
A facile and economical method to synthesize a novel wide gamut fluorescent copolyester with outstanding properties†
Lele
Ma
ab,
Jiajian
Liu
 *a,
Chuncheng
Li
*a,
Yaonan
Xiao
a,
Shaohua
Wu
a and
Bo
Zhang
a
*a,
Chuncheng
Li
*a,
Yaonan
Xiao
a,
Shaohua
Wu
a and
Bo
Zhang
a
aCAS Key Laboratory of Engineering Plastics, Institute of Chemistry Chinese Academy of Sciences (ICCAS), Beijing 100190, P.R. China. E-mail: ljj6609891@iccas.ac.cn; lichch@iccas.ac.cn
bUniversity of the Chinese Academy of Sciences, Beijing 100049, P.R. China
First published on 7th December 2021
Abstract
In recent years, non-traditional intrinsic luminescence (NTIL) polymers have attracted extensive attention because of their interesting fluorescence properties. The traditional polyester polyethylene terephthalate (PET) is well known for its wide application, yet the fluorescence characteristics of this polyester have rarely been reported. Herein, a series of high molecular weight PET modified copolyesters were synthesized by a simple and economical two-step polycondensation method, and for the first time we found that the copolyesters exhibited an unexpected green fluorescence under 365 nm UV light. Therefore, we obtained a series of new PET copolyesters with wide color gamut fluorescence characteristics and excellent mechanical and thermodynamic properties. These copolyesters exhibited aggregation-induced emission (AIE) characteristics in solution. The preliminary results showed that the spatial aggregation of electron-rich groups such as the benzene ring, the carbonyl group and the carbon oxygen bond was the main cause of the unusual fluorescence. This study broadens the application field of the traditional polyester and effectively enriches its fluorescence properties, and has theoretical and practical significance for the research of non-traditional intrinsic luminescent polymer materials.
Introduction
Non-traditional intrinsic luminescence (NTIL) polymers have attracted more and more attention because of their interesting photophysical properties, easy film-forming ability and good mechanical processing properties.1–4 They are widely used in many fields, such as biological imaging,5–7 solar energy conversion, chemical probes, fluorescent sensors,8–11 and drug delivery.12,13Compared with traditional luminescent materials, non-traditional intrinsic luminescent polymer materials generally show the characteristics of aggregation induced luminescence,14 that is the spatial aggregation of electron rich groups such as amino, carbonyl, anhydride, cyano, ester and amide groups and hydrogen bonds in the molecule forms new chromophores with a large conjugated structure, so as to emit fluorescence.15,16 Tang and co-workers17 synthesized a copolymer of maleic anhydride and vinyl acetate by free radical polymerization of maleic anhydride and vinyl acetate. The copolymer only contains carbonyl in its molecular chain; however, it can emit fluorescence under the irradiation with a 365 nm ultraviolet lamp in solution, and the fluorescence color has a significant solvent effect; Du et al.18 performed the polycondensation of glycerol and a linear aliphatic dicarboxylic acid to obtain a partially branched all biological degradable copolyester. The methanol solution of the copolyester can emit a strong blue fluorescence under the irradiation with a 365 nm UV lamp; the highly branched polyamine synthesized by Yang et al.19 contains only non-traditional chromophore amino groups, but it can also produce bright blue fluorescence in solution; Feng et al.20 synthesized a polyhydroxyaminoformate based on covalent crosslinking. By adjusting the degree of crosslinking, the concentration of luminescent substances (the ester group and amine group) produced by polymer crosslinking can be well adjusted, so that the material emits different degrees of blue fluorescence; M. Mahapatra21 and co-workers synthesized aliphatic ternary copolyesters containing N-isopropylacrylamide, methacrylic acid (MAA), acrylic acid (AA), butyl acrylate (BA), hydroxyethyl methacrylate (HEMA) and 2-acrylamide-2-methylpropane sulfonic acid (AMPS) by C–C/C–N coupling solution polymerization. A strong blue fluorescence can be observed under UV excitation through chain entanglement and the interaction within and between molecular chains; Xu et al.22 synthesized unconventional fluorescent polynorbornene containing amino succinimide side groups through the Michael addition and ring opening polymerization, and it showed blue fluorescence at 470–478 nm.
However, the luminous region of the NTIL polymer is usually concentrated in the blue region, which has the disadvantage of a narrow luminous color region. Besides, the molecular weight of the synthesized polymer is generally low, resulting in insufficient mechanical properties of the materials. In addition, the raw materials used are expensive and the synthetic method is generally complex. The above shortcomings limit the application and promotion of this kind of material. Therefore, overcoming the above shortcomings and realizing economical, simple and safe synthesis of new fluorescent polymers with wide color region fluorescence characteristics and excellent mechanical properties have important scientific value and practical application prospects.23,24
As one of the most common engineering plastics in the world, polyethylene terephthalate (PET) has the characteristics of large output and large consumption. It is widely used in many fields such as fibers, films, packaging and so on.25–27 However, due to its single molecular structure and limited performance regulation range, it is mostly used in traditional fields. PET is obtained by esterification and polycondensation of terephthalic acid and ethylene glycol. There is a benzene ring with a conjugated structure in the molecule, and there is a possibility of fluorescence emission from the perspective of the molecular structure. Yet, there are few studies on the fluorescence phenomenon of PET, and most of the studies focus on the fluorescence emission of the terephthalic acid monomer at 342 nm and the fluorescence emission of the ground dimer at 364, 374 and 386 nm.28–33 There are few reports on the fluorescence emission in the visible region of 400 nm–800 nm.34,35 Therefore, it is generally believed that pure PET polymers have little application value in the field of fluorescent materials.
