
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cooperative photoredox/gold catalysed cyclization of 2-alkynylbenzoates with arenediazonium salts: synthesis of 3,4-disubstituted isocoumarins†
Valentina
Pirovano
 *,
Elisa
Brambilla
,
Giorgia
Fanciullacci
and
Giorgio
Abbiati
*
*,
Elisa
Brambilla
,
Giorgia
Fanciullacci
and
Giorgio
Abbiati
*
Dipartimento di Scienze Farmaceutiche, Sezione di Chimica Generale e Organica “A. Marchesini”, Università degli Studi di Milano, Via Venezian, 21, 20133 Milano, Italy. E-mail: valentina.pirovano@unimi.it; giorgio.abbiati@unimi.it
First published on 29th September 2022
Abstract
Several isocoumarins have been synthesised in good to excellent yields starting from 2-alkynylbenzoates and arenediazonium salts. The strategy involves a domino arylation/oxo-cyclization catalysed by a dual photoredox/gold catalytic system. The reactions run under mild conditions at room temperature in wet acetonitrile under irradiation with a blue-LED lamp, in the presence of a cationic gold catalyst and a cheap organic photocatalyst. The scope is quite broad and allows the preparation of isocoumarins differently disubstituted in positions 3 and 4. A plausible reaction mechanism is proposed.
Introduction
The isocoumarin (1H-isochromen-1-one) structure1 represents the heterocyclic skeleton of several compounds characterised by noteworthy biological activities.2 The synthesis of this nucleus has been accomplished both by metal-free3 and metal-catalysed4 methodologies. Several salts and complexes of transition metals have been successfully used, starting from early (titanium, chromium) to late (iron, ruthenium, rhodium, iridium, nickel, palladium, and zinc) and coinage (copper, silver, gold) ones. Among them, palladium- and copper-based catalysts are probably the most investigated and used.5An intriguing strategy to build up the pyran-2-one moiety of the isocoumarin nucleus is the cyclization of γ-alkynyl carboxylic acids or esters. Many approaches involving Brønsted acid catalysis6 (also in the presence of an additional electrophile)7 and LA/TM catalysis8 have been reported. Moreover, in some papers, this transformation was obtained by a cascade coupling/annulation sequence starting from o-halobenzoic acid/esters and terminal alkynes9 or by a CH bond-activation/annulation path starting from benzoic acids and internal alkynes.10 Our contribution to the development of effective and sustainable methods to prepare 3-substituted isocoumarins starting from 2-alkynyl-arylcarboxylates is represented by two recent papers. In the first, isocoumarins have been obtained by an AgOTf/p-TSA co-catalysed approach, characterised by mild reaction conditions, high yields and selectivity, and a low catalyst loading.11 In the second, we described the use of a p-TSA-based Deep Eutectic Solvent (DES) as an environmental-friendly “active” solvent under microwave heating able to promote this transformation. In this case, the proposed procedure is characterised by a high degree of sustainability, the cleanness of the reactions in reduced times, and the cheapness and reusability of the active solvent.12
In the last few years, photoredox catalysis has become a trendy alternative strategy for several organic transformations.13 In this context, cooperative photoredox catalysis14 – and in particular photocatalysis with transition metal complexes15 – allows the improvement of molecular complexity through the “browsing” among the different oxidation states of the metal catalysts without the requirement for stoichiometric sacrificial reagents. Among the transition metal complexes used in this cooperative fashion, gold complexes represent interesting candidates because when gold catalysis meets photocatalysis, a valence change of the gold center can easily be achieved via electron transfer and radical addition.16
Cooperative photoredox/TM catalysis has been broadly used for the synthesis of different nitrogen-, sulphur- and oxygen-containing heterocycles. Surprisingly, isocoumarins seem to be a bit neglected. To the best of our knowledge, the only example was reported by Alcaide, Almendros, and co-workers in 2017.17 In this paper the authors prepare seven 3,4-diarylisocoumarins starting from methyl 2-((trimethylsilyl)ethynyl)arylcarboxylates and arenediazonium salts (6 equiv.) in the presence of triphenylphosphine gold chloride (10 mol%) and a Ru-based photoredox catalyst (2.5 mol%). The reactions run in a mixture of methanol and acetonitrile (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at rt under irradiation of the light of a fluorescence bulb for 1–6 hours to give the desired 3,4-homosubstituted isocoumarins in yields ranging from 59 to 72%. The main limitations of this approach are (i) the homosubstitution in positions 3 and 4 of the isocoumarin, (ii) the use of an expensive Ru-based photocatalyst, and (iii) a slightly narrow scope. A strongly related cascade approach starting from 2-alkynylphenols and arenediazonium salts for the preparation of 2,3-diarylbenzofurans was reported one year before by Ollivier, Fensterbank, and co-workers.18 The authors used the same dual catalytic system in methanol at rt under visible light. The reactions are slower (16 h), and the yields seem to be strongly affected by the nature of the substituents on both phenols and arenediazonium salts. Inspired by these works, and in connection with our continuous interest in the gold-catalysed synthesis of heterocycles19 and the development of alternative approaches to isocoumarins,11,12 we report here a new approach to 3,4-heterodisubstituted isocoumarins through a dual photoredox/gold catalysed arylative oxo-cyclization of 2-alkynylbenzoates in the presence of arenediazonium salts.
