
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of transesterifications in reversible polycondensations and a reinvestigation of the Jacobson–Beckmann–Stockmayer experiments†
Hans R.
Kricheldorf
*a,
Steffen M.
Weidner
 b and
Jana
Falkenhagen
b
b and
Jana
Falkenhagen
b
aUniversität Hamburg, Institut für Technische und Makromolekulare Chemie, Bundesstrasse. 45, D-20145 Hamburg, Germany
bBundesanstalt für Materialforschung und -prüfung, BAM 6.3, Richard-Willstätter-Strasse 11, D-12489 Berlin, Germany
First published on 1st February 2022
Abstract
The polycondensations of adipic acid and 1,10-decanediol catalyzed by toluene sulfonic acid (TSA) were reinvestigated using MALDI TOF mass spectrometry and NMR spectroscopy. Unexpected reactions of TSA were detected along with incomplete conversion of the monomers. Furthermore, transesterification reactions of end-capped poly(1,10-decanediol adipate) and end-capped poly(ε-caprolactone) catalyzed by TSA were studied. Despite the quite different (ionic) reaction mechanisms, it was found that for polycondensations performed in bulk intermolecular transesterification is more efficient than the intramolecular “back-biting”; this scenario was not considered in the Jacobson–Stockmayer theory of reversible polycondensations. These results also confirm that the Jacobson–Stockmayer explanation of reversible polycondensations solely on the basis of ring chain equilibration is not only devoid of any experimental evidence, but also in contradiction to the results elaborated in this work.
Introduction
Following the footsteps of his former advisor Carothers, in 1946 Flory published the first theory of step growth polymerization with focus on polycondensation.1,2 His theory exclusively concerned irreversible polycondensations, meaning that the growing steps were irreversible and the polycondensates were the result of a kinetically controlled process. He had not been aware that another group of polycondensations might exist, namely reversible polycondensations. Reversible in this context means that each growing step is, in principle, reversible, and when the equilibration reactions responsible for the reversibility were fast, the entire polycondensation was under thermodynamic control at any stage. In two papers published in 1950, Jacobson, Beckmann and Stockmayer presented experimental facts suggesting that in proton catalyzed polyester syntheses reversible formation of cycles occurs via “back-biting”.3 Since a ring-chain equilibrium automatically entails a ring-ring and a chain-chain equilibrium, rapid “back-biting” has the consequence that the entire polycondensation process must be understood as a reversible kind of polymerization. On the basis of their experimental work, Jacobson and Stockmayer (J + S) developed a set of mathematical equations describing the entire course of reversible polycondensations.4 In the following four decades, other research groups, above all Semlyen and co-workers, studied the ring chain equilibria of numerous polymers.5–9 These authors demonstrated that ring-chain equilibration can, indeed, be achieved for a variety of polymers. An application of ring-chain equilibration theory to ring-opening polymerization was later contributed by Szymanski.10 These results were taken as evidence for the correctness of the JS approach as a general theory of reversible polycondensations.The first critique of the JS theory was published by the first author in 200311 and 2014,12 but did not suffice to rebut the widely accepted JS-theory. However, recently, the authors could demonstrate that reversible, like irreversible, polycondensations involve end-to-end (ete) cyclization, the existence of which was denied by J + S.13,14 The existence of ete-cyclization has the consequence that all condensation products must be cycles regardless of the monomer structure and reaction conditions. Even more recently, the authors have found that reversible polycondensations exist, which proceed without the contribution of back-biting up to high conversions (98 ± 1%).15 In such cases the reversibility is based on intermolecular trans-reactions also completely ignored by J + S. However, one must admit that analytical methods allowing for their detection were lacking at that time. This type of reversible polycondensation is, of course, totally outside the JS theory. The experimental work presented by the authors was based on cyclic monomers and various tin catalysts.13–15 However, the experimental work of Jacobson, Beckmann and Stockmayer (JBS) differs largely from that, because it is based on the polycondensations of 1,10-decanediol and adipic acid, and because toluene sulfonic acid (TSA) was used as the catalyst ((a) in Scheme 1). However, proton-catalyzed esterification and transesterification mechanisms (Scheme 1) and tin-carboxylate or phenoxide-catalyzed transesterification mechanisms (coordination–insertion mechanism) are quite different. To find out, to what extent the JBS experiments are comparable to the experiments and conclusions published by the authors, it seemed to be advisable to reinvestigate the JBS experiments by means of modern analytical methods. The focus of this study was on the existence and effectiveness of intermolecular transesterification reactions. If such equilibration reactions exist under the experimental conditions studied by JBS, their theory, and thus their mathematical equations, would have been built on the wrong assumption that back-biting is the only source of reversibility.
 | ||
| Scheme 1 Proton-catalyzed reaction mechanisms: (a) esterification of a carboxylic acid, (b) transesterification, and (c) formation of ether groups. | ||
Experimental
Materials
1,10-Decane diol, dimethyl adipate, adipoyl chloride, diethyl succinate (DESu), ε-caprolactone (εCL) toluene sulfonic acid monohydrate (TSA-H2O), methyl laurate, diethyl succinate (DESu), acetic anhydride and pure pyridine were purchased from Alfa Aesar (Karlsruhe, Germany) and used as received. Dry THF was purchased from Sigma (Taufkirchen, Germany), whereas dichloromethane was distilled over P4O10.Syntheses of telechelic poly(1,10-decane diol adipate)s (Table 1)
| Exp. no. | Code | Method | M n (meas.) | M w (meas.) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a A molar excess of 1,10-decane diol. b A molar excess of adipic acid. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1X | JBS-1 | JBS +5%)a slow bubbling | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 900 |
17700 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1Y | JBS-1R | Repetition of JBS-1X | 12000 |
18500 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2X | JBS-2 | JBS (+5%)a slow bubbl. + stir. | 6100 | 12500 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2Y | JBS-2R | Repetition of JBS-2X | 5800 | 12000 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | JBS-3 | JBS (+5%)a double amount of TSA slow bubbling | 9700 | 19200 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | JBS-4 | JBS (+5%)a fast bubbling | 9800 | 24500 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | JBS-5 | JBS (+5%)b slow bubbling | 3900 | 10500 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | PDA10 | Irrevers. polycondensation (+10%)a | 7100 | 14200 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | PDA5 | Irrevers. polycondensation (+5%)a | 9200 | 19100 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | PDA0 | Irrevers. polycondensation (+0%)a | 31000 |
74000 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In a parallel experiment a magnetic bar was added, and the melt was stirred until the increasing viscosity stopped the stirrer (JBS-2). In a third analogous experiment 750 mg of TSA-H2O were used as a catalyst (JBS-3).