In this paper, poly(terephthalic acid–succinic acid–ethylene glycol) copolyester was synthesized by introducing the flexible aliphatic diacid succinic acid into PET. It was found for the first time that the copolyester could emit green fluorescence under the irradiation with a 365 nm UV lamp. Furthermore, a series of copolyesters with different fluorescence properties were synthesized by introducing the aliphatic diol butanediol into copolyesters and changing the carbon chain length of aliphatic diacids. The fluorescence, thermal and mechanical properties of all copolyesters were characterized in detail. By establishing the corresponding relationship between the molecular structure and fluorescence properties, the fluorescence mechanism of copolyesters was preliminarily explored. We speculate that the fluorescence of copolyesters comes from the 3D spatial electron channel formed by the spatial aggregation of groups such as the benzene ring, the carbonyl group and the carbon oxygen bond, and the conformation of chain segments and the density of the above chromophores become the main factors affecting the fluorescence emission of copolyesters. The new high molecular weight copolyester synthesized in this paper has a simple synthesis method and exhibits fluorescence emission in the visible region that cannot be achieved with the traditional polyester, which can broaden the application field of the traditional polyester. Besides, the copolyester exhibits better mechanical properties, which makes up for the shortcomings of NTIL materials' insufficient mechanical properties. In addition, the establishment of the corresponding relationship between the molecular structure and fluorescence properties is conducive to enriching the fluorescence mechanism of polyester materials, and has major theoretical significance for the research of non-traditional intrinsic luminescent polymer materials.
Experiment
Materials and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) using an Ubbelohde thermostat viscometer at 25 ± 0.1 °C; the 1H NMR spectrum with trifluoroacetic acid-d as the solvent was recorded on a Bruker DMX-300 NMR spectrometer. Tetramethylsilane (TMS) was used as an internal standard for chemical shifts.
:1) using an Ubbelohde thermostat viscometer at 25 ± 0.1 °C; the 1H NMR spectrum with trifluoroacetic acid-d as the solvent was recorded on a Bruker DMX-300 NMR spectrometer. Tetramethylsilane (TMS) was used as an internal standard for chemical shifts.
The fluorescence properties were characterized using a HORIBA fluorescence spectrometer; the excitation wavelength was 350 nm and the slit size was 5 nm. The solution sample was dissolved in trifluoroacetic acid solvent for testing, and the solid sample was pressed into films with the same thickness for testing.
The thermal behavior of polymers was examined by differential scanning calorimetry (DSC) on a DSC Q2000 apparatus from TA Instruments. The thermograms were obtained from 4–5 mg samples at heating and cooling rates of 10 °C min−1 under a nitrogen flow of 50 mL min−1. Thermogravimetric analysis (TGA) of the samples (2.0–3.0 mg) was performed on a PerkinElmer TGA 8000 analyzer under a nitrogen atmosphere with a heating rate of 20 °C min−1 from 50 to 650 °C.
Tensile tests were performed with dumbbell-shaped samples according to ISO 527 (2012) using an Instron 1122 tensile testing machine with a 500 N load cell at a crosshead speed of 50 mm min−1. All testing samples with dimensions of 10.0 cm (length) × 4.0 mm (neck width) × 2.0 mm (thickness) were prepared by injection-molding at 240 °C under a pressure of 800 bar. At least 5 samples were tested to obtain an average value of mechanical properties.
Bending performance tests were performed with oblong-shaped samples using an Instron 1122 tensile testing machine at a speed of 2 mm min−1. All testing samples with dimensions of 10.0 cm(length) × 10.0 mm (width) × 2.0 mm (thickness) were prepared by injection-molding at 200 °C under a pressure of 800 bar. At least 2 samples were tested to obtain an average value of mechanical properties.