:1) at rt under irradiation of the light of a fluorescence bulb for 1–6 hours to give the desired 3,4-homosubstituted isocoumarins in yields ranging from 59 to 72%. The main limitations of this approach are (i) the homosubstitution in positions 3 and 4 of the isocoumarin, (ii) the use of an expensive Ru-based photocatalyst, and (iii) a slightly narrow scope. A strongly related cascade approach starting from 2-alkynylphenols and arenediazonium salts for the preparation of 2,3-diarylbenzofurans was reported one year before by Ollivier, Fensterbank, and co-workers.18 The authors used the same dual catalytic system in methanol at rt under visible light. The reactions are slower (16 h), and the yields seem to be strongly affected by the nature of the substituents on both phenols and arenediazonium salts. Inspired by these works, and in connection with our continuous interest in the gold-catalysed synthesis of heterocycles19 and the development of alternative approaches to isocoumarins,11,12 we report here a new approach to 3,4-heterodisubstituted isocoumarins through a dual photoredox/gold catalysed arylative oxo-cyclization of 2-alkynylbenzoates in the presence of arenediazonium salts.
Results and discussion
We start our investigation by choosing the reaction between methyl 2-(p-tolylethynyl)benzoate 1a and benzenediazonium tetrafluoroborate 2a as a model system. Taking into account the reaction conditions described in the recent related literature,17,18 we tested the activity of different gold complexes, Ru-based and organic photocatalysts, additives, solvents, and light sources. A selection of results of this screening is reported in Table 1.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Gold catalyst [Au] (10 mol%) | Additive | Photo catalyst PC (2.5 mol%) | Solvent | hν | Overall yielda (%) | 3/4 ratio |
| a Yields referred to pure isolated products. b No more starting material, by-products. c Reaction time: 5 h. | |||||||
| 1 | [Au(PPh3)Cl] | — | [Ru(bpy)3(PF6)2] | CH3CN | Blue-LED | 74 | 1:1.7 |
| 2 | [Au(PPh3)Cl] | p-TSA (10 mol%) | [Ru(bpy)3(PF6)2] | CH3CN | Blue-LED | 70 | 1:1 |
| 3 | [Au(PPh3)NTf2] | p-TSA (10 mol%) | [Ru(bpy)3(PF6)2] | CH3CN | Blue-LED | 89 | 2:1 |
| 4 | [Au(PPh3)NTf2] | p-TSA (30 mol%) | [Ru(bpy)3(PF6)2] | CH3CN | Blue-LED | 84 | 1:1 |
| 5 | [Au(PPh3)NTf2] | p-TSA (5 mol%) | [Ru(bpy)3(PF6)2] | CH3CN | Blue-LED | 51 | 1:1.65 |
| 6 | [Au(PPh3)NTf2] | — | [Ru(bpy)3(PF6)2] | CH3OH | Blue-LED | 78 | 1:1.9 |
| 7 | [Au(PPh3)NTf2] | — | [Ru(bpy)3(PF6)2] | CH3OH/CH3CN 1:3 |
Blue-LED | 13b | 1:0 |
| 8 | [Au(PPh3)NTf2] | — | [Ru(bpy)3(PF6)2] | CH3OH/CH3CN 1:9 (=25 equiv. of MeOH) |
Blue-LED | 42b | 1:0 |
| 9 | [Au(PPh3)NTf2] | CH3OH (10 equiv.) | [Ru(bpy)3(PF6)2] | CH3CN | Blue-LED | 93 | 1:0 |
| 10 | [Au(PPh3)NTf2] | H2O (10 equiv.) | [Ru(bpy)3(PF6)2] | CH3CN | Blue-LED | 96 | 1:0 |
| 11 | [Au(JohnPhos)NTf2] | H2O (10 equiv.) | [Ru(bpy)3(PF6)2] | CH3CN | Blue-LED | nr | — |
| 12 | [Au(P(p-CF3Ph)3)NTf2] | H2O (10 equiv.) | [Ru(bpy)3(PF6)2] | CH3CN | Blue-LED | 77 | 1:0 |
| 13 | [Au(PPh3)NTf2] | H2O (10 equiv.) | [Ru(bpy)3Cl2] | CH3CN | Blue-LED | 64 | 1:0 |
| 14 | [Au(PPh 3 )NTf 2 ] | H 2 O (10 equiv.) | Eosin Y | CH 3 CN | Blue-LED | 96 |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0 :0
|
| 15 | [Au(PPh3)NTf2] | H2O (10 equiv.) | Eosin Y | CH3CN | Green-LED | 68 | 1:0.22 |
