DOI:
10.1039/D2PY00405D
(Paper)
Polym. Chem., 2022,
13, 3471-3478
Organocatalytic selective coupling of episulfides with carbon disulfide for the synthesis of poly(trithiocarbonate)s and cyclic trithiocarbonates†
Received
31st March 2022
, Accepted 17th May 2022
First published on 26th May 2022
Abstract
This work reports how to efficiently utilize carbon disulfide (CS2) as a sulfur-containing C1 resource for the purpose of synthesizing both linear poly(trithiocarbonate)s and cyclic trithiocarbonates. When initiated by PPNCl or phosphazene benzoxide, the copolymerization of CS2 with episulfides affords perfectly alternating poly(trithiocarbonate)s in the absence of any catalyst. In contrast, when initiated by tetrabutylammonium halide, the same coupling reaction of CS2 with episulfides results in the formation of cyclic trithiocarbonates. The role played by initiating onium salts in the linear/cyclic selectivity as well as reaction conditions such as the temperature, the type of solvent, and the feeding ratio of CS2 to episulfides was investigated. Lastly, when placed in the presence of a radical source or treated with UV irradiation, poly(trithiocarbonate)s undergo complete unzipping to produce cyclic trithiocarbonates. This remarkable feature has been harnessed to prepare degradable polymers using oligotrithiocarbonates as self-immolating linkers between dithiol alkyl precursors.
Introduction
As an abundant and cheap resource, CS2 is used in the production of rayon, cellulose films and carbon tetrachloride. Incorporation of sulfur atoms affords materials with special properties such as high refractive indices,1,2 heavy metal binding properties,3 conducting4–6 and biological activity.7 However, in comparison with the extensive work carried out on isoelectronic carbon dioxide,8–21 research on CS2 as a sulfur-containing C1 resource has been very limited, including its coupling with epoxides or episulfides. The copolymerization of CS2 with epoxides to produce poly(dithiocarbonate)s and its coupling with the same epoxides to generate cyclic dithiocarbonates have been investigated by Zhang,22–24 Darensbourg24–26 and other groups;27–33 due to O/S scrambling reactions, various products were generated with different structures. In contrast, such O/S scrambling reactions are nonexistent when CS2 is copolymerized with episulfides. However, the control of the chemoselectivity between linear poly(trithiocarbonate)s and cyclic trithiocarbonates generated during the copolymerization of CS2 with episulfides is a real challenge. The first report of copolymerization of CS2 with episulfides can be traced back to the 1970s. Soga et al.34,35 used CdEt2, ZnEt2 and Hg(SBu)2 as catalysts to copolymerize CS2 with episulfides. These catalysts showed low catalytic activity and the resulting polymers contained 93% of polythioether linkages. Recently, Zhu et al.36 using DFT calculations revealed that in spite of a lower bond dissociation energy (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S in CS2 is indeed equal to 105.3 kcal mol−1, compared to 127.2 kcal mol−1 of CO in CO2) of CS compared to CO, a higher energy is required to activate CS2, rendering its copolymerization more challenging.
S in CS2 is indeed equal to 105.3 kcal mol−1, compared to 127.2 kcal mol−1 of CO in CO2) of CS compared to CO, a higher energy is required to activate CS2, rendering its copolymerization more challenging.
In 2007, Nozaki and co-workers first reported the successful copolymerization of CS2 with propylene sulfide (PS) resulting in the formation of a well-defined alternating poly(trithiocarbonate) when initiated by PPNCl and activated by a chromium salen complex.37 The polymerization could be optimized to obtain poly(trithiocarbonate)s up to 42.6 kg mol−1 molar mass with a 92% linear/cyclic selectivity. Zhang et al. on the other hand used an asymmetrical Schiff base chromium complex for such copolymerizations and obtained poly(trithiopropylene carbonate)s with molar masses limited to 10 kg mol−1, a linear/cyclic selectivity of 88% and increased yields.38 Recently, Silvano et al.39 reported the copolymerization of cyclohexene sulfide with CS2, and used the same (salen) CrCl complex as the one reported by Nozaki. In the latter case, poly(trithiocyclohexenecarbonate)s were obtained with a lower linear/cyclic selectivity (76%) and a broader polydispersity. Because of the high thiophilicity of the transition metal used, chances are high for the produced poly(trithiocarbonate)s to be contaminated with metallic residues, which may prevent their use in biomedical applications or as optical materials. In addition, such an approach requires a multistep synthesis of organometallic catalysts.
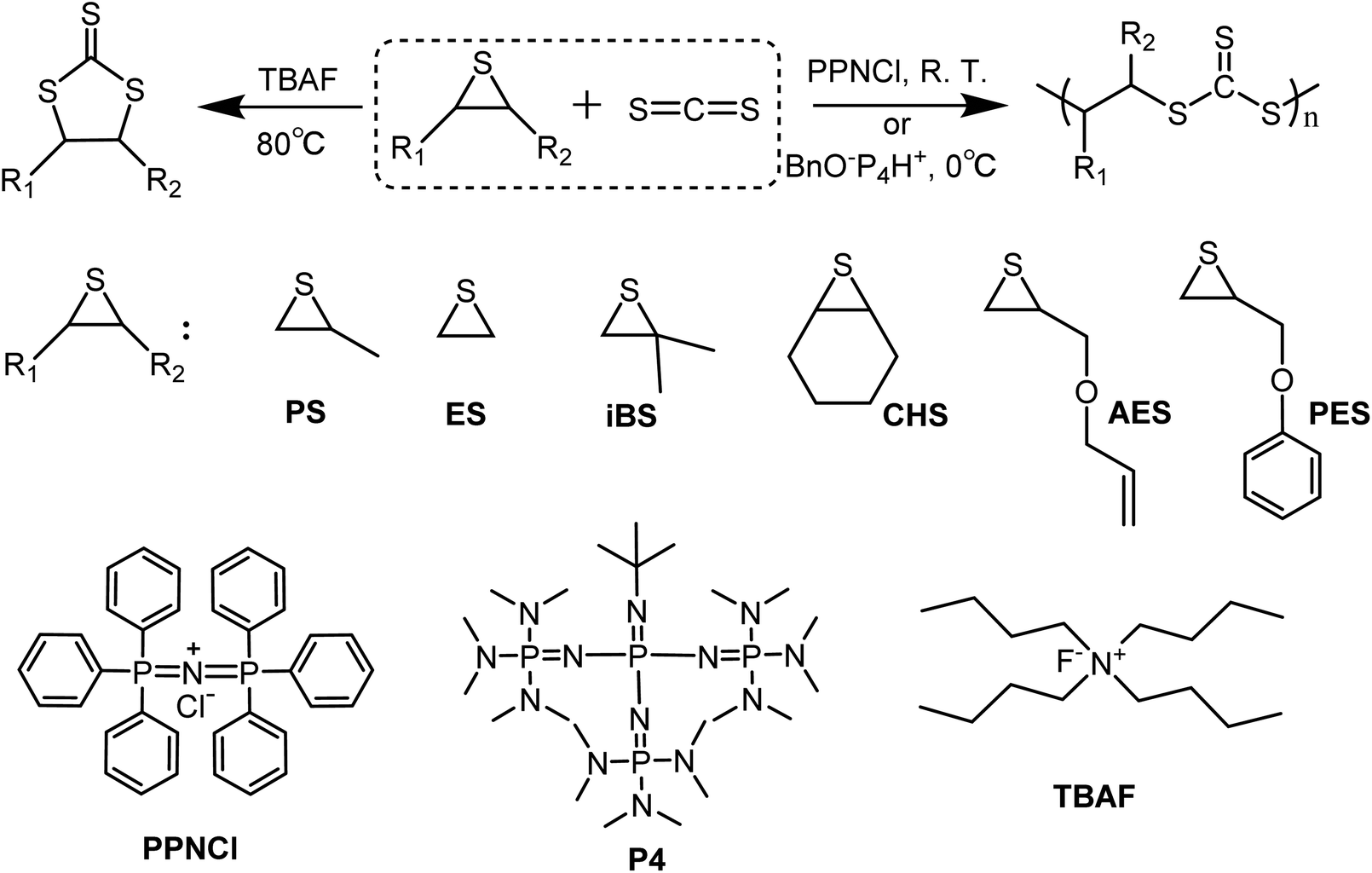
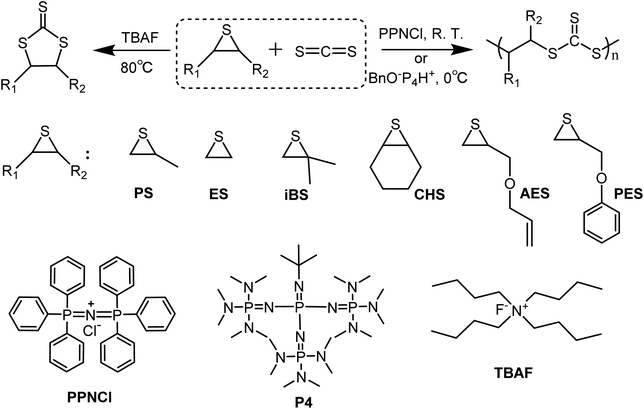
