
Synthesis and depolymerization of self-immolative poly(disulfide)s with saturated aliphatic backbones†
Magnus
Hansen-Felby
 a,
Andreas
Sommerfeldt
ab,
Martin Lahn
Henriksen
c,
Steen Uttrup
Pedersen
*ab and
Kim
Daasbjerg
*ab
a,
Andreas
Sommerfeldt
ab,
Martin Lahn
Henriksen
c,
Steen Uttrup
Pedersen
*ab and
Kim
Daasbjerg
*ab
aDepartment of Chemistry, Aarhus University, Langelandsgade 140, DK-8000 Aarhus C, Denmark. E-mail: sup@chem.au.dk; kdaa@chem.au.dk
bInterdisciplinary Nanoscience Center (iNANO), Aarhus University, Gustav Wieds Vej 14, DK-8000 Aarhus C, Denmark
cDepartment of Engineering, Plastic and Polymer Engineering, Aabogade 40a, 8200 Aarhus N, Denmark
First published on 3rd December 2021
Abstract
Self-immolative polymers (SIPs) are a class of degradable stimuli-responsive polymers, which, upon removal of labile end-caps, depolymerize selectively and stepwise to small molecules. In light of our recent discovery of poly(dithiothreitol) (pDTT), a versatile SIP with a remarkably simple synthesis procedure, we investigated a broader range of unfunctionalized poly(disulfide)s. It is demonstrated that saturated aliphatic backbones can easily be made from 1,4-butanedithiol, 1,5-pentanedithiol, and 1,6-hexanedithiol monomers and, compared with pDTT, these polymers show enhanced stability, solubility, and processability. SIP polymers derived from the smaller 1,3-propanedithiol monomer with end-caps installed could not be synthesized. Polymers of 1,4-butanedithiol and 1,5-pentanedithiol undergo end-to-end depolymerizations upon end-cap removal, taking hours to days under basic conditions and not minutes as for pDTT. Degradation of the polymer of 1,6-hexanedithiol occurs by less well-defined pathways providing a complex product mixture of macrocyclic disulfides.
Introduction
The demand for responsive and degradable polymers is ever-increasing such as in medical detection and treatment,1 and for consumer plastics.2 A profoundly studied functional group in this respect is disulfide, a relatively strong covalent bond with a dynamic nature as it can shuffle between opened thiol and closed disulfide forms through the thiol–disulfide exchange reaction,3 or shuffle substituents through the disulfide metathesis reaction.4 This reversibility of disulfides has been exploited in areas of self-healing materials,5,6 sustained release of biomolecules,7 and degradable polymers.8,9 Common for the materials is the utilization of poly(disulfide)s (pDSs) where disulfide bonds are distributed along the polymeric backbone. Conventional polymerization methods for producing pDSs include step-growth of monomers already containing a disulfide moiety, oxidative step-growth of dithiols, or ring-opening polymerization (ROP) of cyclic disulfides.10Endo et al. published a series of articles on 1,2-dithianes utilized in ROP either through a disulfide metathesis mechanism, initiated by heat,11–13 or a thiol–disulfide exchange mechanism approach, initiated by thiols (Scheme 1a).11–13 The thermally driven ROP produced polycatenane structures with intertwined macrocycles, which degrade to smaller cyclic oligomers by UV exposure. Thiol-initiated ROP produced linear polymers. The product distribution was further characterized by Liu et al., using anionic ROP of lipoic acid and its derivatives (Scheme 1b). They found that the amount of macrocyclic products depended on the bond strength of the end-cap disulfide. A weak disulfide bond at the end-cap, such as aromatic, resulted in macrocyclization, possibly through backbiting reactions, whereas stronger end-cap disulfides, such as aliphatic, remained inert, thus producing linear polymers.14 Furthermore, both architectures showed degradable behavior once diluted and reintroduced to the polymerization conditions at elevated temperatures.