| 16 | [Au(PPh3)NTf2] | H2O (10 equiv.) | Eosin Y | CH3CN | CFL (21 W) | 70 | 1:0.9 |
| 17 | [Au(PPh3)NTf2] | H2O (10 equiv.) | Eosin Y | CH3CN | Dark | 55c | 0:1 |
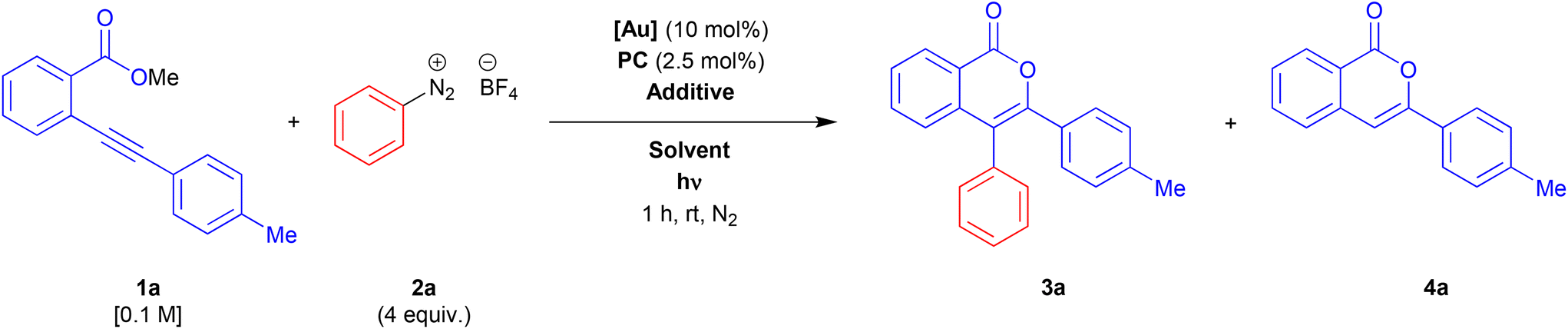
Under Alcaide conditions in acetonitrile and using a blue-LED (465 nm) as a light source, we observed the formation of a mixture of the desired product 3a beside a two-fold amount of the simple cyclization product 4a, in 74% overall yield (Table 1, entry 1). The selectivity toward the arylated product 3a was improved by adding 10 mol% of p-TSA as an additive, with a slight loss in overall yield (Table 1, entry 2). We were pleased to find that changing the counter-ion of the gold complex yield and selectivity were improved to 89% and 2:1, respectively (Table 1, entry 3), while an increase or a reduction of the additive amount had a negative effect on both the parameters (Table 1, entries 4 and 5). Then, we tested the effect of a protic polar solvent as an alternative additive (Table 1, entries 6–9). The reaction in pure methanol resulted in a heavy loss of selectivity (Table 1, entry 6), conversely, the use of methanol as minority co-solvent together with acetonitrile gave 3a as a single product (Table 1, entries 7–9). Interestingly, the yield seemed to grow inversely proportional to the amount of methanol, with the best results in the presence of 10 equiv. of methanol (Table 1, entry 9). A cheaper and more sustainable protic solvent as water gave the same selectivity with a slight improvement in yield (Table 1, entry 10). Next, we test the behaviour of electron-richer and electron-poorer phosphine gold complexes (Table 1, entries 11 and 12). The reaction in the presence of [Au(JohnPhos)NTf2] failed (Table 1, entry 11), whereas [Au(P(p-CF3Ph)3)NTf2] gave great results in terms of selectivity but a worse yield (Table 1, entry 12). We changed also the photocatalyst and the wavelength of the LED light (Table 1, entries 13–15). We were pleased to find that the inexpensive organic photocatalyst eosin Y20 gave the same results as Ru(bpy)3(PF6)2, (Table 1, entry 14), whereas the green-LED lamp (525 nm) gave worse results in both terms of selectivity and yield (Table 1, entry 15). The efficiency of the blue-LED light was confirmed by a test with a 21 W CFL (compact fluorescence light), which induce a loss of both yields and selectivity (Table 1, entry 16). Finally, a control test in the dark confirmed the key contribution of the photocatalytic cycle to the arylation step. If the activation of the photocatalyst is hampered, the reaction follows a simple metal-catalysed cyclization-without-arylation path to give 4a, according to that observed in the already studied reactions of 2-alkynylbenzoates in the presence of a silver catalyst11 (for a plausible mechanism see ESI, Scheme SI1†).
Under the optimised reaction conditions, we explored the scope and limitations of the approach. The 2-alkynylbenzoates 1 were prepared by a Sonogashira cross-coupling21 between 2-bromo-benzoates and terminal alkynes as previously described.11,12,22 Arenediazonium salts were synthesised according to previous literature.23 All unknown compounds have been fully characterised through NMR spectroscopy, mass spectrometry, and elemental analysis. The results are summarised in Scheme 1.