In this work, we investigate the potential of truly metal-free initiators and wish to demonstrate that such an approach is viable to selectively produce either poly(trithiocarbonate)s or cyclic trithiocarbonates (Scheme 1) through the coupling of CS2 with episulfides. When either PPNCl or phosphazenium benzoxide was used as an initiator, fully alternating poly(trithiocarbonate)s could be indeed obtained with a linear/cyclic selectivity up to 95% under conditions different from those considered by Nozaki and without resorting to a transition metal-based activator. On the other hand, when tetrabutylammonium fluoride was used at high temperatures, only cyclic trithiocarbonates were produced with very high selectivity. Lastly, the self-immolative behavior of the obtained poly(trithiocarbonate)s was demonstrated for the first time, a process triggered by a radical source or UV irradiation resulting in the degradation of linear poly(trithiocarbonate)s into cyclic trithiocarbonates.
 |
| | Scheme 1 Selective coupling reaction of CS2 and episulfides: copolymerization of CS2 with propylene sulfide to produce linear poly(trithiopropylene carbonate); coupling CS2 with episulfides to produce cyclic trithiocarbonates. | |
Results and discussion
Copolymerization of CS2 with episulfides to produce linear poly(trithiocarbonate)s
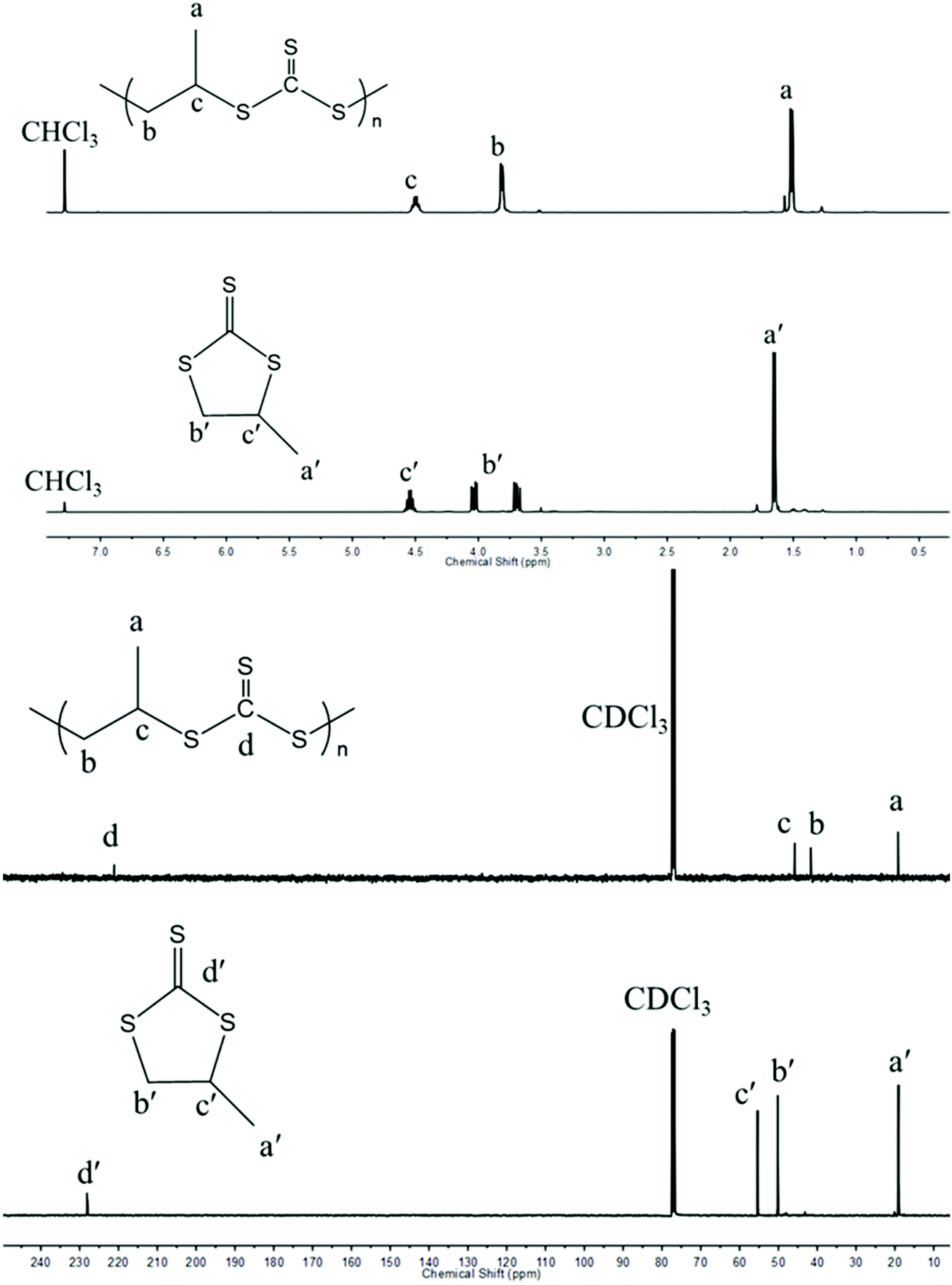
In contrast to Nozaki and coworkers who described the copolymerization of CS2 with PS using PPNCl along with a chromium salen complex,37 we tried to carry out such copolymerizations without any activator and under metal-free conditions. As shown in entry 1, Table 1, PS (Fig. S1 and S2†) could be ring-opened using PPNCl alone as the initiator unlike PO which remained unchanged under similar conditions. When copolymerizing PS with an equal molar ratio of CS2 for a period of 12 h and under similar conditions to those described by Nozaki, a poly(trithiocarbonate) (PPSC) sample was obtained with 59% linear/cyclic selectivity (entry 2, Table 1 and Fig. S3†) and 67% overall yield was obtained. In contrast to our work, Nozaki and coworkers discontinued their copolymerization after 5 h and obtained a lower yield (16%) and a lower linear vs. cyclic selectivity of 37% in the absence of an activator.37 The poly(trithiocarbonate) sample obtained by us showed a perfectly alternating structure in agreement with the reported literature. The structures of PPSC and cyclic trithiocarbonates (CTC) after purification were respectively determined by 1H NMR (Fig. 1) and FTIR (Fig. S4†). Only three groups of peaks at 4.48, 3.79, and 1.49 ppm corresponding to the linear trithiocarbonate enchainment were detected; the integration ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3 and the absence of thioether linkages from 2.6–3.0 ppm (ref. 40) confirmed the perfectly alternating structure. As interestingly noted by Nozaki, changing the feeding ratio of PS to CS2 in favor of the latter compound resulted in a higher selectivity (entries 3–6, Table 1). Molar masses of PPSC reaching values as high as 45.4 kg mol−1 with 67% linear/cyclic selectivity (entry 3, Table 1) could be obtained for the PPSC sample corresponding to entry 3 in Table 1. With a ratio of 1:5 between PS and CS2, a linear/cyclic selectivity of 79% could be obtained (entry 5, Table 1); in contrast, only CTC could be isolated for a ratio of 2:1 between PS and CS2 (entry 6, Table 1). Besides the nature of the anion initiating the polymerization – here Cl− – the above results show that thioanions are mainly responsible for the production of CTCs. Upon using an excess amount of CS2 with respect to PS, the addition of CS2 by growing ether thioanions is favored over back-biting reactions. On the other hand, the trithiocarbanions formed being less nucleophilic than ether thioanions do not cause back-biting by reaction with trithiocarbonate functions.37 Increasing further the amount of CS2 improved slightly the linear vs. cyclic selectivity but slowed down the overall copolymerization. Carrying out copolymerization in a non-coordinating solvent such as toluene which is unable to solvate ion pairs also helped to increase the linear vs. cyclic selectivity up to 87% (entry 7, Table 1). Finally the addition of hydroxyl or thiol-ended transfer agents that can serve as initiating species was also beneficial for the production of linear polymeric trithiocarbonates against cyclic trithiocarbonates. As indicated above, the initiation by Cl− generates halide-terminated chains that favor back-biting reactions because of the electronegativity of the terminal Cl. Upon using thio-terminated transfer agents, such back-biting occurring at the very early stage of the polymerization could almost be avoided. In the presence of thiols, the linear/cyclic selectivity indeed reached up to 84% (entries 10–12 in Table 1); the 1H NMR characterization results confirm that 1-dodecanethiol (DoSH) acted as a