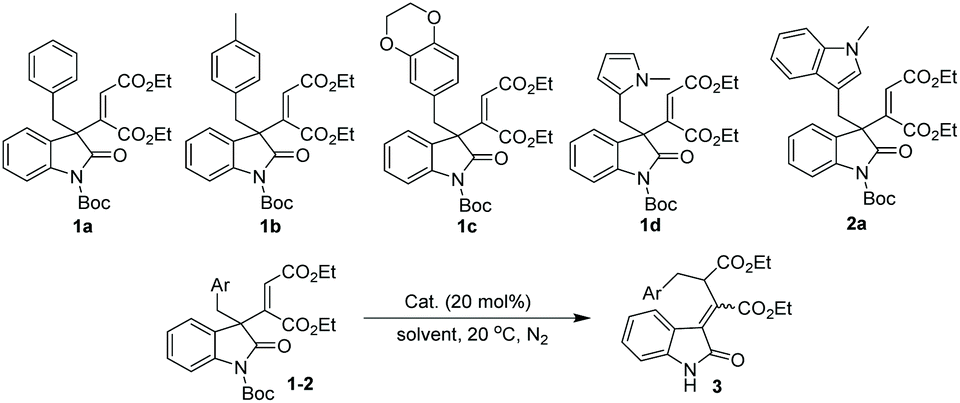
To test our hypothesis on this 1,3 carbon–carbon rearrangement reaction, different 3-alkenyl oxindoles were initially synthesized via our reported method (Table 1).5a With these substrates in hand, the migration ability of aliphatic substituents at the 3-position of oxindoles was subsequently examined by treating 1 and 2 with 20 mol% of Cu(OTf)2 in CH2Cl2 at 20 °C. It was shown that the substituents at the 3-position of oxindoles had an obvious influence on the reactivity. For example, not the desired products but N-deprotected starting materials were obtained when 1a–1d were employed as substrates (Table 1, entries 1–4), while the reaction of (indol-3-yl)methyl substituted 3-alkenyl oxindole 2a successfully afforded a deprotected rearrangement product 3a under the same conditions, although with a poor yield and Z/E selectivity (entry 5, 27% yield of the Z isomer, Z/E = 40:60). Inspired by this result, a careful evaluation of catalysts was next carried out. Reactions with other copper salts such as CuSO4·5H2O or Cu(OAc)2·H2O failed to give any products (entries 6 and 7), while a trace amount of 3a was observed when PdCl2 or FeF2 was employed as a catalyst (entries 8 and 9). Besides, a stronger Lewis acid, AlCl3, could not afford 3a with a satisfactory result either (entry 10, 25% yield of the Z isomer, Z/E = 68:32). Encouragingly, screening of different rare earth metal triflates indicated that Yb(OTf)3 had a good catalytic ability, and provided the desired product 3a in moderate yield and Z/E selectivity (entry 13, 51% yield, Z/E = 63:37). With this optimal catalyst, further optimization of the reaction conditions was performed. Finally, the rearrangement product 3a was obtained with the best yield and Z/E selectivity when THF was employed as the reaction medium (entry 16, 72% yield of the Z isomer, Z/E = 87:13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Solvent | Catalyst | Yieldb | Z/Ec |