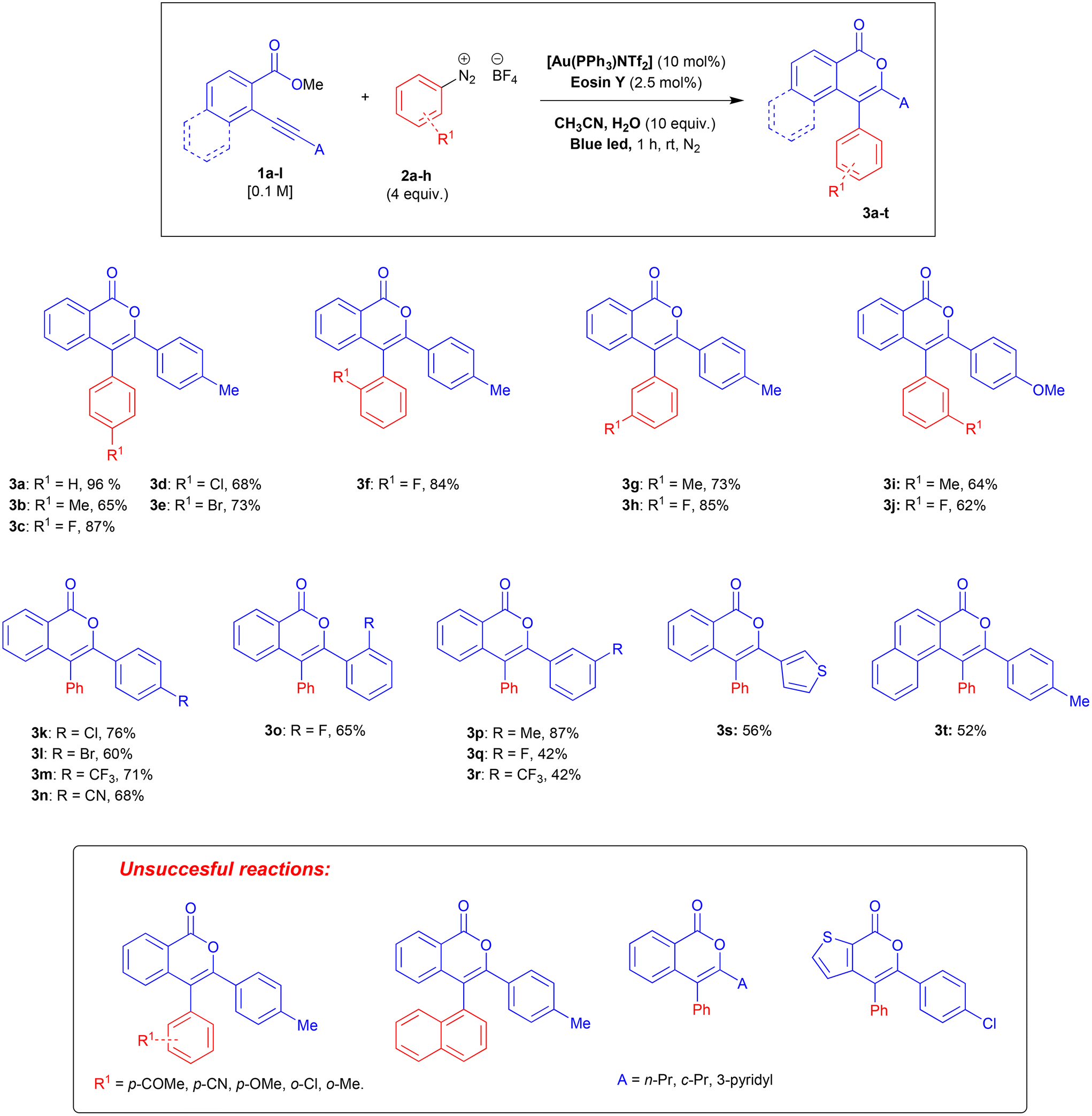 | ||
| Scheme 1 Scope and limitation of the approach. | ||
When the arenediazonium partner is substituted in the ortho-position, only small groups like fluorine are allowed (3f) whereas more bulky substituents (Cl and Me) resulted in mixtures of unidentified by-products. The problems related to this kind of steric hindrance were confirmed by the failure of 2-naphtalenediazonium salt to give the desired isocoumarin.