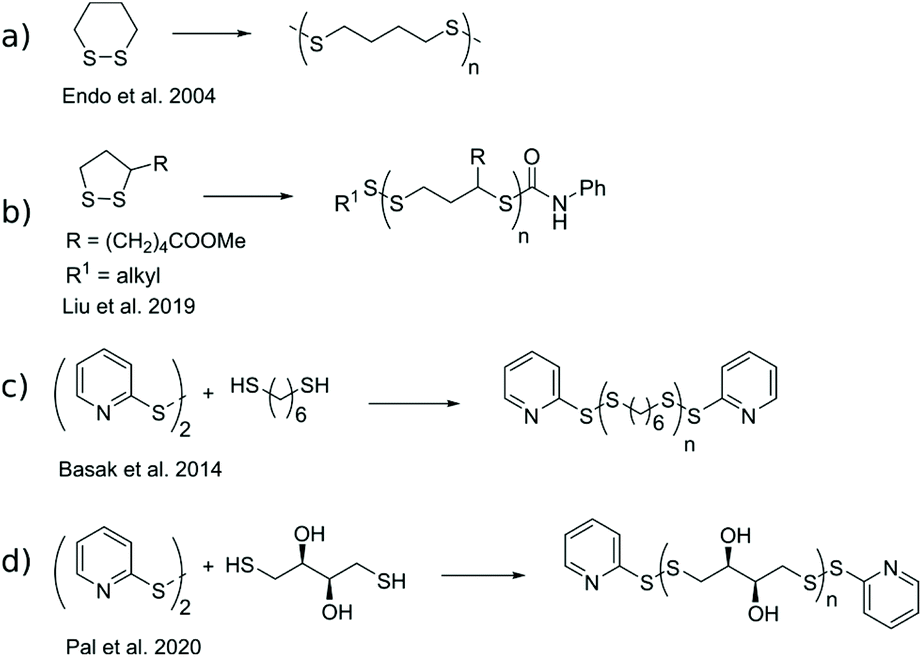 | ||
| Scheme 1 Selected synthetic routes for production of pDSs (stoichiometry not shown). | ||
Basak et al. published a general method for producing telechelic pDSs and they showed the efficacy of the procedure through the mild and efficient polycondensation between 1,6-hexanedithiol and 2,2′-dithiodipyridine (DTDP) (Scheme 1c).15 Recently, Pal et al. extended this procedure with a solid-state synthesis protocol, to develop the first self-immolative poly(disulfide) from monomeric dithiothreitol (DTT), with thiopyridinic end-caps (Scheme 1d).16 The synthesized pDTT exhibited SIP behavior, as kinetic control between random backbone scission and end-terminal pyridinic disulfide scission favored the latter. Consequently, addition of 1 equiv. DTT with respect to (w.r.t.) end-cap resulted in fast end-cap removal followed by cascade of intramolecular backbiting thiol–disulfide exchanges, leading to ring-closed cyclic DTT (cDTT). With catalytic amount of base present (e.g. Et3N) to deprotonate thiol groups, complete depolymerization was observed within minutes (Scheme 2).
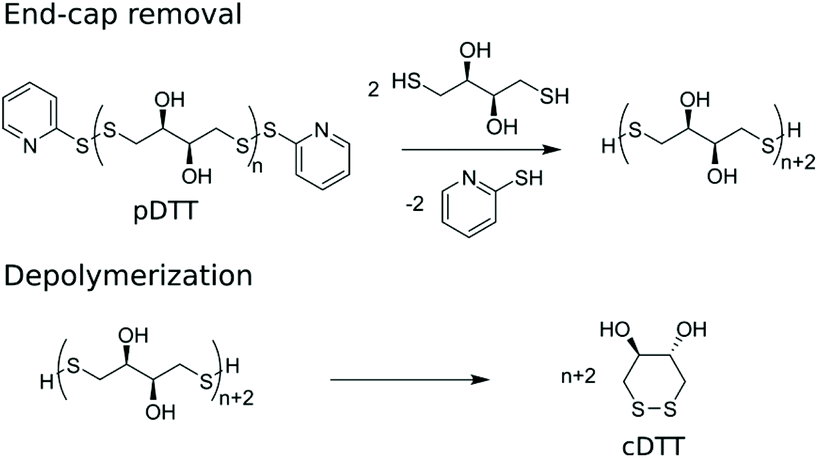 | ||
| Scheme 2 Depolymerization of pDTT through end-cap removal followed by cascade of intramolecular cyclization to produce cDTT. | ||
Recently, Yardley et al.17 published a detailed review on studies pertaining to SIP backbones. In general, depolymerization of SIPs into small monomeric units enables efficient recycling of polymers into pristine virgin materials once the polymers have served their purposes.18 Furthermore, a variety of different applications for SIPs have been discussed, including drug delivery,19,20 reversible adhesives,21 microscale-pumps,22 and chemical detection.23,24
To expand applications of pDS SIPs, more insight into the relationship between structure and properties of the polymers is required. For example, utilizing different backbone structures and side groups has been shown to have huge effect on SIP depolymerization rate of aromatic poly(carbamates) and poly(glyoxylamides),25,26 and the stability and processability of poly(benzyl ethers).27 While the vast array of end-caps for SIPs provides an expansive list of different triggering possibilities,22,28–30 the number of backbones that exhibit SIP behavior remains limited.17 pDTT is the first published SIP utilizing the thiol–disulfide exchange reaction, while the pendant hydroxyl groups provide chemical handles for easy post-polymerization modifications.16 Compared with other SIPs, the synthesis of pDTT is simple, as it requires no solvent and can be conducted under ambient conditions without tedious cleaning procedures. Furthermore, the cyclization reaction utilized for depolymerization is entirely based on thiol–disulfide exchange cyclization. Disadvantageous features of pDTT are its low stability toward base, heat, and UV light. Furthermore, existence of extensive intramolecular hydrogen bonds renders pDTT insoluble in most media.
