
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermal response and thermochromism of methyl red-based copolymer systems – coupled responsiveness in critical solution behaviour and optical absorption properties†
Thorben Gwydion
Jaik
 a,
Betty
Ciubini
b,
Francesca
Frascella
b and
Ulrich
Jonas
*a
a,
Betty
Ciubini
b,
Francesca
Frascella
b and
Ulrich
Jonas
*a
aDepartment of Chemistry and Biology, University of Siegen, Adolf-Reichwein-Strasse 2, D-57076, Siegen, Germany. E-mail: jonas@chemie.uni-siegen.de
bDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, IT-10129, Torino, Italy
First published on 14th January 2022
Abstract
Until now, only limited experimental knowledge and sparse theoretical treatment about the mechanisms of thermochromism of azo dyes in solution has been available. Especially the coupling of thermoresponsiveness of polymers with the inherent thermochromism of azo dyes is attractive to enhance the optical response for applications like polymeric optical pH- and temperature-dual sensors. To elucidate the different mechanisms contributing to the thermochromism of such azo chromophores, we synthesised monomers based on the constitutional isomers of the common pH indicator methyl red. The ortho-isomer was copolymerised with hydrophilic monomers and the photocrosslinker benzophenone acrylamide, with the resulting copolymers being converted to networks by irradiation with UV-light and yielding hydrogels after swelling with water. N-Isopropylacrylamide was used as comonomer to introduce thermoresponsiveness in the polymers in form of a lower critical solution temperature (LCST) behaviour. Three different dye systems with varying protonation states were investigated by temperature-dependant UV-vis spectroscopy: as monomers in solution, as part of copolymers in solution, and as photocrosslinked hydrogels. Consequently, we were able to identify the four different mechanisms of vibronic thermochromism, thermo-solvatochromism, thermo-perichromism and thermo-halochromism. Their interplay was investigated by choosing appropriate combinations of solvents, acid and comonomers. The LCST behaviour of the N-isopropylacrylamide copolymers could be exploited to strongly influence thermochromism, providing insight into the mechanisms of critical solution behaviour of polymers and thermochromism alike. The experimental data suggest that various thermochromic mechanisms act simultaneously and mutually influence each other, specifically with thermo-solvato- and thermo-perichromism affecting thermo-halochromism. These effects are best described by the terms thermo-solvato-halochromism and thermo-peri-halochromism. Notably, on the basis of the identified thermochromic mechanisms prevailing in the monomer solutions, the behaviour of the more complex polymer systems can be elucidated, and consequently, the distinct properties of the dye in combination with polymer-inherent phenomena can be deduced. To our knowledge, this is the first comprehensive study to harmonise the understanding of the different thermochromic mechanisms in azobenzene, their mutual action, and the strong influence of thermoresponse on thermochromism.
1. Introduction
A large number of materials show a change of colour with temperature, which is generally termed thermochromism. This has been observed and exploited to sense temperature in systems like dye-based luminescent sensors for biological and biomedical applications,1–3 nanomaterials,4 supramolecular systems5 and dye aggregates.6 Generally, thermochromism describes the change of colour due to a change of temperature and depending on the scientific community, this is defined as being either purely reversible or both reversible and irreversible processes.7–9 Polymeric materials exhibiting thermochromic behaviour are particularly versatile and can be used in a variety of systems to achieve a change in absorption, emittance, or reflectance of a system. The term thermochromic polymers entails a manifold of systems with either the polymer itself being thermochromic or the polymer being a matrix for thermochromic dyes, complexes or particles.7 The thermochromic mechanism relevant for the work presented in this paper is based on the change in absorption of organic dyes in dependence on temperature.One class of optical thermometers entails solvatochromic dyes coupled to a polymer phase transition. Owing to the change in polarity in the polymer coil during the transition, either the fluorescence or absorption change with temperature.10
Another established thermochromic system consists of a pH-sensitive dye embedded in a hydrogel matrix that changes colour reliant on a thermally dependant protonation equilibrium, notably alkaline PVA-borax hydrogels with cresol red or Reichardt's dye as indicator dyes.11,12
Azo dyes are a particularly interesting class of photochromic molecules that show additional mechanisms for a colorimetric response with respect to external stimuli, besides light, such as thermochromism (responding to a temperature change), halochromism (pH change) and solvatochromism (change of solvent interaction). Thermochromism in azobenzene derivatives is often coupled to a thermochromic matrix, with the azobenzene only enhancing the optical effects in the matrix material. In one example, this was realised by adding azobenzene to an LCST polymer–opal composite to tune the transmission band over the whole visible spectrum via trans–cis-isomerisation and temperature. The polymer volume phase transition changes the distance between the particles in the opals, effectively tuning the optical interference, while the photochromism of the azobenzene influences the base colour of the composite.13 In other studies, azobenzenes were combined with photochromic polyelectrolytes based on polythiophenes or polydiacetylenes (PDA), enhancing the thermochromic behaviour of the polymers or enhancing thermal reversibility, apparently by acting as controlled defects or as stabilising groups, in the case of PDA micelles.14–20
A thermochromic effect solely relying on the azo dye core has been investigated for several phenylazonaphthols.21 The tautomeric equilibrium between the enol and quinone forms was followed in solution in dependence of temperature, with the quinone form absorbing at longer wavelengths and being more stable at low temperatures.
For pH-sensitive and thus halochromic azobenzenes, thermochromism has been observed in different environments. In one of the first examples, the well-known pH indicator methyl red (2-{[4-(dimethylamino)phenyl]diazenyl}benzoic acid, in short here MR) was adsorbed on silica gel. In aqueous solution, the MR chromophore changes from a red protonated form with an absorption maximum at ∼525 nm at pH 4.4 to a yellow, deprotonated form above pH 6.2 (abs. max. ∼430 nm).22,23 Protonated MR exists in two different tautomeric cations, namely an ammonium ion with a protonated tertiary amine, and an azonium structure protonated at the azo bridge. In MR, the azonium form is stabilised via a quinoidal resonance structure and intramolecular hydrogen bonding, rendering it the preferred structure.24 Adsorption on silica gel results in a colour change of MR from red (acidic) to orange (neutral/basic) upon heating. The authors attribute this effect to an increase of basic sites at the silica gel surface at low temperatures due to release of water and an increase in acidity of the dye at higher temperatures.25 Another halochromic azobenzene has been shown to have the opposite behaviour when immobilised in ion-exchange micelles: the protonated form was more stable than the deprotonated form. This was explained by a change in the partition of water between the aqueous bulk and the periphery of the micelle.26 An example for a dual colorimetric sensor to measure simultaneously pH and temperature is established with the azobenzene derivative Disperse Red 1 in thermoresponsive copolymers.27,28
Besides halochromism, MR also shows an intense solvatochromism. An absorption band at ∼425 nm dominates the spectrum in polar solvents, while in apolar solvents an absorption band at ∼500 nm prevails. The coexistence of the two absorption bands characterises the spectra in dependence of several solvent parameters, such as solvent composition, solvent polarity, hydrogen bonding and Kamlet–Taft-parameters.29,30
Two scenarios have been proposed to explain the solvatochromic behaviour of MR.31,32 In both studies, the change in absorption has been attributed to the change in the ratio of intermolecular hydrogen bonding to solvent molecules versus intramolecular hydrogen bonding of the carboxylic acid in ortho-position to the azo bridge. The reasoning for the change in absorption wavelength, however, is vastly different in the two publications, as outlined below. The first study31 assumes solvatochromism to result from a solvent-dependant, intramolecular prototropic equilibrium by relocating a proton between the azobenzene and an azonium form. The azonium ion absorbs at longer wavelengths than the azobenzene, owing to its quinoid character with lower energy of the π* orbital. The author showed that the dominance of either the azonium or the azobenzene band does not only depend on solvent polarity but on several other solvent parameters, like hydrogen bonding and polarizability. Accordingly, prediction of the spectral features is only reliable withing the same solvent family (hydrogen bond donating or accepting, halogenated aliphatic and aromatic solvents). The second study32 attributes the solvatochromic shifts solely to the balance between the hydrogen bond accepting parameter β and donating parameter α of the solvent. According to the authors, these parameters dictate whether intermolecular (to the solvent molecules) or intramolecular hydrogen bonding is preferred in MR. The absorption band at ∼425 nm is considered here as the π → π* transition of MR, while the band at ∼500 nm is considered to be the n → π* transition. Based on the presented calculations, intramolecular hydrogen bonding, which is increased in apolar solvents, spreads the π* orbital over a larger part of the molecule and lowers its energy. With that, the n → π* transition is energetically preferred.
The interpretation of the first study appears more plausible based on the detailed UV-vis absorption behaviour of azobenzenes as discussed in the following.31 Owing to a quinoid orbital structure, the main absorption band of protonated MR lies at longer wavelengths than the absorption band of neutral or anionic MR.23 Both the azonium ion of MR as well as a strongly intramolecularly hydrogen bonded MR show a strong absorption band around 500 nm. The similarity between the absorption behaviour of the azonium ion of MR compared to the intramoleculary hydrogen-bonded MR in certain solvents suggests that an intramolecular hydrogen shift leads to a quinoid character of the zwitterionic molecule, which is accompanied by a redshift. The existence of the long wavelength absorption band of the azonium ion corresponds to the π → π* and conforms to the general observation that the π → π* transition is lowered in energy upon protonation of azobenzenes.33 This is supported by the fact, that MR belongs to the class of pseudostilbenes, in which the n → π* and π → π* transitions are energetically nearly degenerate. Furthermore, the n → π* transition is generally weaker in azobenzenes.34 A large shift in absorption without a considerable decrease of the extinction coefficients involving the non-bonding orbitals is, therefore, unlikely.
Concluding from the various experimental results found in literature, the coexistence of several chromic mechanisms in MR renders this dye a suitable candidate for sensing applications. Yet, a detailed understanding of the mechanistic interplay would be required for effective exploitation, but a comprehensive picture is still lacking. An early application example, which is still in use today, is the determination of acid production by some bacteria during glucose digestion by MR, referred to in microbiology as the Methyl Red test.35 Immobilisation of the MR probe is preferred for many sensing applications, yet direct contact with a liquid medium is mandatory, which can be provided by the open network structure of swollen polymer gels. As such, polymer matrices based on poly-N-isopropylacrylamide (PNiPAAm)36 and poly-N-(2-hydroxyethyl)acrylamide (PHEAm)37 are interesting candidates in biomedicine, as they are known to be biocompatible and even non-fouling in the case of PHEAm.
PNiPAAm is additionally interesting owing to its lower critical solution temperature (LCST)-type thermoresponsive behaviour in solution. It is soluble at low temperatures and undergoes a volume phase transition when increasing the temperature, leading to insolubility of the macromolecule beyond the transition temperature. This transition temperature can be influenced by copolymerisation and added solutes, like salts or small organic molecules that undergo hydrogen bonding.38,39 Recently, a new polymerisation method on the basis of reversible activation-deactivation of the surface of Ni–Co alloy nanoparticles has been proposed for the synthesis of poly-methacrylate, which may become an interesting candidate in the near future as well.40
These research accounts inspired our present study about the thermochromic behaviour of MR-containing polymer systems. Here, the synthesis of a series of MR-based azobenzene monomers with acrylamide functionalities is reported, from which copolymers were prepared, utilising N-(2-hydroxyethyl)acrylamide (HEAm) or N-isopropylacrylamide (NiPAAm) monomers and a benzophenone acrylamide (BPAAm) photocrosslinker. By irradiation with UV-light, crosslinked networks were formed from these polymers, yielding hydrogels after swelling with water. For the monomeric and polymeric systems, the thermochromic behaviour was investigated in alcoholic and aqueous media under neutral, slightly acidic, and strongly acidic conditions.
2. Experimentals
2.1. Materials and equipment
All solvents used were of Milli-Q®, spectroscopic, or HPLC-grade. Absolute ethanol was purchased from VWR Chemicals. Tetrahydrofurane was dried and distilled over potassium. Trifluoroacetic acid was purchased from Carl Roth (Germany) in PEPTIPURE® ≥99.9% quality. Sulfuric acid (≥95%, Fisher Chemical), hydrochloric acid (37%, Anal. Reag. Gr., Fisher Chemical), acetic acid (Anal. Reag. Gr., ChemSolute), methyl red (Alfa Aesar), carbonyldiimidazole (97%, Alfa Aesar), N-(2-hydroxyethyl)acrylamide (97%, Sigma Aldrich), 4-aminobenzoic acid (Merck) and 3-aminobenzoic acid (98%, Merck) were used as received. 1,8-Diazabicyclo[5.4.0]undec-7-ene was dried over calciumchloride (anhydrous, technical, Bernd Kraft) and distilled in vacuo. N-Isopropylacrylamide was recrystallised from n-hexane. Azobisisobutyronitrile was recrystallised from methanol. 4-Benzophenoneacrylamide was synthesised according to literature.41 4-(3-Triethoxysilyl)propoxybenzophenone was synthesised by Mr Daniel John according to literature.42UV-vis measurements were performed on a Thermo Scientific™ Evolution™ 220 UV-Vis-spectrophotometer. If not stated otherwise, the measurements were done with 100 nm min−1 and a resolution of 1 nm.
NMR-measurements were performed on either a Bruker AV 400 or a Jeol EZC 500. Detailed assignments of peaks are given in the ESI† in the corresponding spectra.
GPC/SEC was measured on a PSS GRAM linM column (Polymer Standards Service GmbH, Mainz, Germany) in dimethylacetamide with LiBr (0.1 g L−1) at 60 °C with PMMA-standards as reference.
Only selected monomer and polymer syntheses are shown here to illustrate the synthetic process. Full experimental details and NMR-data are given in the ESI.†
δH(400 MHz; CDCl3) 7.82 (d, J = 9.3 Hz, 2H; Ph–H), 7.74 (dd, J = 7.9, 1.1 Hz, 1H; Ph–H), 7.56 (qd, J = 7.9, 1.6 Hz, 2H; Ph–H), 7.39 (ddd, J = 7.6, 6.7, 1.9 Hz, 1H; Ph–H), 6.72 (d, J = 9.3 Hz, 2H; Ph–H), 6.12 (dd, J = 17.0, 1.5 Hz, 1H; CHCHH(trans)), 5.85 (s, br, 1H; CONH), 5.63 (dd, J = 17.0, 10.4 Hz, 1H; CHCHH), 5.43 (dd, J = 10.4, 1.5 Hz, 1H; CHCHH(cis)), 4.39 (t, J = 5.0 Hz, 2H; COOCH2), 3.59 (q, J = 5.3 Hz, 2H; CONHCH2), 3.07 (s, 6H; N(Me)2).
δC(101 MHz; CDCl3) δ 168.61 (COO), 165.49 (CONH), 152.99 (Ph), 152.54 (Ph), 143.54 (Ph), 132.08 (Ph), 130.59 (COCHCH2), 129.70 (Ph), 128.50 (Ph), 127.72 (Ph), 126.36 (COCHCH2), 125.35 (Ph), 119.50 (Ph), 111.70 (Ph), 64.35 (COOCH2), 40.35 (N(Me)2), 38.79 (CONHCH2).
P1 (poly(HEAm-co-o-MREAm-co-BPAAm)): δH(500 MHz, MeOD) 8.5–6.5 (br, aromatic H), 4.57 (COOCH2), 4.38 (Ar–CONHCH2), 3.66 (CH2CH2OH), 3.51–3.12 (CONHCH2), 2.4–1.25 (backbone).
P2 (poly(NiPAAm-co-HEAm-co-o-MREAm-co-BPAAm)): δH(500 MHz, D2O) 8.0–6.5 (br, aromatic H), 4.41 (COOCH2), 3.91 (NHCH(CH3)2), 3.68 (CH2CH2OH), 3.36 (CONHCH2), 2.3–1.25 (backbone), 1.16 (NHCH(CH3)2).
P2b (poly(NiPAAm-co-HEAm-co-o-MREAm-co-BPAAm)): δH(400 MHz, MeOD) 8.0–6.75 (br, aromatic H), 4.38 (COOCH2), 3.96 (NHCH(CH3)2), 3.65 (CH2CH2OH), 3.12 (N(Me)2), 2.3–1.25 (backbone), 1.16 (NHCH(CH3)2).
P3 (poly(HEAm-co-MAA-co-o-MREAm-co-BPAAm)): δH(400 MHz, D2O) 8.5–6.5 (br, aromatic H), 4.42 (COOCH2), 3.66 (CH2CH2OH), 3.34 (CONHCH2), 2.4–1.25 (backbone), 1.00 (backbone-CH3).
P4 (poly(NiPAAm-co-MAA-co-o-MREAm-co-BPAAm)): δH(500 MHz, MeOD) 8.0–6.5 (br, aromatic H), 4.58 (COOCH2), 4.38 (Ar–CONHCH2), 3.91 (NHCH(CH3)2), 3.12 (N(Me)2), 2.3–1.25 (backbone), 1.16 (NHCH(CH3)2).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hydrogen peroxide, 3:1) and rinsed thoroughly with water. The slides were dried under a nitrogen stream. They were submerged in an ethanolic solution of benzophenone silane (1 mmol L−1) for 24 hours before they were rinsed thrice with absolute ethanol and finally dried under a nitrogen stream.
:hydrogen peroxide, 3:1) and rinsed thoroughly with water. The slides were dried under a nitrogen stream. They were submerged in an ethanolic solution of benzophenone silane (1 mmol L−1) for 24 hours before they were rinsed thrice with absolute ethanol and finally dried under a nitrogen stream.
Polymers were drop-casted on the glass slides from methanolic solution (1 w%, 200 μL on 2.4 cm × 2.4 cm slides). Photocrosslinking was performed at 302 nm with an energy of 20.3 J cm−2. The polymer films of P1 and P2 were annealed at 170 °C prior to photocrosslinking. All films were washed with water until the supernatant remained colourless before thermochromicity measurements.
3. Results and discussion
The synthesis of the MR-based monomers and polymers is briefly discussed first, followed by the analysis of their thermochromic behaviour. The optical characteristics are described for the individual monomers and then elaborated in relation to the more complex thermochromism of the polymers in the following order:(i) The dye monomers in simple solvents.
(ii) The dye monomers in binary solvent mixtures.
(iii) Copolymers bearing the dye in solution.
(iv) Photocrosslinked, water–swollen polymer gels.