| a Unless otherwise noted, all reactions were carried out with 1 or 2a (0.1 mmol) and the catalyst (20 mol%) in 1 mL of the solvent at 20 °C under a N2 atmosphere for 2 days. b Isolated yield of the Z isomer. c Determined by 1H NMR analysis of the Z and E mixture. | |||||
| 1 | 1a | CH2Cl2 | Cu(OTf)2 | — | — |
| 2 | 1b | CH2Cl2 | Cu(OTf)2 | — | — |
| 3 | 1c | CH2Cl2 | Cu(OTf)2 | — | — |
| 4 | 1d | CH2Cl2 | Cu(OTf)2 | — | — |
| 5 | 2a | CH2Cl2 | Cu(OTf)2 | 27 | 40:60 |
| 6 | 2a | CH2Cl2 | CuSO4·5H2O | — | — |
| 7 | 2a | CH2Cl2 | Cu(OAc)2·H2O | — | — |
| 8 | 2a | CH2Cl2 | PdCl2 | Trace | — |
| 9 | 2a | CH2Cl2 | FeF2 | Trace | — |
| 10 | 2a | CH2Cl2 | AlCl3 | 25 | 68:32 |
| 11 | 2a | CH2Cl2 | Bi(OTf)3 | Trace | — |
| 12 | 2a | CH2Cl2 | Lu(OTf)3 | — | — |
| 13 | 2a | CH2Cl2 | Yb(OTf)3 | 51 | 63:37 |
| 14 | 2a | CH2Cl2 | Sc(OTf)3 | 48 | 65:35 |
| 15 | 2a | EtOAc | Yb(OTf)3 | 59 | 92:8 |
| 16 | 2a | THF | Yb(OTf)3 | 72 | 87:13 |
| 17 | 2a | Acetone | Yb(OTf)3 | 72 | 80:20 |
| 18 | 2a | CH3CN | Yb(OTf)3 | 52 | 56:44 |
| 19 | 2a | Toluene | Yb(OTf)3 | 44 | 49:51 |
| 20 | 2a | MeOH | Yb(OTf)3 | — | — |
By adopting the conditions described in Table 1, the substrate scope of this 1,3-rearrangement was investigated for ester, oxindole and indole moieties of substrates 2. As shown in Scheme 1, a range of 3-alkenyl oxindoles 2 participated well in the rearrangement reaction. It was found that a smaller ester moiety on substrate 2 brought a higher yield and Z/E selectivity (3b, 86%, Z/E = 91:9); however, the separation of Z/E isomers was difficult when other substrates bearing a dimethyl maleate moiety were employed as reactants. On the other side, the reaction efficiency was found to be less dependent on the substituent of oxindole in most cases. For example, substrates