In this work, we set out to modify the poly(disulfide) backbone in aiming to increase polymer stability, solubility, and processability, and to study the mechanism at play for degradation. Specifically, this investigation includes 1,3-propanedithiol (3-DT), 1,4-butanedithiol (4-DT), 1,5-pentanedithiol (5-DT), and 1,6-hexanedithiol (6-DT), to test the influence of ring size and to provide unfunctionalized alternatives to pDTT. In addition, this study will shed light on the type of obtainable cyclization products.
Results and discussion
Polymerization and characterization
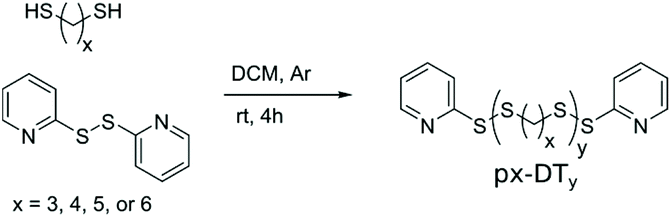
Four potential monomers, x-DT (x = 3, 4, 5, or 6), for SIPs were studied. The synthesis protocol employed was previously developed by Basak et al.15 and only slightly revised for 3-DT and 4-DT (see ESI†). The co-reagent DTDP acts in this protocol both as promoter for the step-growth polymerization and as end-cap when added in excess. The molecular weight of the synthesized polymers was controlled through the stoichiometric imbalance between DTDP and x-DT Thus, the larger this imbalance is in favor of DTDP, the shorter the polymer chains becomes. Said procedure was successfully applied for 4-DT, 5-DT, and 6-DT producing linear polymers with thiopyridinic end-caps (denoted px-DTy). Unfortunately, the procedure did not provide end-capped polymers with 3-DT. Instead, a rubbery white insoluble solid was formed, likely due to the formation of large intertwined catenane structures prevalent for dithiolanes.13,31,32Table 1 summarizes the scope and properties of the synthesized polymers. Yields of p4-DT, p5-DT, and p6-DT are decent to good (43–80%) with PDIs ranging from 1.13–1.68, which is remarkably low for step-growth reactions. In general, p4-DT shows larger PDIs, which, most likely, is the result of a greater tendency for this polymer to depolymerize during synthesis by formation of 1,2-dithiane (c4-DT). Experimentally determined Mn using end-group analysis in 1H NMR agrees reasonably well with the theoretically expected mass with the largest discrepancy seen for the shortest polymers, p4-DT10, p5-DT10, and p6-DT10, where the measured Mn tends to be somewhat higher. This is most likely because shorter polymer chains, which are more polar (from the influence of the thiopyridinic end-cap), are removed during work-up when carrying out precipitation into polar solvents. Note that such a fractionation effect would also lead to less polydisperse samples by narrowing the measured PDI. Furthermore, values of Mn obtained from GPC are consistently higher than from NMR. A plausible explanation is that pDS has a less compact structure than PMMA used as standards for GPC calibration, resulting in relatively shorter elution times and thus overestimation of Mn for pDSs by GPC.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Sample | n DTDP/nx-DT | M n Theoa (kDa) | M n NMRb (kDa) | M n GPCc (kDa) | PDIc | Yield (%) |
T
m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (°C)