Irreversible polycondensation with adipoyl chloride. 1,10-Decane diol (110 mmol) and adipoyl chloride (100 mmol) were weighed into a 250 mL round bottom flask and a magnetic bar was added under a blanket of argon. Dry dichloromethane (120 mL) was added immediately thereafter, and a solution of pyridine (210 mmol) in dichloromethane (40 mmol) was added rapidly, but dropwise under cooling with ice. Approx. 20 h later most of the pyridine hydrochloride was removed by washing with water (250 mL) and the remaining part was concentrated in vacuo to about 150 mL and precipitated into cold methanol (1.6 L). The precipitated telechelic polyester (PDA 10) was isolated after 1 h by filtration and dried in vacuo at 40 °C; yield: 88%. The 1H NMR spectrum indicated an average degree of polymerization of an analogous polycondensation performed with 105 mmol 1,10-decanediol (PDA 5); yield: 83%.
Equilibration of JBS type PDAs (“100% polyesters”) (Tables 2 and 3)
The polyesters JBS-1Y or JBS-3 (20 mmol, for exp. no. 1 and 2 or 10 mmol for exp. no. 3 and 4) were weighed into a 50 mL Erlenmeyer flask, and a magnetic bar and chlorobenzene were added. The reaction vessel was placed into an oil bath thermostated at 130 °C and samples (approx. 1 mL) were taken after 2.5 and 50 h by means of a warm pipette and used for direct characterization by means of MALDI TOF mass spectrometry and gel permeation chromatography.| Exp. no. | Conc. (mol L−1) | Time (h) | Cycles | M n | M w | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Starting material JBS-1Y. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0a | 4.0a | 0 | 0 | 12000 |
18500 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1A | 1.0 | 2.5 | Traces | 10400 |
18500 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1B | 1.0 | 50 | + | 9600 | 18300 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2A | 0.5 | 2.5 | + | 9500 | 17700 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2B | 0.5 | 50 | ++ | 8700 | 16900 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3A | 0.2 | 2.5 | + | 10000 |
17000 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3B | 0.2 | 50 | ++ | 8500 | 14500 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4A | 0.1 | 2.5 | ++ | 10500 |
16500 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4B | 0.1 | 50 | +++ | 6700 | 10000 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Exp. no. | Conc. (mol L−1) | Time (h) | Cycles | M n | M w |
|---|---|---|---|---|---|
| 0 | 4.0 | — | — | 9700 | 19200 |
| 1A | 1.0 | 2.5 | Traces | 10100 |
20100 |
| 1B | 1.0 | 50 | + | 11700 |
20200 |
| 2A | 0.5 | 2.5 | + | 9700 | 17200 |
| 2B | 0.5 | 50 | ++ | 12800 |
19200 |
| 3A | 0.2 | 2.5 | + | 8400 | 16000 |
| 3B | 0.2 | 50 | ++ | 10200 |
16300 |
| 4A | 0.1 | 2.5 | ++ | 6100 | 11200 |
| 4B | 0.1 | 50 | +++ | 6800 | 9300 |
Equilibrations of PDA5 and cPDA (Tables 4 and S1†)
tPDA-5 (2.9 g ∼ 10 mmol of PDA units) was weighed into a 50 mL Erlenmeyer flask; chlorobenzene (6.5 mL), TSA (0.4 mL of a 0.5 M solution in chlorobenzene) and a magnetic bar were added. This reaction mixture corresponded to a ca. 1 M solution of PDA with regard to the repeat units. The reaction vessel was placed into an oil bath thermostated at 130 °C and after 2.5, 25 and 196 h, approx. 1 mL of the reaction mixture was removed for direct characterization with GPC and MALDI TOF measurements.| Exp. no. | Substrate | Conc. (mol L−1) | Temp. (°C) | Time (h) | M n | M w |
|---|---|---|---|---|---|---|
| 0 | PDA5 | 4.0 | — | — | 9200 | 19100 |
| 1 | PDA5 | 1.0 | 130 | 2.5 | 9200 | 14500 |
| 2 | PDA5 | 1.0 | 130 | 25.0 | 8800 | 14000 |
| 3 | PDA5 | 1.0 | 130 | 50.0 | 9800 | 15000 |
| 4 | PDA5 | 0.2 | 109 | 2.5 | 10200 |
14300 |
| 5 | PDA5 | 0.2 | 109 | 25.0 | 10800 |
14500 |
| 6 | PDA5 | 0.2 | 109 | 50.0 | 13100 |
15700 |
Two parallel experiments were performed with 1.48 g of PDA5, 23 mL of chlorobenzene and 0.2 mL of a 0.5 M solution of TSA corresponding to a 0.1 M solution of DA units.
Equilibration of cPDA in 1 M solution was conducted analogously using TSA at a DA/Cat ratio of 50/1.
Acetylation of telechelic poly(decane diol adipate), PDA5
PDA5 (100 mmol) was dissolved in dichloromethane (150 mL); acetic anhydride (50 mmol) and pyridine (0.5 mL) were added, and the reaction mixture was refluxed for 24 h. It was then precipitated into cold methanol and after 2 h the precipitated polyester was isolated by filtration. Complete acetylation was evidenced using an 1H NMR spectrum (acetate signal at 2.23 ppm). Mn = 19500, Mw = 19800.
Transesterification of PDA5/AcPDA5 with methyl laurate (Table 5)
PDA5 or AcPDA5 (40 mmol) and methyl laurate (10 mmol) were weighed into a 50 mL Erlenmeyer flask and a magnetic bar was added. The reaction vessel was placed into an oil bath thermostated at 130 °C. After 1 h and 2 h a sample was taken and characterized by MALDI TOF mass spectrometry and GPC measurements.| Exp. no. | Starting material | Temp. (°C) | Time (h) | M n | M w | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Properties of the staring materials. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0a | PDA5 | — | — | 9200 | 19100 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1A | PDA5 | 130 | 1 | 3000 | 6100 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1B | PDA5 | 130 | 2 | 2900 | 5400 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2A | PDA5 | 109 | 1 | 3300 | 7000 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2B | PDA5 | 109 | 2 | 3000 | 6300 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0a | AcPDA5 | — | — | 9400 | 19800 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3A | AcPDA5 | 130 | 1 | 3200 | 7200 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3B | AcPDA5 | 130 | 2 | 3100 | 6600 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4A | AcPDA5 | 109 | 1 | 3400 | 7500 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4B | AcPDA5 | 109 | 2 | 3200 | 6800 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Synthesis of end-capped PCL: AcPCLUn
AcPCLUn-1: 11-Undecenol (2 mmol) was weighed into a 100 mL flame-dried Erlenmeyer flask; ε-caprolactone (200 mmol) and a magnetic bar were added under a blanket of argon. The reaction vessel was placed into an oil bath thermostated at 130 °C for 3 h. PCL was dissolved in 80 mL of dry dichloromethane, and acetic anhydride (20 mmol) and pyridine (1 mL) were then added. After 6 d at 20–22 °C the reaction mixture was poured into methanol (1 L). After 1 h, the precipitated and crystallized PCL was isolated by filtration and dried at 30 °C in vacuo. Yield: 91%; the MALDI TOF mass spectrum indicated complete acetylation. Mn = 22000, Mw = 32000.