3.1. Synthesis of methyl red-based monomers and polymers
The monomers in this study were synthesised in a one-pot synthesis in two successive steps by first reacting the parent compound methyl red with carbonyldiimidazole (CDI) and subsequent coupling of the resulting imidazolide with the monomer N-(2-hydroxyethyl)acrylamide without intermediate workup (cf.Scheme 1). The imidazolide synthesis was based on a previously reported reaction procedure by Staab.43,45 The subsequent esterification step of the MR imidazolide with an alcohol does not occur spontaneously at room temperature upon mixing, but only after addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as strong base. DBU has previously been reported to be an excellent catalyst for the amidation of aromatic acids employing CDI48 and proved here to be efficient for the esterification with HEAm as well. The yields for the target compounds around 90% were only achieveable upon adding an excess of acetic acid before further purification by extraction with water-dichloromethane. The obtained esters are base labile and extraction with water without prior acidification substantially reduced the yield by hydrolysis. It is worthwhile to note, that synthesis attempts for the target monomers employing either N-hydroxysuccinimide (NHS) active esters, acyl chlorides or carbodiimides were not satisfactory. The active ester- and acyl chloride-routes failed to yield any product. The target compound could be synthesised in reasonable yields around 68% when utilising the coupling reagent 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI) with 4-dimethylaminopyridine as a catalyst. Yet, the more convenient reaction conditions and higher yields, as well as more affordable coupling reagents rendered the imidazolide route as the preferred procedure. Overall, we established a simple, high-yield synthesis for acrylamide monomers carrying an azobenzene chromophore that is equally applicable to all constitutional isomers of the parent compound MR.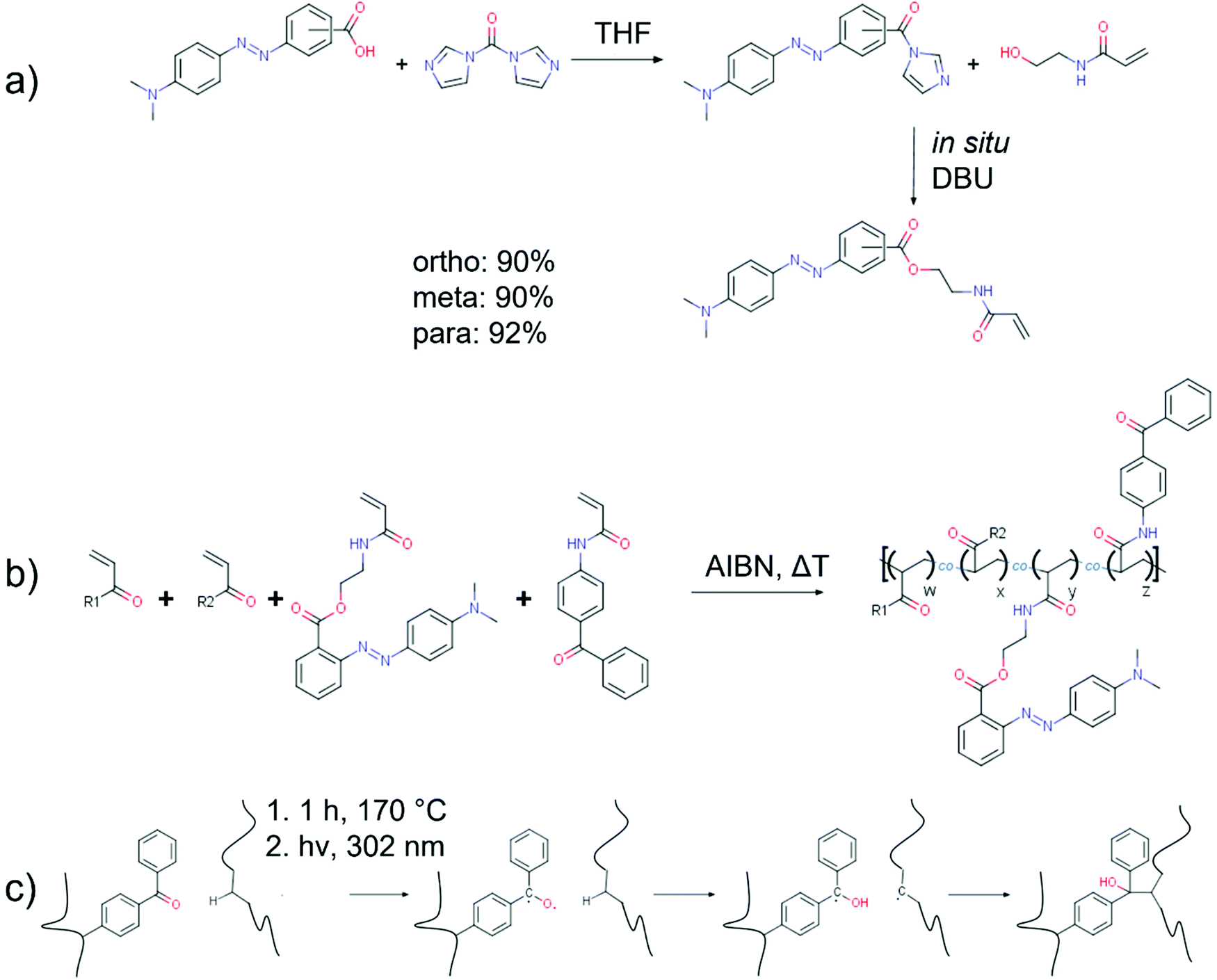 | ||
| Scheme 1 a) Carbonyldiimidazole-mediated one-pot synthesis yielding the constitutional isomers of the methyl red ester of N-(2-hydroxyethyl)acrylamide. (b) Free radical copolymerisation of two generic acrylic monomers, o-MREAm and BPAAm. (c) Benzophenone-based photocrosslinking process optimised for the polymers containing an azobenzene chromophore, involving formation of a biradical, hydrogen abstraction, and crosslinking with a second chain.46,47 | ||
Free radical copolymerisation of the ortho-MR monomer with NiPAAm, HEAm, MAA and BPAAm comonomers in solution was successful, while attempts at homopolymerisation of only the o-MR monomer failed. The obtained copolymers have dispersities Đ > 2 and only about two thirds of the dye monomer feed were built into the copolymers. With a Mn of 15–22 kDa the copolymers remain rather small as well (cf.Table 1). All these observations agree with previous studies about the retardation effect of aromatic azo compounds on free radical polymerisations.49–53 This effect was attributed to the formation of stable radicals at the azo group, increasing the rate of transfer, and lowering the overall polymerisation rate and degree of polymerisation. Furthermore, the rather high initiator concentration of 2 mol% was used to offset the retardation effects and obtain polymers in reasonable time frames. The high initiator concentration, as well as the retardation effects, increase the dispersity of the copolymers.
| Polymer | o-MREAm [w%] | BPAAm [w%] | M n [103 Da] | M w [103 Da] | Đ | Yield [%] |
|---|---|---|---|---|---|---|
| 1 | 4.5 (65%) | 1.9 (94%) | 17.4 | 37.7 | 2.17 | 79 |
| 2 | 4.9 (63%) | 2.0 (91%) | 14.9 | 32.2 | 2.16 | 70 |
| 2b | 5.2 (67%) | 2.1 (96%) | 22.1 | 51.9 | 2.35 | 81 |
| 3 | 4.8 (63%) | 2.0 (95%) | 15.5 | 31.7 | 2.04 | 85 |
| 4 | 4.4 (62%) | 1.8 (88%) | 16.0 | 51.4 | 3.21 | 86 |
The built-in ratio of the photocrosslinker BPAAm, on the other hand, was close to the feed ratio. For the chemical structures provided in the figures, the numbers given after parentheses relate to the nominal feed composition and do not necessarily reflect the built-in ratios.
The preparation of the photocrosslinked gels is discussed further below in the context of their optical characterisation.
3.2. Proton-induced thermochromism of the constitutional isomers of methyl red derivatives in solution
The major incentive of this section is the elucidation of a structure–property relationship for three dye monomers with respect to the influence of their positional isomerism on thermo-halochromism and vibronic thermochromism. For this purpose, we specifically investigated the three isomers of the methyl red ester derived from N-(2-hydroxyethyl)acrylamide (MREAm), that bear the ester linkage in ortho-, meta- or para-position to the azo-bridge. All three derivatives show similar absorption behaviours in the UV-visible range in neat ethanol. The magnitudes of the extinction coefficients follow the order ortho < meta < para. The absorption maxima for ortho- and meta-derivatives lie almost at the same wavelength, while the maximum is slightly red shifted for the para-isomer. These derivatives show only minimal thermochromism in neat ethanol (no acid present). With increasing temperature, the maximum does not shift considerably (∼0.5 nm/10 °C), but the absorption bands slightly broaden and their asymmetric shape decreases (cf. ESI Fig. S17†).In ethanolic trifluoroacetic acid at a concentration of 1 v/v%, the different constitutional isomers of MREAm are partially protonated (cf.Fig. 1). The azonium form is generated upon addition of an acid by protonation of the azobenzene, which acts as the base in the system. With rising temperature, the absorption band of the protonated form, the azonium ion (longer wavelength), diminishes for all isomers. In particular for the ortho-isomer, the band corresponding to the neutral azobenzene (shorter wavelength) visibly increases. This thermochromic phenomenon involves deprotonation of the azonium form (the conjugated acid of the azobenzene) upon increasing the temperature. We refer to this process involving a temperature-dependant ionisation/protonation equilibrium as “thermo-halochromism” in relation to previous reports on salt-concentration dependant thermochromism of betaine dyes54,55 and the IUPAC definition of halochromism.56 The main absorption band of p-MREAm is at 438 nm, with devolving shoulders at around 505 nm and 540 nm upon increasing temperatures. These shoulders correspond to the azonium cation. m-MREAm shows a similar behaviour compared to p-MREAm, with the main absorption band lying at a shorter wavelength of 417 nm. The attenuating shoulders are localised at 506 nm and 537 nm and the bands of the neutral azobenzene and the azonium are better separated for this isomer. In comparison to the para-isomer, the shoulders are overall lower in intensity with respect to the main absorption band. o-MREAm has a reversed ratio of the absorption of the protonated species and the neutral species compared to the other isomers. Here, the main absorption band is that of the azonium ion (522 nm) while the absorption band of the neutral species (∼425 nm) is about a third in intensity.
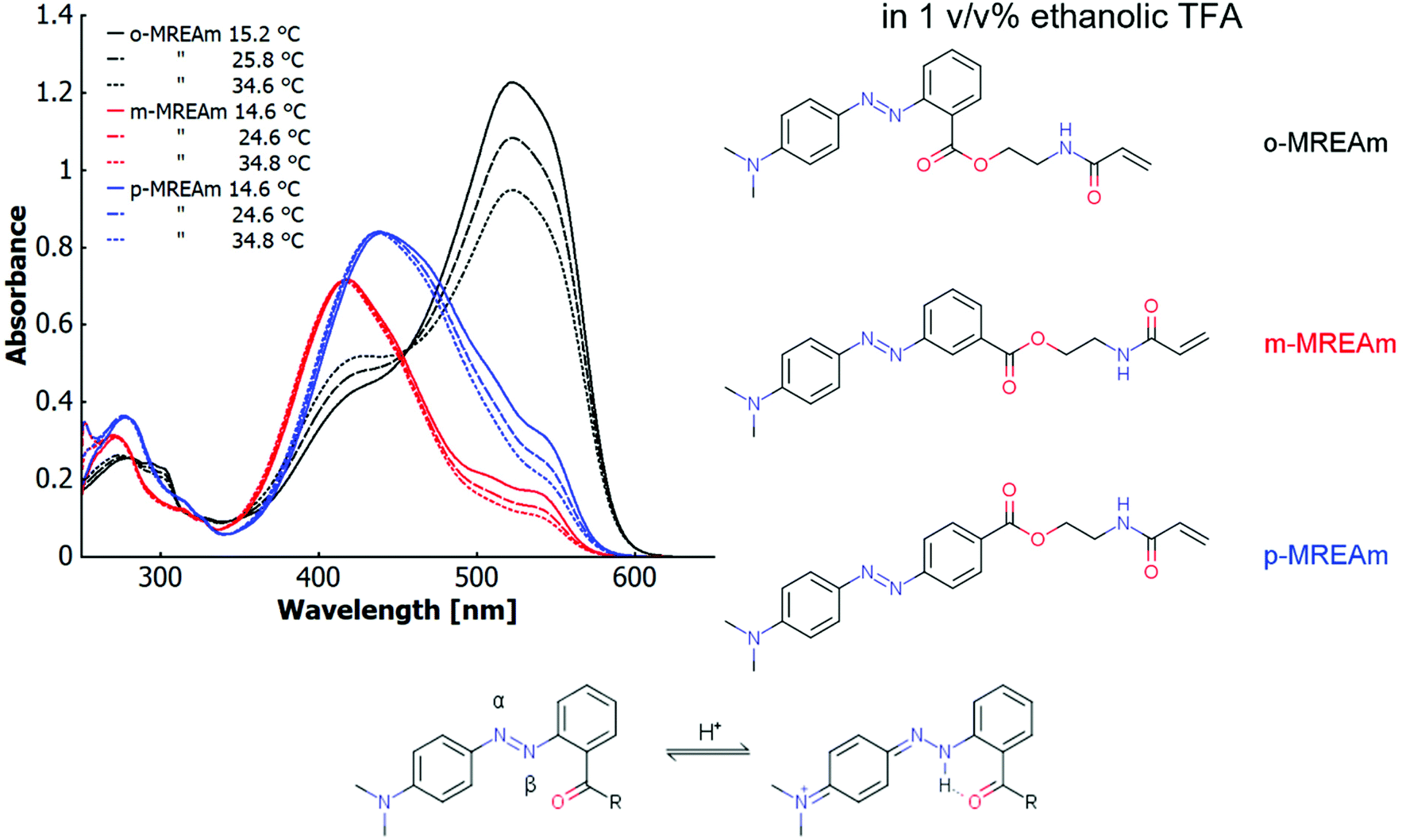 | ||
| Fig. 1 UV-vis spectra showing thermo-halochromism of ortho-, meta- and para-methyl red ester of N-(2-hydroxyethyl)acrylamide (2.7 × 10−5 mol L−1) at different temperatures in ethanolic trifluoroacetic acid (1 v/v%) with a depiction of the underlying azobenzenze–azonium ion equilibrium. | ||
Two analysis tools for UV-vis spectroscopy can be employed to elucidate the origin of the small shift of the maximum in neat ethanol and the spectral behaviour upon the changes of the equilibrium between the neutral and the protonated species with temperature variation. Derivative spectroscopy as the first method allows to identify sub-band structures convoluted under an absorption peak by the appearance of negative peaks in the second and fourth derivative at the position of the sub-band maxima.57 Difference spectroscopy represents the second tool. With this method, spectral differences occurring upon variation of a system parameter are determined by subtracting the UV-vis spectrum of a reference state from all following variants. This has been shown to be effective in determining the vibronic fine-structure in temperature-dependant UV-vis measurements of neat trans-azobenzene.58
In neat ethanol, a slight change of the absorption band asymmetry at 420 nm for ortho, at 419 nm for meta, or at 438 nm for para with temperature variation can be observed, suggesting an underlying equilibrium of transitions between energetically similar states (cf. ESI Fig. S17, S31 and S32†). These transitions are expressed by a sub-band structure, which are convoluted in the observed asymmetric absorption band. The concomitantly observed decrease in absorbance may be ascribed to the temperature-dependant change of the solvent density or in the variation of the population of states with pronounced differences in the extinction coefficients. To analyse this behaviour, the second spectral derivatives were generated for the main UV-vis bands at several temperatures. The wavelengths of the observable minima in these second derivatives are summarised in Table 2. They lie at slighty shorter and longer wavelengths than the convoluted maxima in the parent UV-vis spectra and change in intensity with temperature (cf. red arrows and colourful triangles in ESI Fig. S31 and S32†). The energy difference between these sub-bands is around 1590 cm−1 (ortho) to 1870 cm−1 (meta). A third sub-band may be concealed by overlap with the second sub-band, being visible only as a shoulder at 400 nm in the derivative spectra.
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 01) and wavelength shift (λ-shift) of the absorbance maximum with temperature increase from around 10 °C to around 50 °C of neutral o-, m- and p-MREAm in ethanol or binary water–ethanol mixtures (XEtOH = 0.31)
01) and wavelength shift (λ-shift) of the absorbance maximum with temperature increase from around 10 °C to around 50 °C of neutral o-, m- and p-MREAm in ethanol or binary water–ethanol mixtures (XEtOH = 0.31)
| Derivative | Solvent | λ max[nm] | 0–0max [nm] | 0–1max [nm] |
01 [cm−1] |
λ-shift [nm] |
|---|---|---|---|---|---|---|
| o-MREAm | EtOH | 420 | 450 | 420 | 1590 | 420 → 417 |
| m-MREAm | EtOH | 419 | 451 | 416 | 1870 | 418 → 415 |
| p-MREAm | EtOH | 437 | 470 | 434 | 1760 | 437 → 435 |
| o-MREAm | H2O:EtOH |
441 | 460 | 420 | 2070 | 441 → 430 |
| m-MREAm | H2O:EtOH |
449 | 462 | 417 | 2340 | 449 → 432 |
| p-MREAm | H2O:EtOH |
470 | 483 | 438 | 2130 | 470 → 461 |
For the explanation of the sub-band structure, we consider three possibilities:
First possibility: The observed spectroscopic features correspond to a vibronic fine structure. The vibronic fine structure of azobenzenes strongly depends on the substituents of the aromatic rings. It has been shown before that azobenzene and aminoazobenzene dyes have a more pronounced fine structure than pseudostilbenes (push–pull-type azobenzenes). This vibronic structure has also been demonstrated to be temperature-dependant.59–61 Recently, the temperature-dependence of vibronic transitions has been studied for neat trans-azobenzene.58 For o-MREAm in ethanol, the determined energy difference between the two sub-bands is in accordance with a vibronic origin of the spectral features. Specifically, some stretching modes of the aromatic rings as well as the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– stretching mode are found around that particular energy range (ca. 1400 cm−1) in the parent compound (MR).62,63 Even though the energy difference between the sub-bands is higher than the energy of the –NN– stretching mode, coupling of the involved vibrational modes is likely to influence the observed maximum. This is indicated by significant broadening of the sub-bands and their apparent overlap, which is apparent even in the derivative spectra.
N– stretching mode are found around that particular energy range (ca. 1400 cm−1) in the parent compound (MR).62,63 Even though the energy difference between the sub-bands is higher than the energy of the –NN– stretching mode, coupling of the involved vibrational modes is likely to influence the observed maximum. This is indicated by significant broadening of the sub-bands and their apparent overlap, which is apparent even in the derivative spectra.
Second possibility: A temperature-induced shift of the equilibrium between several stable rotamers causes the sub-band structure. This effect has been ascribed to the features in the fine structure of the optical absorption of isophthalaldehyde,64 but also to absorption65 or fluorescence characteristics of stilbenes66–68 and to azo compounds.69
Third possibility: The thermal peak shift may be associated with a temperature-induced change in the structure of the solvent shell, which has been reported for the related azo dye methyl orange.70
Considering previous studies58,59,61–63 about vibrational and vibronic behaviour of azobenzenes, the first possibility is the most probable explanation. Thus, we attribute these spectral features to vibronic sub-bands.
The spectral changes with temperature are best scrutinised by difference spectroscopy. For this purpose, the spectra at the respective lowest temperature were taken as reference and subtracted from each spectrum at higher temperatures (cf. ESI Fig. S32g–i†). The positive bands in the difference spectra result from an increase in absorbance at wavelengths well below the absorption maximum in the parent UV-vis spectra (∼400 nm) at higher temperatures, which coincides with the shoulder visible in the derivative spectra (cf. ESI Fig. S32d–f†). The negative bands correspond to a decrease in absorbance and vary between the positional isomers. For the ortho- and meta-isomers, the decrease occurs at wavelengths related to both the 0–0 (vibrational ground state of the electronic ground state S0 to vibrational ground state of the first excited electronic state S1) transition as well as the 0–1 transition (vibrational ground state in S0 to first excited vibrational state in S1). Owing to the involvement of at least two vibronic sub-bands, this leads to a considerable asymmetric band structure. For the para-isomer, on the other hand, the negative band is highly symmetric with its minimum at the wavelength of the 0–0 transition, indicating an absorbance change related to a single vibronic sub-band. This suggests a correlation between the symmetry of the molecule and the vibronic changes with temperature.
Summarising these results, we conclude that the observed thermochromism is of vibronic origin, where vibronic sub-bands of lower energy decrease and those of higher energy increase with higher temperature. We call this phenomenon “vibronic thermochromism”.
Besides the vibronic thermochromism of the neutral dyes, as discussed above, thermochromism of the azonium ion must be understood as well to fully embrace the underlying mechanism of the thermo-halochromic behaviour in a partially protonated system. Compared to the neutral isomers of the MREAm dye, the corresponding azonium ions show a red-shifted absorption band at 510–520 nm (cf. ESI Fig. S33a–c†). In the case of 4-aminoazobenzene-derived dyes, protonation primarily occurs at the β-nitrogen of the azo-bridge (cf.Fig. 1 and ESI Scheme S1†). The resulting azonium ion has a partial quinoid structure, lowering the energy of the electronic transition. However, alternatively protonation may occur at the amino substituent, forming an ammonium ion, which absorbs at ∼320 nm. For the parent compound ortho-methyl red, the tautomeric equilibrium between the ammonium and the azonium form lies on the side of the azonium ion owing to intramolecular hydrogen bonding to the carbonyl in ortho-position.71–75 The same can be observed for the MR-monomers discussed here, where the effect is particularly pronounced for the ortho-isomer (cf. ESI Fig. S33a–c†). The maxima of the main absorption band of the azonium ions of all positional isomers of MREAm at 510–520 nm do not shift with temperature. However, the fine structure of these bands, which is more apparent than for the neutral dyes, changes characteristically. Especially in the meta- and the para-isomers, sub-bands are clearly visible in the unprocessed absorption spectra (cf. ESI Fig. S33a–c†).
The derivative and difference spectra of the azonium ions in ethanolic solution can be analysed in the same way as the neutral dye above (Table 3). This detailed analysis can be found in the ESI (ESI below Fig. S33†).