transfer agent and thus as a chain initiator, being effectively incorporated at the α-end of the produced PPSC (Fig. S5†) and the 1H NMR calculated molar mass (5.6 kg mol−1) is in agreement with the one obtained with SEC (6.0 kg mol−1); in contrast, alcohols such as benzyl alcohol (BnOH) and tert-butyl alcohol (tBuOH) were less effective than DoSH at initiating chains probably due to the lower basicity of thioanions and their inability to abstract protons from alcohols (entries 8 and 9 in Table 1) and generate oxanions. In the latter case, the molar masses of the generated chains were little affected by the presence of BnOH or tBuOH. Further investigation of the copolymerization of CS2 with another episulfide, ethylene sulfide (ES), under the same conditions as above (entry 13 in Table 1) afforded a polymer which is insoluble in common organic solvents. Upon characterization of the crude polymerization mixture by IR and gravimetry, the conversion appeared to be almost complete with a yield of 90% for poly(trithioethylene carbonate); the linear/cyclic selectivity reached 95%, which is a much higher value than that obtained for poly(trithiocarbonate) generated from PS (67%, entry 2 in Table 1). The higher reactivity of ES compared to PS and the insolubility of the polymer generated have favored propagation over back-biting.
:2:3 and the absence of thioether linkages from 2.6–3.0 ppm (ref. 40) confirmed the perfectly alternating structure. As interestingly noted by Nozaki, changing the feeding ratio of PS to CS2 in favor of the latter compound resulted in a higher selectivity (entries 3–6, Table 1). Molar masses of PPSC reaching values as high as 45.4 kg mol−1 with 67% linear/cyclic selectivity (entry 3, Table 1) could be obtained for the PPSC sample corresponding to entry 3 in Table 1. With a ratio of 1:5 between PS and CS2, a linear/cyclic selectivity of 79% could be obtained (entry 5, Table 1); in contrast, only CTC could be isolated for a ratio of 2:1 between PS and CS2 (entry 6, Table 1). Besides the nature of the anion initiating the polymerization – here Cl− – the above results show that thioanions are mainly responsible for the production of CTCs. Upon using an excess amount of CS2 with respect to PS, the addition of CS2 by growing ether thioanions is favored over back-biting reactions. On the other hand, the trithiocarbanions formed being less nucleophilic than ether thioanions do not cause back-biting by reaction with trithiocarbonate functions.37 Increasing further the amount of CS2 improved slightly the linear vs. cyclic selectivity but slowed down the overall copolymerization. Carrying out copolymerization in a non-coordinating solvent such as toluene which is unable to solvate ion pairs also helped to increase the linear vs. cyclic selectivity up to 87% (entry 7, Table 1). Finally the addition of hydroxyl or thiol-ended transfer agents that can serve as initiating species was also beneficial for the production of linear polymeric trithiocarbonates against cyclic trithiocarbonates. As indicated above, the initiation by Cl− generates halide-terminated chains that favor back-biting reactions because of the electronegativity of the terminal Cl. Upon using thio-terminated transfer agents, such back-biting occurring at the very early stage of the polymerization could almost be avoided. In the presence of thiols, the linear/cyclic selectivity indeed reached up to 84% (entries 10–12 in Table 1); the 1H NMR characterization results confirm that 1-dodecanethiol (DoSH) acted as a transfer agent and thus as a chain initiator, being effectively incorporated at the α-end of the produced PPSC (Fig. S5†) and the 1H NMR calculated molar mass (5.6 kg mol−1) is in agreement with the one obtained with SEC (6.0 kg mol−1); in contrast, alcohols such as benzyl alcohol (BnOH) and tert-butyl alcohol (tBuOH) were less effective than DoSH at initiating chains probably due to the lower basicity of thioanions and their inability to abstract protons from alcohols (entries 8 and 9 in Table 1) and generate oxanions. In the latter case, the molar masses of the generated chains were little affected by the presence of BnOH or tBuOH. Further investigation of the copolymerization of CS2 with another episulfide, ethylene sulfide (ES), under the same conditions as above (entry 13 in Table 1) afforded a polymer which is insoluble in common organic solvents. Upon characterization of the crude polymerization mixture by IR and gravimetry, the conversion appeared to be almost complete with a yield of 90% for poly(trithioethylene carbonate); the linear/cyclic selectivity reached 95%, which is a much higher value than that obtained for poly(trithiocarbonate) generated from PS (67%, entry 2 in Table 1). The higher reactivity of ES compared to PS and the insolubility of the polymer generated have favored propagation over back-biting.