Substitution on the arenediazonium partner in para- and meta-positions with halo groups or an alkyl group are well-tolerated and the corresponding isocoumarins 3a–j were obtained from good to excellent yields. Interestingly, the reactions with arenediazonium salts bearing groups with enhanced electron-withdrawing (EW) or electron-donating (ED) properties failed and gave complex mixtures of unidentified by-products. A possible reason for these failures should be that the potential of eosin Y may not be competitive for the reduction of some diazonium salts. To overcome this problem, we made two attempts with 4-cyanobenzenediazonium tetrafluoborate in the presence of two photocatalysts characterised by higher excited state reduction potential (i.e., fac-Ir(ppy)3: 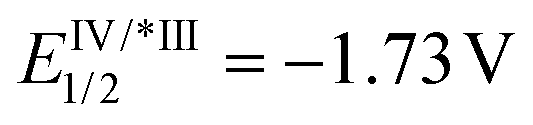 and [Ru(bpy)3(PF6)2]:
and [Ru(bpy)3(PF6)2]: 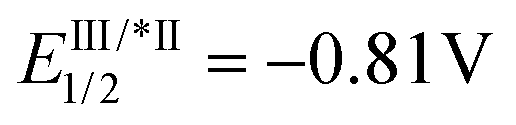 )15 but also in these cases the reaction failed. Furthermore, it has been reported that in dual gold/photoredox catalysis, the reactions of methoxy substituted benzenediazonium salts are better promoted by gold catalysts with electron-poor ligands,24 maybe because the addition of the aryl radical to the gold complex is favourite. For this reason, we try the reaction with 4-methoxybenzenediazonium tetrafluoborate under the optimised condition in the presence of a different gold complex, namely [Au[P(p-CF3-Ph)3]NTf2], but also in this case the main product arises from the metal-catalysed cyclization-without-arylation.
)15 but also in these cases the reaction failed. Furthermore, it has been reported that in dual gold/photoredox catalysis, the reactions of methoxy substituted benzenediazonium salts are better promoted by gold catalysts with electron-poor ligands,24 maybe because the addition of the aryl radical to the gold complex is favourite. For this reason, we try the reaction with 4-methoxybenzenediazonium tetrafluoborate under the optimised condition in the presence of a different gold complex, namely [Au[P(p-CF3-Ph)3]NTf2], but also in this case the main product arises from the metal-catalysed cyclization-without-arylation.
Next, we investigated the effect of the substitution on the alkynyl terminus of the o-alkynyl arylcarboxylates. The presence of a phenyl group bearing in para, ortho, or meta position halo- or alkyl-groups is in general well tolerated (3k–r), although strong EW groups gave sometimes the desired isocoumarins only in modest yields (3q,r). Electron-rich heterocycles at the alkyne terminus are well tolerated (3s) whereas electron-poor ones hamper the reaction as well as alkyl and cycloalkyl substituents. Modifications on the carboxylate moiety seem to be allowed only without perturbation of the electronic properties (3t).