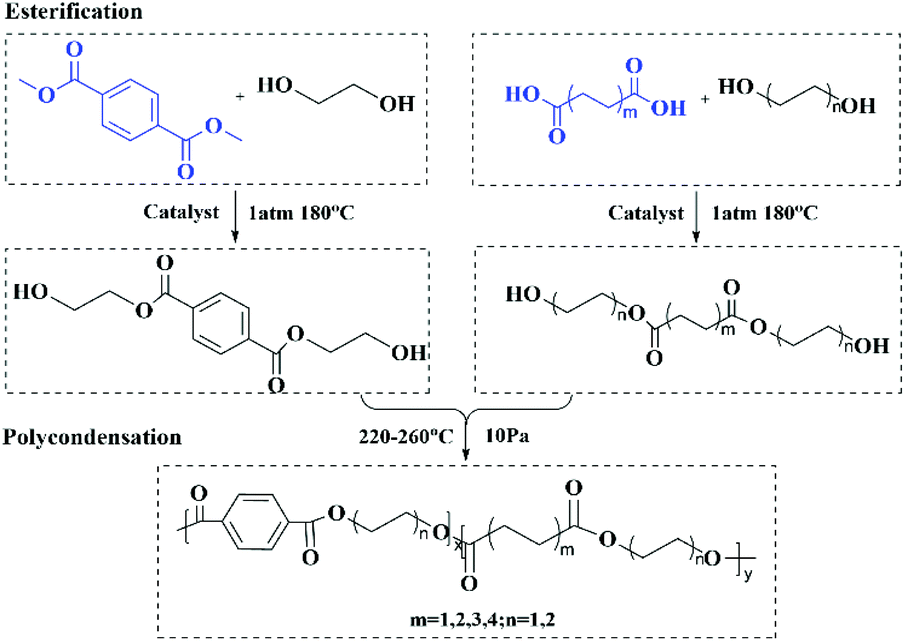 | ||
| Scheme 1 Synthesis of PExBTyAm. | ||
Results and discussion
Composition and structural characterization
A series of high molecular weight copolymers were synthesized by two-step melt polycondensation according to the method shown in Scheme 1. The composition and chemical structure of the synthesized polymer were characterized by NMR spectroscopy. Fig. 1(a) and (b) show the 1H NMR spectrum of PET70A4 and PE60BT70A4 respectively.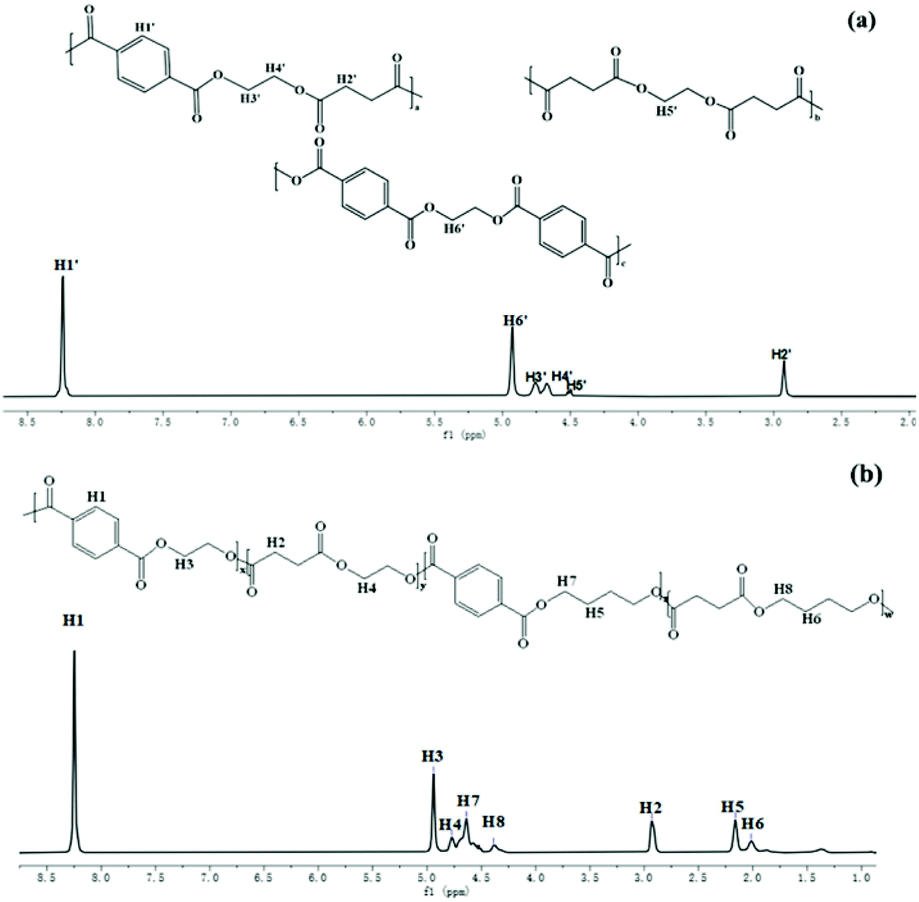 | ||
| Fig. 1 (a) 1HNMR spectra of PET70A4; (b) PE60BT70A4. | ||
As can be seen from Fig. 1(a), the peaks at δ 8.24 and δ 2.84 ppm were attributed to hydrogen atoms H1′ and H2′ on terephthalic acid and succinic acid, respectively. The characteristic peak at δ 4.92 ppm was attributed to the hydrogen atom H6′ on the ethylene glycol unit with terephthalic acid attached to both sides. The characteristic peak at δ 4.45 ppm was attributed to the hydrogen atom H5′ on the ethylene glycol unit connected with succinic acid on both sides. And the characteristic peaks at δ 4.79 ppm and 4.68 ppm were respectively attributed to the hydrogen atoms H3′ and H4′ connected to terephthalic acid and succinic acid.
As can be seen from Fig. 1(b), the peak at δ 4.94 ppm was assigned to the hydrogen atom (H3) on the ethylene glycol unit connected to terephthalic acid; The peak at δ 4.77 ppm belonged to the hydrogen atom (H4) on the ethylene glycol unit connected to succinic acid; The peaks of the butanediol unit at δ 2.16, δ 2.02, δ 4.64, and δ 4.37 ppm were assigned to hydrogen atoms 5, 6, 7, and 8 and the peaks at δ 8.25 and δ 2.93 ppm were assigned to the terephthalic acid unit and succinic acid unit, respectively. The results of 1H NMR showed that we successfully synthesized copolyesters PET70A4 and PE60BT70A4.
Table 1 lists the component composition and viscosity of copolyesters. The molar content of each component of copolyesters was calculated by NMR. The synthesized copolyester was dissolved in the phenol tetrachloroethane solvent, and its intrinsic viscosity was measured using an automatic viscometer. It can be seen from the table that we have successfully prepared high molecular weight copolyesters. The copolymerization composition of the synthesized copolyesters was close to that of the feed.
| Feeding ratio (EG/BDO/TPA/A) | Composition ratio (EG/BDO/TPA/A) | [η] (dl g−1) | |
|---|---|---|---|
| PET70A4 | 100/0/70/30 | 100/0/69.4/30.6 | 1.04 |
| PET50A4 | 100/0/50/50 | 100/0/50/50 | 1.37 |
| PET30A4 | 100/0/30/70 | 100/0/32.6/67.4 | 1.51 |
| PE60BT70A4 | 60/40/70/30 | 66.9/33.1/77/23 | 0.74 |
| PE60BT70A6 | 60/40/70/30 | 55.2/44.8/74.6/25.4 | 1.10 |
| PE60BT70A8 | 60/40/70/30 | 56.8/43.2/74.1/25.9 | 0.81 |
| PE60BT70A10 | 60/40/70/30 | 58.4/41.6/75.8/24.2 | 1.05 |
Fluorescence properties of copolyesters
It is generally believed that pure PET polymers have little application value in the field of fluorescent materials. Through characterization, we found for the first time that copolyesters exhibited fluorescence emission characteristics in bulk and solution. Taking PET70A4 as an example, Fig. 2(c) shows the photo of its bulk fluorescence under the irradiation of a 365 nm UV lamp. The copolyester was dissolved in the trifluoroacetic acid solvent and prepared in PET70A4/TFA solutions with different concentrations of 0.0002 g mL−1, 0.002 g mL−1, 0.02 g mL−1 and 0.2 g mL−1 respectively. Fig. 2(a) and (b) show the color changes of the solution under daylight and a 365 nm ultraviolet lamp, respectively.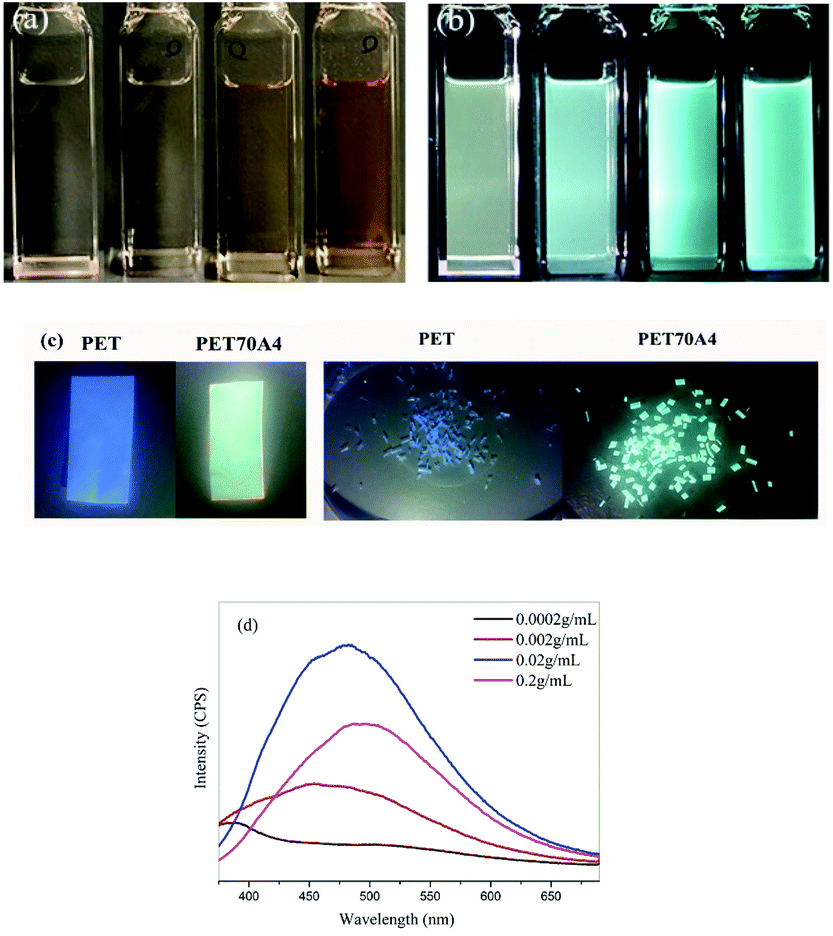 | ||
| Fig. 2 (a) Photographs of different concentrations of PET70A4 /TFA solution under daylight; (b) photographs of different concentrations of PET70A4 /TFA solution under a 365 nm ultraviolet lamp; (c) bulk fluorescence of PET and PET70A4 irradiated with a 365 nm ultraviolet lamp; (d) fluorescence emission spectra of PET70A4 /TFA solutions with different concentrations at 350 nm excitation wavelength. | ||