01, 12) of the protonated o-, m- and p-MREAm in ethanolic sulfuric acid (1 v/v%) or binary water–ethanol mixtures (XEtOH = 0.31) with hydrochloric acid (5 M)
| Derivative | Solvent | λ max [nm] | 0–0max [nm] | 0–1max [nm] | 0–2max [nm] |
01 [cm−1] |
12 [cm−1] |
|---|---|---|---|---|---|---|---|
| o-MREAm | EtOH | 519 | 549 | 513 | 482 | 1280 | 1250 |
| m-MREAm | EtOH | 511 | 544 | 508 | 477 | 1300 | 1280 |
| p-MREAm | EtOH | 516 | 548 | 511 | 480 | 1320 | 1260 |
| o-MREAm | H2O:EtOH |
522 | 553 | 516 | 485 | 1300 | 1240 |
| m-MREAm | H2O:EtOH |
511 | 543 | 508 | 477 | 1270 | 1280 |
| p-MREAm | H2O:EtOH |
514 | 546 | 510 | 479 | 1290 | 1270 |
The temperature-dependant protonation equilibrium between the azonium ion and the neutral dye were also analysed with derivative and difference spectroscopy. As stated above, the fundamental process is a shift of the equilibrium towards the neutral dye with increasing temperature (cf. ESI Fig. S34a–c†). This trend is visible both in the derivative spectra (cf. ESI Fig. S34d–f†) and difference spectra (cf. ESI Fig. S34g–i†) as follows. In dependence of their initial degree of protonation, the shape of the positive and negative peaks in the difference spectra vary between the positional isomers. For the ortho-isomer, the negative bands in the difference spectra resemble the shape of the absorption band of the azonium ion, just with the opposite sign (cf. ESI Fig. S33a and S34g†). The position of the positive band coincides with the one of the main absorption band of the neutral species (cf. ESI Fig. S32a and S34g†). On the other hand, the samples of the initially less protonated meta- and para-isomers exhibit spectral features of thermo-halochromism and additionally of vibronic thermochromism from the neutral population. The negative band in the difference spectra appears like a combination of the negative bands in vibronic thermochromism (cf. ESI Fig. S32h and i†) with the absorption band of the corresponding azonium ions of opposite sign (cf. ESI Fig. S33b and c and S34h and i†). We assume that the positive bands are related to the vibronic thermochromism found for the neutral species (cf. ESI Fig. S32h and i†). The appearance of overlapping features is mainly a consequence of vibronic thermochromism acting simultaneously to thermo-halochromism resulting from the shift of the protonation equilibrium, which affect the absorption spectra with similar magnitudes under the conditions of weak protonation. In the case of the ortho-isomer, thermo-halochromism is prevalent, overshadowing vibronic effects.
It is worthwhile to mention that the difference spectra of the partially protonated meta- and para-isomers in ethanolic TFA show more pronounced vibronic features in the negative peaks than would be expected from a simple subtraction of the azonium ion spectra (cf. ESI Fig. S34h and i†). This suggests a vibronic contribution to thermo-halochromism.
3.3. Relationship of pKa and thermochromicity in MR monomers
To determine a possible correlation between pKa and thermochromicity (meaning the quantification of the extent of thermochromism76), the different constitutional isomers of the MR derivative were titrated in H2O:EtOH (XEtOH = 0.31) with 5 M HCl in H2O:EtOH (XEtOH = 0.31) (cf.Fig. 2). Water/ethanol mixtures were chosen as solvent because the solubility in pure water was too low for UV-vis measurements, and according to literature, the pKa values of azo dyes do not change severely with different percentages of ethanol.77
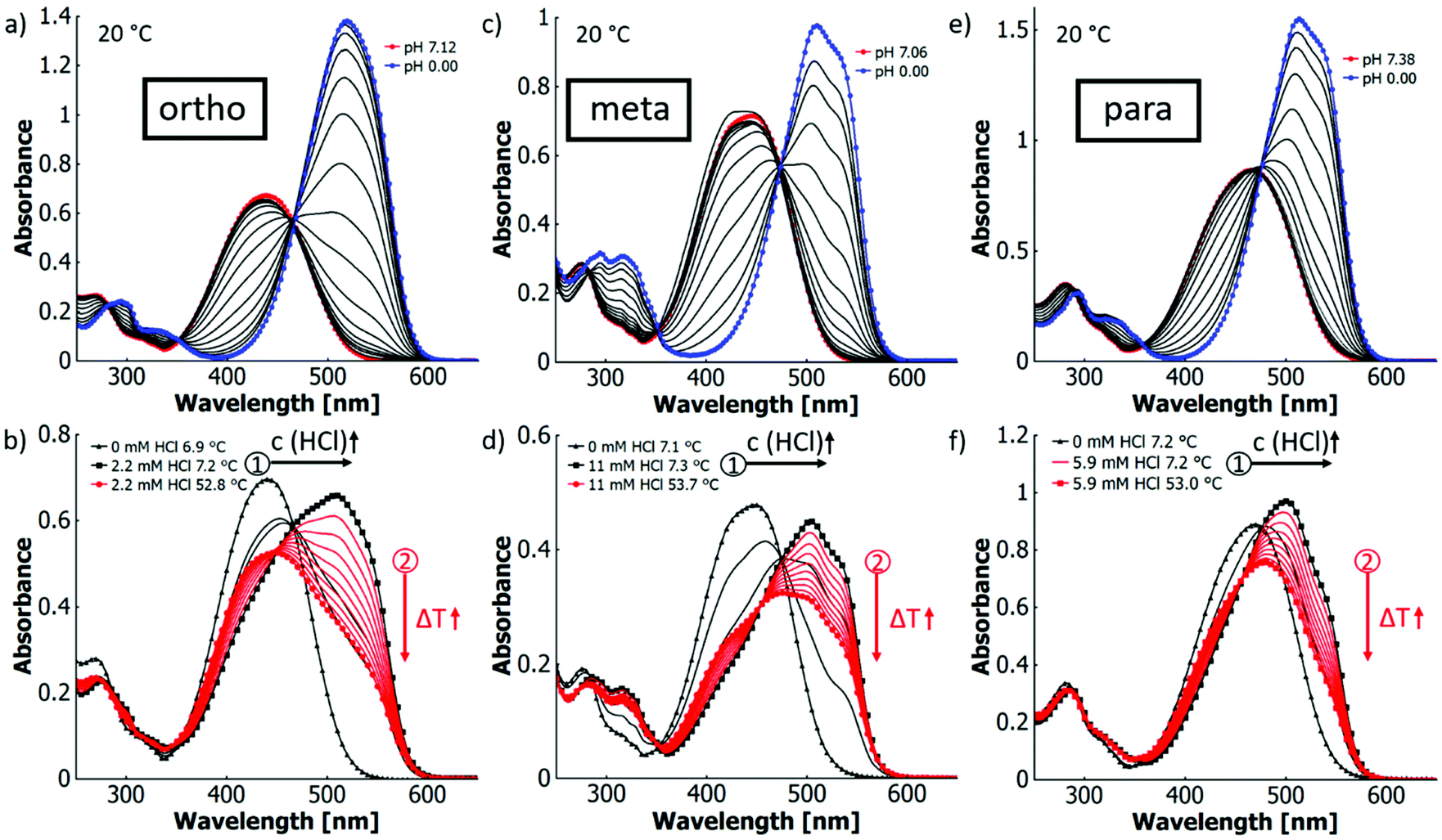 | ||
| Fig. 2 UV-vis spectra in H2O:EtOH (XEtOH = 0.31) at different pH values at 20 °C for (a) o-MREAm, (c) m-MREAm and (e) p-MREAm (all 2.7 × 10−5 mol L−1); and the spectra after titration (black arrow 1) with hydrochloric acid at ∼6–7 °C to ∼50% protonation at different temperatures (red arrow 2) for (b) o-MREAm, (d) m-MREAm and (f) p-MREAm (all 2.7 × 10−5 mol L−1). The black curves in (b), (d) and (e) are spectra of intermediate titration steps to emphasise the isosbestic points. | ||
All three isomers show similar spectral changes upon titration. In the neutral state, they have an absorption maximum at around 450 nm and with lower pH values, shoulders evolve around 515 nm, which eventually result in distinct absorption bands. In all three cases, the extinction coefficients are higher for the azonium ion than for the neutral forms, following the order meta < para < ortho. In contrast, the red shifts of the absorption maxima are in the order para < meta < ortho. Therefore, at similar degrees of protonation the visible change in colour is strongest for the ortho-isomer (cf.Fig. 2a, c and e, Table 4). The pKa values of the configurational isomers of MREAm in H2O:EtOH (XEtOH = 0.31) can be determined from the spectral changes upon titration (ortho-isomer: pKa 2.24, para-isomer: 1.83, meta-isomer: 1.54; cf. ESI Fig. S18† and Table 4). This trend can be explained by two factors: firstly, the mesomeric stabilisation of the azonium cation is higher for the para- and the ortho-than for the meta-isomer. Secondly, the ortho-isomer has the additional option for intramolecular hydrogen bonding of the β-protonated azo bridge with the carbonyl oxygen, further stabilising the cation as observed for the parent MR.73 The same tendency can be found for the pKa values previously reported in the parent isomers in water (cf. ESI Table S2†).23,24,78,79 In the ortho-isomer (ΔpKa 0.14), the difference between the parent compound and the derivative is small and may be explained with the change in solvent. This difference, however, increases with the acidity of the isomer (para: ΔpKa 0.25; meta: ΔpKa 0.45). This suggests that intramolecular hydrogen bonding is the dominating influence that stabilises the ortho-azonium ion, while mesomeric and inductive effects lower the pKa changing from the free acid (MR) to an ester (MREAm).
:EtOH (XEtOH = 0.31) acified with HCl
| Isomer | pKa | ε(dye) [L mol−1 cm−1] | ε(H+dye) [L mol−1 cm−1] | ε(H+dye)/ε(dye) | λ max(dye) [nm] | λ max(H+dye) [nm] | Δλmax [nm] |
|---|---|---|---|---|---|---|---|
| ortho | 2.2 | 24600 |
50600 |
2.06 | 438 | 519 | 81 |
| meta | 1.6 | 26300 |
35900 |
1.37 | 445 | 510 | 65 |
| para | 1.8 | 31800 |
56800 |
1.79 | 467 | 513 | 46 |
In Fig. 2(b), (d) and (f), the dyes were first titrated (black arrow 1) at low temperatures (6–7 °C) to a degree of 50% protonation followed by a successive temperature increase (red arrow 2) in order to observe maximal thermochromic variations. We refer to this process as “thermotitration”. Under these conditions, all isomers of MREAm show similar thermochromic behaviour. The decrease in absorbance of the protonated species upon heating is in the same order of magnitude. With increasing temperatures, the absorption bands around 515 nm decrease in intensity while those around 450 nm increase, leading to an overall blueshift (cf.Fig. 2b, d and f).
To determine the correlation between thermochromicity and pKa, the thermochromicity must be quantified. For this purpose, van't Hoff analysis was chosen. Fig. 4(d) shows for the ortho-isomer the van't Hoff plot of the natural logarithm of the absorbances ratio of the azonium ion and the neutral azobenzene R versus the inverse of the absolute temperature. The specifically selected absorbance wavelengths were chosen to avoid a large spectral overlap.
While van't Hoff plots of lnK vs. T−1 usually provide a linear relationship, the plots in the present examples are non-linear, suggesting a significant temperature dependence of the reaction enthalpy. In order to account for this non-linearity, the plots were fitted by eqn (1) according to previously reported procedures for protein titrations and complexation studies.80,81
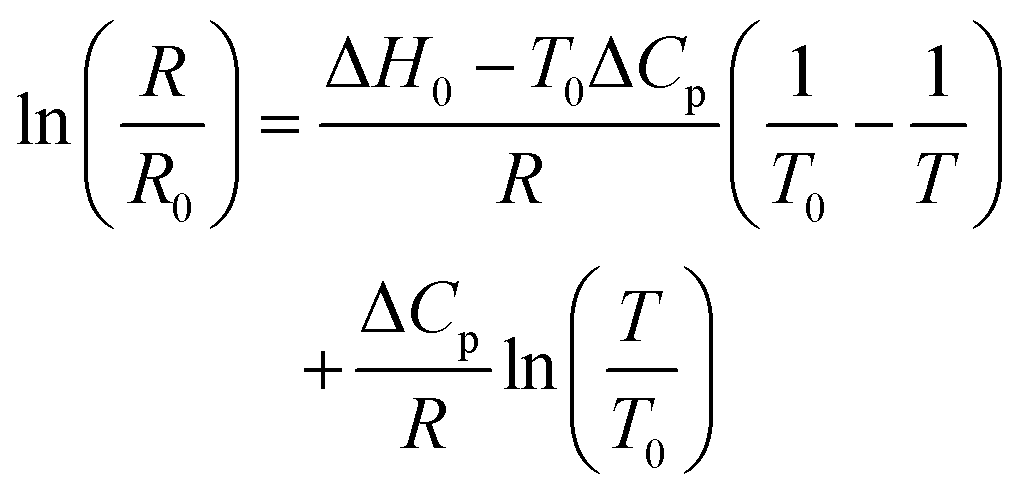 | (1) |
A simplified representation of this equation introduces the parameters “a”, “b” and “c” as follows: ln(R) = a − b × 1/T + c × ln(T). These fit parameters together with the extracted values for ΔH0 and ΔCp are summarised in Table 7 and will be discussed in further detail after introducing all systems. The enthalpy ΔH0 may be used as a measure of the extent of protonation (and in turn deprotonation) at 25 °C. It also tells whether the process is endothermic or exothermic. The heat capacity of ionisation ΔCp quantifies how temperature-dependant the enthalpy is. This provides information about whether deprotonation becomes more favourable or disfavourable with increasing temperatures. Both values may be considered as thermochromicity parameters.
Plotting ΔH0 and ΔCp for the different consitutional isomers vs. their pKa shows an almost linear correlation with both thermochromicity parameters (cf. ESI Fig. S19†). As direct consequences, both the magnitude of the thermochromic effects as well as the pH, at which they can be observed, depend on the pKa.
3.4. Variants of thermochromism for the MREAm monomers in binary solvent mixtures
In order to analyse the individual thermochromic mechanisms that contribute to the overall optical behaviour, we investigate the effect of the different permutations for the combination of ethanol, water, and acid as constituents for the liquid medium. Isosbestic points can be observed in UV-vis absorption spectra when titrating the MREAm monomers in water–ethanol mixtures with hydrochloric acid, which shifts the azobenzene protonation equilibrium between the neutral dye (around 455 nm) and the azonium cation (around 515 nm). At ∼20 °C these isosbestic points (cf.Fig. 2a, c and e) are at the same wavelength as those found for thermotitration (following the black arrow 1 in Fig. 2b, d and f) at low temperature (6–7 °C) when partially titrating to 50% protonation. Surprisingly, when raising the temperature from 6–7 °C to above 50 °C for the partially protonated sample in the binary solvent mixture, no isosbestic point is visible. Instead, the intersections of the corresponding absorption spectra continuously blueshift, which is particularly pronounced at lower temperatures (cf.Table 5).:EtOH (XEtOH = 0.31) acidified with HCl, and for o-MREAm in ethanol acidified with TFA
| Isomer | Isosbestic point ΔpH 19.8 °C [nm] | Isosbestic point ΔpH 6–7 °C [nm] | Regions of intersections [nm] |
|---|---|---|---|
| ortho | 465 | 465 | 455–434 |
| meta | 474 | 475 | 457–440 |
| para | 474 | 476 | 463–448 |
Since chemical equilibria are established between the free proton, the azobenzene and the azonium ion upon titration as well as during temperature variation, isosbestic points would be expected for both cases. Apparently, a fundamental difference must exist between the proton concentration-influenced and temperature-controlled equilibria to cause the blueshift. To elucidate this phenomenon, we separately expatiate on the individual mechanisms of thermochromism that coexist for the dye in acidified water–ethanol-mixtures. For this purpose, the following three scenarios are contrasted with each other: (1) vibronic thermochromism (neat ethanol), (2) thermo-halochromism (one solvent with added acid) and (3) thermo-solvatochromism (binary solvent mixture without acid). The additional fourth scenario (4) combines the case of a binary solvent mixture with that of added acid. It will be discussed with respect to the findings of the first three scenarios.
In the first scenario (1) of neat ethanolic solution, the azobenzene monomers show only minimal thermochromism (cf.Fig. 3a). This phenomenon has been discussed above and involves changes in the sub-band structure that is likely of vibronic origin. Thus, we call it vibronic thermochromism.
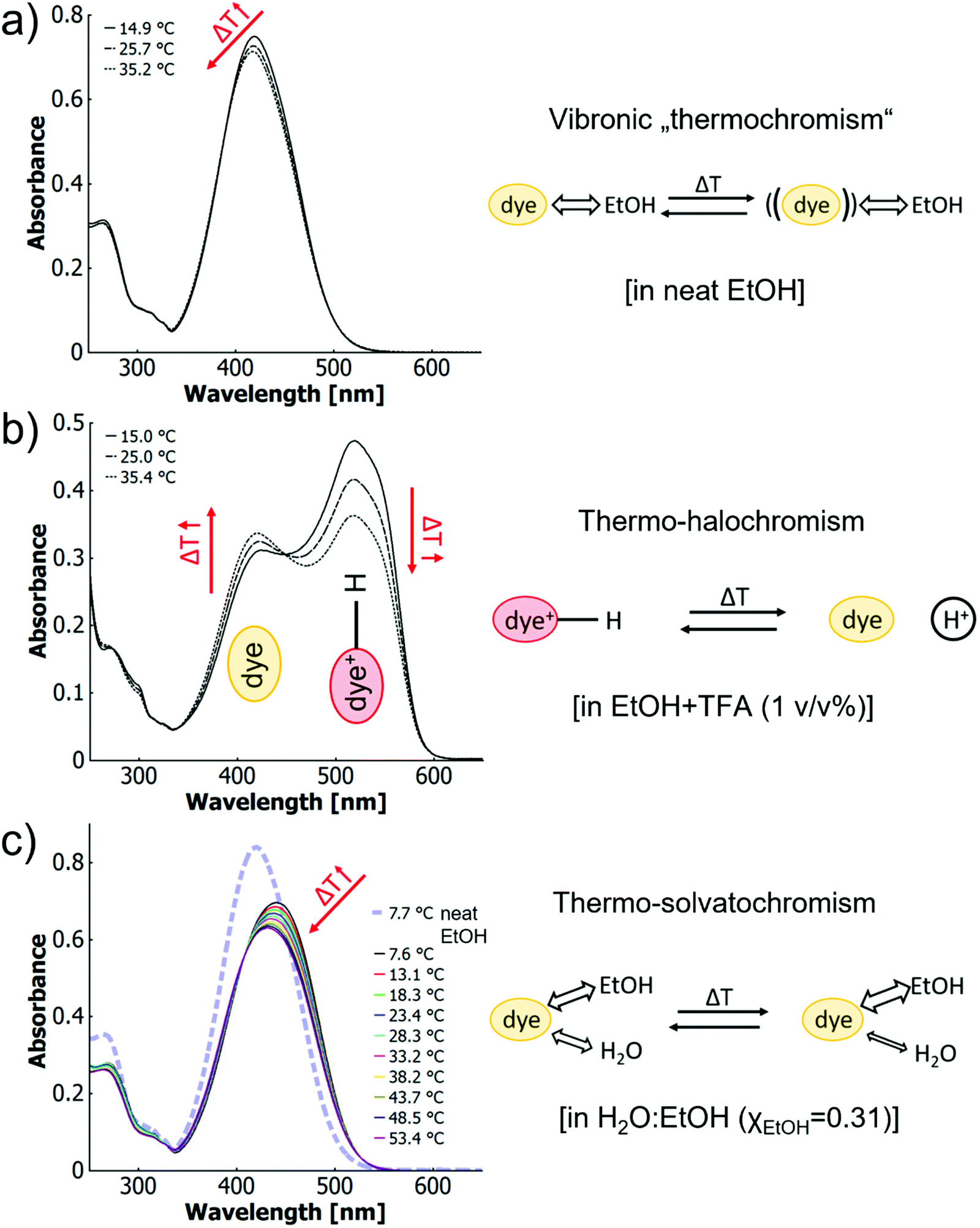 | ||
| Fig. 3 UV-vis spectra of o-MREAm (2.7 × 10−5 mol L−1) at different temperatures with cartoons showing the corresponding thermochromic processes involving (a) vibronic thermochromism in neat ethanol; (b) thermo-halochromism in ethanolic trifluoroacetic acid (1 v/v%); and (c) thermo-solvatochromism in H2O:EtOH (XEtOH = 0.31). | ||
The second scenario (2) of the monomer in a single solvent with added acid (here ethanol with trifluoroacetic acid, cf. Fig. 3b) has already been discussed above. As no appreciable wavelength shift of the maxima with temperature is observed for neither the protonated species (522 nm) nor the neutral species (419 nm), a pure protonation equilibrium is more likely than a change in the quality of solvation.