with either electron-withdrawing or electron-donating groups at the C5 position on the benzene ring of the oxindole moiety were well tolerated, affording the corresponding rearrangement products 3c–3j with 73%–85% yields and 78:22–>99:1 Z/E selectivity. Notably, when Fmoc-N substituted 2i was employed as the substrate, the desired product 3i was obtained with high yield and Z/E selectivity (85% yield, >99:1 Z/E). Besides, 3-alkenyl oxindoles with electron-donating groups at C4, C5, C6, and C7 on the indole ring also smoothly participated in the target reaction, affording products with good yields and Z/E selectivity (3k–3n, 64%–89%, Z/E 83:17–92:8).
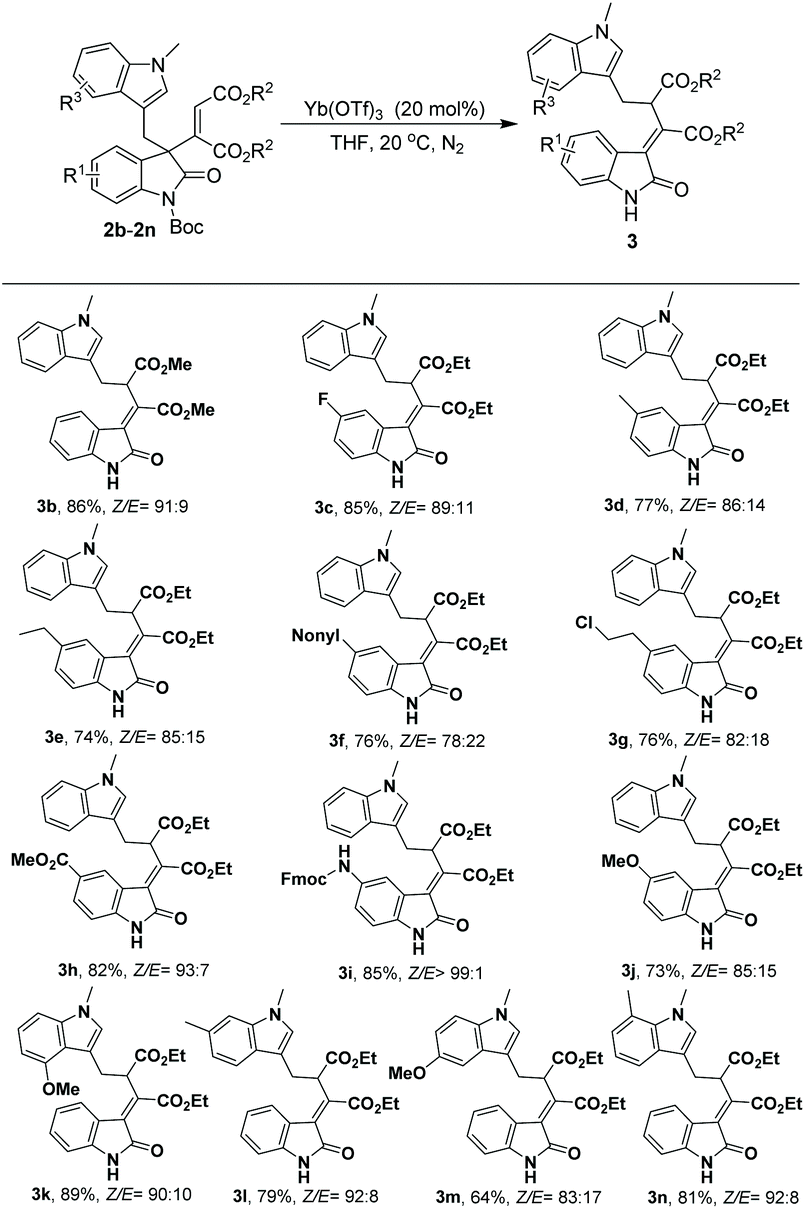 | ||
| Scheme 1 Yb(OTf)3 catalyzed [1,3]-rearrangement of 3-alkenyl oxindoles: unless otherwise noted, all reactions were carried out with 2 (0.1 mmol) and Yb(OTf)3 (20 mol%) in THF (1.0 mL) at 20 °C under an N2 atmosphere for 3 days; reactions to produce 3f, 3h, 3j, 3l and 3m were carried out for 2 days; isolated yields of the Z isomer are given; Z/E ratios were determined by 1H NMR analysis of the mixture. | ||
In an effort to explore the synthetic facility of our method, an operationally simple, one-pot sequential catalytic synthesis of 3-ylideneoxindoles was also developed. As shown in Scheme 2, after sequential additions of 1 mol% of DABCO and 20 mol% of Yb(OTf)3 to the mixture of diethyl but-2-ynedioate 4 and (indol-3-yl)methyl substituted oxindoles 5 in 1 mL EtOAc, the starting materials were smoothly converted into the corresponding rearrangement products 3 in one pot with good Z/E selectivities and overall yields (Scheme 2, 55%–72% yield of the Z isomer, Z/E 86:14–>92:8). The relative configuration of the major isomer of 3a was assigned as the Z configuration by X-ray analysis (Scheme 2),10 and the minor isomer was also assigned by X-ray analysis.