(°C) |
| a Number average molecular weight calculated using Mn = (MDT − 2MH) /δ + MDTDP, where δ = nDTDP/nx-DT − 1 with n denoting molar amount.16 Note that y = 1/δ. b Number average molecular weight calculated using end-group analysis in 1H NMR. c Molecular weights and PDI determined using GPC (gel permeation chromatography) are reported relative to poly(methylmethacrylate) (PMMA) standards (see ESI†). d Determined using differential scanning calorimetry (DSC). | |||||||
| p4-DT10 | 1.1 | 1.4 | 2.5 | 3.6 | 1.18 | 43 | 36 |
| p4-DT20 | 1.05 | 2.6 | 3.2 | 5.4 | 1.35 | 65 | 38 |
| p4-DT40 | 1.025 | 5.0 | 5.8 | 7.3 | 1.67 | 71 | 44 |
| p5-DT10 | 1.1 | 1.6 | 2.6 | 4.4 | 1.13 | 40 | 34 |
| p5-DT20 | 1.05 | 2.9 | 3.2 | 5.0 | 1.25 | 80 | 36 |
| p5-DT40 | 1.025 | 5.6 | 4.5 | 5.7 | 1.28 | 71 | 39 |
| p6-DT10 | 1.1 | 1.7 | 2.9 | 4.6 | 1.19 | 72 | 40 |
| p6-DT20 | 1.05 | 3.2 | 3.6 | 5.0 | 1.23 | 68 | 41 |
| p6-DT40 | 1.025 | 6.2 | 6.7 | 7.0 | 1.38 | 79 | 43 |
None of the synthesized pDS polymers show discernible glass transition temperature (Tg) when analyzed with DSC. Instead, unlike the amorphous pDTT, they exhibit crystalline behavior with distinct melting temperatures (Tm).16 For each of the p4-DT, p5-DT, and p6-DT series, a small increase is seen in Tm with increasing molecular weight (or chain length), although all Tm values are close to physiological temperature. Comparing polymers across the three series, while keeping the same degree of polymerization, reveals no trend in Tm as both p4-DT and p6-DT have higher Tm than p5-DT. Note that p4-DT samples exhibited a large melting peak, indicating fast nucleation, even at fast scan rates (40 °C min−1). Thermograms are collected in ESI.†
The px-DTy polymers are all soluble in a range of solvents, including dichloromethane, chloroform, diethyl ether, and tetrahydrofuran while being insoluble in solvents such as pentane, methanol, dimethylsulfoxide, and N,N-dimethylformamide. The polymers exhibit good base resistance with no visible degradation in the presence of 1,8-diazabi-cyclo[5.4.0]undec-7-ene (DBU) (<50 mM) and Et3N (<32 mM). Thus, the unfunctionalized pDS polymers all show very different properties from pDTT, as they are not only crystalline but also soluble in a variety of solvents while being stable toward base. In addition, they possess an enhanced processability with access to more volatile solvents. These properties enable other applications than the amorphous pDTT does i.e. where rigidity and processability are in focus.
End-cap removal
Next, the self-immolative behavior was studied of p4-DT20, using an identical procedure as reported for the structurally similar pDTT.16 First, end-cap removal is accomplished using a stoichiometric amount of DTT w.r.t. the pyridinic end-caps, resulting in complete substitution of 2-mercaptopyridine within 5 min as monitored by 1H NMR in CDCl3 (Fig. S1†). Within the same time range, two new peaks (3.65 and 2.99 ppm) arise from released cDTT. The