From the figure, we found that the solution emitted a blue-green fluorescence of different intensities under the irradiation of a 365 nm UV lamp. Copolyesters exhibited aggregation induced luminescence characteristics. In order to characterize the relationship between fluorescence and solution concentration more accurately, the prepared PET70A4/TFA solution was characterized using the fluorescence emission spectrum. All samples were measured at the same excitation wavelength (350 nm) and the same slit size (5 nm). The characterization results are shown in Fig. 2(d) and the fluorescence emission wavelengths are listed in Table 2. When the solution concentration was 0.0002 g mL−1, the fluorescence emission curve was close to a straight line with a very weak fluorescence intensity and the fluorescence emission wavelength was at 387 nm, which was located in the invisible region. With the increase of solution concentration, the fluorescence emission wavelength red-shifted, and the fluorescence emission intensity increased significantly.
| Concentration(g mL−1) | λ ex (nm) | λ em (nm) |
|---|---|---|
| 0.0002 | 350 | 387 |
| 0.002 | 350 | 463 |
| 0.02 | 350 | 483 |
| 0.2 | 350 | 496 |
According to the reports in the literature,34 the fluorescence emission peak below 400 nm corresponds to the fluorescence of the benzene ring single molecule or the benzene ring dimer. When the solution concentration reached 0.2 g mL−1, the fluorescence emission wavelength red-shifted to 496 nm. The fluorescence intensity of the 0.2 g mL−1 solution in Fig. 2 was lower than that of the 0.02 g mL−1 solution. By looking up the references, we speculate that the self-absorption phenomenon which generally existed in a high concentration leads to a lower fluorescence intensity. We have added the detailed explanation in the ESI, as shown in Fig. S4.†36,37 According to the literature reports, this type of fluorescence mechanism was generally attributed to the clustering triggered emission (CTE) mechanism; that is, the fluorescence emission center comes from the three-dimensional aggregation of chromophores such as carbonyl and amino groups, the benzene ring and the carbon oxygen bond in the molecule. With the increase of aggregation degree, the molecular fluorescence emission increases. C. Du and co-workers synthesized the alternating copolymer of vinyl benzene and carbon monoxide. The results showed that the copolymer could emit blue fluorescence in concentrated solution and solid powder. Through the theoretical calculation of the small molecule model and single crystal analysis, it was confirmed that the cluster luminescent body was formed between carbonyl and phenyl through intramolecular and intermolecular interactions.38 The copolyester materials we have synthesized possess a similar luminescent group, the benzene ring and the carbonyl group, to those in the above literature. Therefore, it is reasonable to speculate that the fluorescence mechanism of copolyesters is the conjugation caused by the spatial aggregation of electron-rich groups such as the intramolecular benzene ring, the carbonyl group and the carbon oxygen bond.
Relationship between the fluorescence properties and molecular structure of copolyesters
In order to preliminarily explore the luminescence mechanism of copolyesters, we synthesized poly(cyclohexanedicarboxylic acid–succinic acid–ethylene glycol) copolyester PEC70A4 with the same copolymerization ratio by replacing terephthalic acid with 1,4-cyclohexanedicarboxylic acid. At the same excitation wavelength of 350 nm, the fluorescence spectra of PET70A4 and PEC70A4 were obtained. The characterization results are shown in Fig. 3(a). PEC70A4 exhibited weak fluorescence emission at 435 nm, and its intensity was much weaker than that of PET70A4, which proved that strong fluorescence emission couldn't be realized only by carbonyl. In addition, the strongest fluorescence emission wavelength of PET was 386 nm, which proved that the fluorescence emission in the visible region couldn't be realized only by the benzene ring. The fluorescence of copolyesters comes from the combined effect of electron rich groups such as benzene rings, carbonyls and carbon oxygen bonds in the molecule.