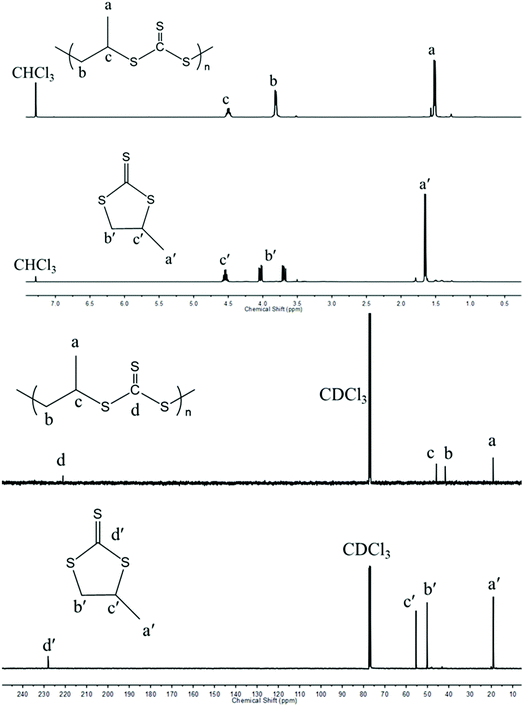 |
| | Fig. 1
1H (top) and 13C NMR (bottom) spectra of PPSC and CTC in CDCl3. | |
Table 1 Ammonium salt-initiated copolymerization of episulfides and CS2a
| Entry |
PS:CS2:I |
CTA:I |
Conv.b (%) |
Sel.b (%) |
M
n(theo.)b (kg mol−1) |
M
n(SEC)c (kg mol−1) |
Đ
|
|
The polymerizations were performed at 25 °C for 12 h unless otherwise noted; PPNCl was used as the initiator (I).
Determined by 1H NMR spectroscopy.
Determined by GPC in THF and with linear polystyrene as the standard.
Not analyzed.
Toluene (equal volume to monomers) was used as the solvent.
Ethylene sulfide was used as the monomer; conversion and selectivity were calculated by gravimetry.
|
| 1 |
100:0:1 |
|
5 |
— |
— |
NA |
NAd |
| 2 |
500:500:1 |
|
67 |
59 |
29.6 |
35.0 |
1.4 |
| 3 |
500:1000:1 |
|
49 |
67 |
24.6 |
45.4 |
1.4 |
| 4 |
500:1500:1 |
|
40 |
74 |
22.2 |
31.0 |
1.3 |
| 5 |
500:3000:1 |
|
9 |
79 |
— |
NA |
NA |
| 6 |
1000:500:1 |
|
62 |
0 |
— |
NA |
NA |
| 7e |
500:500:1 |
|
45 |
87 |
29.4 |
28.0 |
1.4 |
| 8 |
500:1000:1 |
BnOH (10/1) |
32 |
79 |
20.0 |
37.5 |
1.4 |
| 9 |
500:1000:1 |
tBuOH (10/1) |
47 |
75 |
26.4 |
34.6 |
1.5 |
| 10 |
500:1000:1 |
DoSH (1/1) |
57 |
80 |
17.1 |
21.3 |
1.5 |
| 11 |
500:1000:1 |
DoSH (5/1) |
52 |
82 |
5.3 |
9.3 |
1.5 |
| 12 |
500:1000:1 |
DoSH (10/1) |
64 |
84 |
3.7 |
6.0 |
1.6 |
| 13f |
500:1000:1 |
|
90 |
95 |
— |
NA |
NA |
Besides increasing the ratio of CS2 to PS and using appropriate transfer agents, we considered another factor, namely the temperature of the polymerization to favor propagation over back-biting. Using phosphazenium alkoxide obtained by deprotonation of benzyl alcohol with the superbase P4, we carried out the copolymerization of CS2 with PS at various temperatures. At 60 °C only cyclic trithiocarbonates were produced whereas at 0 °C and below (entries 3 and 6 Table 2) the level of linear vs. cyclic selectivity reached values (90%) comparable to those claimed in Nozaki's work where a salen chromium complex was used as an activator. However, lower temperatures caused a slower rate of polymerization. All the PPSC samples obtained at low temperatures exhibited narrow molar distributions with molar masses around 10 kg mol−1. Because of the slow rate of polymerization at 0 °C, we did not attempt to obtain samples of higher molar masses.
Table 2 P4 catalyzed copolymerization of PS and CS2a
| Entry |
T (°C) |
PS:CS2:I |
Conv.b (%) |
Selectivityb (%) |
M
n, theo.b (kg mol−1) |
M
nc (kg mol−1) |
Đ
|
|
The polymerizations were performed under neat conditions for 12 h and the ratio of P4 to BnOH was set as 1:1 unless otherwise noted.
Determined by 1H NMR spectroscopy.
Determined by GPC in THF and with linear polystyrene as the standard.
The polymerizations were performed for 24 h.
|
| 1 |
30 |
100:600:1 |
80 |
77 |
9.2 |
10.1 |
1.4 |
| 2 |
60 |
100:600:1 |
100 |
0 |
|
|
|
| 3 |
0 |
100:600:1 |
53 |
90 |
7.2 |
8.2 |
1.3 |
| 4 |
−10 |
100:600:1 |
45 |
92 |
6.1 |
7.3 |
1.3 |
| 5 |
−20 |
100:600:1 |
21 |
90 |
2.8 |
3.2 |
1.3 |
| 6 |
−40 |
100:600:1 |
9 |
92 |
|
|
|
| 7d |
0 |
200:1200:1 |
84 |
83 |
20.9 |
21.5 |
1.4 |
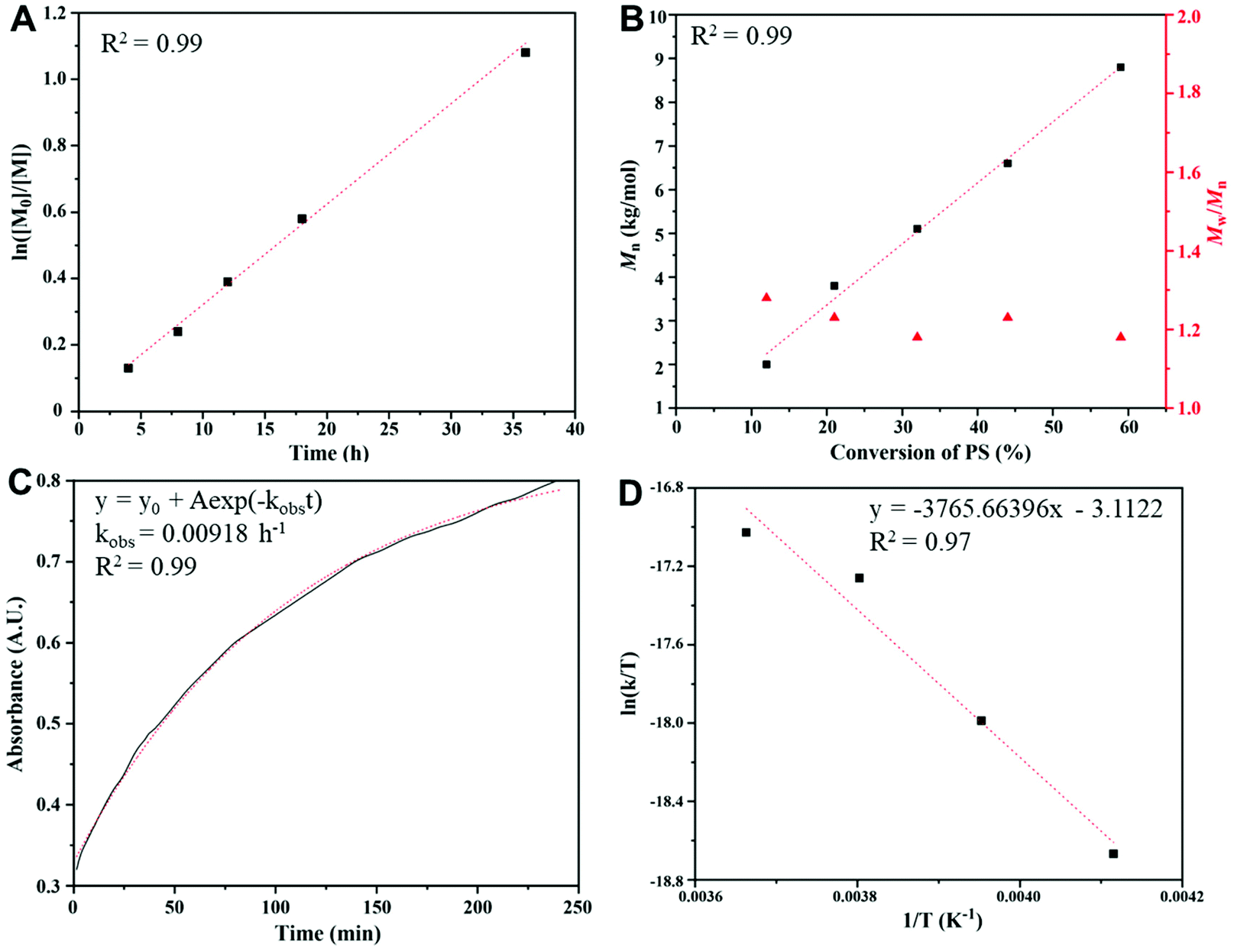