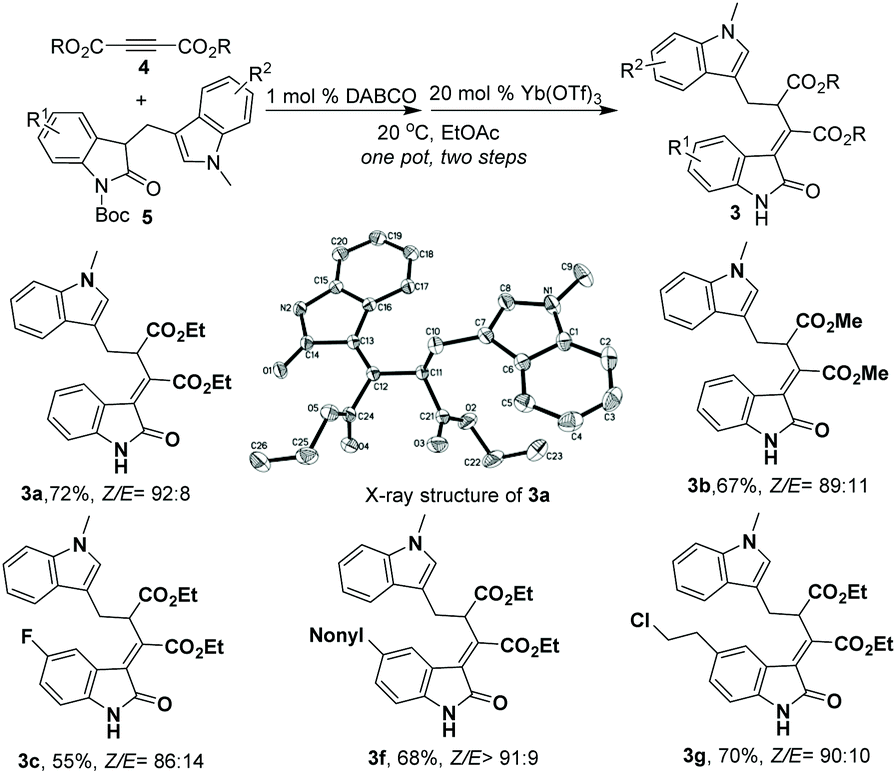 | ||
| Scheme 2 One-pot sequential catalytic synthesis of 3-ylideneoxindoles: unless otherwise noted, all reactions were carried out with 5 (0.1 mmol), alkyne ester 4 (0.15 mmol) and DABCO (1 mol%) in 1 mL of EtOAc at 20 °C under a N2 atmosphere. After the completion of the addition reaction between 5 and 4 (monitored by TLC), Yb(OTf)3 (20 mol%) was added to the resulting mixture under a N2 atmosphere and stirred for 3 days at 20 °C; isolated yields of the Z isomer are given; Z/E ratios were determined by 1H NMR analysis of the mixture. | ||
Importantly, the multifunctional 3-ylideneoxindoles obtained in this transformation can be readily transformed into valuable building blocks by conventional reactions. For example, the rearrangement product 3a can be converted into multi-substituted carbazole106 with 45% yield in the presence of 1.5 equiv. of DDQ and 0.2 mL of MeOH (Scheme 3). The structure of 6 was assigned by X-ray analysis (Scheme 3).11 With regard to the mechanism of this interesting transformation,12 we supposed that 3a might firstly undergo cyclization to form intermediate (A), which then afforded an isocyanate intermediate (B) via DDQ mediated deprotonation and ring-opening reaction; next, oxidative aromatization and subsequent addition of methanol to isocyanate (B) afforded carbazole 6 in moderate yield.
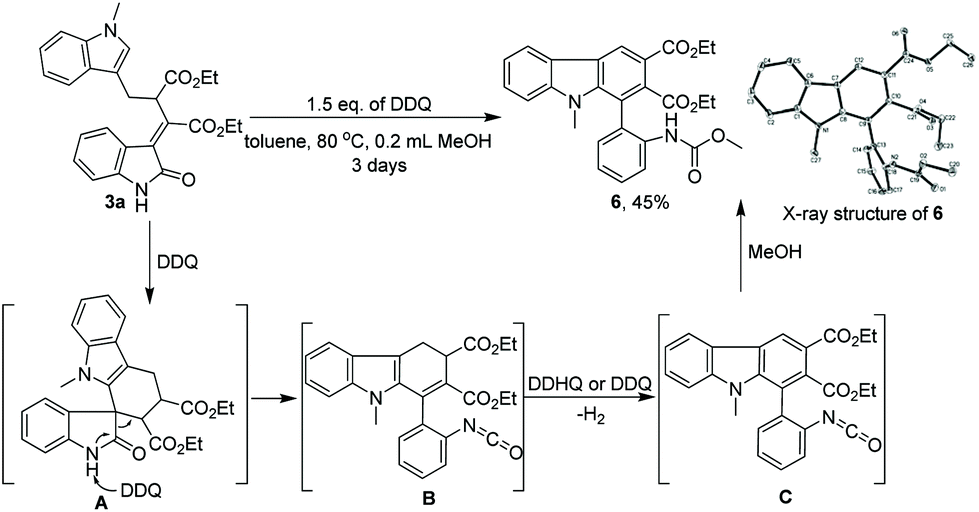 | ||
| Scheme 3 Synthesis of multi-substituted carbazole 6 from the rearrangement product 3a. | ||
To demonstrate the synthetic utility of our protocol, a larger scale catalytic procedure was performed. As shown in Scheme 4, 3 mmol 2b (1.5 g) was smoothly converted into corresponding 3b with 72% yield and 82:18 Z/E selectivity under standard reaction conditions.