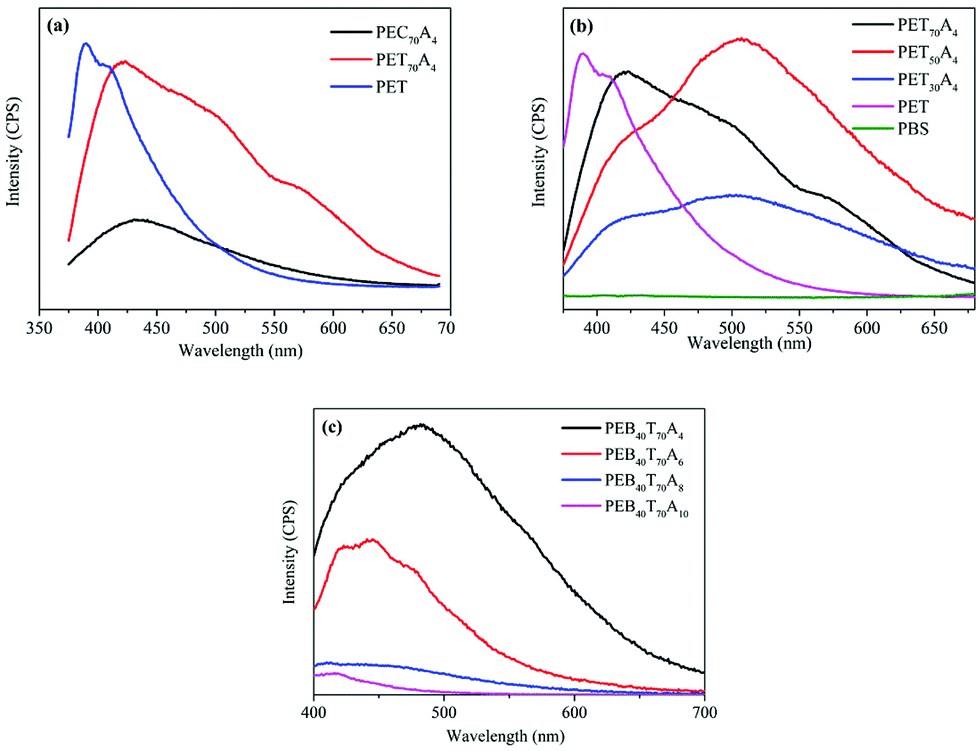 | ||
| Fig. 3 (a) Fluorescence emission spectra of PEC70A4 and PET70A4 at 350 nm excitation wavelength; (b) fluorescence emission spectra of PETA4 with different molar contents of succinic acid; (c) fluorescence emission spectra of PEBTA with aliphatic diacids with different carbon chain lengths. | ||
PETA4 copolyesters with different succinic acid contents were synthesized by two-step melt polycondensation. The succinic acid contents in the copolyesters were 30 mol%, 50 mol% and 70 mol%, respectively, which were then compared with that of pure PET. All copolyesters were characterized by fluorescence spectroscopy at 350 nm excitation wavelength. The fluorescence spectra are shown in Fig. 3(b) and fluorescence emission wavelengths are listed in Table 3. It can be seen from the chart that the fluorescence emission wavelength of pure PET was 386 nm, which is basically located in the invisible region. This was consistent with the report in the literature. Under the excitation of 350 nm ultraviolet light, pure PET mainly shows the single molecule fluorescence of the benzene ring and the face-to-face dimer fluorescence caused by the benzene ring. With the increase of the succinic acid content in the copolyester, the fluorescence emission wavelength red-shifted, and the copolyester could emit an unreported green fluorescence visible to the naked eye. The strongest fluorescence emission wavelength of PET70A4 was 421 nm and that of PET50A4 was 507 nm. With the further increase of succinic acid content, the strongest fluorescence emission wavelength of PET30A4 changed to 504 nm. However, the fluorescence emission intensity decreased significantly. Different from the single molecule fluorescence and dimer fluorescence below 400 nm, the copolyester showed a longer fluorescence emission wavelength. Therefore, we speculated that the addition of the aliphatic diacid made the molecular chain flexible, which promoted the aggregation of electron-rich groups such as the benzene ring, the carbonyl group and the carbon oxygen bond, and formed an electron interaction channel in three-dimensional space. The formation of a large conjugated structure between molecules led to the phenomenon of fluorescence emission in the visible region. PET didn't show this fluorescence characteristic because of its rigid molecular chain, which was not conducive to the aggregation of chromophores.
| λ ex (nm) | λ em (nm) | |
|---|---|---|
| PET | 350 | 386 |
| PBS | 350 | — |
| PET70A4 | 350 | 421 |
| PET50A4 | 350 | 507 |
| PET30A4 | 350 | 504 |
| PE60BT70A4 | 350 | 481 |
| PE60BT70A6 | 350 | 441 |
| PE60BT70A8 | 350 | 439 |
| PE60BT70A10 | 350 | 417 |
The fluorescence spectrum is shown in Fig. 3(c), and the fluorescence emission wavelength is listed in Table 3. The characterization results suggested that the fluorescence emission wavelength of copolyesters blue-shifted and the fluorescence emission intensity decreased with the increase of the carbon chain length of the aliphatic diacid. The fluorescence emission wavelength of PE60BT70A4 was 481 nm; the strongest fluorescence emission wavelength of PE60BT70A6 was 441 nm; the fluorescence emission wavelength of PE60BT70A8 was 439 nm, and that of PE60BT70A10 shifted to 417 nm. We speculated that the change of the segment length led to the change of the polymer segment flexibility. The segment flexibility affected the aggregation degree of the chromophore and the density of the chromophore at the same time, which would influence the fluorescence emission wavelength of the polymer. When the aggregation degree of the chromophore played a decisive role, the fluorescence emission wavelength increased with the increase of segment length. When the density of the chromophore played a decisive role, the fluorescence emission wavelength decreased with the increase of segment length.