To validate the protocol for the synthesis of functional molecules, we prepare by this way 7-hydroxy-3-(4-hydroxyphenyl)-4-phenyl-1H-isochromen-1-one 3v, which has been tested as a potential estrogen receptor with a slight selectivity for α-subtype.25 The desired product 3v was obtained in good yield (91%) in a two-step process. The reaction of methyl 5-methoxy-2-((4-methoxyphenyl)ethynyl)benzoate 1m with benzenediazonium salt 2a under the optimised reaction conditions gave the intermediate 3u which was transformed in 3v by treatment with an excess of BBr3 (Scheme 2).
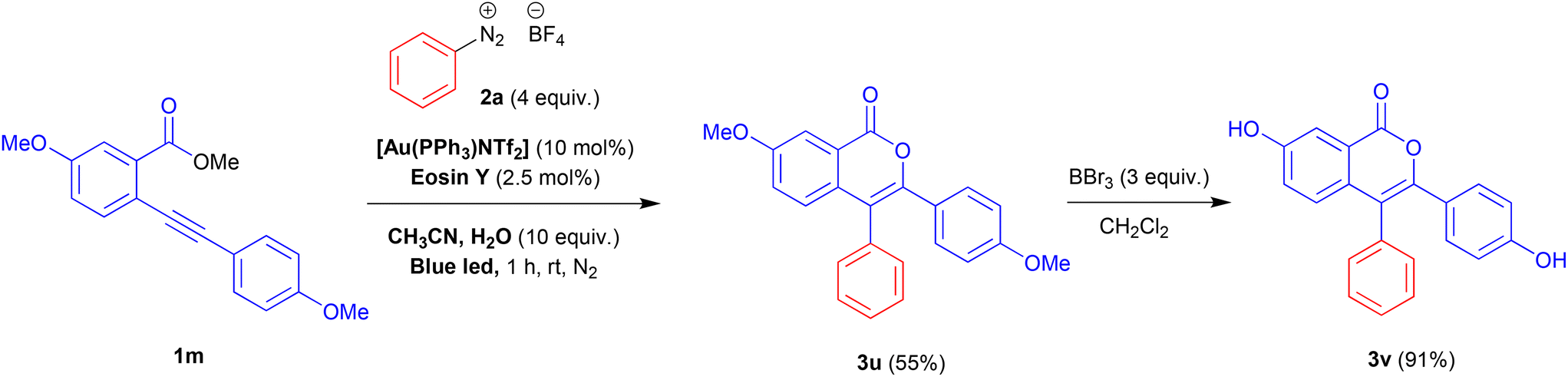 | ||
| Scheme 2 Synthesis of 7-hydroxy-3-(4-hydroxyphenyl)-4-phenyl-1H-isochromen-1-one 3v. | ||
Based on experimental evidence and previous findings on cooperative Au/photoredox catalysis,16–18 a reasonable mechanism involving a photoredox-induced homogeneous Au(I)/Au(III) redox cycle was proposed (Scheme 3). The photoexcitation of eosin Y with the blue-LED promotes the generation of the aryl radical from the arenediazonium salt 2 by a single electron transfer (SET). The aryl radical reacts with the gold(I) catalyst to initially generate a gold(II)–aryl complex A that is further oxidized by the radical-cation of eosin Y to give a gold(III)–aryl complex B thus regenerating the photocatalyst. The redox potential for the oxidation of gold(II) to gold(III) in the specific intermediate is still unknown.16 Moreover, the reduction potentials of eosin Y radical cation and [Ru(bpy)3(PF6)2]3+ (i.e. the PC used with gold in ref. 17) are quite different (+0.7820 and +1.2915 in V vs. SCE, respectively). However, the ability of eosin Y to oxidize gold(II) to gold(III) has been already reported.26 Gold complex B coordinates the triple bond of substrate 1 to generate the π-complex I. The following regiospecific 6-endo-dig intramolecular nucleophilic attack of the carbonyl oxygen to the activated alkyne forms the isochromenylium cation (II/III) stabilized by resonance.11 The water, used as an additive, promotes the removal of the methyl group from intermediate III to give the gold σ-complex IV and a molecule of protonated methanol.6b The successive reductive elimination generates the desired isocoumarin 3 and regenerates the gold(I) catalyst. The by-product 4a, resulting from simple cyclization without arylation, was sometimes obtained under unoptimised conditions (Table 1, entries 1–6), perhaps when the reaction conditions made the protodemetallation path competitive with reductive elimination.
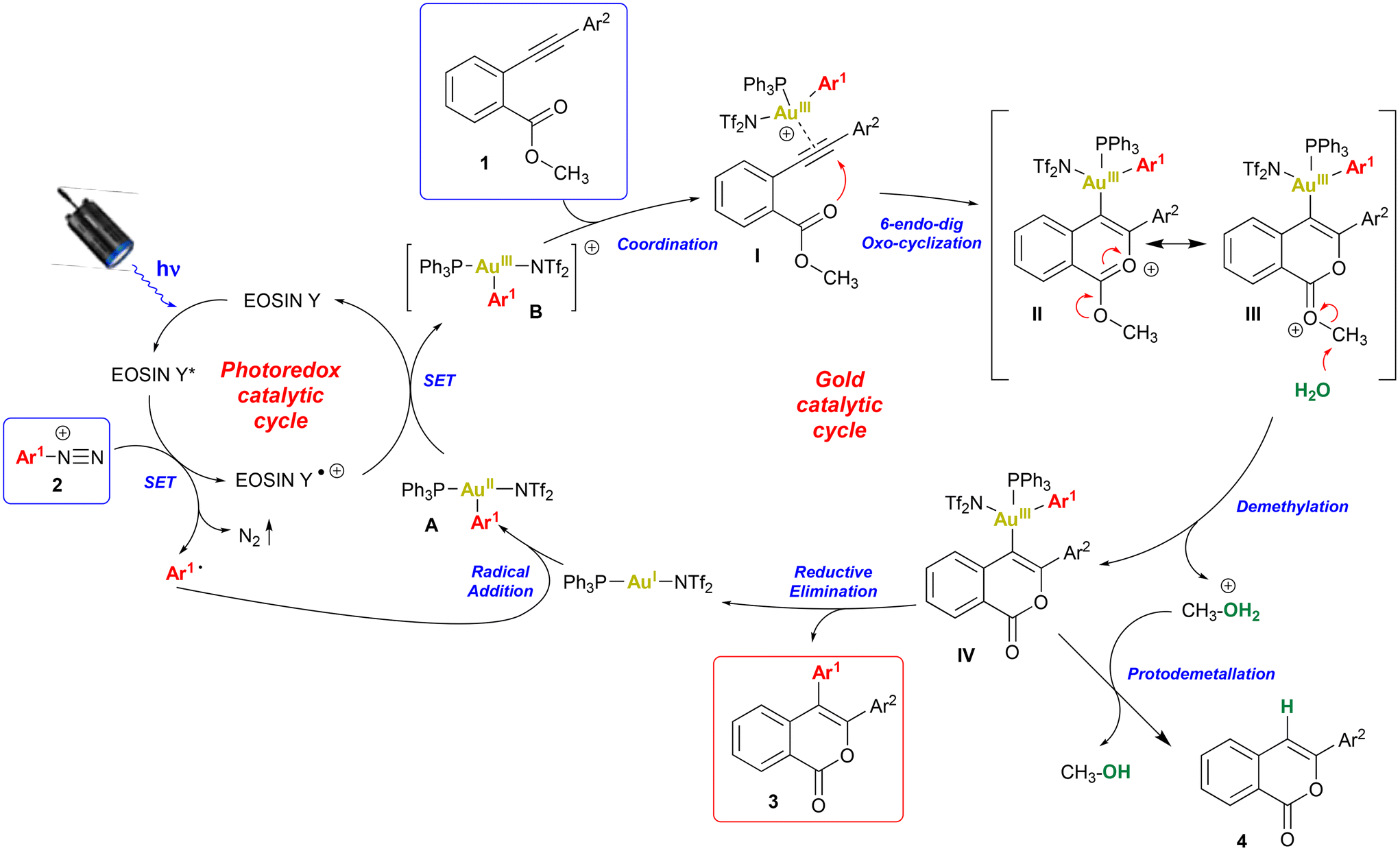 | ||
| Scheme 3 Suggested reaction mechanism. | ||
Conclusions