 | ||
| Scheme 4 A gram-scale procedure of the Yb(OTf)3 catalyzed 1,3-rearrangement. | ||
To gain an insight into the mechanism of the [1,3]-rearrangement, a catalytic cross rearrangement was next carried out. As shown in Scheme 5, 0.05 mmol of 2h and 0.05 mmol of 2n were combined together in THF, and then treated with 20 mol% of Yb(OTf)3 under standard conditions. The fact that cross rearrangement products were not observed while 3h and 3n were obtained (52% and 43% yield, respectively) revealed that the present transformation may undergo an intramolecular, concerted rearrangement pathway; however, a tight ion pair mechanism is also possible.
 | ||
| Scheme 5 A cross rearrangement experiment of the Yb(OTf)3 catalyzed 1,3-rearrangement. | ||
A further attempt to explore more details about the reaction mechanism was carried out as shown in Scheme 6; to obtain a nonracemic rearrangement substrate, 20 mol% of cinchonine was used as a catalyst in the addition reaction of 4 and 5a in THF, affording 2a in 67% yield and 25% ee. Upon treatment with 20 mol% of Yb(OTf)3, nonracemic 2a was converted into 3a in 84% yield and 16% ee under standard conditions. The observation of decreased chirality on 3a indicated that a concerted rearrangement mechanism could be more reasonable.
 | ||
| Scheme 6 A nonracemic [1,3]-rearrangement reaction. | ||
Based on these results, a plausible concerted rearrangement mechanism of [1,3]-rearrangement was proposed (Scheme 7): by the promotion of 20 mol% of Yb(OTf)3, substrates 2 were efficiently activated via Yb3+-carbonyl chelation (Scheme 7, I), thus facilitating the electron transfer from nitrogen of the indole moiety to the maleate part, resulting in concerted migrations of the (1H-indol-3-yl)methyl group and the carbon–carbon double bond with good Z/E selectivity. Subsequently, a six-membered chelate between Boc amides and the Yb cation was formed, which promoted the breakage of the tertiary butyl C–O bond, and thus enabled the cleavage of the Boc group;13 finally, the [1,3]-rearrangement products 3 bearing a more stable conjugate system were produced with good yields and stereoselectivity. It was proposed that the good catalytic ability of Yb(OTf)3 exhibited in the [1,3]-rearrangement transformation may be attributed to its high Lewis acidity and good coordination abilities.14