latter originates from cyclization and release of the newly introduced DTT end-groups, to reveal the decapped p4-DT backbone. The two steps involved in end-cap removal, i.e. end-cap exchange and cyclization of DTT, never become rate-controlling as they are much faster than the subsequent depolymerization of the backbone (Scheme 3). The entire process is base catalysed, as each individual thiol–disulfide exchange step, including end-cap removal, can occur through thiolates rather than thiols with enhanced kinetics.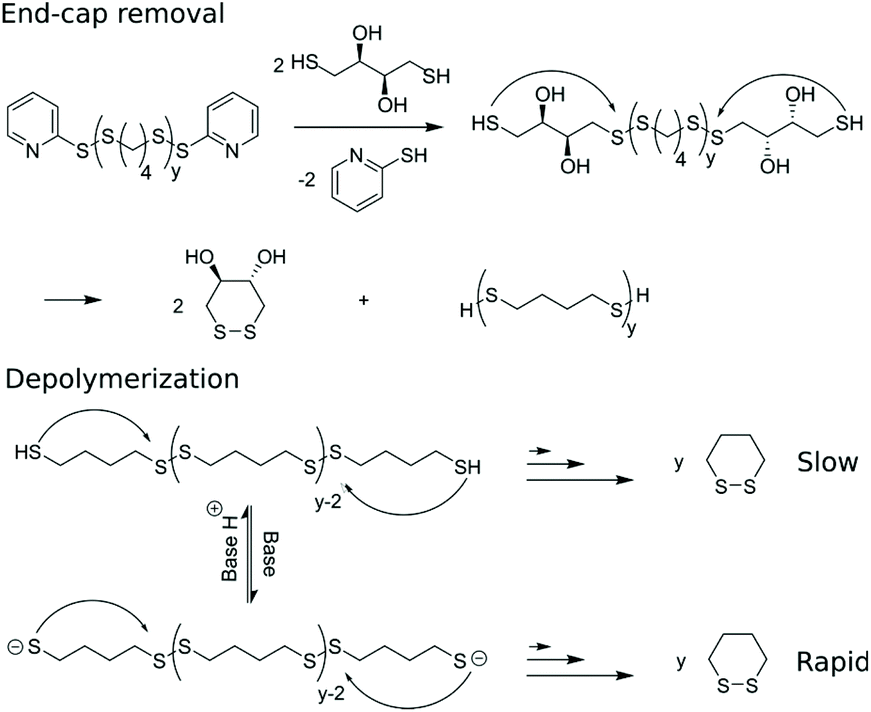 | ||
| Scheme 3 End-cap substitution at p4-DT using DTT followed by cyclization to reveal the decapped p4-DT backbone. End-to-end depolymerization occurs through cascade of intramolecular cyclizations to produce c4-DT. | ||
Depolymerization
Depolymerization of p4-DT in the presence of 1.1 equiv. DTT under ambient conditions provides c4-DT as degradation product as evidenced by appearence of signals at 2.84 and 1.97 ppm in 1H NMR. The process is slow with only 15% 1,2-dithiane (c4-DT) produced in 13 days (Fig. S2†). To enhance depolymerization rate, DBU may be added to promote formation of more reactive thiolates under basic conditions (Scheme 3). Fig. 1a displays 1H NMR spectra before and after addition of DTT/BDU with all relevant peaks assigned.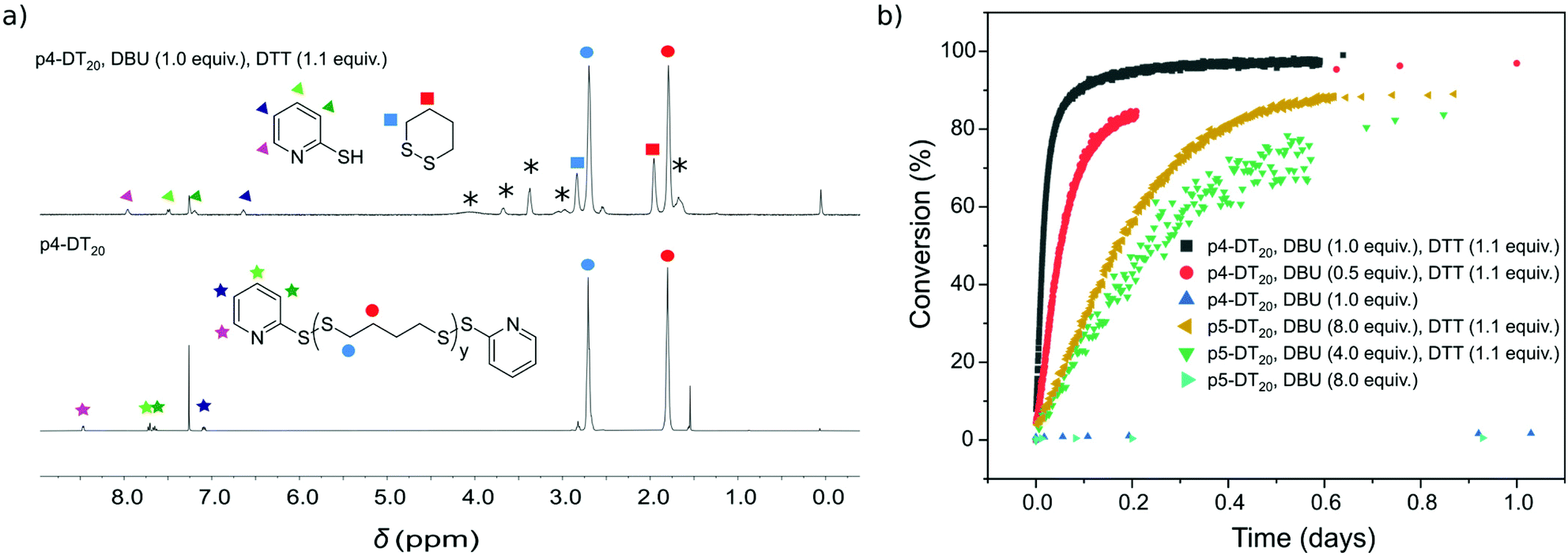 | ||
| Fig. 1 (a) 1H-NMR spectra of p4-DT20 (3.1 mM) before (bottom) and after (top) addition of DTT (6.9 mM; 1.1 equiv.) and DBU (6.3 mM; 1.0 equiv.) under Ar at rt in CDCl3; peaks attributed to DTT and DBU are noted with asterisks. (b) Kinetic traces of conversion of p4-DT20 (3.1 mM) and p5-DT20 (3.1 mM) to c4-DT and c5-DT, respectively, in the presence of varying concentrations of DBU (3.1–50 mM; 0.5–8.0 equiv.) and in the presence or absence of DTT (used for end-cap removal) as measured from integrals at 1.80 and 1.95 ppm for p4-DT, and at 1.50 and 2.01 ppm for p5-DT, in 1H NMR (see Fig. S7† for full kinetic traces). All experiments were conducted under Ar atmosphere at rt in CDCl3 and, in the case of p5-DT, in ampules as well; equivalents are reported w.r.t. end-caps. | ||
Kinetic traces extracted from NMR measurements show for all p4-DT samples initial zero reaction order with a constant rate until ∼60% depolymerisation (Fig. 1b). Zero-order kinetics is expected for SIPs as long as the concentration of reactive termini remains constants, i.e. until the shortest polymer chain reaches a length of two units. At this point further depolymerization decreases the concentration of active termini with the kinetics transitioning into first order. Using 0.5 equiv. DBU shows formation of c4-DT with initial rate of 8.9 × 10−6 s−1, resulting in complete depolymerization of p4-DT20 within two days. Increasing the amount of DBU to 1 equiv. enhances the rate three-fold to 2.8 × 10−5 s−1, enabling full depolymerization within 15 h. Note that end-cap removal is essential in these degradations, as they do not occur without DTT added.