In conclusion, a new cooperative photoredox/gold catalysed approach to isocoumarins starting from 2-alkynylbenzoates and arenediazonium salts has been developed. The methodology is characterised by mild reaction conditions, broad scope, and yields ranging from good to excellent. The strategy involves a cascade arylation/oxo-cyclization promoted by PPh3AuNTf2 and eosin Y. In comparison with the previously reported method,17 this approach allows the preparation of isocoumarins with a different pattern of substitution in positions 3 and 4. Moreover, the use of an inexpensive organic photocatalyst such as eosin Y represents an additional benefit.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Mrs Donatella Nava for the NMR analyses and Mr Stefano Pandini for MS analyses. This research was supported by a grant from Università degli Studi di Milano (Italy), Linea 2 “Dotazione Annuale per Attività Istituzionali” 2022.References
- R. D. Barry, Chem. Rev., 1964, 64, 229–260 CrossRef CAS.
- A. Saeed, Eur. J. Med. Chem., 2016, 116, 290–317 CrossRef CAS.
- A. Saeed and F. A. Larik, Chem. Heterocycl. Compd., 2016, 52, 450–452 CrossRef CAS.
- Z. Ashraf, Chem. Heterocycl. Compd., 2016, 52, 149–151 CrossRef CAS.
- A. Saeed, M. Haroon, F. Muhammad, F. A. Larik, E.-S. Hesham and P. A. Channar, J. Organomet. Chem., 2017, 834, 88–103 CrossRef CAS.
- Some representative examples: (a) G. Le Bras, A. Hamze, S. Messaoudi, O. Provot, P.-B. Le Calvez, J.-D. Brion and M. Alami, Synthesis, 2008, 1607–1611 CAS; (b) G. Zhao, L.-Z. Yuan, M. Roudier, J.-F. Peyrat, A. Hamze, J.-D. Brion, O. Provot and M. Alami, Synthesis, 2016, 3382–3392 CAS.
- Some representative examples: (a) M. Biagetti, F. Bellina, A. Carpita, P. Stabile and R. Rossi, Tetrahedron, 2002, 58, 5023–5038 CrossRef CAS; (b) T. Yao and R. C. Larock, J. Org. Chem., 2003, 68, 5936–5942 CrossRef CAS PubMed; (c) D. J. Faizi, A. Issaian, A. J. Davis and S. A. Blum, J. Am. Chem. Soc., 2016, 138, 2126–2129 CrossRef CAS PubMed; (d) R. Mancuso, C. C. Pomelli, F. Malafronte, A. Maner, N. Marino, C. Chiappe and B. Gabriele, Org. Biomol. Chem., 2017, 15, 4831–4841 RSC.
- Some representative examples: (a) T. Yoshlkawa and M. Shindo, Org. Lett., 2009, 11, 5378–5381 CrossRef PubMed; (b) A. Speranca, B. Godoi, S. Pinton, D. F. Back, P. H. Menezes and G. Zeni, J. Org. Chem., 2011, 76, 6789–6797 CrossRef CAS PubMed; (c) A. S. K. Hashmi, C. Lothschuetz, R. Doepp, M. Ackermann, J. De Buck Becker, M. Rudolph, C. Scholz and F. Rominger, Adv. Synth. Catal., 2012, 354, 133–147 CrossRef CAS; (d) N. A. Mallampudi, G. S. Reddy, S. Maity and D. K. Mohapatra, Org. Lett., 2017, 19, 2074–2077 CrossRef CAS PubMed; (e) P. Zhao, D. Chen, G. Song, K. Han and X. Li, J. Org. Chem., 2012, 77, 1579–1584 CrossRef CAS PubMed; (f) H. Wang, X. Han and X. Lu, Tetrahedron, 2013, 69, 8626–8631 CrossRef CAS; (g) J. Zhang, X. Han and X. Lu, J. Org. Chem., 2016, 81, 3423–3429 CrossRef CAS PubMed; (h) R. Umeda, S. Yoshikawa, K. Yamashita and Y. Nishiyama, Heterocycles, 2015, 91, 2172–2179 CrossRef CAS.