In conclusion, for realizing the fluorescence emission of copolyesters, three conditions should be satisfied as follows:
Firstly, the molecules should contain electron rich chromogenic groups such as the benzene ring, the carbonyl group and the carbon oxygen bond, which are the cause of polymer luminescence; besides, the molecular chain should possess certain flexibility to realize the spatial aggregation of the large conjugated structure formed by the above groups. If the molecular chain is too rigid, such as PET, it is not conducive to the winding of the molecular chain and the aggregation of chromogenic groups. In addition, the benzene ring, carbonyl and other conjugating groups should have sufficient spatial density. The increase of the length of the flexible diacid carbon chain will reduce the spatial density of the above groups and weaken the conjugation. For example, for copolyesters containing adipic acid, octanoic acid and sebacic acid, the spatial density of the intramolecular benzene ring, carbonyl and other groups is lower than that of copolyesters containing succinic acid; therefore, their λem is blue-shifted.
Thermal performance characterization
The study on the thermal properties of copolyesters is of great significance for the processing and application of copolyesters. The change of copolyester copolymerization components will directly affect the glass transition temperature and thermal stability of materials, so that the materials show different states at room temperature and have different applications. In this paper, copolyesters with different copolymerization components were characterized by DSC and TGA.Differential scanning calorimetry (DSC)
The DSC curve of copolyesters at secondary temperature rise is shown in Fig. 4. As can be seen from the figure, all copolyesters only showed one glass transition temperature and no crystallization and melting peaks, revealing that they all had the features of a completely amorphous polymer. The glass transition temperature data (Tg) are listed in Table 4. The Tg values of PET70A4 and PE60BT70A4 were 48.7 °C and 37.2 °C respectively, above room temperature, which could already satisfy the requirements of general materials. However, with the further increase of the content of succinic acid in the copolymerization component, the flexibility of the copolyester molecular chain increased, resulting in the decrease of the glass transition temperature of copolyesters. The Tg of copolyesters decreased from 48.7 °C of PET70A4 to 7.3 °C of PET30A4; besides, due to the same reason, the Tg of copolyesters decreased from 37.2 °C of PE60BT70A4 to 9.4 °C of PE60BT70A10. From the characterization results, it can be suggested that the glass transition temperature of copolyesters would be reduced to some extent after the further addition of flexible segments. Therefore, in future work, it should be considered to introduce a small amount of rigid segments to improve the glass transition temperature of the material.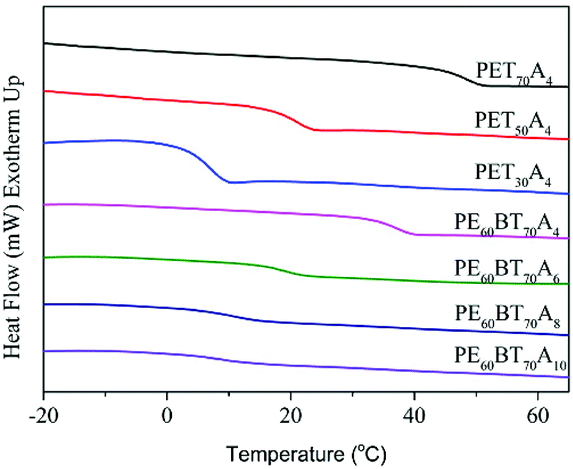 | ||
| Fig. 4 DSC curves of the 2nd heating of PExBTyAm. | ||
| T g(°C) | T d-5%(°C) | T d-max(°C) | T 1/2d(°C) | W res(650 °C) | |
|---|---|---|---|---|---|
| PET70A4 | 48.7 | 408.4 | 462.2 | 462.9 | 12.2 |
| PET50A4 | 23.4 | 384.9 | 476.9 | 466.2 | 12.0 |
| PET30A4 | 7.3 | 369.6 | 480.4 | 450.3 | 9.4 |
| PE60BT70A4 | 37.2 | 394.9 | 447.0 | 447.4 | 10.3 |
| PE60BT70A6 | 17.7 | 395.1 | 434.7 | 439.7 | 10.2 |
| PE60BT70A8 | 10.8 | 401.5 | 439.1 | 442.4 | 10.0 |
| PE60BT70A10 | 9.4 | 401.0 | 436.0 | 442.6 | 8.8 |
Thermogravimetric analysis (TGA)
The thermal weight loss curve and weight loss rate curve of copolyesters under a nitrogen atmosphere are shown in Fig. 5. The parameters such as thermal decomposition temperature (Td-5%), maximum weight loss rate temperature (Td-max), half decomposition temperature (T1/2d) and residual weight at 650 °C are summarized in Table 4. The initial decomposition temperature of all the synthesized copolyesters was close to 400 °C; the half decomposition temperature (T1/2d) was about 450 °C; the maximum weight loss rate temperature was over 430 °C, and when the temperature exceeded nearly 650 degrees, the copolyesters could still retain about 10% of the mass. Thus, the copolyester showed excellent heat-resistant decomposition performance.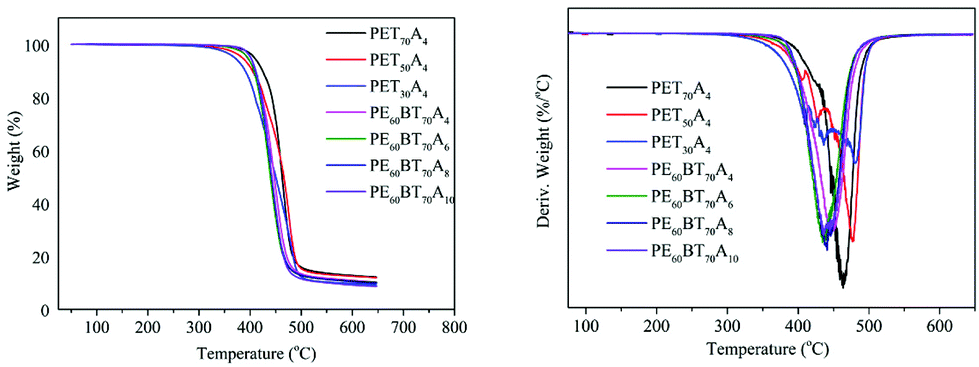 | ||
| Fig. 5 Remaining weight and the corresponding derivatives versus temperature of PExBTyAm. | ||
Mechanical properties
The existing non-traditional intrinsic luminescent polymer materials generally have the problems of low molecular weight and insufficient mechanical properties, which limit the application of materials. In order to broaden the application field of fluorescent copolyesters, in this paper, the mechanical properties of the synthesized copolyesters were characterized in detail.The tensile properties and flexural property curves of copolyesters are shown in Fig. 6. The performance parameters such as tensile strength, elastic modulus, elongation at break, flexural strength and flexural modulus are summarized in Table 5. According to the characterization results, the maximum tensile strength of PET70A4 was 51 MPa, the elastic modulus was 905 MPa, the flexural strength was 59.7 MPa and the flexural modulus was 2065 MPa. Both PET70A4 and PE60BT70A4 exhibited good comprehensive mechanical properties and had good application prospects. With the increase of aliphatic dicarboxylic acid content and the increase of aliphatic dicarboxylic acid carbon chain length, the glass transition temperature of copolyesters decreased, and the tensile and flexural strength decreased to some extent. Thus, some rigid segments will be introduced to further improve the mechanical properties of copolyesters in the follow-up work.