AcPCLUn-2: 11-Undecenol (30 mmol) and εCL (180 mmol) were polymerized as described above. Crude poly(εCL) was dissolved in 80 mL of dichloromethane, and acetic anhydride (20 mmol) and pyridine (5 mL) were added. After 1 h of reflux the reaction mixture was allowed to cool to 20 °C and worked up after 20 h. Yield 86%; the MALDI TOF mass spectrum indicated 50% acetylation; Mn = 15000, Mw = 23500.
Transesterification of AcPCLUn (Table 6)
AcPCLUn-1 (4.6 g, 40 mmol εCL units) and DESu (20 mmol) were weighed into a 50 mL Erlenmeyer flask, a magnetic bar was added, and the reaction vessel was then placed into an oil bath thermostated at 130 °C. After stirring with a spatula under a blanket of argon, TSA (0.4 mL of a 0.5 M solution in warm chlorobenzene) was added. After approx. 30 min the magnetic bar began to rotate indicating a significant loss of viscosity. After 1 h or 2 h samples (1 mL) were removed for direct characterization.| Exp. no. | Substrate | Temp. (°C) | Time (h) | M n | M w |
|---|---|---|---|---|---|
| 0 | AcPCLUn-1 | — | — | 13000 |
37000 |
| 1A | AcPCLUn-1 | 130 | 1.0 | 3400 | 6700 |
| 1B | AcPCLUn-1 | 130 | 2.0 | 3200 | 6800 |
| 0 | AcPCLUn-2 | — | — | 15000 |
23500 |
| 2A | AcPCLUn-2 | 130 | 1.0 | 4900 | 6700 |
| 2B | AcPCLUn-2 | 130 | 2.0 | 3000 | 3400 |
| 4A | AcPCLUn-2 | 109 | 1.0 | 6500 | 9800 |
| 4B | AcPCLUn-2 | 109 | 2.0 | 5300 | 6500 |
AcPCLUn-2 (4.6 g, 40 mmol εCL units) and DESu (20 mmol) were reacted as described above using an oil bath thermostated either at 130 °C or at 109 °C.
Measurements
400 MHz 1H NMR spectra were recorded with a Bruker Avance 400 in 5 mm sample tubes. CDCl3 containing TMS served as the solvent and shift reference.MALDI TOF mass spectrometry was performed using an Autoflex max (Bruker Daltonik GmbH, Bremen) using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) dissolved in chloroform (20 mg mL−1) as a matrix in the linear positive mode. The solution was doped with 5 μL of potassium trifluoroacetate dissolved in THF (3 mg mL−1) to promote potassium adduct ion formation. The matrix solution was premixed with the analyte solutions (chloroform, 4 mg mL−1) in a ratio of 5/2 (v/v). 1 μL of the resulting solution was spotted using a pipette on the stainless-steel sample target. Typically, 8000 single spectra recorded at 4 randomly chosen positions within one spot were accumulated. The spectrometer was previously calibrated with PEO standards.
For the GPC experiments a modular LC system running at 40 °C (isocratic pump, 1 mL min−1, Optilab reX, Wyatt, Germany) was used. Samples were manually injected (100 μL, 2–4 mg mL−1). For instrument control and data calculation Astra 6.1 (Wyatt) was used. The calibration was performed using polystyrene standard sets (Polymer Standards Service – PSS, Mainz, Germany). All Mn and Mw data presented in this work are uncorrected. For the determination of the Mark–Houwing–Sakurada (MHS) relationship a viscometer (Viscostar, Wyatt, Germany) and a multiangle laser light scattering (MALS) detector (Dawn EOS, Wyatt, Germany) were used. Astra 6.1 software (Wyatt) was used for calculating the MHS curves.
Results and discussion
Syntheses of telechelic PDAs (tPDAs) according to the JBS method
JBS was used to prepare a telechelic PDA from 1,10-decanediol and adipic acid with a 5 mol% excess of diol using TSA-H2O as a catalyst. This polycondensation was conducted in bulk at 109 °C, and when the viscosity did not change anymore (after 5 d), the authors concluded that 100% conversion of the adipic acid was reached. They called this PDA (shown in Scheme 2) “100% polyester”. This polyester was then used to study ring-chain equilibration in dichlorobenzene. Therefore, it played a key role in the work of JBS and reproduction of its synthesis was the first step of the present work. Unfortunately, the description of the experiments by JBS is not satisfactory for example with regard to the catalyst. The commercial form of TSA is the monohydrate and water-free TSA must be prepared, usually by azeotropic distillation with solvents such as toluene or chlorobenzene. JBS did not mention, if the monohydrate or neat TSA was used, but because water was eliminated during the polycondensation it does not matter whether neat or hydrated TSA was added. Since only the monohydrate is commercially available, the authors used TSA-H2O. Concerning the stoichiometry, JBS mentioned that 2.4 weight% were used, but they did not define the basis of this calculation. For the first two experiments of the present study and their repetition (no. 1X, Y and 2X, Y, Table 1), the authors used 2.4 weight% relative to adipic acid, because protonation of the COOH group is the key step of the esterification process ((a) in Scheme 1). In the third experiment 2.4 weight% of TSA-H2O relative to the sum of both monomers were used (no. 3, Table 1). | ||
| Scheme 2 Syntheses of telechelic poly(1,10-decane diol adipate) (a) via proton catalyzed reversible polycondensation and (b) via pyridine-catalyzed irreversible polycondensation. | ||
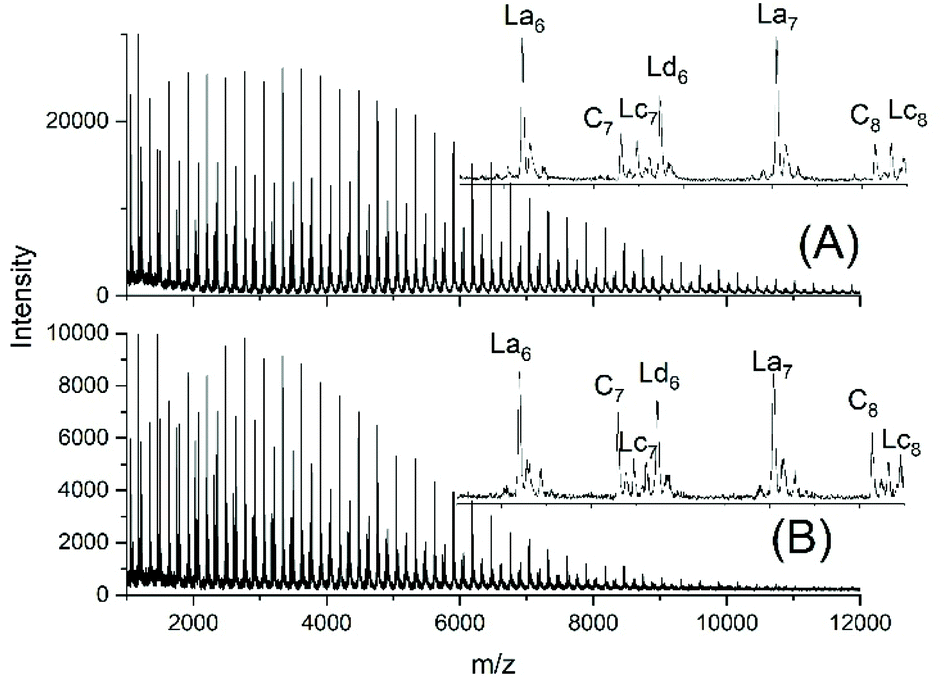
Another problem for an exact repetition of the JBS experiments was the method of “stirring”. JBS did not use a stirrer but achieved motion of the molten oligoesters just by bubbling purified nitrogen through the reaction mixture. The shape of the reactor and the rate of bubbling were not defined. The authors performed the first three experiments with relatively slow bubbling (no. 1X, Y, XY, 3). The first experiment resulted in a mass spectrum with three unexpected features (Fig. 1A). Firstly, it showed a trimodal distribution of peak intensities. Secondly, it displayed a peak of Lc chains, (Scheme 3) indicating incomplete conversion and it displayed peaks of Ld chains having one tosylate end group in addition to a CH2OH group. The reproducibility was confirmed by repetition of the experiment.