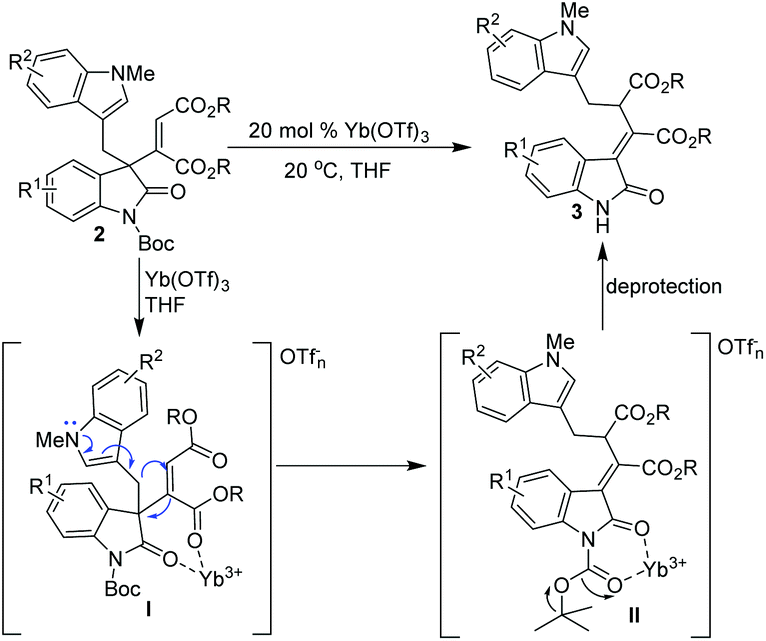 | ||
| Scheme 7 Proposed mechanism for the Yb(OTf)3 catalyzed 1,3-rearrangement of 3-alkenyl oxindoles. | ||
Conclusions
In conclusion, we developed an efficient [1,3]-rearrangement transformation of 3-alkenyl oxindoles by the promotion of Yb(OTf)3, affording diverse 3-ylideneoxindoles with good yields and Z/E selectivities. Besides, a one pot and sequential synthesis of 3-ylideneoxindoles was also achieved, which enhanced the synthetic utility of our methodology. Inspired by this success, further studies on the catalytic asymmetric 1,3-rearrangement are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21573161 and 21571144), the Zhejiang Provincial Natural Science Foundation of China (Y18B020031 and LQ19B020004) and the Foundation of Wenzhou Basic Scientific Research Project (G20180015) for generous financial support.Notes and references
- P. Wipf, in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon, Oxford, 1991, vol. 5, pp. 827–873 Search PubMed.
- L. Claisen, Chem. Ber., 1912, 45, 3157–3166 CrossRef CAS.
- A. C. Cope and E. M. Hard, J. Am. Chem. Soc., 1940, 62, 441–444 CrossRef CAS.
- For selected reviews on rearrangement reactions, see: (a) R. P. Lutz, Chem. Rev., 1984, 84, 205–247 CrossRef CAS; (b) L. E. Overman, Angew. Chem., Int. Ed. Engl., 1984, 23, 579–586 CrossRef; (c) T. G. Schenck and B. Bosnich, J. Am. Chem. Soc., 1985, 107, 2058–2066 CrossRef CAS; (d) B. Ganem, Angew. Chem., Int. Ed. Engl., 1996, 35, 936–945 CrossRef CAS; (e) A. M. M. Castro, Chem. Rev., 2004, 104, 2939–3002 CrossRef CAS; (f) A. Moyano, N. El-Hamdouni and A. Atlamsani, Chem. – Eur. J., 2010, 16, 5260–5273 CrossRef CAS; (g) A. C. Jones, J. A. May, R. Sarpong and B. M. Stoltz, Angew. Chem., Int. Ed., 2014, 53, 2556–5291 CrossRef CAS; (h) T. H. West, S. S. M. Spoehrle, K. Kasten, J. E. Taylor and A. D. Smith, ACS Catal., 2015, 5, 7446–7479 CrossRef CAS; (i) H. Wu, Q. Wang and J. Zhu, Eur. J. Org. Chem., 2019, 1964–1980 CrossRef CAS.
- (a) G. Kang, Q. Wu, M. Liu, Q. Xu, Z. Chen, W. Chen, Y. Luo, W. Ye, J. Jiang and H. Wu, Adv. Synth. Catal., 2013, 355, 315–320 CrossRef CAS; (b) G. Kang, Z. Luo, C. Liu, H. Gao, Q. Wu, H. Wu and J. Jiang, Org. Lett., 2013, 15, 4738–4741 CrossRef CAS; (c) H. Gao, Z. Luo, P. Ge, J. He, F. Zhou, P. Zheng and J. Jiang, Org. Lett., 2015, 17, 5962–5965 CrossRef CAS.