The third scenario (3) is the monomer in the binary solvent system (H2O:EtOH, XEtOH = 0.31) without addition of acid (cf.Fig. 3c). In this case, a blueshift from 441 nm to 431 nm can be observed over a temperature range of 46 K (7.5–53.5 °C). With higher temperatures, the spectra shift towards that of the dye in neat ethanol (cf.Fig. 3a and c). This effect may be attributed to a temperature-dependant change of the solvent–dye interactions at different rates for the two solvents. Such behaviour was first described for betaine– and other zwitterionic dyes in binary water–organic solvent mixtures and has been termed “thermo-solvatochromism”.82–86 In contrast to the present MR systems, the literature suggests for the zwitterionic structures with large dipole moments a quicker change of the balance between hydrogen bonding and hydrophobic interactions for various alcohols than for water, leading to a depletion of the alcohol in the solvation complex.84 The inverse behaviour in our case apparently results from the less polar MR dye being more soluble in ethanol, while being practically insoluble in water. Thus, as the interactions of both solvents change with temperature and water being a non-solvent, the net result will be dominated by the good solvent ethanol. The temperature-induced change in solvent composition in the solvation shell also affects the vibronic characteristics of the dye and as such thermo-solvatochromism resembles features of vibronic thermochromism (1) for the neutral MR dye (cf. ESI Fig. S32 and S35†).
Detailed analysis was performed with derivative and difference spectroscopy to show the exact differences between vibronic thermochromism and thermo-solvatochromism. Interestingly, the thermochromic properties of the azonium ion are not affected by the particular solvent systems in contrast to the neutral azo dye. The detailed analysis can be found in the ESI (below Fig. S35†).
In the fourth scenario (4) a more complex behaviour occurs for the binary solvent mixture water–ethanol (XEtOH = 0.31) when adding free acid, for which the results are shown in Fig. 4(a). Upon heating, the maximum of the azonium ion at around 510 nm gradually decreases, while the absorption band corresponding to the neutral species around 450 nm increases in intensity. However, no isosbestic point is observed, but rather a region of intersecting spectra, which shift to shorter wavelengths. In the ratiometric approach shown in Fig. 4(c), the ratio of the absorbance of the azonium ion and the neutral azobenzene R versus temperature is characterised by a non-linear (exponential) decrease. This characteristic signifies a temperature dependence of the protonation equilibrium between the azonium form and the neutral form. The equilibrium shifts at different rates for different temperatures. In essence, more of the azonium ion is deprotonated per temperature increment at lower temperatures, as indicated by the red slope triangles.
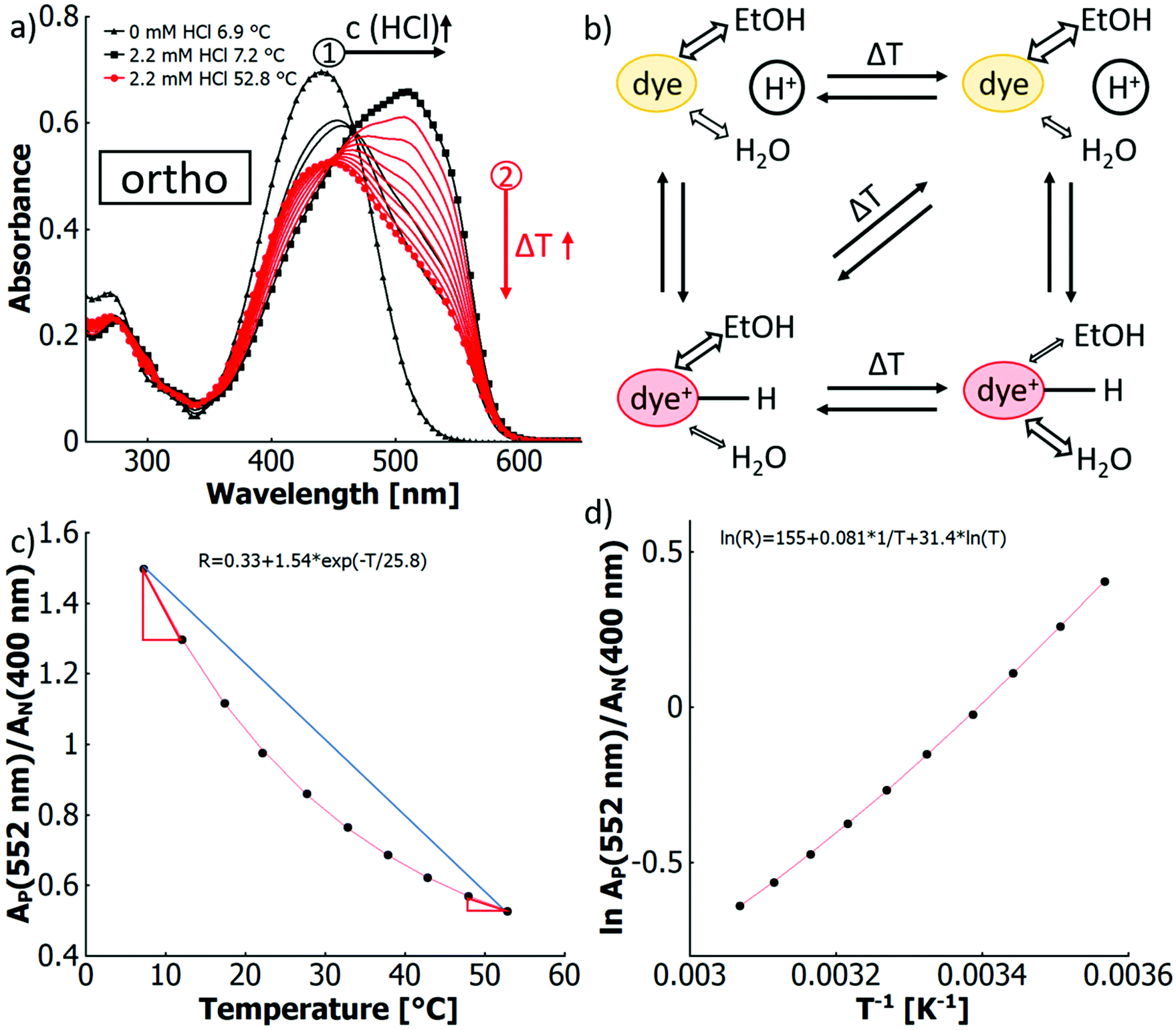 | ||
| Fig. 4 (a) UV-vis spectra demonstrating thermo-solvato-halochromism of o-MREAm (2.7 × 10−5 mol L−1) in H2O:EtOH (XEtOH = 0.31) after titration (black arrow 1) with hydrochloric acid at ∼6–7 °C to ∼50% protonation at different temperatures (red arrow 2) with; (b) cartoon of competing processes; (c) ratio of the absorption of the protonated species (552 nm) and the neutral species (400 nm) (“R” in the equation) vs. temperature, fitted with a first order exponential decay function. The blue line along with the red slope triangles serve as a reference to emphasise the non-linearity of the graphs; (d) natural logarithm of the ratio of the absorption of the protonated species (552 nm) and the neutral species (400 nm) vs. the inverse of the absolute temperature, fitted with a function ln(R) = a − b × 1/T + c × ln(T). | ||
To explain this behaviour mechanistically, the coexisting species and their equilibria in the system must be analysed separately and then harmonised. Owing to the presence of free acid, two species exist: the neutral azobenzene and the positively charged azonium ion. Each species has its distinct interaction with the binary solvent mixture. In the binary solvent mixture, the protonation equilibrium follows the same trend as in a single solvent with added acid (cf.Fig. 3b). With rising temperature, the protonation equilibrium shifts towards the neutral azobenzene.
However, the thermally dependant equilibrium of the water–ethanol composition in the solvent shell of each dye species gains a new aspect. The blueshift indicative for the negative thermo-solvatochromism of the neutral species has been described previously in the third scenario (cf.Fig. 3c), with the effective influence of the solvent shell resembling that of neat ethanol at higher temperatures. In contrast, the polar azonium ion does not show any significant thermochromism neither in ethanol with sulfuric acid nor water with HCl nor in the binary mixture of water and ethanol with HCl (cf. ESI Fig. S20†). The effective influence of the solvation shell and its stabilisation of the ground and excited states does not change. On the other hand, the ethanol–water ratio in the solvation shell does most likely change, as has been described in the literature for zwitterionic dyes.84 This effect may be explained by the fact that both water and ethanol are good solvents for the azonium ion.
The thermal equilibrium between all solvated species is reflected in the combined emergence of thermo-solvatochromism and thermo-halochromism. This equilibrium and the competing processes guiding their interconversion are schematically depicted in Fig. 4(b). The temperature effects on both the protonation equilibrium as well as on the composition of the solvation shell of the neutral azobenzene can be followed by changes of spectral features. However, for the azonium ion no spectroscopic signature is observed for a conceivable temperature effect on the solvation shell, since in this case water and ethanol are both good solvents. Yet, as with increasing temperature water dominates the interactions in the solvation shell for the binary solvent mixture,84 the solvated and less polar azobenzene is destabilised in relation to the more polar azonium ion. Consequently, the two species have a different proton affinity in dependence of their solvation shell composition, which in turn is differently affected by temperature. This means that also the shift of the protonation equilibrium within a given temperature interval is differently affected at low compared to high temperatures. Thus, at higher temperatures, the protonated species (azonium ion) shows a lower incremental concentration decrease by the halochromic pathway, which is characterised by the exponential dependence in the non-linear reduction of its absorbance in Fig. 4(c).
Additionally, the difference in the extent of thermo-solvatochromism between the neutral and the protonated species results in the region of intersections of absorption curves for different temperatures instead of an expected isosbestic point, as visible in Fig. 4(a). An isosbestic point could be observed if at least two absorbing species have an equal absorbance upon variation of a system parameter like temperature.87–90 Yet, in our present system, the absorbing species is not simply represented by only an isolated molecular framework of the dye, but the chromophore being strongly influenced in its conformation and electronic structure by its solvation shell in dependence of the temperature. This fact is expressed by the observed thermo-solvatochromism. Consequently, the protonation equilibrium does not simply comprise two static absorbing species, which would result in only thermo-halochromism, but the equilibrium exists between the azobenzene with a temperature-variable solvation shell and the azonium ion, whose absorption is temperature invariant. We define the here witnessed, combined effects of thermo-solvatochromism and thermo-halochromism as thermo-solvato-halochromism, which leads to the above-mentioned continuous shift of the curve intersections with the absence of an isosbestic point.
As a further consequence, the graphs of A(protonated)/A(neutral) vs. temperature in Fig. 4c and ESI Fig. S42† are non-linear insofar that the shift of the protonation equilibrium becomes smaller with increasing temperatures. Consequently, the magnitude of thermochromism is reduced and we refer to this process as diminishing thermo-solvato-halochromism.
The difference spectra from the thermotitrations in ESI Fig. S37g–i† indicate distinct reaction pathways that establish the protonation/deprotonation equilibrium in dependence of either the proton concentration or temperature. Such spectra exhibit a significantly different vibronic structure for several proton concentrations compared to those resulting from temperature variation. This suggests that a change in the molecular gestalt, defined here as the conformation of the dye and the structure of its solvent shell, is differently influenced by titration or temperature variation.
In essence, the results above corroborate the existence of three different mechanisms of thermochromism of methyl red (MR)-based azobenzene monomers in solution. The first and simplest thermochromism on the basis of vibronic transitions was identified via derivative and difference spectroscopy (cf.Fig. 3a and ESI Fig. S31†). The second mechanism is thermo-halochromism. Solutions of partially protonated MR-monomers (e.g., in ethanolic trifluoroacetic acid) show a temperature-dependant equilibrium between the neutral azobenzene and the ionic azonium species. The equilibrium shifts towards the neutral species at elevated temperatures (cf.Fig. 3b). The third mechanism is thermo-solvatochromism in binary solvent mixtures like H2O:EtOH (XEtOH = 0.31). Here, the spectra shift towards that in pure ethanol at higher temperatures (cf.Fig. 3c).
The obtained data suggest that these mechanisms do coexist and influence each other in binary solvent mixtures containing free acid. Under these conditions, thermo-solvatochromism affects substantially the extent of thermo-halochromism. In other words, the extent of deprotonation upon heating is different at low and at high temperatures (cf.Fig. 4). As such and stated above, this process may be best described by the term thermo-solvato-halochromism.
3.5. Thermochromism of methyl red-containing copolymers in solution
When transitioning from the individual dye monomer to a macromolecular architecture, the polymer chain provides a further contribution to the microenvironment of the polymer-bound dye in addition to the solvent, which is reflected in the concept of thermo-perichromism. The particular effects resulting from this transition will be elaborated for different polymer systems in the following. Various copolymers were prepared from the MR-based monomers by free radical polymerisation. By proper choice of the comonomers a thermoresponsive behaviour can be imparted and with this an associated polarity change upon temperature variation. Furthermore, copolymerisation of carboxylic acid-containing comonomers provide the possibility of an intrinsic proton source.As a reference, the first system (P1) comprises a non-thermoresponsive, acid-free copolymer made from the azo-chromophore o-MREAm, the hydrophilic main monomer HEAm, and the photocrosslinker benzophenone acrylamide (BPAAm). HEAm imparts a polar character to the copolymer, and thus the polymer facilitates dissolution of the attached hydrophobic dye in water. BPAAm is a versatile photocrosslinker that forms covalent bonds between the polymer chains via C,H-insertion upon UV irradiation.46,91 In the second copolymer system (P2), NiPAAm was copolymerised together with HEAm, o-MREAm and BPAAm to introduce thermoresponsive solution behaviour. In the third polymer (P3), carboxylic acid functions were introduced into the HEAm-based P1 system by copolymerisation with methacrylic acid (MAA). For the fourth system (P4), o-MREAm, NiPAAm, BPAAm, and MAA were copolymerised, yielding an intrinsically acidic, thermoresponsive polymer.
The thermochromism of all copolymers P1–P4 was investigated in aqueous and in ethanolic solution. Furthermore, all four copolymers were also studied after transformation into hydrogels by photocrosslinking and swelling with water. In the following, the four different copolymers are discussed in order of increasing complexity: (P1) non-thermoresponsive without intrinsic acid moieties; (P2) thermoresponsive without intrinsic acid moieties; (P3) non-thermoresponsive with intrinsic acid moieties and (P4) thermoresponsive with intrinsic acid moieties.
The description will be provided in two parts. First, the thermochromic behaviour of the polymers will be described phenomenologically for each polymer before the systems are quantified via van't Hoff analysis and compared to each other.
The P1 solutions both in ethanol and in water show a thermochromic effect visible as a blueshift in Fig. 5 with temperature increase. In ethanol, the wavelength of the maximum shifts from 423 nm at 7.5 °C to 420 nm at 48.5 °C (cf.Fig. 5a). The behaviour of the copolymer and the free dye are essentially identical under these conditions. In aqueous solution, the maximum blue-shifts from 454 nm at 7.4 °C to 448 nm at 48.7 °C (cf.Fig. 5b). This observation is reminiscent of thermo-solvatochromism in the binary solvent mixture. However, the more general term thermo-perichromism should be used here, as perichromism describes a colour shift related to a change in the local molecular environment (here, the macromolecular structure behaves analogously to a solvent molecule interacting with the dye).92
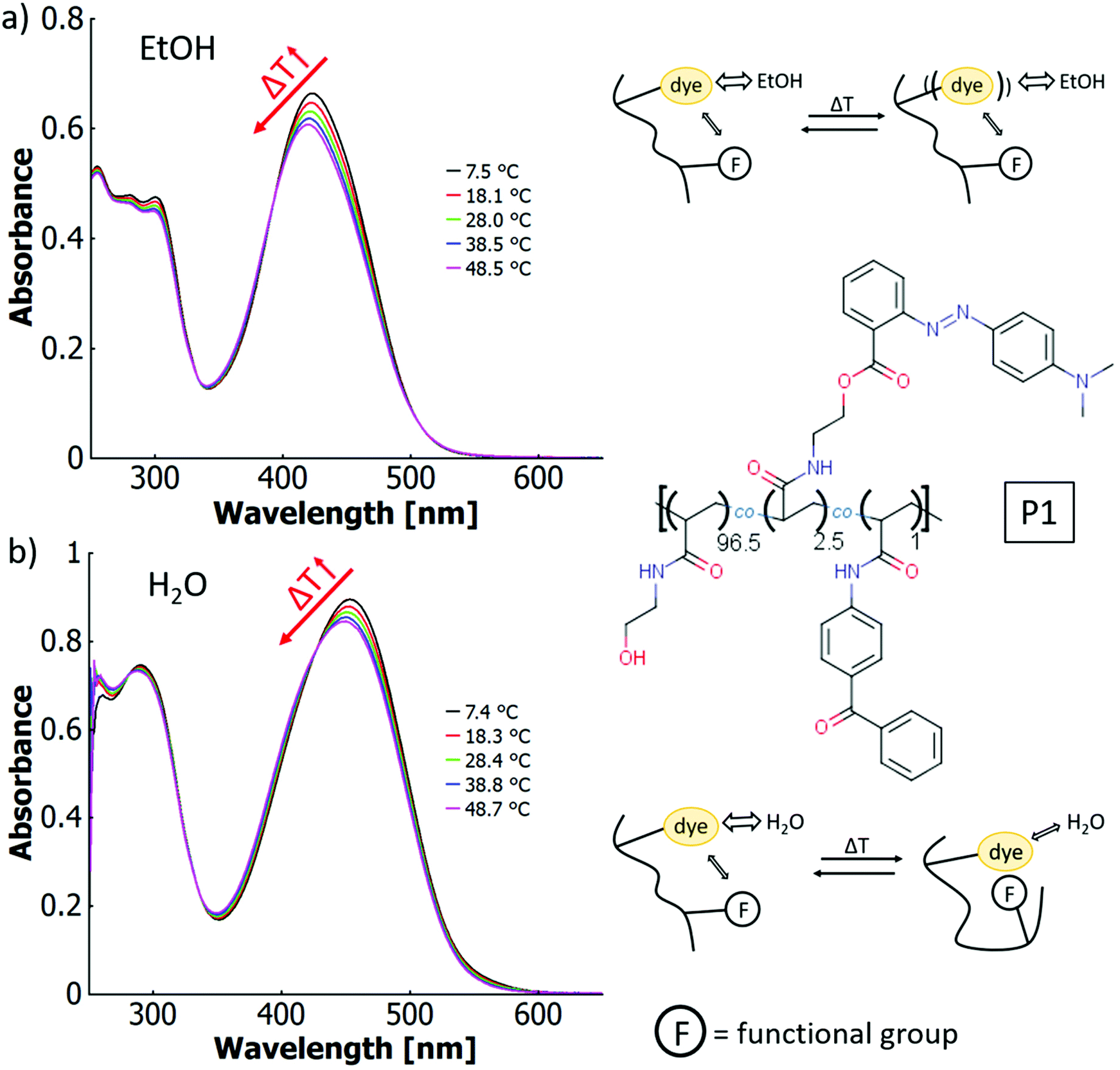 | ||
| Fig. 5 UV-vis spectra of a poly(HEAm-co-o-MREAm-co-BPAAm) copolymer P1 at different temperatures (a) in neat ethanol (0.02 w% polymer) and (b) in pure water (0.03 w% polymer), with the chemical structure of the copolymer and thermo-perichromic mechanism sketched on the right side. | ||
By addition of TFA as acid to solutions of P1, which does not contain any intrinsic acid-moieties in the polymer backbone, thermo-halochromism can be induced. This was demonstrated by partial protonation leading to the coexistence of neutral dye and azonium ion (cf.Fig. 6). The general behaviour is similar to the isolated dye monomer discussed above (cf.Fig. 3 and 4). The absorbance related to the azonium ion decreases and the one of the neutral azobenzene correspondingly increases with higher temperatures. While an analogous trend is observed for ethanol and for water, the details of thermochromism vary considerably between the two solvents.