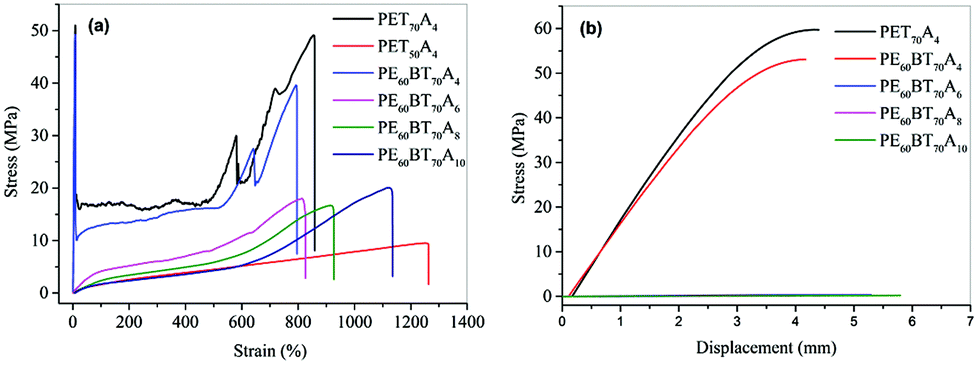 | ||
| Fig. 6 Tensile stress–strain curves of PExBTyAm (a); flexural stress–strain curves of copolyester(b). | ||
| Elastic modulus (MPa) | Tensile strength (MPa) | Elongation at break (%) | Flexural strength (MPa) | Flexural modulus (MPa) | |
|---|---|---|---|---|---|
| PET | — | 55–78 | 50 | ||
| PET70A4 | 905 | 51.0 ± 1.3 | 856 ± 100.4 | 59.7 | 2065 |
| PET50A4 | 1.3 | 9.5 | 1259 | — | — |
| PET30A4 | — | — | — | — | — |
| PE60BT70A4 | 973 | 49.2 ± 1.5 | 882 ± 121 | 53.1 | 1828 |
| PE60BT70A6 | 5.3 | — | 823 ± 13 | 0.4 | 11 |
| PE60BT70A8 | 3.6 | — | 907 ± 32 | 0.2 | 5 |
| PE60BT70A10 | 3.1 | — | 1070 ± 46 | 0.2 | 5 |
Conclusions
In conclusion, we have synthesized high molecular weight PExBTyAm copolyesters by simple and economical two-step polycondensation, and for the first time we found that the copolyesters can emit visible green fluorescence under the irradiation with a 365 nm UV lamp, and exhibit PET fluorescence in the fluorescence region. This kind of copolyester shows an obvious aggregation induced luminescence. In addition, the fluorescence emission wavelength of copolyesters can be adjusted by adjusting the carbon chain length and the content of the aliphatic dibasic acid and diol in the flexible segment of the copolymerization component. At the same time, the copolyester has excellent mechanical and thermodynamic properties. The synthesis of the new fluorescent copolyester effectively enriches the optical properties of the traditional polyester and effectively makes up for the shortcomings of non-traditional intrinsic luminescent materials (NTILs), such as the narrow luminescence region, generally low molecular weight, insufficient mechanical properties and complex synthesis methods. It has important theoretical significance for the research of non-traditional intrinsic luminescent polymer materials, and it has the prospect of being used for chemical probes, fluorescent sensors, solar energy conversion and other luminescence fields.Author contributions
Lele Ma: conceptualization, investigation, writing – original draft, writing – review & editing, formal analysis, and visualization; Jiajian Liu: conceptualization, writing – review & editing, methodology, and supervision; Chuncheng Li: funding acquisition, supervision, writing – review & editing, and project administration; Yaonan Xiao: supervision; Shaohua Wu: validation; Bo Zhang: validation and resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are greatly thankful for the help from teachers and students of the research group.Notes and references
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A. M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35–117 CrossRef CAS.
- Z. Zhang, H. Zhang, M. Kang, Na Li, D. Wang and B. Z. Tang, Sci. China: Chem., 2021, 64, 1990–1998 CrossRef CAS.
- Y. Meng, S. Guo, B. Jiang, X. Zhang, L. Zou, C. Wei, Y. Gong, S. Wu and Y. Liu, J. Mater. Chem. C, 2021, 9, 8515–8523 RSC.
- S. Tang, T. Yang, Z. Zhao, T. Zhu, Q. Zhang, W. Hou and W. Z. Yuan, Chem. Soc. Rev., 2021, 50, 12616–12655 RSC.
- L. Shao, K. Wan, H. Wang, Y. Cui, C. Zhao, J. Lu, X. Li, L. Chen, X. Cui, X. Wang, X. Deng, X. Shi and Y. Wu, Biomater. Sci., 2019, 7, 3016–3024 RSC.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 1–22 CrossRef.
- B. Saha, K. Bauri, A. Bag, P. K. Ghorai and P. De, Polym. Chem., 2016, 7, 6895–6900 RSC.
- K. Bauri, B. Saha, J. Mahanti and P. De, Polym. Chem., 2017, 8, 7180–7187 RSC.