- For reviews on [1,3]-sigmatropic rearrangement reactions see: (a) P. A. Leber and J. E. Baldwin, Acc. Chem. Res., 2002, 35, 279–287 CrossRef CAS PubMed; (b) D. Lee and M. Kim, Org. Biomol. Chem., 2007, 5, 3418–3427 RSC; (c) C. G. Nasveschuk and T. Rovis, Org. Biomol. Chem., 2008, 6, 240–254 RSC For selected examples on [1,3]-sigmatropic rearrangement reactions, see: (d) E. Tayama, S. Otoyama and W. Isaka, Chem. Commun., 2008, 35, 4216–4218 RSC; (e) C. K. De, N. Mittal and D. Seidel, J. Am. Chem. Soc., 2011, 133, 16802–16805 CrossRef CAS; (f) C.-H. Chen, G. S. Yellol, P.-T. Lin and C.-M. Sun, Org. Lett., 2011, 13, 5120–5123 CrossRef CAS; (g) N.-A. Harada, T. Nishikata and H. Nagashima, Tetrahedron, 2012, 68, 3243–3252 CrossRef CAS; (h) K. C. Guérard, A. Guérinot, C. Bouchard-Aubin, M.-A. Ménard, M. Lepage, M. A. Beaulieu and S. Canesi, J. Org. Chem., 2012, 77, 2121–2133 CrossRef; (i) E. Tayama, K. Horikawa, H. Iwamoto and E. Hasegawa, Tetrahedron, 2013, 69, 2745–2752 CrossRef CAS; (j) D. V. Vidhani, J. W. Cran, M. E. Krafft, M. Manoharan and I. V. Alabugin, J. Org. Chem., 2013, 78, 2059–2073 CrossRef CAS PubMed; (k) Q. Zhou and Y. Li, J. Am. Chem. Soc., 2014, 136, 1505–1513 CrossRef CAS; (l) C. N. Kona and C. V. Ramana, Chem. Commun., 2014, 50, 2152–2154 RSC; (m) C. N. Kona, M. N. Patil and C. V. Ramana, Org. Chem. Front., 2016, 3, 453–456 RSC; (n) P. Mi, R. Kiran Kumar, P. Liao and X. Bi, Org. Lett., 2016, 18, 4998–5001 CrossRef CAS PubMed; (o) I. Nakamura, M. Owada, T. Jo and M. Terada, Org. Lett., 2017, 19, 2194–2196 CrossRef CAS; (p) O. A. Ivanova, A. O. Chagarovskiy, A. N. Shumsky, V. D. Krasnobrov, I. I. Levina and I. V. Trushkov, J. Org. Chem., 2018, 83, 543–560 CrossRef CAS; (q) S. Gandhi and B. Baire, J. Org. Chem., 2019, 84, 3904–3918 CrossRef CAS.
- L. Claisen, Ber. Dtsch. Chem. Ges., 1896, 29, 2931–2933 CrossRef.
- For selected examples of the synthesis of 3-ylideneoxindoles, see: (a) S. Couty, B. Liégault, C. Meyer and J. Cossy, Org. Lett., 2004, 6, 2511–2514 CrossRef CAS PubMed; (b) R. Yanada, S. Obika, T. Inokuma, K. Yanada, M. Yamashita, S. Ohta and Y. Takemoto, J. Org. Chem., 2005, 70, 6972–6975 CrossRef CAS; (c) D. M. D'Souza, F. Rominger and T. J. J. Müller, Angew. Chem., Int. Ed., 2005, 44, 153–158 CrossRef; (d) R. Yanada, S. Obika, Y. Kobayashi, T. Inokuma, M. Oyama, K. Yanada and Y. Takemoto, Adv. Synth. Catal., 2005, 347, 1632–1642 CrossRef CAS; (e) T.-S. Jiang, R.-Y. Tang, X.-G. Zhang, X.-H. Li and J.-H. Li, J. Org. Chem., 2009, 74, 8834–8837 CrossRef CAS PubMed; (f) Y. Yu, K. J. Shin and J. H. Seo, J. Org. Chem., 2017, 82, 1864–1871 CrossRef CAS.