Replacing DBU with a weaker base, Et3N (4 equiv.), still allows depolymerization of p4-DT10, p4-DT20, and p4-DT40, although incomplete (<50%) and with a slow depolymerization that depends on the backbone length with shorter polymers degrading faster. Importantly, excluding oxygen using an argon atmosphere results in faster and much higher conversion (95%) of p4-DT10 (Fig. S3†). Conducting degradation of p4-DT20 in sealed glass ampules with argon enabled full depolymerization within one year when utilizing only 1 equiv. Et3N (Fig. S4†).
A closer inspection of 1H NMR spectra (see Fig. S5†) reveals a partial reattachment of the mercaptopyridinic end-caps during the sluggish depolymerization as seen by reappearance of pyridinic end-cap related peaks at 8.47, 7.72, 7.65, and 7.09 ppm. A likely explanation is that influxing air oxidizes pyridinethiol to DTDP which reinstalls the end-caps. In principle, oxidative recombination of thiol-terminated chains would exert a similar stalling effect on depolymerization, although this is less likely to occur as such thiols/thiolates would be less prone to oxidation than pyridylthiolate. In particular, degradations of the long chain p4-DT20 and p4-DT40 with their relatively low amount of end-caps are prone to be involved in the recapping pathway, leading to overall slower and incomplete depolymerizations. This is especially relevant when a weak base such as Et3N is applied in low excess. Nevertheless, the kinetics observed in these exceedingly slow depolymerization reactions using Et3N as base does not exhibit the expected transition from zero order to first order kinetics as seen with DBU. Initial rates estimated within the first day are in the range of 1.3–14 × 10−8 s−1 for the three different p4-DT samples (Table S1†), independent of whether a protective argon atmosphere is present or not.
Similar depolymerization of p5-DT20 with 1.1 equiv. DTT to the 7-membered ring, 1,2-dithiepane (c5-DT), occurs as expected much slower than for p4-DT20, and even with 1 and 2 equiv. DBU full depolymerizarion is not achievable within 56 days (Fig. S6†). However, increasing the number of DBU equivalents to 4 and 8 leads to >90% conversion within 13 and 2 days, respectively (see Fig. S7† for kinetic traces beyond 1 day). Kinetic traces for the initial 24 h degradation of p5-DT20 show a zero reaction order until ∼60% depolymerisation. With 4 equiv. of DBU the rate constant is estimated to be 1.7 × 10−6 s−1 and with 8 equiv. of DBU it is 2.6 × 10−6 s−1. This development is in line with the mechanism shown in Scheme 3 where the role of the base is to facilitate the thiolate pathway in the depolymerization reaction through deprotonation of thiols. Note also that degradation of p5-DT is significantly slower than that of p4-DT, despite using much more base (DBU). In fact, p5-DT could not be depolymerized with Et3N.
At first sight, the observation that changing DBU concentration affects depolymerization kinetics is surprising as DBU should be basic enough to deprotonate all thiol groups. However, we attribute this effect to the use of chloroform as reaction medium. While chloroform works well for NMR analysis, it is not optimal for kinetic studies involving bases because of its own relatively high acidity that will prevent a full conversion of thiol groups to thiolates. Under these conditions, the intrinsic depolymerization rate cannot easily be extracted. A less acidic solvent such as tetrahydrofuran would make a better choice as reaction medium for a detailed kinetic study.
Upon exposing p6-DT20 to DTT/DBU, degradation of the polymer initiated, but in this case, a complex mixture of smaller compounds is detected by 1H NMR and thin-layer chromatography (TLC) (Fig. S8†). None of the prominent TLC spots could be identified as 1,2-dithiocane (c6-DT), i.e. the 8-membered ring cyclic disulfide expected had the polymer been self-immolative. Instead, the largest isolated fraction (∼30%) is found to be the dimeric and cyclic 1,2,9,10-tetra-thiacyclohexadecane identified by reference to literature values (Fig. S9†).33 This is a 16-membered macrocyclic compound consisting of two C6 disulfides. We suppose that the extended reaction times, in connection with the strong DBU base, allow p6-DT to depolymerize in other ways than simple unzipping, such as random backbiting and metathesis as reported for lipoates by Liu et al.14 No depolymerization of p6-DT is achieved with Et3N.