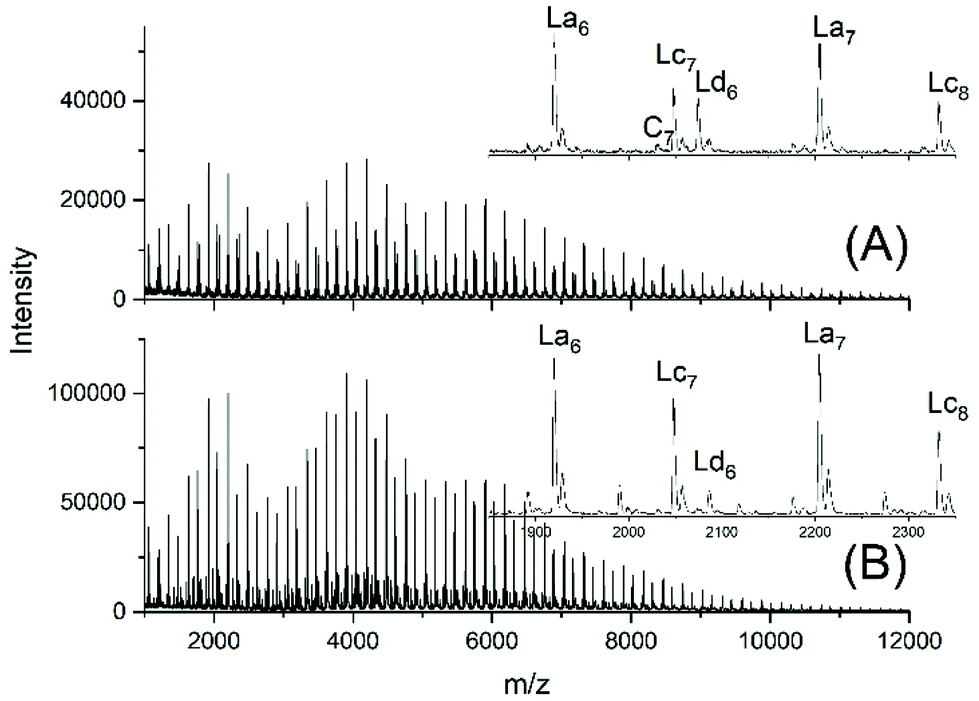 | ||
| Fig. 1 MALDI TOF mass spectra of telechelic PDA prepared with 5 mol% excess of 1,10-decane diol via the JBS method with 2.4 weight% of TSA-H2O relative to adipic acid: (A), slow bubbling (JBS-1R, Table 1) and (B) slow bubbling with stirring (JBS-2, Table 1). | ||
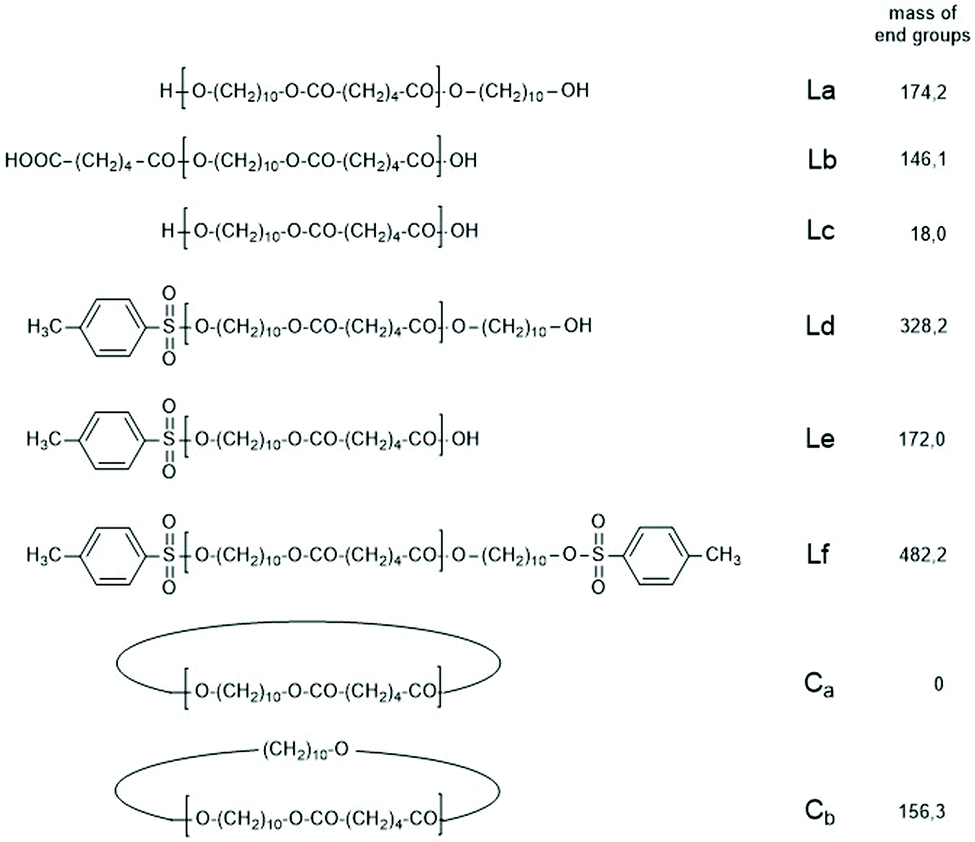 | ||
| Scheme 3 Potential reaction products of the reversible polycondensation of 1,10-decane diol and adipic acid catalyzed by 4-toluene sulfonic acid (TSA). | ||
To improve homogenization and conversion, a second experiment was conducted using a small magnetic bar for stirring (no. 2X, Y, Table 1). However, the trimodal distribution of peak intensities did not vanish, the molecular weights were significantly lower than those obtained without stirring and the mass spectra indicated more small peaks of unidentified byproducts (Fig. 1B). These poor results proved again the reproducibility, but a more detailed study of this unexpected phenomenon was not of interest for the purpose of this work. A third experiment was conducted with 2.4 weight% of TSA relative to the sum of both monomers accompanied by slow bubbling (no. 3, Table 1). The MALDI TOF mass spectrum (Fig. S1†) displayed the same features as those of the JBS-2 samples. A significant improvement of the conversion was not achieved.