A kinetic study (Fig. 2) of the copolymerization of CS2 with PS was performed under the following conditions: a feeding ratio of 600:100 for CS2:PS at −10 °C with BnOH/P4 as the initiator. The ln([M0]/[M]) vs. time plot (Fig. 2A) shows a first order character in the studied range of conversions. The molar masses of PPSC samples (Fig. 2B) increase linearly with monomer conversion with narrow polydispersity indexes (Đ ≤ 1.3), indicating a “living” character. The in situ FT-IR (Fig. S6†) real-time online monitoring at 1180 cm−1, assigned to the CS bond, allowed us to follow the buildup of trithiocarbonate functions. The adsorption vs. time plot (Fig. 2C) showed an exponential fit that also demonstrated a first order polymerization character. Based on the in situ FT-IR monitoring results at different temperatures (0, −10, −20, −30 °C, Fig. 2D), the enthalpy and entropy of the copolymerization of PS and CS2 could be derived (ΔH = 7.48 kcal mol−1, ΔS = −53.37 cal mol−1 K−1) and an activation energy for the formation of linear PPSC equal to 21.5 kJ mol−1 was deduced. This value of the activation energy is much lower than the one determined by Darensbourg et al. for the copolymerization of CO2 with epoxides when catalyzed by the salen Cr catalyst (67.6 kJ mol−1).41 This confirms that the formation of linear poly(trithiocarbonate)s is easier than that of linear polycarbonates and that it can be carried out at lower temperatures.
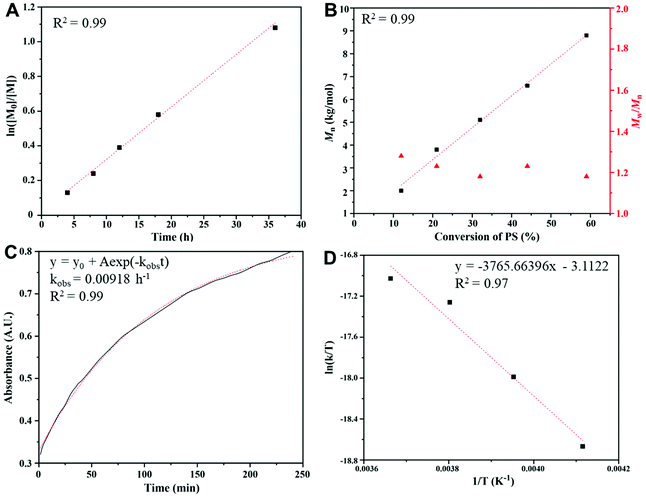 |
| | Fig. 2 Kinetics study of copolymerization of CS2 with propylene sulfide initiated by phosphazenium benzoxide at low temperature: (A) plot of ln([M0]/[M]) vs. time; (B) plots of Mn and Đ vs. monomer conversion; (C) the absorption intensity of trithiocarbonates at 1180 cm−1 measured by in situ FT-IR spectroscopy; and (D) plot of ln(k/T) vs. 1/T for the formation of PPSC. | |
Coupling of CS2 with episulfides to form cyclic trithiocarbonates
Cyclic trithiocarbonate (CTC) derivatives are key intermediates for the synthesis of tetrathiafulvalene derivatives.4–6 Because of S/O scrambling reactions, the coupling of CS2 with epoxides generates a mixture of cyclic trithiocarbonates and dithiocarbonates that is tedious to separate.29–33 Upon direct coupling of CS2 with episulfide catalyzed by cyclic and linear amidines,42 it is possible to isolate cyclic trithiocarbonates as shown by Aoyagi and Endo. Choi and coworkers immobilized amine into a silica support in an attempt at recycling the catalyst but the cyclic trithiocarbonates were contaminated with linear polymers.43 Here we demonstrate that using an appropriate initiator with a more electronegative anion than Cl− and higher temperatures, the coupling of CS2 with PS essentially yields cyclic trithiocarbonates. The activation energy for the formation of cyclic trithiocarbonates is such that above 60 °C with F− as the anion, back-biting reactions largely predominate over propagation. Whatever the episulfides tried, the outcome of their coupling with CS2 was the exclusive formation of the corresponding cyclic trithiocarbonates as shown by 1H NMR characterization (entries 9–13, Table 3; Fig. S7–15†).
Table 3 Coupling reaction of episulfides and CS2a
| Entry |
Catalyst |
Monomer |
T (°C) |
PS:CS2:Cat |
Conv.b (%) |
TOFc (h−1) |
Selectivityb (%) |
|
The coupling reaction was performed under neat conditions for 12 h.
Determined by 1H NMR spectroscopy.