- M. Mitra, M. Mahapatra, A. Dutta, P. K. Chattopadhyay, M. Deb, J. S. D. Roy, C. Roy, S. Banerjee and N. R. Singha, ACS Omega, 2020, 5, 3333–3345 CrossRef CAS PubMed.
- Y. Wang, L. Feng and S. Wang, Adv. Funct. Mater., 2019, 29, 1–20 Search PubMed.
- A. Dutta, M. Mahapatra, M. Deb, M. Mitra, S. Dutta, P. K. Chattopadhyay, S. Banerjee, P. C. Sil, D. K. Maiti and N. R. Singha, ACS Biomater. Sci. Eng., 2020, 6, 1397–1407 CrossRef CAS PubMed.
- P. Li, J. Xu, B. Qi, K. Li, X. Gu, X. Niu and Y. Fan, Nanosci. Nanotechnol. Lett., 2018, 10, 1287–1291 CrossRef.
- B. Saha, N. Choudhury, S. Seal, B. Ruidas and P. De, Biomacromolecules, 2019, 20, 546–557 CrossRef CAS PubMed.
- H. Zhang, Z. Zhao, P. R. McGonigal, R. Ye, S. Liu, J. W. Y. Lam, R. T. K. Kwok, W. Z. Yuan, J. Xie, A. L. Rogach and B. Z. Tang, Mater. Today, 2020, 32, 275–292 CrossRef CAS.
- B. Saha, B. Ruidas, S. Mete, C. Das Mukhopadhyay, K. Bauri and P. De, Chem. Sci., 2020, 11, 141–147 RSC.
- W. Z. Yuan and Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 560–574 CrossRef.
- E. Zhao, J. W. Y. Lam, L. Meng, Y. Hong, H. Deng, G. Bai, X. Huang, J. Hao and B. Z. Tang, Macromolecules, 2015, 48, 64–71 CrossRef CAS.
- Y. Du, H. Yan, W. Huang, F. Chai and S. Niu, ACS Sustainable Chem. Eng., 2017, 5, 6139–6147 CrossRef CAS.
- W. Yang and C.-Y. Pan, Macromol. Rapid Commun., 2009, 30, 2096–2101 CrossRef CAS PubMed.
- Z. Feng, W. Zhao, Z. Liang, Y. Lv, F. Xiang, D. Sun, C. Xiong, C. Duan, L. Dai and Y. Ni, ACS Appl. Mater. Interfaces, 2020, 12, 11005–11015 CrossRef CAS PubMed.
- M. Mahapatra, A. Dutta, J. S. D. Roy, M. Deb, U. Das, S. Banerjee, S. Dey, P. Chattopadhyay, D. K. Maiti and N. R. Singha, Langmuir, 2020, 36, 6178–6187 CrossRef CAS PubMed.
- J. Xu, D. Luo, X. Yin, H. Zhang, L. Wang and H. Wang, Macromol. Chem. Phys., 2017, 218, 1–9 Search PubMed.
- C. Shang, N. Wei, H. Zhuo, Y. Shao, Q. Zhang, Z. Zhang and H. Wang, J. Mater. Chem. C, 2017, 5, 8082–8090 RSC.
- R. Ye, Y. Liu, H. Zhang, H. Su, Y. Zhang, L. Xu, R. Hu, R. T. K. Kwok, K. S. Wong, J. W. Y. Lam, W. A. Goddard, III and B. Z. Tang, Polym. Chem., 2017, 8, 1722–1727 RSC.
- H. Zhang and Z.-G. Wen, Waste Manage., 2014, 34, 987–998 CrossRef CAS PubMed.
- F. Welle, Resour., Conserv. Recycl., 2011, 55, 865–875 CrossRef.
- G. Albini, V. Brunella, B. Placenza, B. Martorana and V. G. Lambertini, J. Ind. Text., 2019, 48, 992–1008 CrossRef CAS.
- D. Mary, M. Albertini and C. Laurent, J. Phys. D: Appl. Phys., 1997, 30, 171–184 CrossRef CAS.
- K. Kojima, Y. Takai and M. Ieda, Jpn. J. Appl. Phys., Part 1, 1982, 21, 860–864 CrossRef CAS.
- K. Kojima, Y. Takai and M. Ieda, Jpn. J. Appl. Phys., Part 1, 1983, 22, 1436–1438 CrossRef CAS.
- I. Ouchi, I. Nakai, M. Ono and S. Kimura, Jpn. J. Appl. Phys., Part 1, 2004, 43, 8107–8114 CrossRef CAS.
- H. Itagaki, Y. Inagaki and N. Kobayashi, Polymer, 1996, 37, 3553–3558 CrossRef CAS.
- H. Itagaki, J. Lumin., 1997, 72–4, 435–436 CrossRef.
- X. Chen, Z. He, F. Kausar, G. Chen, Y. Zhang and W. Z. Yuan, Macromolecules, 2018, 51, 9035–9042 CrossRef CAS.
- X. He, X. Zhou, K. Jia, D. Zhang, H. Shou and X. Liu, Mater. Lett., 2016, 182, 367–371 CrossRef CAS.
- G. Henderson, J. Chem. Educ., 1977, 54, 57–59 CrossRef CAS.
- K. Yamasaki, T. Kimura, H. Masuya and I. Tokue, Rev. Sci. Instrum., 1997, 68, 3697–3701 CrossRef CAS.
- C. Du, H. Chu, Z. Xiao, L. Zhong, Y. Zhou, W. Qin, G. Liang and H. Gao, Macromolecules, 2020, 53, 9337–9344 CrossRef CAS.
- W. Huang, H. Yan, S. Niu, Y. Du and L. Yuan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3690–3696 CrossRef CAS.
- D. Liu, C. Li, Y. Xu, D. Zhou, H. Wang, P. Sun and H. Jiang, Polymer, 2017, 113, 274–282 CrossRef CAS.
- H.-Q. Wang, H.-F. Wang, H.-B. Lu, C.-L. Lu and X.-Y. Li, Chem. J. Chin, Univ. Chin., 2007, 28, 388–390 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01222c |
| This journal is © The Royal Society of Chemistry 2022 |