This unsatisfactory situation resulted in a fourth experiment with relatively fast bubbling of argon, whereas all other experimental conditions were identical to those of experiment no. 1. Now a quite different mass spectrum was obtained (Fig. 2A).
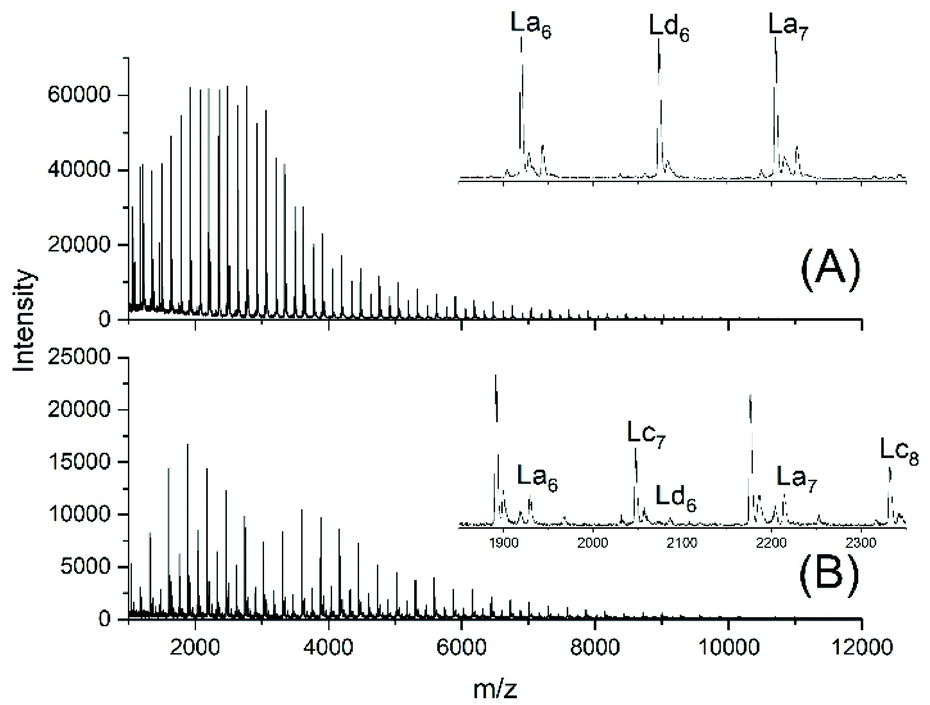 | ||
| Fig. 2 MALDI TOF mass spectra of telechelic polyLAs prepared via the JBS method at 109 °C: (A) 5 mol% excess of 1,10-decanediol, fast bubbling (JBS-4, Table 1) and (B) 5 mol% excess of adipic acid, slow bubbling (JBS-5, Table 1). | ||
The trimodal distribution had vanished and the conversion of adipic acid was nearly complete, so that, at first glance, this experiment complied with what the authors had expected from the JBS experiment. However, the peaks of Ld chains having one tosylate end group were particularly intensive, suggesting that almost all TSA was esterified, whereas the peaks of the doubly tosylated chains (Lf) were barely detectable. A tosylate group may, in principle, be formed by esterification of TSA with CH2OH groups or by acidolytic cleavage of ester groups in the PDA backbone. The large predominance of the mono-tosylated Ld chains suggests that the tosylate groups were mainly formed by esterification during the first stage of the polycondensation (<50% conversion) when the reaction mixture contained large amounts of 1,10-decane diol and CH2OH terminated oligomers. This suggestion was supported by a fourth type of polycondensation where a 5 mol% excess of adipic acid was used (no. 5). Such a polycondensation was not performed by JBS, but it had the benefit that it helped to explain the formation of tosylate end groups. The mass spectrum displayed the peaks of Lc chains, indicating incomplete conversion in analogy to all other experiments conducted with slow bubbling, whereas the peak representing tosylate terminated (Ld) chains was extremely weak (Fig. 2B). Since the concentration of CH2OH groups in that polycondensation experiment was much lower than those in polycondensations with an excess of diol, the mass spectrum of JBS-5 supports the hypothesis that the tosylate groups are mainly formed by esterification of CH2OH groups. Further experimental results confirming this hypothesis are discussed in the following section. Regardless of the mechanism, the formation of tosylate end groups has three consequences relevant to the course of the polycondensation. First, it reduces the number of CH2OH groups, second, it produces water and third, it reduces the concentration of acidic protons and thus, the catalytic activity of the system.
Finally, it should be noted that the shapes of the GPC curves of samples JBS-1 through JPB-5 (listed in Table 1) were also reflected by the peak distribution in the MALDI mass spectra. This means that when the mass spectrum was trimodal the corresponding GPC curve also revealed tri- or even multimodal mass distribution. Likewise, monomodal GPC curves were obtained when the mass spectra showed the monomodal distribution of mass peaks. Fig. S2† illustrates this analogy.