Turnover frequency (TOF) = (mole of episulfide consumed)/(mole of ammonium × h). TBAF, TBABr, TBAI, and TBAAc are tetraammonium fluoride, bromide, iodide, and acetate, respectively; PS: propylene sulfide, ES: ethylene sulfide, CHS: cyclohexene sulfide, AES: allyl ether episulfide, PES: phenyl ether episulfide, iBS: isobutylene sulfide. |
| 1 |
TBABr |
PS |
25 |
500:500:1 |
<1 |
<1 |
|
| 2 |
TBAI |
PS |
25 |
500:500:1 |
<1 |
<1 |
|
| 3 |
TBAF |
PS |
25 |
500:500:1 |
45 |
18.8 |
80 |
| 4 |
TBAAc |
PS |
25 |
500:500:1 |
78 |
32.5 |
63 |
| 5 |
TBAF |
PS |
60 |
500:500:1 |
>99 |
41.7 |
>99 |
| 6 |
TBAF |
PS |
60 |
5000:5000:1 |
91 |
379 |
>99 |
| 7 |
TBAF |
PS |
80 |
10000:10000:1 |
83 |
691 |
>99 |
| 8 |
TBAF |
PS |
80 |
20000:20000:1 |
57 |
950 |
>99 |
| 9 |
TBAF |
ES |
80 |
20000:20000:1 |
>99 |
1667 |
>99 |
| 10 |
TBAF |
CHS |
80 |
5000:5000:1 |
90 |
375 |
>99 |
| 11 |
TBAF |
AES |
80 |
5000:5000:1 |
32 |
133 |
>99 |
| 12 |
TBAF |
PES |
80 |
5000:5000:1 |
30 |
125 |
>99 |
| 13 |
TBAF |
iBS |
80 |
5000:5000:1 |
92 |
383 |
>99 |
Thermal behavior of poly(trithiocarbonate)s and their degradation triggered by a radical source
The thermal behavior of PPSC was characterized by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) under a nitrogen flow. As shown in Fig. S16,† PPSC exhibited a glass transition temperature (Tg) of about 25 °C in agreement with the reported value.37 Its TGA analysis showed the onset of degradation at around 170 °C (Fig. S17†), a profile that reminds that of poly(propylene carbonate). On the other hand, PESC which is obtained by copolymerization of ethylene sulfide with CS2 showed a lower Tg value (below 20 °C, Fig. S18†) than PPSC but a similar decomposition profile (Fig. S19†). As for the refractive index (nD), it is known that sulfur-containing polymers exhibit large values; indeed, the nD value for PPSC was found to be equal to 1.78 (via an Abbe refractometer, cast film, 25 °C), which is one of the largest nD values among the reported sulfur-containing polymers.
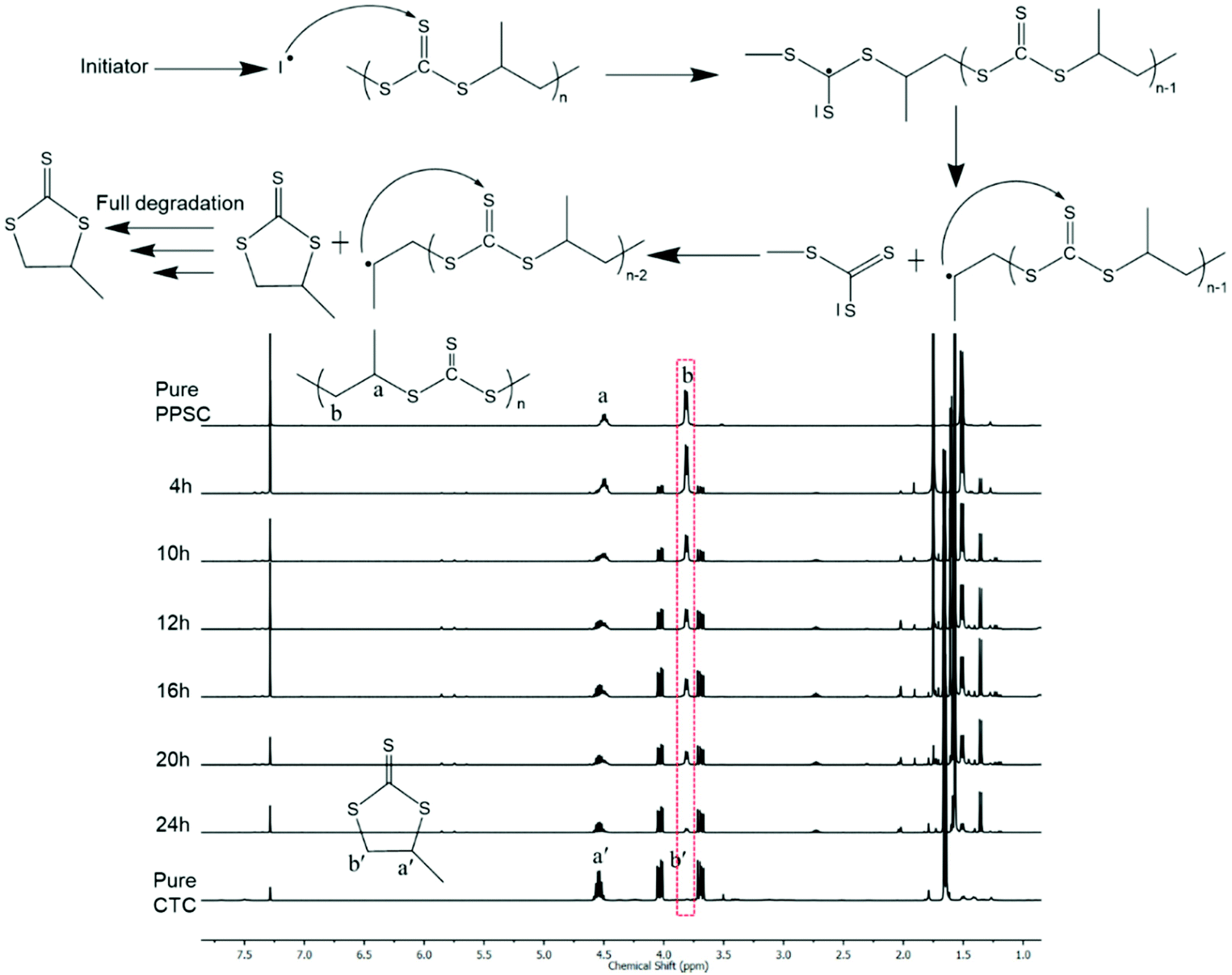
Unlike polycarbonates that can be chemically degraded or depolymerized only under anionic or enzymatic conditions,44,45 poly(trithiocarbonate)s are vulnerable to radical attack. Yet attempts at utilizing poly(trithiocarbonate)s as RAFT platforms to grow under controlled conditions polymer chains or blocks by radical means are scarce.46,47 The examples found of such polymeric RAFT agents that were used to control radical polymerization of vinyl monomers were all prepared by polycondensation through the coupling of ditrithiocarbonate anions with a difunctional electrophilic reagent. With these works in mind, we subjected our poly(trithiocarbonate)s to radical reactions. In the presence of 2,2′-azobis(2-methylpropionitrile) (AIBN) used as a radical source, poly(trithiocarbonate)s undergo complete unzipping that proceeded via radical mechanisms, resulting in the formation of cyclic trithiocarbonates (CTC). As shown in Fig. 3, PPSC degraded very fast in the presence of 5% (wt/wt) of AIBN at 70 °C and almost fully degraded into CTC in 24 h. As a comparison, PPSC remains unchanged in a control experiment under the same conditions in the absence of AIBN. Knowing that PPSC can be radically degraded by AIBN, we further explored its degradation under UV irradiation. As shown in Fig. S20,† PPSC also undergoes degradation under UV irradiation but at a much slower rate than that observed in the presence of AIBN: under UV irradiation, 75% of PPSC was decomposed into CTC after 60 h.