NMR spectra comparing p4-DT20, p5-DT20, and p6-DT20 with their respective monomers and depolymerization products are available in ESI (Fig. S10–S15†). Notably, all cyclic disulfides formed can, in principle, be reductively transformed into starting materials, thus highlighting the good chemical resuablity of all components.
Although the cleavable nature of disulfide bonds is well known, utilization of this dynamic bond for cascade ring-closing reaction resulting in amplified response has only recently been established.34 In this report, we find that the alkyl spacing between disulfide bonds plays an important role in how easily the polydisulfide can depolymerize as a result of continuous depolymerization or as a dynamic bond exchanging agent. In other words, the ring size of the released cyclic disulfide has, as expected, a major influence both on depolymerization kinetics and thermodynamics. While a 6-membered ring (c4-DT) is able to attain full or near-complete depolymerization, the 7-membered ring (c5-DT) reaches a maximum of ∼90% under the utilized conditions. Formation of 8-membered rings (c6-DT) is not observed but instead, a myriad of degradation products is formed, where the most prominent is a dimer of c6-DT. In contrast, p4-DT, and p5-DT depolymerize through a similar self-ïmmolative mechanism as pDTT by the formation of a small cyclic disulfides.
Both p4-DT and p5-DT are interesting SIPs, as they have tuneable degradation rates depending on the base applied and its concentration. With a weak base such as Et3N, the time scale for p4-DT is months or even years, while it is days or weeks with the stronger DBU base. The previously published SIP, pDTT, had complete depolymerization within minutes, thus being less stable toward base. We attribute the faster depolymerization of pDTT to increased acidity promoted by the hydroxyl groups located on the backbone chain on the associated thiol group, thereby increasing the concentration of its more reactive thiolate in the presence of base. This feature that depolymerization can be accomplished at different time scales for these SIPs, dependent both on the backbone structure and base, will provide a basis for material selection in potential applications, e.g. for the design of slow or targeted drug delivery systems. The role of the thiol/thiolate equilibrium on the kinetics of depolymerization will be investigated in detail in a future publication.
Conclusions
Two new self-immolative polydisufides, p4-DT and p5-DT, and a degradable polymer, p6-DT, with saturated aliphatic backbones were synthesized with remarkably low PDI values ranging from 1.13–1.67. The new unfunctionalized polymers showed good solubility in common apolar solvents and a high degree of crystallinity with melting temperatures close to physiological temperature, modestly increasing with chain length. The new polymers were found to be more tolerant toward bases and exhibited higher thermal stability than pDTT while maintaining the self-immolative properties when treated with DTT and DBU/Et3N. The ring size of the released cyclic disulfide degradation product proved to be an important factor for the rate of depolymerization as formation of the 6-membered ring was observed to be significantly faster (hours/days) than the 7-membered (days/weeks), and, in particular, 8-membered one which was not formed at all. In addition, the rate of depolymerization was tuneable through use of different bases. Notably, all cyclic disulfides might be reductively transformed into starting materials, if desired, thus highlighting the good chemical reusablity of all components in a circular economy perspective. Besides the listed benefits found in p4-DT, we expect the most important feature to be the improved solubility in a broad range of organic solvents, making p4-DT a more forgiving SIP to work with than pDTT.Author contributions
M.H.F. and A.S. conceived and designed the experiment; M.H.F. and A.S. performed the experiment; S.U.P. and K.D. supervised the project; M.H.F., A.S. and M.L.H. analyzed the data; M.H.F., A.S., S.U.P., and K.D. wrote the paper. All the authors contributed to the realization of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to financial support from Independent Research Fund Denmark (9041-00096B).Notes and references
- E. Cabane, X. Zhang, K. Langowska, C. G. Palivan and W. Meier, Biointerphases, 2012, 7, 9 CrossRef CAS PubMed.
- S. Gao, G. Tang, D. Hua, R. Xiong, J. Han, S. Jiang, Q. Zhang and C. Huang, J. Mater. Chem. B, 2019, 7, 709–729 RSC.
- Z. A. Liang, Y. Z. Meng, L. Li, X. S. Du and A. S. Hay, Macromolecules, 2004, 37, 5837–5840 CrossRef CAS.
- M. R. Krause, T. A. Daly, P. F. Almeida and S. L. Regen, Langmuir, 2014, 30, 3285–3289 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- Y. Xu and D. Chen, Macromol. Chem. Phys., 2016, 217, 1191–1196 CrossRef CAS.