Synthesis of PDAs by irreversible polycondensations
For comparison, two telechelic PDA diols based on an excess of 10 or 5 mol% of 1,10-decanediol were prepared by irreversible polycondensation of the diol with adipoyl chloride and pyridine in dry solvents (PDA10 and PDA5, Table 1). The Mn and Mw values of the PDA5 sample were quite similar to those of the JBS-1 samples. The mass spectrum revealed a characteristic difference, because as expected the peak of tosylated chains (Ld) and the peaks were absent (Fig. 3A). In contrast, small peaks of cyclics were observable. Their intensities were lower for the PDA10 sample. When a polycondensation was performed with equimolar amounts of both monomers, their MALDI mass spectra almost exclusively displayed the peaks of cycles (Fig. S3†). These samples were used for equilibration studies (see below). | ||
| Fig. 3 MALDI TOF mass spectra of telechelic PDA prepared with 5 mol% excess of 1,10-decanediol via irreversible polycondensation: (A) virgin product (PDA5, Table 1) and (B) after treatment with TSA for 24 h at 130 °C (no. 2, Table 4). | ||
Equilibration of telechelic PDAs
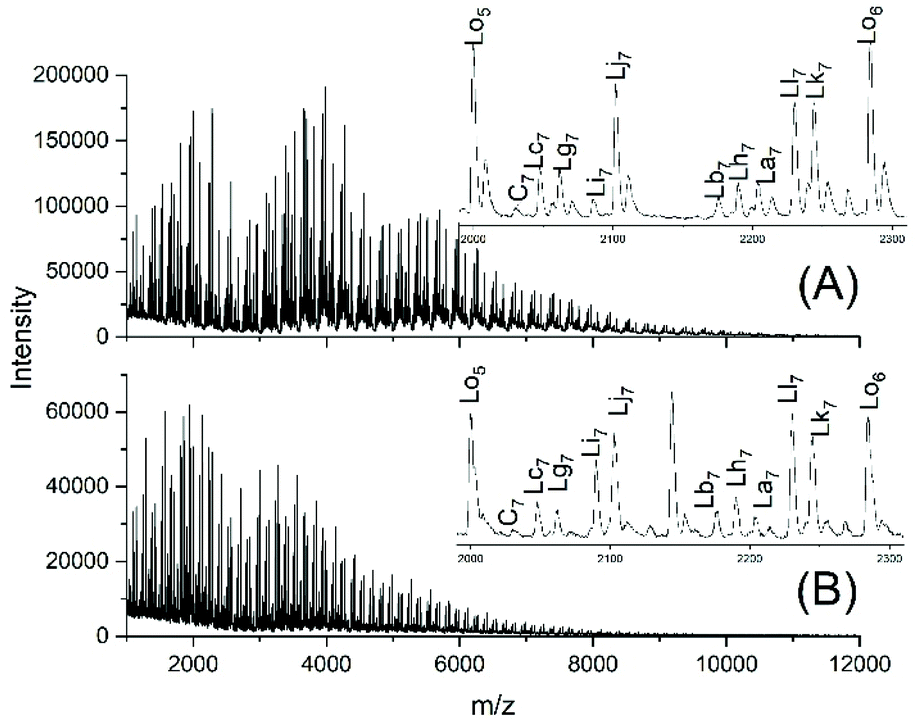
JBS diluted their “100%” polyesters with dichlorobenzene and thermostated these solutions at 130 °C for several hours or days. They observed a decreasing viscosity with time but an increasing viscosity upon concentration of the dilute solutions and concluded that these phenomena result from a reversible formation of small cycles via “back-biting”. Direct spectroscopic evidence for the formation of cycles was not available at that time. Again, JBS delivered a poor description of their experiments, because the solution with the maximum concentration was given as “weight fraction 1.000” (Table 1 in ref. 3). We must confess that the meaning of this term was not clearly understandable. Neat PDA has a molarity around 4 M and, thus, in our experiments neat PDA was diluted in four steps by a factor of 40 (Tables 2 and 3), assuming that this procedure covers the concentrations studied by JBS. The MALDI TOF mass spectra (Fig. 4 and 5) demonstrated that, as expected by JBS, cycles were formed the fraction of which increased with time and with decreasing concentration of the JBS sample. Hence, the existence of ring-chain equilibration assumed by JBS just on the basis of viscosity measurements was confirmed by the results of the experiments listed in Tables 2 and 3.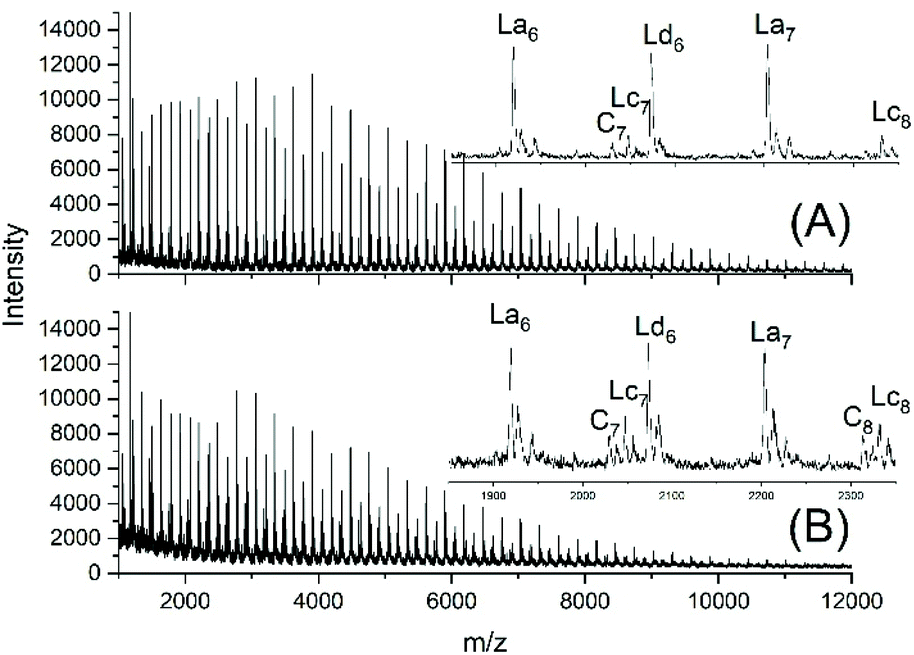 | ||
| Fig. 4 MALDI TOF mass spectra of equilibrated JBS-1R (no. 1Y, Table 1): (A) 1 M solution after 2.5 h (no. 1A, Table 2), (B) 1 M solution after 50 h (no. 1B, Table 2). | ||
 | ||
| Fig. 5 MALDI TOF mass spectra of equilibrated JBS-1R (no. 1Y, Table 1): (A) 0.1 M solution after 2.5 h (no. 4A, Table 3), (B) 0.1 M solution after 50 h (no. 4B, Table 3). | ||
For comparison with JBS samples characterized by incomplete conversion and having tosylate end groups, further equilibration experiments were conducted with PDA5 (Table 4). Water-free TSA was used, and the time was varied (Fig. 3B and Fig. S4†). In the absence of the solvent, several side reactions occurred (Fig. 3B). However, Fig. S4† clearly demonstrates that the fraction of cycles increased when the concentration was lowered. Therefore, these experiments confirm the existence of a ring chain equilibration under the JBS conditions. Only tiny peaks of tosylated Ld chains were observable.