- Some recent representative examples: (a) V. Subramanian, V. R. Batchu, D. Barange and M. Pal, J. Org. Chem., 2005, 70, 4778–4783 CrossRef CAS PubMed; (b) M. Sun, L. Su, J. Dong, L. Liu, Y. Zhou and S.-F. Yin, Tetrahedron Lett., 2017, 58, 2433–2437 CrossRef CAS; (c) R. G. Chary, G. R. Reddy, Y. S. S. Ganesh, K. V. Prasad, S. K. P. Chandra, S. Mukherjeec and M. Pal, RSC Adv., 2013, 3, 9641–9644 RSC; (d) M. R. Kumar, F. M. Irudayanathan, J. H. Moon and S. Lee, Adv. Synth. Catal., 2013, 355, 3221–3230 CrossRef CAS.
- Some representative examples: (a) S. Warratz, C. Kornhaaß, A. Cajaraville, B. Niepçtter, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513–5517 CrossRef CAS PubMed; (b) L. Ackermann, J. Pospech, K. Graczyk and K. Rauch, Org. Lett., 2012, 14, 930–933 CrossRef CAS PubMed; (c) D. A. Frasco, C. P. Lilly, P. D. Boyle and E. A. Ison, ACS Catal., 2013, 3, 2421–2429 CrossRef CAS; (d) G. Jiang, J. X. Li, C. Zhu, W. Wu and H. Jiang, Org. Lett., 2017, 19, 4440–4443 CrossRef CAS PubMed.
- J. Gianni, V. Pirovano and G. Abbiati, Org. Biomol. Chem., 2018, 16, 3213–3219 RSC.
- F. Curti, M. Tiecco, V. Pirovano, R. Germani, A. Caselli, E. Rossi and G. Abbiati, Eur. J. Org. Chem., 2019, 1904–1914 CrossRef CAS.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS; (c) R. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 335, 2727–2744 CrossRef; (d) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- X. Lang, J. Zhaob and X. Chen, Chem. Soc. Rev., 2016, 45, 3026–3038 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- S. Witzel, A. S. K. Hashmi and J. Xie, Chem. Rev., 2021, 121, 8868–8925 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros, A. Busto, F. Herrera, C. Lázaro-Milla and A. Luna, Adv. Synth. Catal., 2017, 359, 2640–2652 CrossRef CAS.
- Z. Xia, O. Khaled, V. Mouriès-Mansuy, C. Ollivier and L. Fensterbank, J. Org. Chem., 2016, 81, 7182–7190 CrossRef CAS.
- (a) E. Brambilla, V. Pirovano, M. Giannangeli, G. Abbiati, A. Caselli and E. Rossi, Org. Chem. Front., 2019, 6, 3078–3084 RSC; (b) V. Pirovano, E. Brambilla, S. Rizzato, G. Abbiati, M. Bozzi and E. Rossi, J. Org. Chem., 2019, 84, 5150–5166 CrossRef CAS PubMed; (c) V. Pirovano, M. Borri, G. Abbiati, S. Rizzato and E. Rossi, Adv. Synth. Catal., 2017, 359, 1912–1918 CrossRef CAS; (d) V. Pirovano, M. Negrato, G. Abbiati, M. Dell'Acqua and E. Rossi, Org. Lett., 2016, 18, 4798–4801 CrossRef CAS PubMed.
- (a) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC; (b) V. Srivastavaa and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC.
- (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 50, 4467–4470 CrossRef For some recent reviews see: (b) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS; (c) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- X. Lin, Z. Fang, C. Zeng, C. Zhu, X. Pang, C. Liu, W. He, J. Duan, N. Qin and K. Guo, Chem. – Eur. J., 2020, 26, 13738–13742 CrossRef CAS PubMed.
- J. Liu, E. Xu, J. Jiang, Z. Huang, L. Zheng and Z.-Q. Liu, Chem. Commun., 2020, 56, 2202–2205 RSC.
- X.-z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844–5847 CrossRef CAS PubMed.
- M. De Angelis, F. Stossi, M. Waibel, B. S. Katzenellenbogen and J. A. Katzenellenbogen, Bioorg. Med. Chem., 2005, 13, 6529–6542 CrossRef CAS.
- A. Tlahuext-Aca, M. N. Hopkinson, R. A. Garza-Sanchez and F. Glorius, Chem. – Eur. J., 2016, 22, 5909–5913 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01371a |
| This journal is © The Royal Society of Chemistry 2022 |