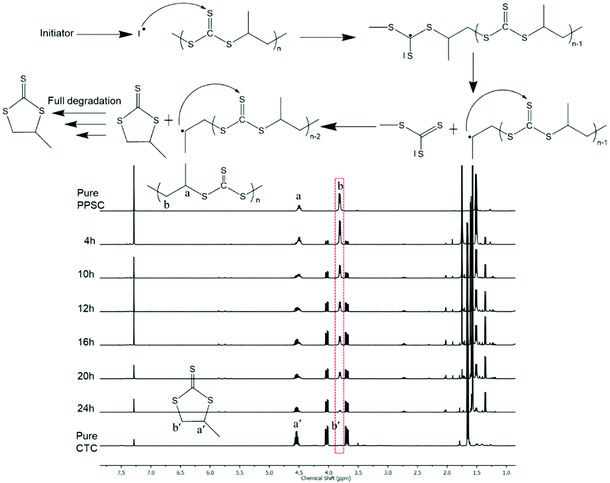 |
| | Fig. 3
1H NMR monitoring degradation of PPSC using AIBN at 70 °C and a plausible radical induced degradable process. | |
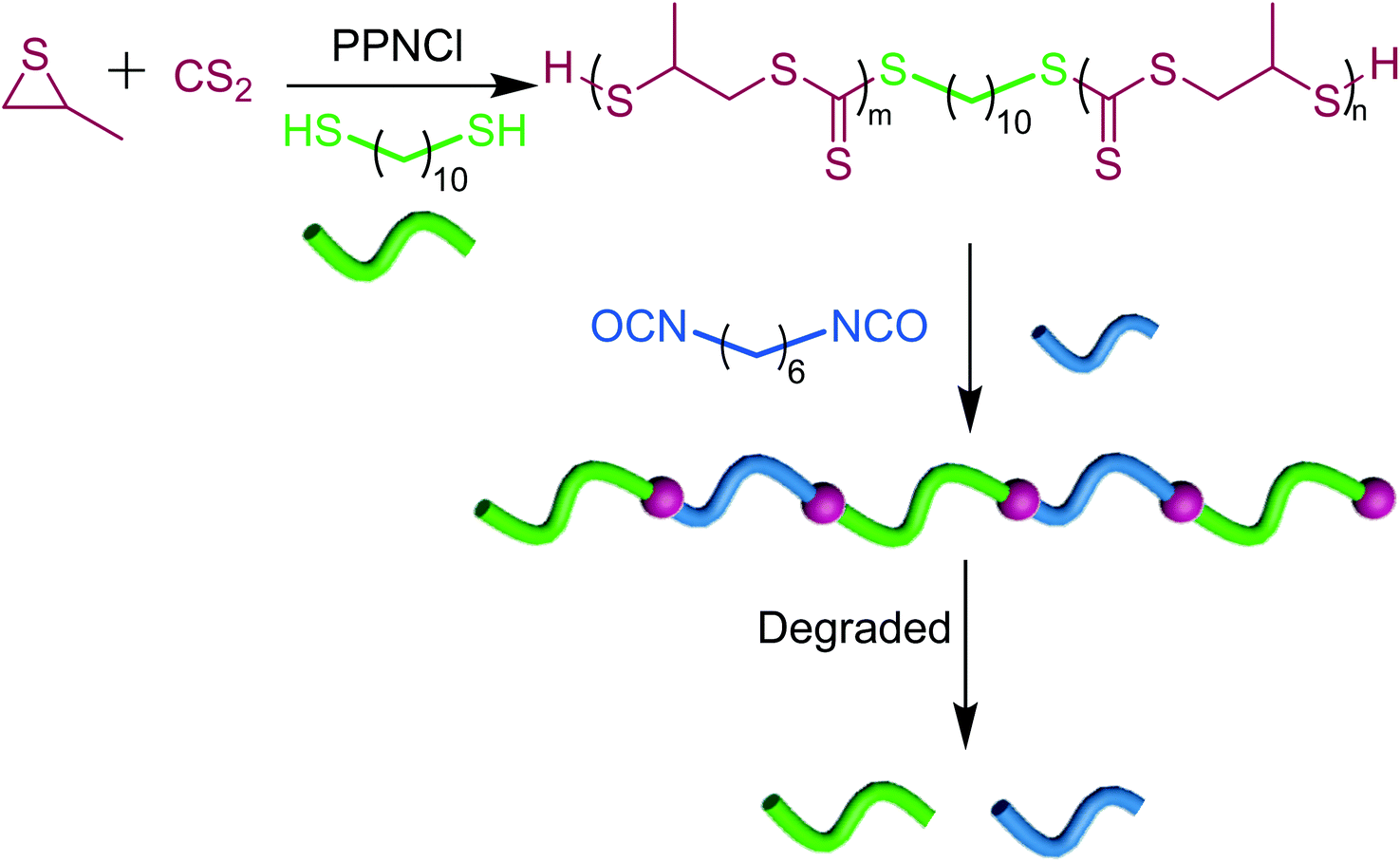
Self-immolating polymers using poly(trithiocarbonate)s as linkers
Since poly(trithiocarbonate)s exhibit self-immolation properties and undergo unzipping in the presence of a radical trigger, we thought of utilizing them as degradable linkers in an attempt to harness such a remarkable feature (Scheme 2). Low molar mass α-SH/ω-SH poly(trithiocarbonate)s (0.8 kg mol−1) were synthesized using PPNCl as the catalyst in the presence of 100 eq. of 1,10-decanedithiol serving as a difunctional CTA. As shown in Fig. S21,† the amount of trithiocarbonate units in each polymer chain was determined as 4 which was in agreement with the GPC results. The introduction of a few trithiocarbonate units at the 1,10-decane chain ends followed by their polycondensation with diisocyanates generates a polyethylene-type chain including degradable linkers. By condensation between the thiol function carried by end-standing trithiocarbonates and hexamethylene diisocyanate, a thiourethane-containing polymer was prepared up to 35 kg mol−1 molar mass. The 1H NMR (Fig. S22†) and FTIR (Fig. S23†) results confirm the successful polycondensation reaction. In such a thiourethane-containing polymer, the onset of decomposition occurs at 220 °C (Fig. S24†). Due to the presence of trithiocarbonate units, such a thiourethane-containing polymer exhibits facile degradability under a radical trigger (Fig. S25†). In short, the degradability of oligotrithiocarbonates can serve to prepare all kinds of degradable polymers provided trithiocarbonate moieties are incorporated into them.