Analogous experiments were performed with cyclic PDA (Table S1†). The most interesting result was that the peaks of tosylated chains (Ld, Le, Lf) were barely detectable. Since CH2OH groups were present in PDA5, but not in cPDA, these results indicate that the vast majority of tosylated chains are formed by esterification of CH2OH groups and not by acidolytic transesterification of ester groups. Hence, these results and those mentioned above for PDA5 equilibration confirm the suggestion presented in the first section in connection with the samples JBS-4 and JBS-5. The tosylate end groups are primarily or exclusively formed by esterification of TSA with CH2OH groups. Since the mechanism of this reaction was not relevant for the purpose of this work, a more detailed study in this direction was not intended.
Transesterification experiments
The first series of experiments was performed with telechelic PDAs. For this purpose, PDA5 (no. 6, Table 1) was acetylated with an excess of acetic anhydride and pyridine. The resulting AcPDA5 was characterized by mass spectrometry and GPC measurements. Methyl laurate served as a reaction partner for three reasons: low volatility, good miscibility with the molten PDA and unambiguous identification of the transesterification products by MALDI TOF mass spectrometry. Potential reaction products of the transesterification reactions are summarized in Scheme 3 along with the symbols used for peak assignments in the mass spectra. In addition to the acetylated PDA5, the starting material PDA5 having two OH end groups was used for the transesterification experiments. Furthermore, two temperatures were used, 109 and 130 °C, according to the temperatures used by JBS for the synthesis of telechelic PDA5 and for equilibration measurements. Moreover, the time was varied from 1 h to 2 h. All these experiments are summarized in Table 5.Regardless of the temperature, time and starting material, the mass peaks of the transesterification products having lauroyl or methyl ester end groups (Scheme 4) were found in the mass spectra of all experiments.
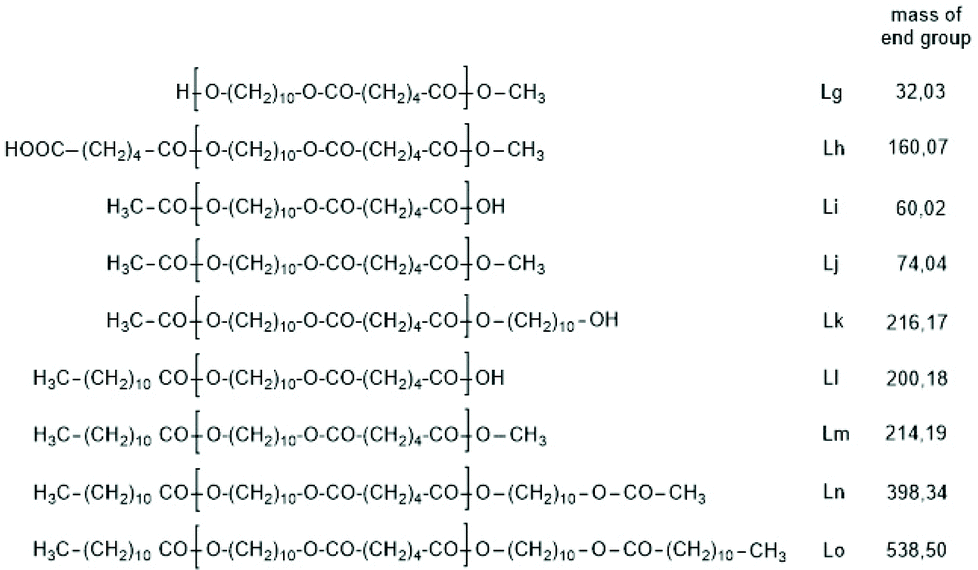 | ||
| Scheme 4 Potential reaction products of the transesterifications of PDA5 or AcPDA-5 with methyl laurate catalyzed by neat TSA. | ||
Fig. 6A and B show the complexity of the reaction products. The appearance of the intensive peaks of transesterification products such as Lh, Lj and Li-Lo after 1 h, even at 109 °C, indicates that the intermolecular transesterifications were fast and efficient. Since tiny peaks of cycles became only detectable after 2.5 h and since the equilibration experiments described in Tables 2 and 3 demonstrated that ring chain equilibration requires more than 2.5 h (in agreement with the original JBS experiments), the results of these transesterification experiments clearly demonstrate that intermolecular transesterifications were considerably faster.
 | ||
| Fig. 6 MALDI TOF mass spectra of the transesterification products of PDA5 or AcPDA5 with methyl laurate: (A) PDA5 after 1 h/109 °C (no. 2A, Table 5), (B) AcPDA5 after 2 h/130° (no. 3B, Table 5). | ||
A second series of transesterification experiments was conducted with end-capped poly(εCL) to make clear that the results obtained from end-capped PDA are not unique, but also valid for other aliphatic polyesters.
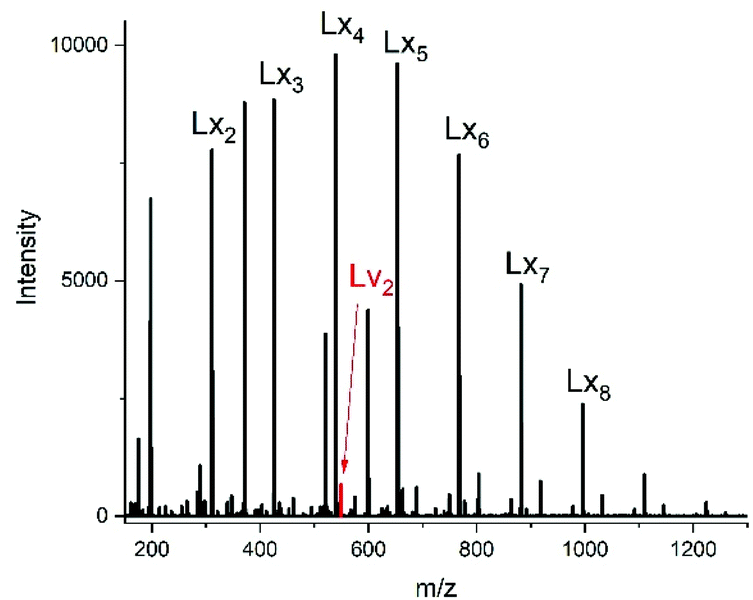
For this purpose, two ROPs of εCL catalyzed by SnOct2 were initiated with 11-undecenyl alcohol (Un) at a LA/In ratio of 100/1 (yielding PCLUn-1) and at a LA/In ratio of 60/1 (yielding PCLUn-2). The CH2OH end groups were fully acetylated in the case of PCLUn-1 yielding (AcPCLUn-1) or acetylated by 50% (yielding AcPCLUn-2). The resulting AcPCLUn samples were characterized by MALDI TOF mass spectrometry (Fig. S5†) and by GPC measurements. Diethyl succinate was used as a reaction partner because of its low volatility and its good miscibility with the Ac-PCL-Un samples and because MALDI TOF mass spectrometry allowed for unambiguous identification of the transesterification products. The six transesterification experiments listed in Table 6 were catalyzed by water-free TSA, with variation of temperature and time. The interpretation of the results proved to be quite simple. All experiments gave, in principle, the same result, namely almost complete transesterification with two consequences. First, a significant reduction of the molecular weight as evidenced by the GPC data and by the mass spectra presented in Fig. 7 and Fig. S6† was found. A comparison with Fig. S5† illustrates this point. The second consequence was the formation of poly(εCL) containing a succinate group and two ethyl ester end groups (Lx, Ly, Scheme 5) Lx and Ly are isomers and thus not distinguishable by mass spectrometry) as the main reaction product (Fig. 7 and S5†). These structures result from the fact that an εCL unit/DESu ratio of 2/1 was used corresponding to a 100/1 or 50/1 ratio of ethyl ester/undecenyl ester end groups. Due to this high molar ratio, almost all Ac and Un end groups were replaced by succinyl and ethyl end groups respectively. These data indicate, in turn, that intermolecular transesterifications were quite rapid and efficient even at 109 °C. The peaks of cycles were not detectable even in the ESI TOF mass spectra, a typical example of which is displayed in Fig. 8. In summary, all these transesterification experiments demonstrate that catalysis with TSA and with tin compounds agree in that intermolecular transesterification reactions are more efficient than equilibration via back-biting. In polymer melts intermolecular transesterifications may be slower than in the experiments with liquid esters conducted in this work. However, even when the efficiency of intermolecular transesterification is not higher than that of back-biting, the conclusions presented below are still valid.