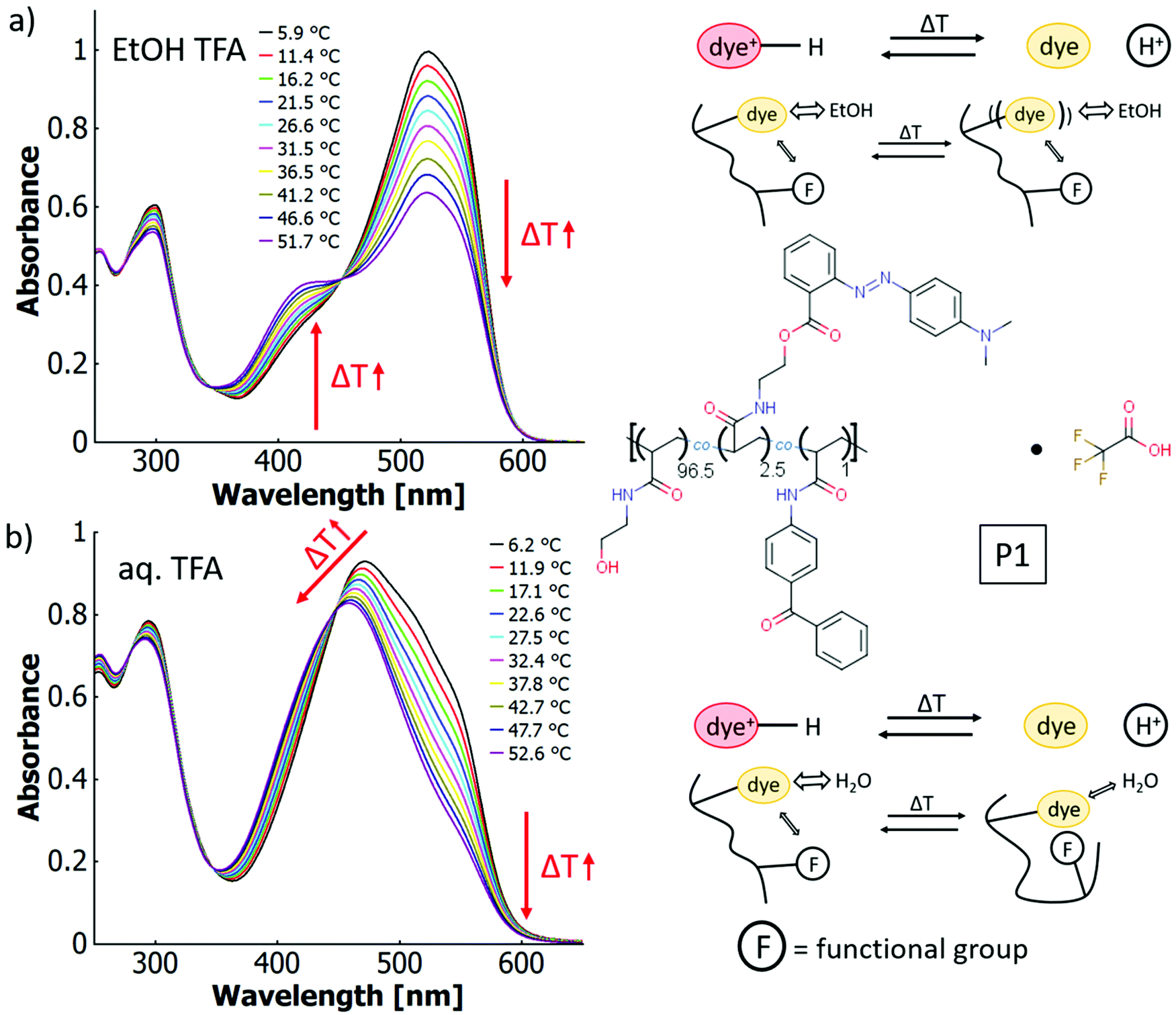 | ||
| Fig. 6 UV-vis spectra of a poly(HEAm-co-o-MREAm-co-BPAAm) copolymer P1 at different temperatures (a) 0.02 w% in ethanolic trifluoroacetic acid (1 v/v%) and (b) 0.03 w% in aqueous trifluoroacetic acid (0.001 v/v%), with the chemical structure of the copolymer and thermo-peri-halochromic mechanism sketched on the right side. | ||
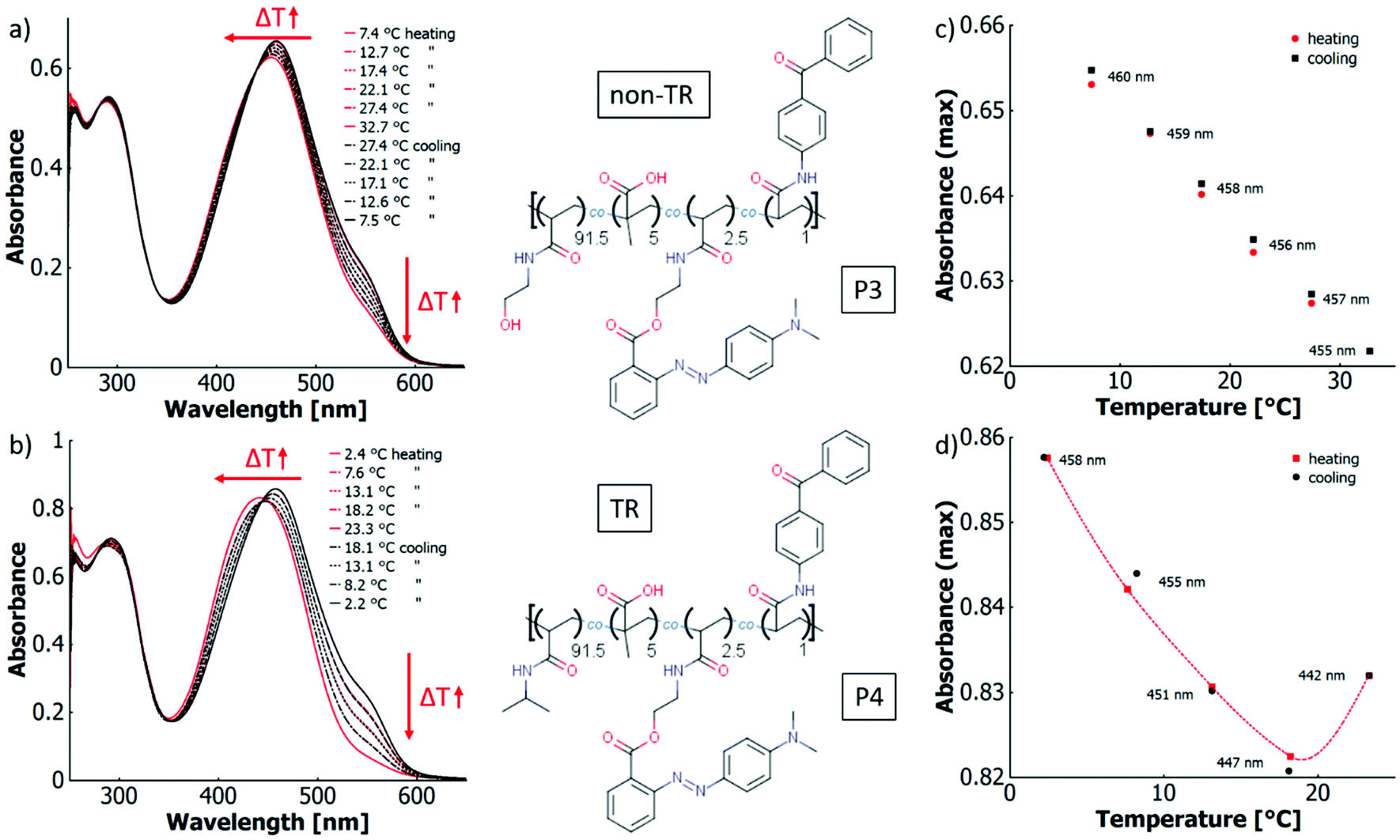
In ethanol with added acid (TFA), the absorption bands of the neutral species at ∼423 nm and the protonated species at ∼525 nm are well separated (cf.Fig. 6a), which facilitates calculation of the peak ratios (cf. ESI Fig. S43†). The occurrence of an isosbestic point at 452 nm instead of a region of intersections suggests that the influence of the thermo-perichromic mechanism on the thermo-halochromic mechanism is negligible in ethanolic solution. Under these conditions of partial protonation, the wavelength maximum of the absorption band of the azonium ion does not shift appreciably upon temperature variation. Also, the polymer-bound, pure azonium ion present in ethanolic sulfuric acid does not show a wavelength shift upon temperature increase (cf. ESI Fig. S24a†), yet the measurement is affected by a stronger scattering background because of reduced solubility.
In aqueous solutions of the same polymer P1 with added acid (TFA) the overlap between the absorption bands of the neutral dye at ∼453 nm and the protonated species at ∼500 nm is high, which leads to only a shoulder in the main absorption band (cf.Fig. 6b). Similar to the spectral behaviour of the parent dye monomer in binary solvent mixtures of water and ethanol (cf.Fig. 4), a region of intersections exists instead of an isosbestic point. The intersections of the curves between adjacent temperature steps gradually blue-shift from 452 nm at the lowest temperatures (6.2 °C) to 443 nm at the highest temperatures (52.6 °C, cf. Fig. 6b and ESI Fig. S44†). The existence of the region of intersections for the dye monomer was attributed to the mutual influence of thermo-solvatochromism and thermo-halochromism, with the former being only observable for the neutral azobenzene and not the azonium ion.
Interestingly, the completely protonated dye monomer (azonium ion) shows no thermo-solvatochromism, while the same azonium unit incorporated into the polymer is characertised by a change in absorption upon temperature variation. The wavelength maximum for the completely protonated polymer P1 in aqueous HCl red shifts from 500 nm at 7.8 °C to 507 nm at 59.4 °C, while the absorbance of the shoulder at around 530 nm increases slightly (cf. Fig. S25a†). Furthermore, the P1 absorption maximum is considerably blue-shifted compared to the one of the dye monomer at around 520 nm in different solvent systems (cf.Table 6). With the redshift upon heating, the spectroscopic features of the polymer approach those of the parent dye monomer. This significant difference in thermochromic behaviour must result from specific interactions between the polymer-bound azonium ion and the macromolecular framework. In particular, the hydrophilic OH– and amide side groups in the polymer can form polar and hydrogen bonding interactions while the (CH2–CHR) backbone provides a hydrophobic character.
| Neutral aq. | Neutral EtOH | Neutral binary mixture | Azonium aq. | Azonium EtOH | Azonium binary mixture | |
|---|---|---|---|---|---|---|
| o-MREAm | — | 420 nm | 441 nm | 522 nm | 519 nm | 519 nm |
| P1 | 453 nm | 423 nm | — | 500 nm | 520 nm | — |
| P2 | 452 nm | 422 nm | — | 499 nm | 518 nm | — |
| P3 | — | 422 nm | — | 501 nm | 520 nm | — |
| P4 | — | 421 nm | — | 501 nm | 517 nm | — |
This difference is also signified in derivative spectroscopy, where the monomer shows three vibronic sub-bands in all solvents, while the polymer only shows two vibronic sub-bands (cf. ESI Fig. S39†). The balance of the individual contributions from the water–dye, water–macromolecule and dye–macromolecule interactions is affected by temperature and with this the associated thermochromic mechanisms. Apparently, the solvation of the dye substituent by water increases at higher temperatures.
The similarity between thermo-solvatochromic and thermo-perichromic behaviour can be revealed with derivative spectroscopy. In ethanolic solution, the second derivative of P1 evolves similarly to that of the dye monomer, with both sub-bands decreasing uniformly (cf. ESI Fig. S32d and S38c†). In aqueous solution, the 0–0 sub-band of the copolymer shows a stronger decrease compared to the other sub-bands, as in the case of the dye monomer in the binary solvent mixture (cf. ESI Fig. S35d and S38d†).
In ethanol, the temperature-induced changes in the absorption spectra are virtually the same for the polymer systems P1 and P2 both for the neutral and the partially protonated form. Thus, the same arguments as discussed above apply here (cf. ESI Fig. S43, S23 and S45†). Yet, the behaviour of the completely protonated polymer in ethanolic sulfuric acid deviates slightly between P1 and P2 (cf. ESI Fig. S24†). Under these conditions, polymer system P2 has a higher solubility than polymer system P1 and thus shows no scattering effects (opposed to the inverse wavelength dependence of the baseline in P1 due to scattering, cf. ESI Fig. S24a and b†). Upon increasing the temperature of the P2 solution, the absorption maximum slightly red-shifts from 517 nm at 5.7 °C to 520 nm at 44.8 °C.
In aqueous solutions, the thermochromic behaviour of P1 and P2 differs considerably. For P2 and without the addition of acid, the wavelength of the absorption maximum blue-shifts from 450 nm at 7.0 °C to 443 nm at 53.4 °C. However, the absolute value of absorbance at the peak maximum decreases until ∼43 °C to then sharply increase again upon further heating (cf.Fig. 7a and c). This can be explained by the LCST phase transition of the P2 copolymer in aqueous solution. When the macromolecule passes the coil-to-globule transition, the system starts to scatter, which increases the overall absorbance. As large agglomerates are absent at these low mass concentrations of the polymer,93 the absorbance increase can be attributed to stronger Rayleigh scattering (λ−4 dependence) due to the change of refractive index between coil and globule.94 When adding acid to these aqueous P2 solutions, thermo-halochromism is induced. Because of the large spectral overlap between the neutral azobenzene and the azonium ion in aqueous solutions, the presence of the azonium ion is reflected by a shoulder of the main absorption band at around 550 nm. The absorbance decreases over a large temperature range (7.1 °C to 51.4 °C) before scattering effects dominate, leading to an overall absorbance increase again (cf.Fig. 7 and ESI Fig. S46a and b†). In comparison to the non-protonated P2, the absorption maximum of the partially protonated polymer shifts more strongly to shorter wavelengths upon heating (465 nm at 7.1 °C to 449 nm at 56.8 °C). Owing to the large spectral overlap, this blueshift occurs when the azonium ion content decreases by the thermo-halochromic pathway. The absorbance at maximum shows the same general behaviour as the neutral polymer, since it decreases until the phase transition temperature, to then drastically increase again by scattering upon the coil-to-globule transition. Yet, the associated minimum occurs at higher temperatures in the partially protonated case (∼48–49 °C) compared to the neutral case.
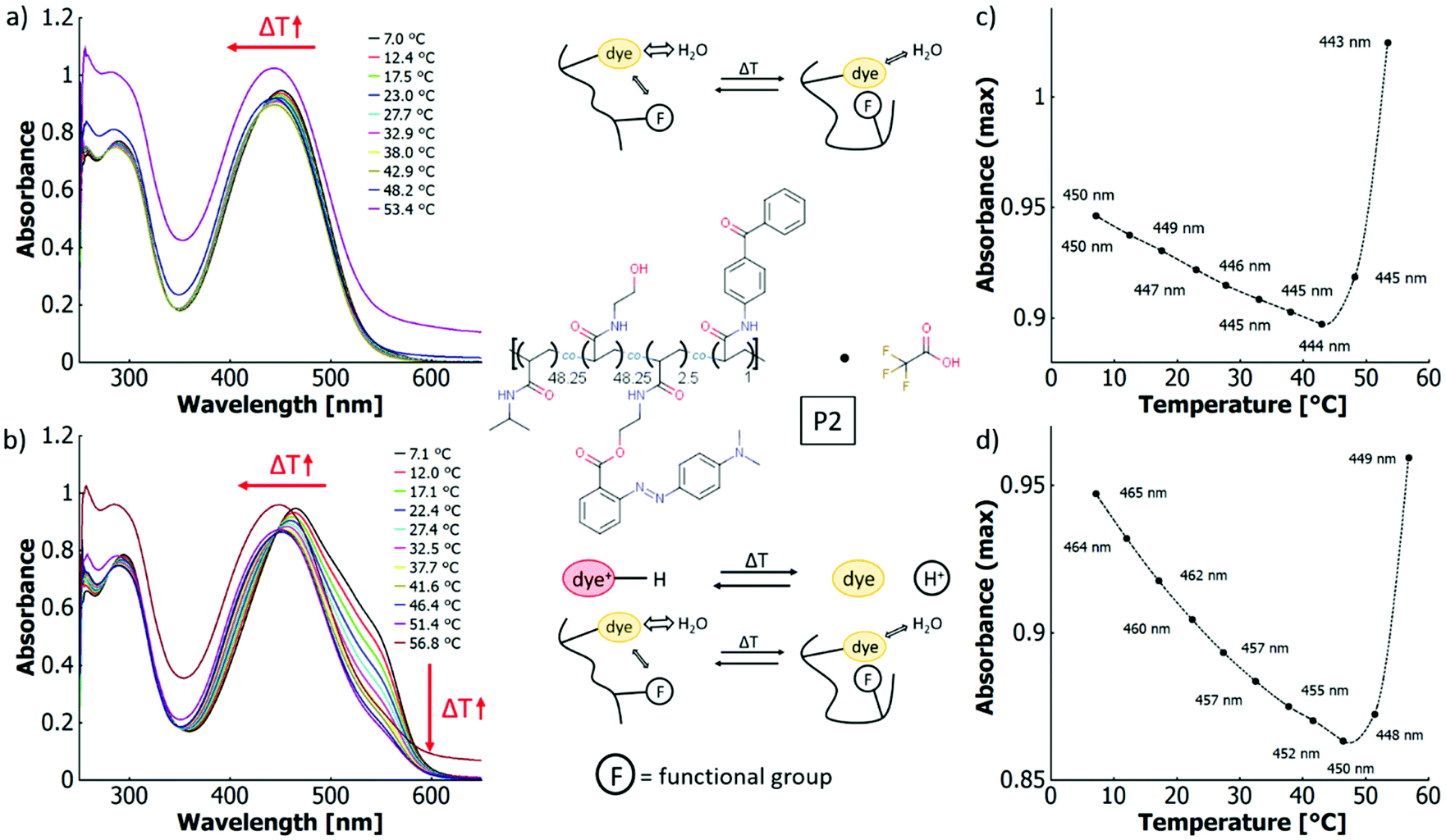 | ||
| Fig. 7 UV-vis spectra of a poly(NiPAAm-co-HEAm-co-o-MREAm-co-BPAAm) copolymer (P2) 0.03 w% in (a) pure water, and (b) aqueous trifluoroacetic acid (0.001 v/v%) at different temperatures (direction of shifts in absorbance upon heating marked with red arrows); absorbance at maximum (wavelength indicated in graph) vs. temperature for (c) pure water and (d) aqueous trifluoroacetic acid (0.001 v/v%). The lines are generated by a non-rounded Akima interpolation and only serve as guide to the eye. The structure of the copolymer (acid optional) and cartoons of the ongoing thermochromic processes are shown in the middle section. | ||
According to the literature, the cloud point of neat polyNiPAAm homopolymer solutions is constant or decrease with lower pH values and higher anion concentrations.95,96 Therefore, addition of trifluoroacetic acid should not influence the phase transition significantly. Thus, the observed disparate behaviour of the neutral and the partially protonated P2 copolymer is attributed to the charge effect of the azonium ion. As a direct consequence, the protonation equilibrium of thermo-halochromism affects the LCST behaviour since the cloud point temperature depends on the residual azonium content, which directly accounts for the charge of the polymer and with this its degree of hydrophilicity. The more hydrophilic the polymer, the higher the transition temperature.97,98
With the thermoresponsive transition, the polarity of the polymer changes and with this the interaction with the dye. Consequently, the thermo-perichromism is directly affected by the thermoresponsiveness. To investigate this effect in further detail, a more hydrophobic copolymer P2b with the same constitutents but a reduced HEAm content was studied in aqueous solutions. For this copolymer P2b without added acid, the minimum in the graph of the absorbance maximum vs. temperature lies at around 21 °C (cf. ESI Fig. S22b†), which is about 20 K lower than that of the more hydrophilic P2. This trend follows the decrease of the cloud point temperature being affected by a reduction in hydrophilicity. At the same time, the absorption maxima shift from 447 nm at 7.7 °C to 436 nm at 28.0 °C (cf. ESI Fig. S22†). Here, the blueshift of the wavelength (Δλ = −11 nm) for P2b in the temperature interval of ΔT = 20 K is larger compared to copolymer P2 (ΔT = 46 K, Δλ = −7 nm).
These data clearly support the hypothesis that the thermo-perichromic shift directly depends on the thermoresponse. The range of the wavelength shift is related to the difference in hydrophobicity of the microenvironment of the dye below and above the coil-to-globule transition. This difference is larger in more hydrophobic polymers that show a lower cloud point temperature, as in these systems the polarity change between the hydrophilic coil and the hydrophobic globule is expected to be larger.
When acid (TFA, 0.13 mM) is added to the P2b solution, thermo-halochromism dominates the temperature-dependant spectral changes in analogy to P2 and P1. At the same acid concentration, the degree of protonation at low temperatures is roughly the same for both copolymers P2 and P2b. Yet, the temperature dependence of the protonation equilibrium is different for the two copolymers. The lowest degree of protonation is reached around the minimum in the graph of the absorbance maximum vs. temperature, which we associate with the coil-to-globule transition (cf.Fig. 7b and ESI Fig. S22b†). Apparently, the temperature influence on the protonation equilibrium levels off around the cloud point and the corresponding coil-to-globule transition. Inside the collapsed globule, the water content is reduced and hydrophobic interactions dominate.98,99 As the neutral species partitions into the hydrophobic regions, it is depleted from the protonation equilibrium with the azonium ion in the aqueous phase. Consequently, the concentration of the azonium ion, which favours a polar environment, decreases. Therefore, the characteristic LCST behaviour for a given polymer determines the temperature range of thermo-halochromism.