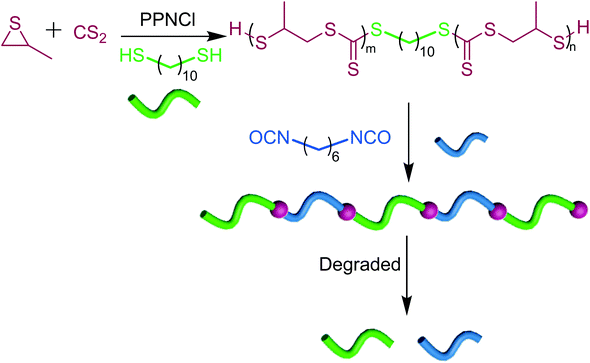 |
| | Scheme 2 Trithiocarbonates as degradable linkers for the preparation of degradable polymers (m + n = 4). | |
Conclusion
In this work, we have successfully demonstrated a way to utilize CS2 as a C1 resource through highly selective coupling of CS2 with episulfides under metal-free conditions. Poly(trithiocarbonates) could be obtained with 95% linear/cyclic selectivity initiated by onium salts in the presence of an excess amount of CS2; on the other hand, CTC could be only produced with an unprecedented activity of a TOF value up to 1667 h−1 at 80 °C when catalyzed by TBAF. In contrast to polycarbonates, poly(trithiocarbonate)s could be decomposed in a radical way in the presence of AIBN or by UV irradiation. This work presented a green and sustainable method for the preparation of sulfur-containing polymers and CTC with great potential as biomedical and optical materials. Taking advantage of the presence of thiocarbonylthio moieties in poly(trithiocarbonate)s, attempts are currently being made in our laboratory to control the radical polymerization of some vinyl monomers.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This research work is supported by KAUST under baseline funding (BAS/1/1374-01-01).
References
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K. J. Kim, J. Van Der Laan and N. A. Nguyen, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS PubMed.
- E. Marianucci, C. Berti, F. Pilati, P. Manaresi, M. Guaita and O. Chiantore, Polymer, 1994, 35, 1564–1566 CrossRef CAS.
- S. Wu, M. Luo, D. J. Darensbourg and X. Zuo, Macromolecules, 2019, 52, 8596–8603 CrossRef CAS.
- D. Cortizo-Lacalle, S. Arumugam, S. E. Elmasly, A. L. Kanibolotsky, N. J. Findlay, A. R. Inigo and P. J. Skabara, J. Mater. Chem., 2012, 22, 11310–11315 RSC.
- N. M. Tucker, A. L. Briseno, O. Acton, H.-L. Yip, H. Ma, S. A. Jenekhe, Y. Xia and A. K. Y. Jen, ACS Appl. Mater. Interfaces, 2013, 5, 2320–2324 CrossRef CAS.
- F. Otón, R. Pfattner, N. S. Oxtoby, M. Mas-Torrent, K. Wurst, X. Fontrodona, Y. Olivier, J. Cornil, J. Veciana and C. Rovira, J. Org. Chem., 2011, 76, 154–163 CrossRef.
- Y. Robbe, J. P. Fernandez and R. Dubief, Eur. J. Med. Chem., 1982, 17, 235–243 CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2013, 114, 1709–1742 CrossRef PubMed.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS.
- K. Huang, C. L. Sun and Z. J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- B. Yu and L. N. He, ChemSusChem, 2015, 8, 52–62 CrossRef CAS.
- M. Honda, M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2014, 4, 2830–2845 RSC.
- I. Omae, Catal. Today, 2006, 115, 33–52 CrossRef CAS.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS.
- H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS.
- Y. Xu, L. Lin, M. Xiao, S. Wang, A. T. Smith, L. Sun and Y. Meng, Prog. Polym. Sci., 2018, 80, 163–182 CrossRef CAS.
- Y. Li, Y.-Y. Zhang, L.-F. Hu, X.-H. Zhang, B.-Y. Du and J.-T. Xu, Prog. Polym. Sci., 2018, 82, 120–157 CrossRef CAS.
- X. B. Lu, W. M. Ren and G. P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS.
- X.-H. Zhang, F. Liu, X.-K. Sun, S. Chen, B.-Y. Du, G.-R. Qi and K. M. Wan, Macromolecules, 2008, 41, 1587–1590 CrossRef CAS.
- C.-J. Zhang and X.-H. Zhang, Macromolecules, 2019, 53, 233–239 CrossRef.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Macromolecules, 2015, 48, 5526–5532 CrossRef CAS.
- D. J. Darensbourg, J. R. Andreatta, M. J. Jungman and J. H. Reibenspies, Dalton Trans., 2009, 8891–8899 RSC.
- D. J. Darensbourg, S. J. Wilson and A. D. Yeung, Macromolecules, 2013, 46, 8102–8110 CrossRef CAS.
- J. Diebler, H. Komber, L. Häußler, A. Lederer and T. Werner, Macromolecules, 2016, 49, 4723–4731 CrossRef CAS.
- Y. Liu, X. Zhang, Z. Zhang, L. Liu, D. Fan and X. Lü, Inorg. Chem. Commun., 2015, 55, 132–134 CrossRef CAS.
- J. Cao, M. Yu, H. Li, L. Wang, X. Zhu, G. Wang, Y. Shi and C. Cao, Res. Chem. Intermed., 2015, 41, 5323–5330 CrossRef CAS.
- J. Diebler, A. Spannenberg and T. Werner, Org. Biomol. Chem., 2016, 14, 7480–7489 RSC.
- C. Mei, X. Li, L. Liu, C. Cao, G. Pang and Y. Shi, Tetrahedron, 2017, 73, 5706–5714 CrossRef CAS.
- R. Khalifeh, M. Sorouri, E. K. Damirchi and M. Rajabzadeh, J. Taiwan Inst. Chem. Eng., 2020, 115, 229–241 CrossRef CAS.
- C. Beattie and M. North, ChemCatChem, 2014, 6, 1252–1259 CAS.
- K. Soga, M. Sato, H. Imamura and S. Ikeda, J. Polym. Sci., Polym. Lett. Ed., 1975, 13, 167–171 CrossRef CAS.
- K. Soga, H. Imamur, M. Sato and S. Ikeda, J. Polym. Sci., Polym. Lett. Ed., 1976, 14, 677–684 CrossRef CAS.
- K. An and J. Zhu, Organometallics, 2014, 33, 7141–7146 CrossRef CAS.
- K. Nakano, G. Tatsumi and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 15116–15117 CrossRef CAS PubMed.
- X. Zhang, P. Su, Y. Liu, Q. Shi and X. Lü, Inorg. Chem. Commun., 2017, 85, 59–61 CrossRef CAS.
- S. Silvano, C. F. Carrozza, A. R. de Angelis, I. Tritto, L. Boggioni and S. Losio, Macromolecules, 2020, 53, 8837–8846 CrossRef CAS.
- C.-J. Zhang, T.-C. Zhu, X.-H. Cao, X. Hong and X.-H. Zhang, J. Am. Chem. Soc., 2019, 141, 5490–5496 CrossRef CAS.
- D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586–7591 CrossRef CAS PubMed.
- N. Aoyagi and T. Endo, Tetrahedron Lett., 2018, 59, 1702–1704 CrossRef CAS.
- Y. Takenaka, N. Fukaya, S. J. Choi, G. Mori, T. Kiyosu, H. Yasuda and J.-C. Choi, Ind. Eng. Chem. Res., 2018, 57, 891–896 CrossRef CAS.
- D. J. Darensbourg, Polym. Degrad. Stab., 2018, 149, 45–51 CrossRef CAS.
- H. Cao and X. Wang, SusMat, 2021, 1, 88–104 CrossRef.
- Y. Z. You, C. Y. Hong and C. Y. Pan, Chem. Commun., 2002, 2800–2801 RSC.
- S. Motokucho, A. Sudo, F. Sanda and T. Endo, Chem. Commun., 2002, 1946–1947 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, 1H and 13C NMR, FTIR, DSC, TGA and GPC characterization data Fig. S1–S24. See DOI: https://doi.org/10.1039/d2py00405d |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Xiaoshuang
Feng
* and
Xiaoshuang
Feng