- For a review on the bioactivity of 3-ylideneoxindoles, see: (a) A. Brancale and R. Silvestri, Med. Res. Rev., 2007, 27, 209–238 CrossRef CAS PubMed For selected examples of studies on the bioactivity of 3-ylideneoxindoles, see: (b) M. S. C. Pedras, J. L. Sorensen, F. J. Okanga and I. L. Zaharia, Bioorg. Med. Chem. Lett., 1999, 9, 3015–3020 CrossRef CAS; (c) C. L. Woodard, Z. Li, A. K. Kathcart, J. Terrell, L. Gerena, M. Lopez-Sanchez, D. E. Kyle, A. K. Bhattacharjee, D. A. Nichols, W. Ellis, S. T. Prigge, J. A. Geyer and N. C. Waters, J. Med. Chem., 2003, 46, 3877–3882 CrossRef CAS; (d) T. Jiang, K. L. Kuhen, K. Wolff, H. Yin, K. Bieza, J. Caldwell, B. Bursulaya, T. Tuntlad, K. Zhang, D. Karanewsky and Y. He, Bioorg. Med. Chem. Lett., 2006, 16, 2105–2108 CrossRef CAS PubMed; (e) Q. Zhu, C.-P. Tang, C.-Q. Ke, X.-Q. Li, J. Liu, L.-S. Gan, H.-C. Weiss, E.-R. Gesing and Y. Ye, J. Nat. Prod., 2010, 73, 40–44 CrossRef CAS PubMed; (f) A. Millemaggi and R. J. K. Taylor, Eur. J. Org. Chem., 2010, 4527–4547 CrossRef CAS; (g) H. B. Zou, L. Zhang, J. F. Ouyang, M. A. Giulianotti and Y. P. Yu, Eur. J. Med. Chem., 2011, 46, 5970–5977 CrossRef CAS PubMed; (h) C. Klöck, X. Jin, K. Choi, C. Khosla, P. B. Madrid, A. Spencer, B. C. Raimundo, P. Boardman, G. Lanza and J. H. Griffin, Bioorg. Med. Chem. Lett., 2011, 21, 2692–2696 CrossRef; (i) C.-T. Chiou, W.-C. Lee, J.-H. Liao, J.-J. Cheng, L.-C. Lin, C.-Y. Chen, J.-S. Song, M.-H. Wu, K.-S. Shia and W.-T. Li, Eur. J. Med. Chem., 2015, 98, 1–12 CrossRef CAS PubMed.
- For selected examples of the synthesis of carbazoles, see: (a) J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184–16186 CrossRef CAS PubMed; (b) C. Zhu and S. Ma, Org. Lett., 2014, 16, 1542–1545 CrossRef CAS; (c) K. Paul, K. Bera, S. Jalal, S. Sarkar and U. Jana, Org. Lett., 2014, 16, 2166–2169 CrossRef CAS; (d) J. R. Stepherson, C. E. Ayala, T. H. Tugwell, J. L. Henry, F. R. Fronczek and R. Kartika, Org. Lett., 2016, 18, 3002–3005 CrossRef CAS; (e) S. Chen, L. Wang, J. Zhang, Z. Hao, H. Huang and G.-J. Deng, J. Org. Chem., 2017, 82, 11182–11191 CrossRef CAS PubMed; (f) S. Maiti and P. Mal, Org. Lett., 2017, 19, 2454–2457 CrossRef CAS; (g) S. Yadav, R. Hazra, A. Singh and S. S. V. Ramasastry, Org. Lett., 2019, 21, 2983–2987 CrossRef CAS PubMed.
- CCDC 1815799 for 3a and 1815800 for 6 contain the supplementary crystallographic data for this paper.†.
- For a similar ring-opening transformation, see: J. Cao, J. Sun and C.-G. Yan, Org. Biomol. Chem., 2019, 17, 9008–9013 RSC.
- For a similar mechanism for the cleavage of N-Boc amines, see: (a) J. N. Hernández, M. A. Ramίrez and V. S. Martίn, J. Org. Chem., 2003, 68, 743–746 CrossRef; (b) J. M. López-Soria, S. J. Pérez, J. N. Hernández, M. A. Ramίrez, V. S. Martίn and J. I. Padrón, RSC Adv., 2015, 5, 6647–6651 RSC.
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W.-L. Lam, Chem. Rev., 2002, 102, 2227–2302 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1815799 for 3a and 1815800 for 6. For ESI and crystallographic data in CIF or other electronic formats see DOI: 10.1039/d0ob02032j |
| ‡ These authors made equal contribution to this study. |
| This journal is © The Royal Society of Chemistry 2022 |