These measurements suggest that apart from the rather abrupt cloud point transition during the LCST-type collapse, the polymer polarity changes gradually with temperature. This gradual change is signified by the following two observations. Firstly, the steady decrease in concentration of the azonium species with increasing temperature for partial protonation in acidic solutions, and secondly, by the gradual blueshift of the main absorption band upon heating in acid-free aqueous solutions (cf.Fig. 7 and ESI Fig. S22†). This continuous transition has been shown in the literature for other PNiPAAm-based systems with several methods, e.g., EPR (electron paramagnetic resonance spectroscopy),99 AFM (atomic force microscopy),100 and fluorescence/NRET (non-radiative energy transfer).101
 | ||
| Fig. 8 UV-vis spectra in water at different temperatures of (a) poly(HEAm-co-MAA-co-o-MREAm-co-BPAAm) (P3, 0.02 w%) and (b) poly(NiPAAm-co-MAA-co-o-MREAm-co-BPAAm) (P4, 0.03 w%); absorbance at maximum (wavelength indicated in plot) versus temperature of (c) P3 and (d) P4 (“TR” = thermoresponsive). The lines are generated by a non-rounded Akima interpolation and only serve as guide to the eye. The structures of the copolymers are displayed in the middle section. | ||
Compared to the non-thermoresponsive system P3, the thermoresponsive copolymer P4 shows a stronger decrease in absorbance per temperature increment (cf.Fig. 8a and b). Apparently, the LCST transition in P4 augments both the thermo-perichromic and thermo-halochromic effect. Upon thermal collapse, the macromolecule provides a more hydrophobic environment in which the neutral dye is preferentially accommodated over the charged azonium ion. Consequently, the protonation equilibrium shifts towards the neutral dye, as described above for P2 and P2b. Since the cloud point depends on the polymer concentration, thermochromism of P4 was measured at different concentrations. Upon decreasing the polymer fraction below the concentration at the LCST point, the cloud point, respectively the coil-to-globule transition temperature increases.102,103 Accordingly, with lower polymer concentration the position of the minimum in the absorbance-maximum-vs.-temperature plot shifts to higher temperatures (cf. ESI Fig. S26a–f†), which we associate with a shift of the coil-to-globule transition. In the globular state the hydrophobicity of the polymer increases substantially, modifying the interactions with the dye and therefore serving as a measure for thermo-perichromism as well.
To understand whether polymer concentration affects the thermo-halochromism, the evolution of two features upon temperature variation can be considered. First, the change in the degree of protonation can be determined by the ratio of absorbance at 552 nm (azonium ion) and at 420 nm (neutral dye), referred to as ratiometric approach. Second, the relative variation in azonium ion content per temperature interval can be determined via the change in absorbance at 552 nm. For this purpose, the band is normalised by dividing the absorbance at each temperature by the absorbance at the lowest temperature, termed normalisation approach. These parameters were determined for three concentrations (0.15 g L−1, 0.2 g L−1 and 0.3 g L−1; cf. ESI Fig. S26g–i†). There are at least five components in the acid–base equilibrium of this system comprising the neutral dye, the azonium ion, methacrylic acid, its anion and H3O+ as a proton shuttle (ignoring low concentrations of OH− from self-dissociation of water). The equilibrium between these components depends on temperature and on polymer concentration, with higher concentrations leading to higher degrees of protonation. At all concentrations, the degree of protonation decreases with increasing temperatures and the plots of the ratio of absorbances vs. temperature converge around the coil-to-globule transition (cf. ESI Fig. S26g†). Simultaneously, the normalised absorbances overlap and decrease linearly up to 20 °C to then level off at around 25 °C (cf. ESI Fig. S26e†), indicating almost complete conversion of the azonium ion into the azobenzene.
It follows, that the ratiometric approach shows a dependence of thermochromism on polymer concentration, while the normalisation approach is independent of polymer concentration. The seeming discrepancy between these approaches may be explained by the effects of both the thermo-perichromic and the thermoresponsive behaviour of the copolymer P4. To rationalise this apparent discrepancy, the position of the reference wavelengths, at which the ratiometric approach is performed, as well as the respective protonation state of the chromophore in the spectrum of a partially protonated copolymer in water must be discussed (cf. ESI Fig. S27†). The spectra of P4 result from a combination of the absorption of the neutral azobenzene and the azonium ion with considerable overlap of the absorption bands. This overlap leads to an apparent redshift of the maximum in the partially protonated case compared to the neutral copolymer. On the other hand, the pronounced shoulder at long wavelengths in these spectra results from the absorption of the azonium ion with only minor contributions of the neutral species. The wavelength selected for the normalisation approach is located in this shoulder. Even though minor contributions of the neutral species do change the absorbance here, these changes are small, and we assume that they are caused by density fluctuations. Therefore, the variations are, relative for both species, independent of the concentration. As the absorption of both the neutral and the protonated species is barely affected by temperature changes at this long wavelength (cf. ESI Fig. S27†), thermo-perichromism plays apparently a negligible role.
Accordingly, the decrease in normalised absorbance at 552 nm upon temperature increase for the partially protonated case (cf. ESI Fig. S26h†) is solely caused by thermo-halochromism, and consequently unaffected by concentration. On the other hand, the azonium ion has a low absorbance at the shorter wavelength of 420 nm and is almost unaffected by temperature variation. Thus, the observed thermal effect is attributed to thermo-perichromism in consequence of the thermoresponse of the polymer in solution, which primarily affects the absorbance of the neutral species (cf. Fig. S26i†). Since the thermoresponse is concentration-dependent, the ratiometric approach must be concentration-dependant as well.
Notably, the concentration dependence of the absorbance ratio between 552 nm and 420 nm (i.e., the slopes in the curves of ESI Fig. S26g†) is smaller for higher concentrations than for lower concentrations. This trend follows the variation of the cloud point at low volume fractions in the phase diagramme of LCST-type polymers in solution before the critical point. In this phase diagramme, the slope increases with decreasing polymer concentration.
Based on the observations above, the thermochromic behaviour of the thermoresponsive P4 with intrinsic acid moieties can be explained by the interplay of two mechanisms: thermo-halochromism and thermo-perichromism. Thermo-halochromism reflects the temperature-dependant protonation equilibrium between the azobenzene and the azonium ion and is polymer concentration independant. Thermo-perichromism is a consequence of the modulation of the immediate environment of the dye that varies with temperature. This effect is enhanced by the LCST-behaviour of the copolymer, which steadily decreases its polarity upon heating towards the phase transition. As the polymer phase transition is concentration-dependant, the thermo-perichromic effect is as well.
In their concerted action, thermo-perichromism strongly affects thermo-halochromism via the thermoresponsiveness of the polymer. Thus, upon the induced polarity shift in the microenvironment of the dye, the susceptibility for protonation is greatly altered. As a consequence, thermo-halochromism is strongly enhanced below the cloud point and vanishes above it, as the neutral dye is protected from protonation by the collapsed polymer.
3.6. Thermochromism in photocrosslinked films of methyl red copolymers
Irradiating an unannealed, dry film of P1 reduced the absorbance of the azobenzene chromophore by about 20% (cf. Fig. S54a†). After irradiation, the films were sufficiently crosslinked to swell in water, but their mechanical integrity and surface attachment were low, deeming measurements in the swollen state impossible. As a remedy, annealing above the glass temperature was found to result in a substantial increase in film stability to allow such measurements. This annealing procedure leads to a slight decrease in absorbance and a small blueshift (1 nm) of the absorption maximum alongside a change in band asymmetry. Irradiation of such annealed films reduced the absorbance by just about 10% instead of 20% in the untreated films (cf. Fig. S54b†). For P2 the same increase in film stability was observed after annealing. Both P3 and P4 showed sufficiently high intrinsic film stability after solvent casting, drying, and crosslinking, thus no heat treatment was required prior to photocrosslinking. Upon irradiation, the azobenzene content in the dry film of P3 decreased by about 20% as for unannealed P1. Washing the crosslinked P3 film until the supernatant remained colourless decreased the azobenzene content in the film by about 70%, indicating polymer leaching as a consequence of a less efficient photocrosslinking process (cf. ESI Fig. S55†). The analogous leaching behaviour was observed for the other polymer samples.
A factor that likely influences the crosslinking is the cis–trans isomerism of the azobenzene dye. Although the dye itself does not strongly absorb at the crosslinking wavelength (302 nm, cf. ESI Fig. S56†), there is still residual absorption. As cis–trans isomerisation occurs regardless of the wavelength at which the chromophore is excited, albeit at varying efficacies,34 the dye will use up a certain amount of UV-light either in the isomerisation process or in radiationless decay. The cis-isomer thermally relaxes relatively fast to the trans-isomer even in a rigid matrix,104 which means that absorption processes can be repeated. This secondary pathway of energy consumption may decrease the crosslinking efficiency. Yet, sufficient crosslinking is observed with the given conditions.
The most prominent effect that can be identified in the gel systems is the strong halochromic effect of the integrated carboxylic acid function at much lower concentrations compared to the cases of externally added acid, which is required in much higher concentrations. This effect is much less pronounced in polymer solutions and may be explained by a proximity effect of the integrated carboxylic acid functions on the neighbouring dye moieties, which apparently results in a considerably more efficient protonation process. This pecularity originating from the characteristic network architecture will be elucidated in the frame of the individual polymer systems.
As already described for the dye monomers and the polymers in solution, addition of acid to the polymer systems leads to an “induced thermo-halochromism” (P1 and P2) upon temperature variation, while in the copolymer systems with integrated acid moieties we speak about “intrinsic thermo-halochromism” (P3 and P4).
From an experimental point of view, two general differences between the temperature-dependant measurements of the swollen gels compared to the polymer solutions have to be considered. First, the gels may show substantial scattering even in the swollen state, for which we applied a simplified baseline correction by subtracting the absorbance at 750 nm (neglecting the wavelength dependence of scattering as a first approximation). Second, since the chromophore is directly bound to the surface-attached polymer network the absorbance does not inherently decrease with temperature, which is the case for the corresponding solutions due to a reduced dye concentration by the thermal volume expansion of the solvent. Owing to the confinement by surface-attachment, the average number of chromophores in the illumination volume remains essentially constant independent of potential temperature effects on the swelling state.
In the partially protonated gel system thermo-halochromism dominates. While the azonium ion content in the gel as well as in solution decreases with increasing temperatures, the degree of protonation differs considerably between both systems. An order-of-magnitude higher acid concentration is required in the gel to reach similar degrees of protonation as in solution. Concomitantly, the decrease in the degree of protonation per temperature increment is lower for the gel compared to the solution (cf. ESI Fig. S30a†). This may result from the substantially higher polymer volume fraction in the gel network compared to the polymer chains in solution, increasing the local dye concentration in the network. Higher degrees of protonation amplify the Coulomb repulsion between the like-charged azonium ions, in turn raising the osmotic pressure, which is thermodynamically unfavourable. Consequently, a higher acid concentration is required for protonation of the gel.
For the thermoresponsive copolymer P2, thermo-perichromism of the neutral and the completely protonated gel is similar to that in solution (cf.Fig. 7a; ESI Fig. S25b and S28c and d†). At low temperatures, the partially protonated P2gel and the P2 solution show a lower degree of protonation at the same acid concentration, and a higher variation in the degree of protonation per temperature increment compared to the P1gel and P1 solution (cf. ESI Fig. S29†). Partially replacing HEAm of P1 by the less polar NiPAAm comonomer decreases the overall hydrophilicity of P2. Assuming a weaker hydration, a higher polymer volume fraction of P2 owing to a more compact coil in solution and a lower swelling ratio in the gel is expected compared to P1. At the same acid concentration, the degree of protonation in P2 is thus lower because of the higher proximity of the charged groups and the resulting stronger Coulomb repulsion.
The differences in behaviour of thermoresponsive P2 in solution and in the gel state are more pronounced than for the non-responsive P1 system. While an absorbance increase because of scattering is observed for the P2 solution upon the coil-to-globule transition (cf.Fig. 7), this scattering effect is absent in the P2gel (cf. ESI Fig. S51†). Yet, based on the thermoresponsiveness of P2 in water, a variation of hydrophilicity with temperature should still occur for the P2gel. This is experimentally supported by the aforementioned higher variation of the degree of protonation per temperature increment compared to the P1gel (cf. Fig. S29b†). This variation is similar for both the P2gel and P2 in solution (cf. ESI Fig. S30b†). The absence of scattering in the P2gel may be explained by the higher polymer volume fraction in the gel compared to the solvated coil. Thus, upon collapse, the refractive index variation in the gel is smaller than in the polymer solution.
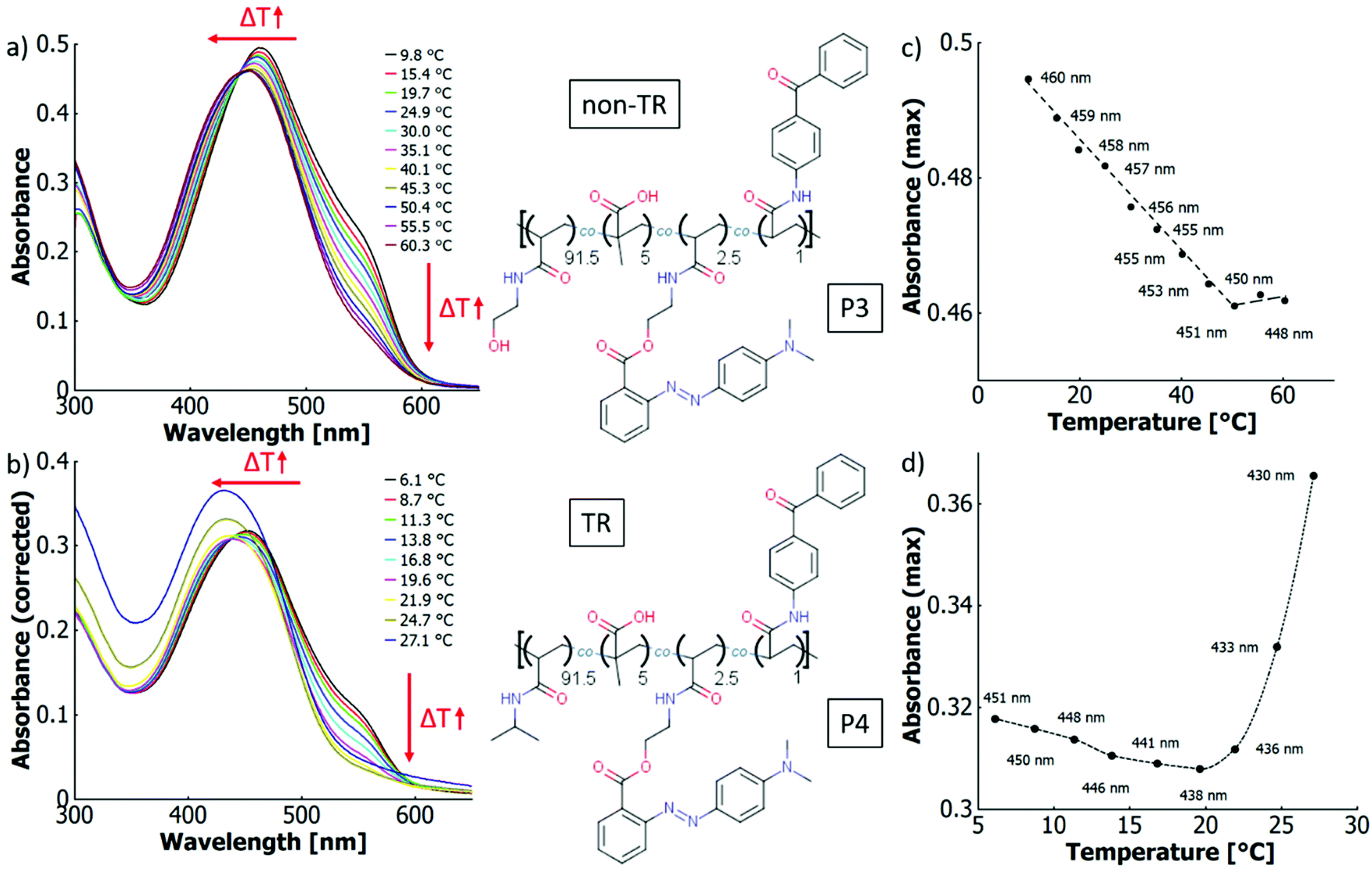 | ||
| Fig. 9 UV-vis spectra at different temperatures of water–swollen, photocrosslinked films of (a) poly(HEAm-co-MAA-co-o-MREAm-co-BPAAm) P3 and (b) poly(NiPAAm-co-MAA-co-o-MREAm-co-BPAAm) P4. Plot of the absorbance at maximum (wavelength indicated in plot) versus temperature for (c) P3 and (d) P4 (“TR” = thermoresponsive). The line in (c) was generated by hand, the one in (d) by a non-rounded Akima interpolation. Both serve as guide to the eye. The absorbance was baseline-corrected by subtracting the absorbance at 750 nm. | ||
For the P3gel, the absorbance at maximum decreases linearly from 10 °C to 50 °C. At temperatures beyond, the absorbance stagnates and the absorption band broadens significantly (cf.Fig. 9a and c). The reason for this behaviour may lie in fluctuations of the film thickness due to an increased gel mobility by Brownian motion at higher temperatures. The degree of protonation at low temperatures is higher in the P3gel than in P3 solution (cf.Fig. 8a and 9a). Two factors may be considered to explain this observation. On the one hand, photobleaching upon UV-irradiation during the crosslinking step already leads to a relative decrease in the number of the dye side groups in the network compared to the acid moieties. On the other hand, for P1gel and P2gel without intrinsic acid much higher acid concentrations are required to protonate the non-bleached dye compared to their aqueous solutions. This completely opposite behaviour suggests that the increase in the degree of protonation for the P3gel is not solely due to photobleaching. We hypothesise that the proximity between the intrinsic carboxylic moieties and dye side groups in the P3gel and P4gel network offsets the requirement of higher free acid concentration observed in P1gel and P2gel. The temperature range in which thermochromic behaviour may be observed, is also larger in the P3gel than in the P3 solution. Temperature-induced deprotonation leads to the same degree of protonation at ∼40 °C in solution and ∼60 °C in the gel (cf. ESI Fig. S48 and S52†). As the initial degree of protonation in the gel system is higher but the extent of deprotonation per temperature increment is similar, the larger dynamic range cannot be attributed to the network architecture itself but is a consequence of the acid proximity effects.
In the thermoresponsive P4gel, the temperature dependence of the absorbance at maximum changes from a negative to a positive slope between 20 °C and 21 °C (cf.Fig. 9d). This point assumably matches the transition temperature, as it corresponds to the thermoresponsive behaviour in more concentrated solutions of P4 (0.3 g L−1, cf. ESI Fig. S26h†). The degree of protonation in P4 at low temperature is similar for the solution and the gel. However, the deprotonation behaviour differs considerably between the two (cf. ESI Fig. S26e, S53b and S30d†).
Except for the P4gel, in all other systems (monomers, polymers P1–P4, and P1–P3gels) the protonation equilibrium shifts stronger to the deprotonated form at lower temperatures than at higher temperatures, especially for the monomers in the binary mixture of water and ethanol. This is documented in Fig. 4c and ESI Fig. S52c† by the decrease in slope of A(protonated)/A(neutral) vs. temperature curves at higher temperature as indicated by the red slope triangles. On the other hand, specifically P4gel exhibits the opposite behaviour with a stronger reduction of the degree of protonation per temperature interval with increasing temperature (red slope triangles in ESI Fig. S53c†). With the P4gel collapse at higher temperatures, the polymer environment becomes more hydrophobic and thus more unfavourable for the polar azonium ion. This drives deprotonation towards the neutral and less polar azobenzene form, which is better accommodated in the hydrophobic environment. Yet, the formation of hydrophobic compartments from collapsing polymer chain segments is no abrupt process. The gradual change in the degree of protonation with temperature suggests that the hydrophobicity increases over a large temperature range. The same effect has been observed for the solution of P4 (cf.Fig. 7 and ESI Fig. S26†). In LCST hydrogels, EPR measurements involving spin probe decomposition also suggest the formation of hydrophobic nanocompartments.99
The rising hydrophobicity with temperature involving the formation and growth of hydrophobic compartments, eventually leading to the gel collapse, can explain the differences between the polymer in solution and as hydrogel. In solution, the polymer chains have sufficient conformational freedom for the azonium ion side groups to orient towards solvent rich regions via chain rearrangement. In contrast, with gels, movement of chain segments is considerably reduced by the crosslinked polymer network. Under these conditions the dye substituent cannot partition freely between the solvent-rich regions and the hydrophobic compartments. Thus, with the volume fraction of the collapsed gel growing upon heating, a larger fraction of the dye is surrounded by a hydrophobic environment, which increasingly shifts the protonation equilibrium from the azonium ion to the neutral azobenzene.