 | ||
| Fig. 7 MALDI TOF mass spectra of the transesterification products obtained from AcPCLUn-1 and DESu at130 °C (A) after 1 h (no. 1A, Table 6) and (B) after 2 h (no. 1B, Table 6). | ||
 | ||
| Scheme 5 Potential reaction products of the transesterifications of AcPCLUn with DESu catalyzed by neat TSA. | ||
 | ||
| Fig. 8 ESI mass spectra of the transesterification of Ac-PCL-Un with DESu at 130 °C/2 h (no. 1B, Table 6). | ||
Discussion
The results of the transesterification experiments are in good agreement with those reported previously for tin-catalyzed experiments. The coordination–insertion mechanism involved when catalysts containing Sn–O–R groups and other covalent metal oxide compounds are used, or the proton catalyzed reactions (Scheme 1, (a) and (b)) are the reaction mechanisms underlying almost all syntheses of polyesters via reversible polycondensations. Therefore, it is unlikely that a reversible polycondensation exists, which obeys the hypothesis of JBS. Even if such “JBS polycondensation” will be found, it has the status of an exotic exception and cannot serve as a basis of a general theory of polycondensations. The reasons for the fact that the JS concept of reversible polycondensations has survived for many decades is mainly the JS theory of ring-chain equilibration with the calculation of equilibrium constants. This part of the JS Theory is per se correct and suggested that when this part of the JS work is correct all other parts must be correct too. The results of the present work and two previous publications demonstrate that this conclusion is not justified. At this point the authors wish to emphasize that they fully respect the work of J, B and S, since in the years before 1950 detailed structural analysis was not feasible due to the lack of suitable analytical methods. However, this does not mean that incorrect hypotheses should be preserved for ever. The failure of the JS hypothesis of reversible polycondensations is, of course, not a unique case in the history of science, and the famous biologist Thomas Huxley has summarized such scenarios with the following words: “It is a [typical] tragedy of science, when a beautiful theory is slayed by an ugly fact”. In the present case the ugly fact is simply the existence of efficient intermolecular transesterification in combination with end-to-end cyclization.Conclusions
The results of this work allow for the following three conclusions. First, a quantitative esterification of adipic acid heated with an excess of 1,10-decanediol is difficult to achieve, mainly because the esterification of the catalyst TSA severely reduces its catalytic efficiency. Second, the ring-chain equilibration assumed for dilute solutions of the telechelic polyesters can be confirmed. Third, intermolecular transesterification reactions are more efficient than ring-chain equilibration, regardless of whether an acid or a tin compound is used as a catalyst. This means, in turn, that the JS hypothesis4 which explains the course of reversible polycondensations exclusively on the basis of ring-chain equilibration is in contradiction to the results of this work. Together with the results of previous studies it may now be concluded that reversible polycondensations obeying the JS theory do not exist.Author contributions
HRK-conceptualization, investigation, supervision, and writing-original draft; SMW-investigation, methodology, visualization, writing-review & editing; JF-investigation and visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank BAM for technical support and A. Myxa (BAM, Berlin) for the numerous GPC measurements. Dr F. Scheliga (TMC, Hamburg) kindly provided the chemicals.Notes and references
- W. H. Carothers, Chem. Rev., 1931, 8, 353–426 CrossRef CAS.
- P. J. Flory, Chem. Rev., 1946, 39, 137–197 CrossRef CAS PubMed.
- H. Jacobson, C. O. Beckmann and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1607–1612 CrossRef CAS.
- H. Jacobson and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1600–1606 CrossRef CAS.
- J. A. Semlyen, in Large Ring Molecules, ed. J. A. Semlyen, J. Wiley & Sons, Chichester, 1996, ch. 1 Search PubMed.
- J. A. Semlyen, Cyclic Polymers, Springer, 2000, pp. 1–46 Search PubMed.
- J. A. Semlyen, in Cyclic Polymers, ed. J. A. Semlyen, Elsevier, Oxford, 1986, p. 1 Search PubMed.
- S. J. Clarson, in Cyclic Polymers, ed. J. A. Semlyen, Kluver Academic Publ., Dordrecht, 2nd edn, 2000, ch. 5 Search PubMed.
- J. B. Carmichael, D. J. Gordon and F. J. Isackson, J. Phys. Chem., 1967, 71, 2011–2015 CrossRef CAS.
- R. Szymanski, in Comprehensive Polymer Science, ed. K. Matyjaszewski and M. Möller, Elsevier BV, Amsterdam, 2012, vol. 4, pp. 31–49 Search PubMed.
- H. R. Kricheldorf, M. Al Masri and G. Schwarz, Macromolecules, 2003, 36, 8648–8651 CrossRef CAS.
- H. R. Kricheldorf, Polycondensation – History and New Results, Springer, 2014, ch. 11 Search PubMed.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Polym. Chem., 2020, 11, 2595–2604 RSC.
- H. R. Kricheldorf and S. M. Weidner, J. Polym. Sci., 2021, 59, 439–450 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and J. Falkenhagen, Polym. Chem., 2021, 12, 5003–5016 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01679b |
| This journal is © The Royal Society of Chemistry 2022 |