- N. D. Bansode, K. R. Sindhu, C. Morel, M. Remy, J. Verget, C. Boiziau and P. Barthelemy, Biomater. Sci., 2020, 8, 3186–3192 RSC.
- D. J. Phillips and M. I. Gibson, Biomacromolecules, 2012, 13, 3200–3208 CrossRef CAS PubMed.
- S. Kim, K. I. Wittek and Y. Lee, Chem. Sci., 2020, 11, 4882–4886 RSC.
- E.-K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752–1763 RSC.
- K. Endo, T. Shiroi and N. Murata, Polym. J., 2005, 37, 512–516 CrossRef CAS.
- K. Endo, T. Shiroi, N. Murata, G. Kojima and T. Yamanaka, Macromolecules, 2004, 37, 3143–3150 CrossRef CAS.
- K. Endo and T. Yamanaka, Macromolecules, 2006, 39, 4038–4043 CrossRef CAS.
- Y. Liu, Y. Jia, Q. Wu and J. S. Moore, J. Am. Chem. Soc., 2019, 141, 17075–17080 CrossRef CAS PubMed.
- D. Basak, R. Kumar and S. Ghosh, Macromol. Rapid Commun., 2014, 35, 1340–1344 CrossRef CAS PubMed.
- S. Pal, A. Sommerfeldt, M. B. Davidsen, M. Hinge, S. U. Pedersen and K. Daasbjerg, Macromolecules, 2020, 53, 4685–4691 CrossRef CAS.
- R. E. Yardley, A. R. Kenaree and E. R. Gillies, Macromolecules, 2019, 52, 6342–6360 CrossRef CAS.
- B. Fan, J. F. Trant, R. E. Yardley, A. J. Pickering, F. Lagugné-Labarthet and E. R. Gillies, Macromolecules, 2016, 49, 7196–7203 CrossRef CAS.
- R. J. Amir and D. Shabat, Chem. Commun., 2004, 1614–1615, 10.1039/b404946b.
- B. Fan, R. E. Yardley, J. F. Trant, A. Borecki and E. R. Gillies, Polym. Chem., 2018, 9, 2601–2610 RSC.
- H. Kim, H. Mohapatra and S. T. Phillips, Angew. Chem., Int. Ed., 2015, 54, 13063–13067 CrossRef CAS PubMed.
- H. Zhang, K. Yeung, J. S. Robbins, R. A. Pavlick, M. Wu, R. Liu, A. Sen and S. T. Phillips, Angew. Chem., Int. Ed., 2012, 51, 2400–2404 CrossRef CAS.
- G. Liu, G. Zhang, J. Hu, X. Wang, M. Zhu and S. Liu, J. Am. Chem. Soc., 2015, 137, 11645–11655 CrossRef CAS PubMed.
- K. Yeung, H. Kim, H. Mohapatra and S. T. Phillips, J. Am. Chem. Soc., 2015, 137, 5324–5327 CrossRef CAS PubMed.
- J. S. Robbins, K. M. Schmid and S. T. Phillips, J. Org. Chem., 2013, 78, 3159–3169 CrossRef CAS PubMed.
- Q. E. A. Sirianni, A. Rabiee Kenaree and E. R. Gillies, Macromolecules, 2019, 52, 262–270 CrossRef CAS.
- Y. Xiao, Y. Li, B. Zhang, H. Li, Z. Cheng, J. Shi, J. Xiong, Y. Bai and K. Zhang, ACS Macro Lett., 2019, 8, 399–402 CrossRef CAS.
- B. Fan, R. Salazar and E. R. Gillies, Macromol. Rapid Commun., 2018, 39, e1800173 CrossRef.
- B. Fan, J. F. Trant and E. R. Gillies, Macromolecules, 2016, 49, 9309–9319 CrossRef CAS.
- M. F. Nichol, K. D. Clark, N. D. Dolinski and J. Read de Alaniz, Polym. Chem., 2019, 10, 4914–4919 RSC.
- A. D. Shaller, W. Wang, H. Gan and A. D. Li, Angew. Chem., Int. Ed., 2008, 47, 7705–7709 CrossRef CAS PubMed.
- F. B. Cougnon, H. Y. Au-Yeung, G. D. Pantos and J. K. Sanders, J. Am. Chem. Soc., 2011, 133, 3198–3207 CrossRef CAS PubMed.
- H. Abul-Futouh, L. R. Almazahreh, M. K. Harb, H. Gorls, M. El-Khateeb and W. Weigand, Inorg. Chem., 2017, 56, 10437–10451 CrossRef CAS.
- R. Bej and S. Ghosh, Bioconjugate Chem., 2019, 30, 101–110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01412a |
| This journal is © The Royal Society of Chemistry 2022 |