The particular thermochromic behaviour of this P4 gel system manifests in a thermo-halochromism that is amplified along the temperature axis by a contribution from the thermo-perichromic mechanism. In other words, the temperature-dependant change of the microenvironment as the underlying mechanism of thermo-perichromism does modulate the protonation equilibrium of thermo-halochromism. The resulting augmenting thermo-peri-halochromism of the thermoresponsive P4gel behaves inversely to the diminishing thermo-solvato-halochromism of the monomers in the binary solvent mixture. The term “augmenting” indicates an increase in the magnitude of thermo-halochromism with temperature.
3.7. Comparison of thermochromicity in different systems
In order to allow the quantification of the phenomenological observations for all different combinations of solvents, dyes, and dye-containing polymers, temperature-dependant van't Hoff analysis was applied to the temperature-dependant UV-vis measurements of acidified, thermochromic solutions. For this, the natural logarithm of the ratio of the absorbances of the azonium ion and the azobenzene ln(R) was plotted against the inverse of the absolute temperature K−1 and fitted according to eqn (1) to extract ΔH0 and ΔCp, as described above in the context of pKa values of the dye monomers. In the case of thermoresponsive systems, only the data below the coil-to-globule transition was fitted because scattering effects prevent accurate fitting. This analysis yields the enthalpy of protonation under standard conditions and the heat capacity of ionisation, which is connected to the interconversion between the azonium ion and the neutral azobenzene and the corresponding optical changes. It is important to note that this analysis is only viable to determine the magnitude of thermo-halochromism, as only in this thermochromic mechanism an equilibrium between different chemical species is of main relevance. The enthalpy ΔH0 may be regarded here as a measure of the extent of protonation at 25 °C. It also tells whether the process is endothermic or exothermic. A negative ΔH0 means that the equilibrium shifts away from the product, which refers to the deprotonation of the azonium ion to yield the neutral azobenzene. The heat capacity of ionisation ΔCp quantifies the temperature dependence of the enthalpy change. As such, it provides information about whether deprotonation becomes more favourable (negative value) or unfavourable (positive value) with increasing temperatures. Both values can be considered as thermochromicity parameters.We define here the linearity factor TL as a third quantity, which is associated with the linearity of the thermo-halochromic process. To obtain this value, the ratio of the absorbances of the azonium ion to that of the azobenzene R was plotted against the temperature in °C. The resulting plot was fitted with an exponential decay function
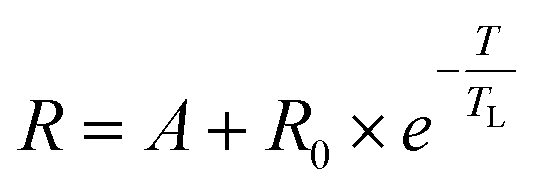 | (2) |
Based on these analyses the systems can be categorised into four sub-groups highlighted by a colour code in Table 7, in accordance with their tendencies regarding the enthalpy ΔH0 and the heat capacity of ionisation ΔCp.
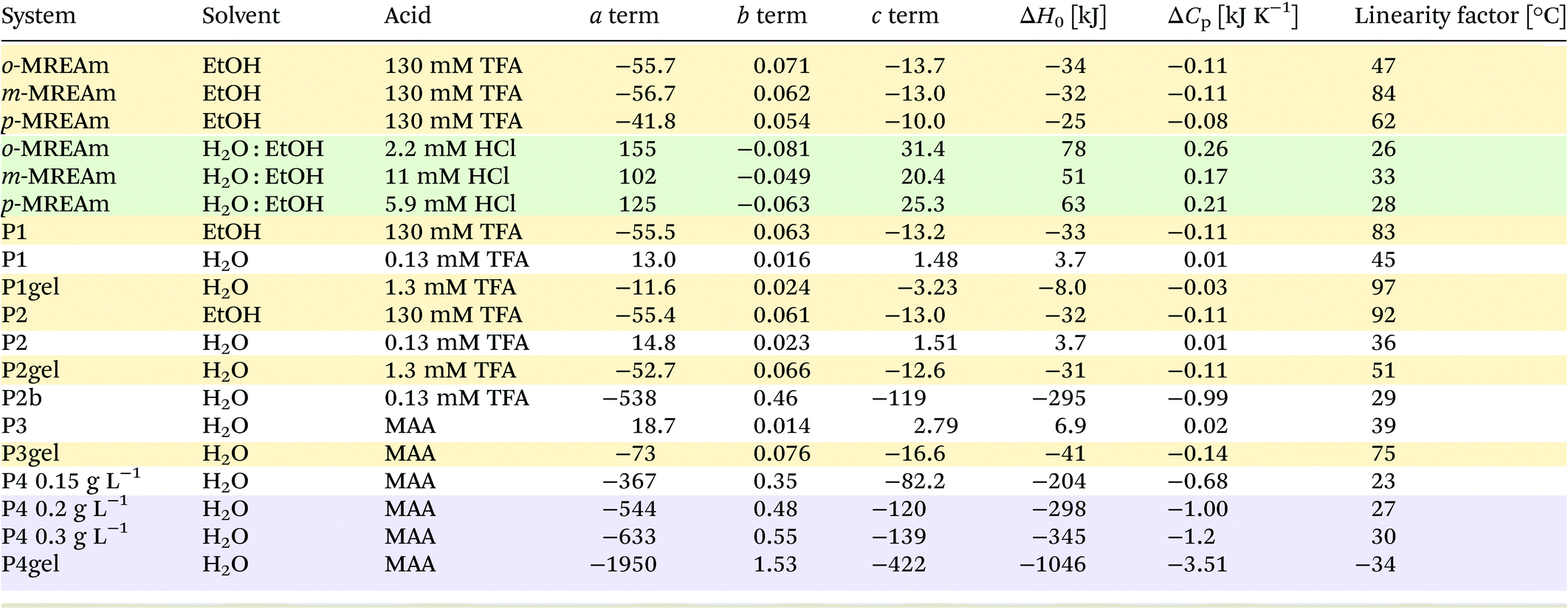
|
o-MREAm, m-MREAm, p-MREAm, P1, and P2 all in ethanol, as well as the photocrosslinked and water swollen gels P1gel, P2gel, and P3gel have a negative ΔH0 and negative ΔCp. This category has been highlighted by an orange hue in Table 7.
P1, P2, and P3 in water have a small positive value both for ΔH0 and ΔCp, represented by the white entries in Table 7.
The constitutional isomers of the MREAm monomers in the binary solvent mixture of water and ethanol have large positive values both in ΔH0 and ΔCp. These entries are marked in green.
The thermoresponsive systems P2b, and P4 for all measured concentrations in aqueous solution, as well as the photocrosslinked and water swollen P4gel have large negative values of ΔH0 and ΔCp, shaded in blue.
The linearity factor TL shows less variation between the different dye monomer and polymer systems than the corresponding thermochromicity parameters ΔH0 and ΔCp. In general, TL is larger for the systems with a low influence of thermo-solvatochromism and thermo-perichromism on thermo-halochromism for the two categories with small negative (orange) or positive (white) ΔCp. Consequently, TL is smaller for those systems with a strong influence on thermo-halochromism, as represented by the two categories green with moderate positive ΔCp and blue with strongly negative ΔCp.
Several conclusions can be drawn from this categorisation. First, incorporation of the dye by copolymerisation has little to no influence on thermochromism in ethanolic solution with added acid. Under these conditions both polymers, the non-thermoresponsive P1 and the thermoresponsive P2, yield the same negative thermochromicity parameters as the free dye monomer. The water swollen gels P1gel, P2gel and P3gel also have similar negative values for the thermochromicity parameters ΔH0 and ΔCp, while the corresponding polymers in solution have positive values for both parameters. The thermo-halochromic behaviour of these gel networks is reminiscent to that of the free dye monomers dissolved in acidified ethanol. Yet, this behaviour is contrary to the thermo-halochromism of the constituting, non-crosslinked polymers in aqueous solution in a partially protonated state, which is affected by their thermo-perichromism as reflected in the change of sign to negative values of ΔH0 and ΔCp. Apparently, this strong effect of crosslinking is a consequence of the more uniform microenvironment in the gel network and the reduced mobility of the dye attached to the crosslinked polymer segments. In contrast to the free polymer coil, no sufficient conformational rearrangement of the macromolecular segments in the network is possible, inhibiting favourable interactions with the surrounding medium. As the heat capacity of ionisation ΔCp is negative for this category (orange in Table 7), deprotonation becomes more favourable at higher temperatures.
Considering the similar thermochromicity values for aqueous P3 (intrinsic acid) and for P1 and P2 (no intrinsic acid) in acidic aqueous solution, there is no obvious difference between intrinsic and induced thermo-halochromism. Additionally, below the transition temperature the thermoresponsive polymer P2 with high cloud point behaves the same as the non-thermoresponsive P1. All three polymers P1, P2, and P3 in water show almost no temperature dependence, with their heat capacity of ionisation ΔCp being close to 0.
The next category comprising the positional isomers of MREAm in H2O:EtOH (XEtOH = 0.31) is highlighted in green in Table 7. For these systems, the influence of thermo-solvatochromism on thermo-halochromism in the binary solvent mixture of water and ethanol has been discussed above in the context of Fig. 4. With increasing temperature, their thermo-solvatochromism results in a reduction of the extent of deprotonation per temperature increment, which is reflected in the positive heat capacity of ionisation ΔCp in these systems. Both thermochromicity parameters ΔH0 and ΔCp correlate linearly with the pKa of the constitutional isomers of MREAm, increasing from meta over para to ortho (cf. ESI Fig. S19†). Even though the difference spectra from thermotitration (cf. ESI Fig. S37g–i†) show considerable deviation in the protonation mechanism between the positional isomers, the linear relationship between ΔCp and pKa corroborate the dominance of the acidity (i.e. pKa) of the dye monomers on their thermo-halochromism in the binary solvent mixture.
Comparing the thermochromicity parameters of P1, P2, and P3 in acidic aqueous solutions (white category in Table 7) and the configurational isomers of the dye monomer in the binary solvent mixture suggests that thermo-perichromism in these particular polymers is analogous to thermo-solvatochromism of the monomers. All these systems share positive values for ΔH0 and ΔCp, which evidences stabilisation of the azonium ion by the respective microenvironment with increasing temperature.
The strongest impact of either thermo-solvatochromism or thermo-perichromism on thermo-halochromism can be found in the category of thermoresponsive polymers in aqueous solution and hydrogels, coloured blue in Table 7, for which the absolute values of both thermochromicity parameters are the highest among all categories. These parameters become more negative with decreasing cloud point temperatures. This has been observed for solutions of P4 with a concentration increase, as well as for solutions of P2 (higher cloud point) in comparison to those of P2b (lower cloud point), where even the sign of ΔH0 and ΔCp switches from positive to negative. With lower cloud point temperatures, the coil-to-globule transition takes place in a narrower temperature interval, accompanied by a more pronounced change in interactions of the dyes with their microenvironment. This, in turn, results in larger absolute values of the thermochromicity parameters with lower cloud points. This hypothesis is further substantiated by the P4gel, for which the absolute values of the thermochromicity parameters are yet larger by an order of magnitude compared to the respective thermoresponsive polymer solutions, agreeing well with the phenomenological discussion above about thermo-halochromism of immobilised dyes in thermoresponsive gels containing intrinsic acid.
The obtained knowledge can be combined into the following conclusions:
1. Varying the microenvironment of a dye via the liquid medium or polymer composition provides the possibility to control the protonation equilibrium by the corresponding azonium ion concentration in dependence of the temperature for partially protonated systems.
2. When the dye microenvironment shifts to a more hydrophilic state upon a temperature increase, which is the case for the binary solvent mixture of water and ethanol, the azonium ion persists against thermally driven deprotonation owing to a better solubilisation.
3. Vice versa, when the dye microenvironment becomes more hydrophobic at higher temperatures, occurring with LCST-type polymers, the azonium ion is deprotonated more easily to yield the less polar azobenzene. This effect becomes particularly obvious for a thermoresponsive polymer in solution, when the azonium ion concentration vanishes close to the coil-to-globule transition at the cloud point.
4. Summary and conclusion
A synthesis route to novel azo dye monomers by esterification of methyl red to N-(2-hydroxyethyl)acrylamide (MREAm) via carbonyldiimidazole coupling could be established for all three positional isomers of the dye. From these monomers, copolymers were obtained by free radical polymerisation employing N-(2-hydroxyethyl)acrylamide (HEAm), N-isopropylacrylamide (NiPAAm), methacrylic acid (MAA), and 4-benzophenoneacrylamide (BPAAm). Here, HEAm provides water-solubility, NiPAAm thermoresponsivity in aqueous solution, MAA the intrinsic acid group as proton donor, and BPAAm the photocrosslinker unit for network formation. Thermochromism was studied for solutions of the monomer and polymer systems in pure water, ethanol, and the mixture thereof. For hydrogels prepared from these polymers by photocrosslinking and subsequent swelling in water, the thermochromic behaviour was investigated.In these experiments three different mechanisms of thermochromism, namely vibronic thermochromism, thermo-solvatochromism, and thermo-halochromism, were identified for the constitutional isomers of the azo dye monomer MREAm. Depending on the particular system and the experimental conditions, various combinations of these thermochromic mechanisms were found. In neat ethanol, the MREAm dyes show vibronic thermochromism by a blueshift of a few nanometers upon temperature increase that can be attributed to a change in the vibronic sub-bands, as observed via derivative spectroscopy and difference spectroscopy. In the binary solvent mixture of ethanol and water, the observed thermo-solvatochromism shows similarities to the vibronic mechanism as a consequence of the competition of the two solvent species in the solvation shell, also suggested by temperature-dependant derivative and difference spectra. By addition of extrinsic acid (trifluoroacetic acid) to a MREAm dye solution in ethanol, thermo-halochromism can be induced. When such a system is titrated to partial protonation with coexisting neutral azobenzene and azonium ion, the protonation equilbrium shifts towards the neutral azobenzene at higher temperatures. However, thermo-halochromism is strongly influenced by the nature of the solvent. Particularly, in the binary solvent mixture with added acid, two phenomena are observed with increasing temperatures: 1. instead of an isosbestic point, a blue-shifting region of intersections exists for consecutive spectra of adjacent temperature steps; 2. the extent of deprotonation per temperature increment decreases. Both phenomena result from the influence of thermo-solvatochromism on thermo-halochromism, which we termed in this particular combination “diminishing thermo-solvato-halochromism”.
Similarly, solutions of copolymers bearing o-MREAm units show thermo-perichromism as the polymer-related analogon to thermo-solvatochromism observed for solutions of the free monomers. The thermo-perichromic effect is stronger in water than in ethanol, which is most pronounced in LCST-type thermoresponsive polymers that undergo a coil-to-globule transition alongside strong changes in the hydrophobicity of the polymers in aqueous solution. The coil-to-globule transition for these systems can be best followed via the change of absorbance at maximum by an increase in scattering. No major differences in thermochromism can be identified for the polymers in solution by the van't Hoff analysis, neither with added (extrinsic) nor with copolymerised (intrinsic) acid. Thermoresponsive LCST behaviour in solution strongly affects thermo-halochromism, which scales with the cloud point, while in non-thermoresponsive polymers thermo-halochromism is almost identical to that of the isolated dye monomers. As the hydrophobicity of the LCST-type polymers increases with temperature and reaches its maximum at the coil-to-globule transition, the attached dyes are completely deprotonated at this point. For polymers with a lower cloud point, a smaller temperature interval is available during this transition for the deprotonation process, thus increasing the extent of deprotonation per temperature increment. Consequently, thermo-halochromism can be conveniently tailored by the design of the molecular architecture of the copolymer.
All hydrogels prepared from the different copolymers also show thermochromic behaviour. Remarkably, the thermoresponsive hydrogels from the LCST polymers with intrinsic acid exhibit the strongest thermochromic behaviour of all examined systems. Here, the extent of deprotonation per temperature increment increases strongly with temperature, and we termed the resulting effect “augmenting thermo-peri-halochromism”. We ascribe this behaviour to an azonium-destabilising change of the microenvironment towards a more hydrophobic character of the polymer.
In conclusion, we identified multiple mechanisms of thermochromism for various azobenzene systems of moderate pKa in solution and in the gel state, and elaborated the influence of solvent, polymer architecture and especially LCST behaviour. Employing van't Hoff analysis, a scaling of thermo-halochromism with the pKa could be demonstrated and the contributing effects were categorised with respect to thermo-solvatochromism and thermo-perichromism. Detailed understanding of the underlying mechanisms is of great value for potential applications of these dye–polymer systems, for example as macromolecular optical thermometers, or in any other scenario involving azobenzenes and temperature variation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Faculty IV of the University of Siegen for funding, Dr Thomas Paululat for NMR measurements, and Silvia Freese for extensive discussions.References
- X. Wang, O. S. Wolfbeis and R. J. Meier, Luminescent probes and sensors for temperature, Chem. Soc. Rev., 2013, 42, 7834–7869 RSC.
- A. Steinegger, I. Klimant and S. M. Borisov, Purely Organic Dyes with Thermally Activated Delayed Fluorescence-A Versatile Class of Indicators for Optical Temperature Sensing, Adv. Opt. Mater., 2017, 5, 1700372 CrossRef.
- L. Yin, C. He, C. Huang, W. Zhu, X. Wang, Y. Xu and X. Qian, A dual pH and temperature responsive polymeric fluorescent sensor and its imaging application in living cells, Chem. Commun., 2012, 48, 4486–4488 RSC.
- X. Wang, R. J. Meier and O. S. Wolfbeis, A Fluorophore-Doped Polymer Nanomaterial for Referenced Imaging of pH and Temperature with Sub-Micrometer Resolution, Adv. Funct. Mater., 2012, 22, 4202–4207 CrossRef CAS.
- A. Malfait, F. Coumes, D. Fournier, G. Cooke and P. Woisel, A water-soluble supramolecular polymeric dual sensor for temperature and pH with an associated direct visible readout, Eur. Polym. J., 2015, 69, 552–558 CrossRef CAS.
- H. Sun, J. Xiang, X. Zhang, H. Chen, Y. Shi, L. Yu, Q. Yang, Q. Li, A. Guan and Y. Tang, A colorimetric temperature sensor of a cyanine dye supramolecule and its application in reversible switch, Appl. Phys. Lett., 2014, 105, 71914 CrossRef.
- A. Seeboth, D. Lötzsch, R. Ruhmann and O. Muehling, Thermochromic polymers–function by design, Chem. Rev., 2014, 114, 3037–3068 CrossRef CAS PubMed.
- W. Theilacker, G. Kortüm and G. Friedheim, Zur Frage der Thermochromie; ein Beispiel für eine echte Valenztautomerie, Chem. Ber., 1950, 83, 508–519 CrossRef CAS.
- J. H. Day, Thermochromism, Chem. Rev., 1963, 63, 65–80 CrossRef CAS.
- C. Pietsch, U. S. Schubert and R. Hoogenboom, Aqueous polymeric sensors based on temperature-induced polymer phase transitions and solvatochromic dyes, Chem. Commun., 2011, 47, 8750–8765 RSC.
- A. Seeboth, J. Kriwanek and R. Vetter, The first example of thermochromism of dyes embedded in transparent polymer gel networks, J. Mater. Chem., 1999, 9, 2277–2278 RSC.
- J. Kriwanek, D. Lötzsch, R. Vetter and A. Seeboth, Influence of a zwitterionic surfactant on the chromogenic behavior of a dye-containing aqueous PVA-polyether gel network, Polym. Adv. Technol., 2003, 14, 79–82 CrossRef CAS.
- K. Matsubara, M. Watanabe and Y. Takeoka, A thermally adjustable multicolor photochromic hydrogel, Angew. Chem., Int. Ed., 2007, 46, 1688–1692 CrossRef CAS PubMed.
- X. Zhao, Synthesis, characterization and structure dependence of thermochromism of polythiophene derivatives, J. Mater. Sci., 2005, 40, 3423–3428 CrossRef CAS.
- X. Y. Zhao and M. Z. Wang, Structure dependence of photochromism and thermochromism of azobenzene-functionalized polythiophenes, eXPRESS Polym. Lett., 2007, 1, 450–455 CrossRef CAS.
- X. Yu, Y. Luo, W. Wu, Q. Yan, G. Zou and Q. Zhang, Synthesis and reversible thermochromism of azobenzene-containing polydiacetylenes, Eur. Polym. J., 2008, 44, 3015–3021 CrossRef CAS.
- S. Wu, L. Niu, J. Shen, Q. Zhang and C. Bubeck, Aggregation-Induced Reversible Thermochromism of Novel Azo Chromophore-Functionalized Polydiacetylene Cylindrical Micelles, Macromolecules, 2009, 42, 362–367 CrossRef CAS.
- S. Wu, Q. Zhang and C. Bubeck, Solvent Effects on Structure, Morphology, and Photophysical Properties of an Azo Chromophore-Functionalized Polydiacetylene, Macromolecules, 2010, 43, 6142–6151 CrossRef CAS.
- M. Leclerc and K. Faid, Electrical and optical properties of Processable Polythiophene Derivatives: Structure-Property relationships, Adv. Mater., 1997, 9, 1087–1094 CrossRef CAS.
- I. Lévesque and M. Leclerc, Novel Dual Photochromism in Polythiophene Derivatives, Macromolecules, 1997, 30, 4347–4352 CrossRef.
- H. Joshi, F. S. Kamounah, G. van der Zwan, C. Gooijer and L. Antonov, Temperature dependent absorption spectroscopy of some tautomeric azo dyes and Schiff bases, J. Chem. Soc., Perkin Trans. 2, 2001, 2303–2308 RSC.
- J. A. Dean and N. A. Lange, Lange's handbook of chemistry, McGraw-Hill; [Distributed by] Knovel, New York, [Norwich, N.Y.], 15th edn, 1999 Search PubMed.
- K. M. Tawarah and H. M. Abu-Shamleh, A spectrophotometric study of the acid-base equilibria of o-methyl red in aqueous solutions, Dyes Pigm., 1991, 17, 203–215 CrossRef CAS.
- T. Kuwabara, A. Nakamura, A. Ueno and F. Toda, Inclusion Complexes and Guest-Induced Color Changes of pH-Indicator-Modified beta.-Cyclodextrins, J. Phys. Chem., 1994, 98, 6297–6303 CrossRef CAS.
- M. Tajima, M. Sugai, K. Matsunaga, T. Yamashita, H. Inoue and M. Hida, Thermochromism of dyes on silica gel, Dyes Pigm., 1998, 39, 97–109 CrossRef CAS.
- X. Du, C. Zhu and X. Xie, Thermochromic Ion-Exchange Micelles Containing H + Chromoionophores, Langmuir, 2017, 33, 5910–5914 CrossRef CAS PubMed.
- C. Pietsch, R. Hoogenboom and U. S. Schubert, Soluble polymeric dual sensor for temperature and pH value, Angew. Chem., Int. Ed., 2009, 48, 5653–5656 CrossRef CAS PubMed.
- L. Liu, W. Li, K. Liu, J. Yan, G. Hu and A. Zhang, Comblike Thermoresponsive Polymers with Sharp Transitions: Synthesis, Characterization, and Their Use as Sensitive Colorimetric Sensors, Macromolecules, 2011, 44, 8614–8621 CrossRef CAS.
- R. W. Taft, J.-L. M. Abboud, M. J. Kamlet and M. H. Abraham, Linear solvation energy relations, J. Solution Chem., 1985, 14, 153–186 CrossRef CAS.
- A. R. Katritzky, D. C. Fara, H. Yang, K. Tämm, T. Tamm and M. Karelson, Quantitative measures of solvent polarity, Chem. Rev., 2004, 104, 175–198 CrossRef CAS PubMed.
- G. Seu, Spectrophotometric study of the prototropic equilibrium of methyl red in organic solvents, Dyes Pigm., 1995, 29, 227–240 CrossRef CAS.
- L. Zhang, J. M. Cole and X. Liu, Tuning Solvatochromism of Azo Dyes with Intramolecular Hydrogen Bonding in Solution and on Titanium Dioxide Nanoparticles, J. Phys. Chem. C, 2013, 117, 26316–26323 CrossRef CAS.
- H. Jaffé, S.-J. Yeh and R. Gardner, The electronic spectra of azobenzene derivatives and their conjugate acids, J. Mol. Spectrosc., 1958, 2, 120–136 CrossRef.
- H. M. D. Bandara and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- S. T. Cowan, Micromethod for the methyl red test, J. Gen. Microbiol., 1953, 9, 101–109 CrossRef CAS PubMed.
- Z. Guo, S. Li, C. Wang, J. Xu, B. Kirk, J. Wu, Z. Liu and W. Xue, Biocompatibility and cellular uptake mechanisms of poly(N -isopropylacrylamide) in different cells, J. Bioact. Compat. Polym., 2017, 32, 17–31 CrossRef CAS.
- H. Chen, C. Zhao, M. Zhang, Q. Chen, J. Ma and J. Zheng, Molecular Understanding and Structural-Based Design of Polyacrylamides and Polyacrylates as Antifouling Materials, Langmuir, 2016, 32, 3315–3330 CrossRef CAS PubMed.
- F. Doberenz, K. Zeng, C. Willems, K. Zhang and T. Groth, Thermoresponsive polymers and their biomedical application in tissue engineering - a review, J. Mater. Chem. B, 2020, 8, 607–628 RSC.
- S. Chen, K. Wang and W. Zhang, A new thermoresponsive polymer of poly(N-acryloylsarcosine methyl ester) with a tunable LCST, Polym. Chem., 2017, 8, 3090–3101 RSC.
- S. A. Mohammad, S. Dolui, D. Kumar, S. R. Mane and S. Banerjee, Facile access to functional polyacrylates with dual stimuli response and tunable surface hydrophobicity, Polym. Chem., 2021, 12, 3042–3051 RSC.
- J. Jia, M. Sarker, M. G. Steinmetz, R. Shukla and R. Rathore, Photochemical elimination of leaving groups from zwitterionic intermediates generated via electrocyclic ring closure of alpha,beta-unsaturated anilides, J. Org. Chem., 2008, 73, 8867–8879 CrossRef CAS PubMed.
- M. Gianneli, R. F. Roskamp, U. Jonas, B. Loppinet, G. Fytas and W. Knoll, Dynamics of swollen gel layers anchored to solid surfaces, Soft Matter, 2008, 4, 1443–1447 RSC.
- H. A. Staab and A. Mannschreck, Synthese von Carbonsäureestern nach der Imidazolidmethode, Chem. Ber., 1962, 95, 1284–1297 CrossRef CAS.
- D. E. Bergbreiter, P. L. Osburn and C. Li, Soluble Polymer-Supported Catalysts Containing Azo Dyes, Org. Lett., 2002, 4, 737–740 CrossRef CAS PubMed.
- H. A. Staab, M. Lüking and F. H. Dürr, Darstellung von Imidazoliden. Synthese von Amiden, Hydraziden und Hydroxamsäuren nach der Imidazolidmethode, Chem. Ber., 1962, 95, 1275–1283 CrossRef CAS.
- G. Dormán, H. Nakamura, A. Pulsipher and G. D. Prestwich, The Life of Pi Star: Exploring the Exciting and Forbidden Worlds of the Benzophenone Photophore, Chem. Rev., 2016, 116, 15284–15398 CrossRef PubMed.
- Marvin was used for drawing and displaying chemical structures, substructures and reactions, Marvin 18.30, ChemAxon (https://www.chemaxon.com).
- C. Larrivée-Aboussafy, B. P. Jones, K. E. Price, M. A. Hardink, R. W. McLaughlin, B. M. Lillie, J. M. Hawkins and R. Vaidyanathan, DBU catalysis of N,N’-carbonyldiimidazole-mediated amidations, Org. Lett., 2010, 12, 324–327 CrossRef PubMed.
- O. F. Olaj, J. W. Breitenbach and I. Hofreiter, Über einen bimolekularen Mechanismus der Radikalbildung bei der durch Di-(3-benzolazobenzoyl-)peroxid angeregten Polymerisation von Styrol, Monatsh. Chem., 1967, 98, 997–1016 CrossRef CAS.
- O. F. Olaj, J. W. Breitenbach and I. Hofreiter, Die Einführung von Starterbruchstücken in das Polymere bei der durch Di-(3-benzolazobenzoyl-)peroxyd angeregten Styrolpolymerisation, Makromol. Chem., 1966, 91, 264–280 CrossRef CAS.
- O. F. Olaj, J. W. Breitenbach and I. Hofreiter, Der Einfluß aromatischer Azoverbindungen auf die Polymerisation von Vinylacetat, Vinylchlorid und Methylmethacrylat, Makromol. Chem., 1967, 110, 72–83 CrossRef CAS.
- D. Braun, G. Arcache, R. J. Faust and W. Neumann, Darstellung und Polymerisationsverhalten von einigen neuen Vinylverbindungen, Makromol. Chem., 1968, 114, 51–69 CrossRef CAS.
- D. Braun and G. Arcache, Über den Einfluß aromatischer Azoverbindungen auf die radikalische Polymerisation von Styrol, Makromol. Chem., 1971, 148, 119–129 CrossRef CAS.
- M. C. Rezende and A. Aracena, In search of the thermo/halochromism of the E(T)(30) pyridinium-N-phenolate betaine dye, Spectrochim. Acta, Part A, 2012, 98, 18–22 CrossRef CAS PubMed.
- L. Hermosilla, M. C. Rezende, V. G. Machado and R. I. Stock, Thermohalochromism of phenolate dyes conjugated with nitro-substituted aryl groups, Spectrochim. Acta, Part A, 2017, 173, 556–561 CrossRef CAS PubMed.
- V. Gold, The IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 2019 Search PubMed.
- L. Antonov and D. Nedeltcheva, Resolution of overlapping UV–Vis absorption bands and quantitative analysis, Chem. Soc. Rev., 2000, 29, 217–227 RSC.
- Š. Sršeň, J. Sita, P. Slavíček, V. Ladányi and D. Heger, Limits of the Nuclear Ensemble Method for Electronic Spectra Simulations: Temperature Dependence of the (E)-Azobenzene Spectrum, J. Chem. Theory Comput., 2020, 16, 6428–6438 CrossRef PubMed.
- H. Mustroph, Studies on the UV-vis Absorption Spectra of Azo Dyes: Part 25. Analysis of the Fine Structure of the π1 -> π1* Band of 4′-Donor-Substituted 4-N,N-Diethylaminoazobenzenesa, Dyes Pigm., 1991, 15, 129–137 CrossRef CAS.
- M. Nepraš, S. Luňák, R. Hrdina and J. Fabian, Electronic excited states of azo compounds: Strong ππ* fluorescence of bis-4,4′-diethylaminoazobenzene, Chem. Phys. Lett., 1989, 159, 366–370 CrossRef.
- S. Lunák, M. Nepraš, R. Hrdina and H. Mustroph, Excited states of azo compounds. II. Vibrational structure of the electronic absorption spectra of 4,4′-di-substituted azobenzene derivatives, Chem. Phys., 1994, 184, 255–260 CrossRef.
- S.-K. Park, C.-K. Lee, K.-C. Min and N.-S. Lee, Fourier Transform Raman Studies of Methyl Red Adsorbed on γ-Alumina and Silica-Alumina, Bull. Korean Chem. Soc., 2004, 25, 1817–1821 CrossRef CAS.
- S. Bell, A. Bisset and T. J. Dines, Ab initio and density functional study of the resonance Raman spectra of Methyl Red, Ethyl Red and their protonated derivatives, J. Raman Spectrosc., 1998, 29, 447–462 CrossRef CAS.
- T. Itoh, Ground-state, excited-singlet-state, and excited-triplet-state energy levels and photophysics of the three rotational isomers of isophthalaldehyde vapor, J. Phys. Chem. A, 2007, 111, 8439–8444 CrossRef CAS PubMed.
- A. Cesaretti, B. Carlotti, F. Elisei, C. G. Fortuna, G. Consiglio and A. Spalletti, A cationic naphthyl derivative defies the non-equilibrated excited rotamers principle, Phys. Chem. Chem. Phys., 2017, 19, 5262–5272 RSC.
- M. Quick, A. L. Dobryakov, I. N. Ioffe, F. Berndt, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, Rotamer-Specific Photoisomerization of Difluorostilbenes from Transient Absorption and Transient Raman Spectroscopy, J. Phys. Chem. B, 2018, 122, 1049–1059 CrossRef CAS PubMed.
- E. Fischer, Emission spectroscopy evidenvce for the existence of rotamers in solutions of trans-diarylethylenes and related compounds, J. Photochem., 1981, 17, 331–340 CrossRef CAS.
- N. Castel and E. Fischer, Frozen-in non-equilibrium rotamer mixtures in 1,2-diarylethylenes obtained by low-temperature irradiation, J. Mol. Struct., 1985, 127, 159–166 CrossRef CAS.
- N. Castel and E. Fischer, Rotamers in azonaphthalenes? A combined photochemical and UV-absorption spectrophotometric study in rigid media, J. Mol. Struct., 1986, 140, 365–369 CrossRef CAS.
- J.-F. Boily and T. M. Seward, On the Dissociation of Methyl Orange: Spectrophotometric Investigation in Aqueous Solutions from 10 to 90 °C and Theoretical Evidence for Intramolecular Dihydrogen Bonding, J. Solution Chem., 2005, 34, 1387–1406 CrossRef CAS.
- E. Sawicki, Physical Properties of the Aminoazobenzene Dyes. IV. The Position of Proton Addition 1, J. Org. Chem., 1957, 22, 365–367 CrossRef CAS.
- E. Sawicki, The Physical Properties of the Aminoazobenzene Dyes. III. Tautomerism of 4-Aminoazobenzene Salt Cations in Acid Solution, J. Org. Chem., 1956, 21, 605–609 CrossRef CAS.
- E. Sawicki, Physical Properties of Aminoazobenzene Dyes. V. The C ∈ /A ∈ Ratio, J. Org. Chem., 1957, 22, 621–625 CrossRef CAS.
- G. E. Lewis, Structures of the mono-acid cations of 4-aminoazobenzene and its derivatives, Tetrahedron, 1960, 10, 129–134 CrossRef CAS.
- I. Y. Bershtein and O. F. Ginzburg, Tautomerism of Aromatic Azo-compounds, Russ. Chem. Rev., 1972, 41, 97–110 CrossRef.
- D. G. Hafeman, K. L. Crawford and L. J. Bousse, Fundamental thermochromic properties of buffered pH-indicator solutions and the formulation of “athermochromic” systems, J. Phys. Chem., 1993, 97, 3058–3066 CrossRef CAS.
- R. K. Srour and L. M. McDonald, Determination of the Acidity Constants of Methyl Red and Phenol Red Indicators in Binary Methanol– and Ethanol–Water Mixtures, J. Chem. Eng. Data, 2008, 53, 116–127 CrossRef CAS.
- K. Tawarah, The tautomeric and acid-base equilibria of p-methyl red in aqueous solutions, Dyes Pigm., 1992, 20, 261–270 CrossRef CAS.
- S. J. Khouri, I. A. Abdel-Rahim, E. M. Alshamaileh and A. M. Altwaiq, Equilibrium and Structural Study of m-Methyl Red in Aqueous Solutions: Distribution Diagram Construction, J. Solution Chem., 2013, 42, 1844–1853 CrossRef CAS.
- H. Naghibi, A. Tamura and J. M. Sturtevant, Significant discrepancies between van't Hoff and calorimetric enthalpies, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5597–5599 CrossRef CAS PubMed.
- Y. Liu and J. M. Sturtevant, Significant discrepancies between van't Hoff and calorimetric enthalpies. II, Protein Sci., 1995, 4, 2559–2561 CrossRef CAS PubMed.
- E. B. Tada, L. P. Novaki and O. A. El Seoud, Solvatochromism in pure and binary solvent mixtures: effects of the molecular structure of the zwitterionic probe, J. Phys. Org. Chem., 2000, 13, 679–687 CrossRef CAS.
- M. S. Antonious, E. B. Tada and O. A. E. Seoud, Thermo-solvatochromism in aqueous alcohols: effects of the molecular structures of the alcohol and the-solvatochromic probe, J. Phys. Org. Chem., 2002, 15, 403–412 CrossRef CAS.
- E. B. Tada, P. L. Silva and O. A. El Seoud, Thermo-solvatochromism of betaine dyes in aqueous alcohols: explicit consideration of the water-alcohol complex, J. Phys. Org. Chem., 2003, 16, 691–699 CrossRef CAS.
- E. B. Tada, P. L. Silva, C. Tavares and O. A. El Seoud, Thermo-solvatochromism of zwitterionic probes in aqueous aliphatic alcohols and in aqueous 2-alkoxyethanols: relevance to the enthalpies of activation of chemical reactions, J. Phys. Org. Chem., 2005, 18, 398–407 CrossRef CAS.
- P. L. Silva, E. L. Bastos and O. A. El Seoud, Solvation in binary mixtures of water and polar aprotic solvents: theoretical calculations of the concentrations of solvent-water hydrogen-bonded species and application to thermosolvatochromism of polarity probes, J. Phys. Chem. B, 2007, 111, 6173–6180 CrossRef CAS PubMed.
- C. Chylewski, Verallgemeinerung des isosbestischen Punktes, Angew. Chem., Int. Ed. Engl., 1971, 83, 214–215 CrossRef.
- J. R. Morrey, Fused Salt Spectrophotometry. III. Isosbestic Points Generated By Variation In Temperature, J. Phys. Chem., 1962, 66, 2169–2173 CrossRef CAS.
- J. Brynestad and G. P. Smith, Isosbestic points and internally linear spectra generated by changes in solvent composition or temperature, J. Phys. Chem., 1968, 72, 296–300 CrossRef CAS.
- M. D. Cohen and E. Fischer, 588. Isosbestic points, J. Chem. Soc., 1962, 3044 RSC.
- S. K. Christensen, M. C. Chiappelli and R. C. Hayward, Gelation of Copolymers with Pendent Benzophenone Photo-Cross-Linkers, Macromolecules, 2012, 45, 5237–5246 CrossRef CAS.
- C. Reichardt, Solvatochromic Dyes as Solvent Polarity Indicators, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- A. V. Gorelov, A. Du Chesne and K. A. Dawson, Phase separation in dilute solutions of poly (N-isopropylacrylamide), Phys. A, 1997, 240, 443–452 CrossRef CAS.
- P. W. Atkins and J. de Paula, Atkins’ physical chemistry, Oxford Univ. Press, Oxford u.a, 9th edn, 2010 Search PubMed.
- M. J. Tiera, G. R. dos Santos, V. A. O. Tiera, N. A. B. Vieira, E. Frolini, R. C. da Silva and W. Loh, Aqueous solution behavior of thermosensitive (N-isopropylacrylamide-acrylic acid-ethyl methacrylate) terpolymers, Colloid Polym. Sci., 2005, 283, 662–670 CrossRef CAS.
- L. Liu, Y. Shi, C. Liu, T. Wang, G. Liu and G. Zhang, Insight into the amplification by methylated urea of the anion specificity of macromolecules, Soft Matter, 2014, 10, 2856–2862 RSC.
- M. L. Ohnsorg, J. M. Ting, S. D. Jones, S. Jung, F. S. Bates and T. M. Reineke, Tuning PNIPAm self-assembly and thermoresponse: roles of hydrophobic end-groups and hydrophilic comonomer, Polym. Chem., 2019, 10, 3469–3479 RSC.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Thermoresponsive polymers with lower critical solution temperature: from fundamental aspects and measuring techniques to recommended turbidimetry conditions, Mater. Horiz., 2017, 4, 109–116 RSC.
- M. J. N. Junk, U. Jonas and D. Hinderberger, EPR spectroscopy reveals nanoinhomogeneities in the structure and reactivity of thermoresponsive hydrogels, Small, 2008, 4, 1485–1493 CrossRef CAS PubMed.
- M. J. N. Junk, R. Berger and U. Jonas, Atomic force spectroscopy of thermoresponsive photo-cross-linked hydrogel films, Langmuir, 2010, 26, 7262–7269 CrossRef CAS PubMed.
- F. M. Winnik, Phase transition of aqueous poly-(N-isopropylacrylamide) solutions: a study by non-radiative energy transfer, Polymer, 1990, 31, 2125–2134 CrossRef CAS.
- T. Kawaguchi, Y. Kojima, M. Osa and T. Yoshizaki, Cloud Points in Aqueous Poly(N-isopropylacrylamide) Solutions, Polym. J., 2008, 40, 455–459 CrossRef CAS.
- T. Kawaguchi, K. Kobayashi, M. Osa and T. Yoshizaki, Is a “cloud-point curve” in aqueous poly(N-isopropylacrylamide) solution binodal?, J. Phys. Chem. B, 2009, 113, 5440–5447 CrossRef CAS PubMed.
- G. J. Lee, D. Kim and M. Lee, Photophysical properties and photoisomerization processes of Methyl Red embedded in rigid polymer, Appl. Opt., 1995, 34, 138–143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01361k |
| This journal is © The Royal Society of Chemistry 2022 |