
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Current-controlled ‘plug-and-play’ electrochemical atom transfer radical polymerization of acrylamides in water†
Mahir
Mohammed
,
Bryn A.
Jones
and
Paul
Wilson
 *
*
University of Warwick, Department of Chemistry, Coventry, CV4 7AL, UK. E-mail: p.wilson.1@warwick.ac.uk
First published on 18th May 2022
Abstract
Aqueous electrochemical atom transfer radical polymerisation (eATRP) can be challenging due to deleterious side reactions leading to the loss of the ω-chain end, increased rates of activation (kact) leading to higher [Pn˙], increased rates of termination, and the lability of the X–CuII/L bond to hydrolysis leading to poor control. Herein, we build on recent advances in eATRP methodology to develop a simplified current-controlled eATRP of acrylamides in water. The simplification arises from the use of commercial, standardised reaction hardware which enables the polymerisations to be performed in a 2-electrode, ‘plug-and-play’, undivided electrochemical cell configuration. Further simplification is afforded by the design of a single stepwise current profile (Iappvs. time) capable of mediating current-controlled eATRP of N-hydroethylacrylamide (HEAm). At room temperature, polymerisation of HEAm to target degrees of polymerisation (DPn,th) of 20–100 proceeds with good control (Đ ≤ 1.50). Loss of control when targeting higher DPn at room temperature is circumvented by lowering the reaction temperature (RT to 0 °C), increasing the stirring rate (400 rpm to 800 rpm) and increasing the catalyst concentration. Using the best conditions, a linear increase in Mn,SEC with DPn (up to DPn = 320) and low dispersity values (DPn,th = 40–160; Đ = 1.26–1.38) were obtained. Furthermore, the current profile and reaction conditions can support the polymerisation of other primary and secondary acrylamides and the retention of the ω-Br chain end is exemplified by a short in situ chain extension. Overall, this represents further simplification of aqueous eATRP with respect to reaction set up and experimental parameters (single current profile) which has been employed to synthesise polyacrylamides with good efficiency and control.
Introduction
Atom transfer radical polymerisation (ATRP)1,2 is a popular technique for polymer synthesis as it enables excellent control over polymer composition, end groups and architecture.3 Through careful consideration of the reaction conditions and catalyst choice, it has been applied to polymerise a variety of functional monomers and to prepare polymers with a range of architectures, including telechelic,4 star,5 brush,6 and graft copolymers,7 for instance.In early ATRP,8,9 the active catalysts (typically a copper (Cu) complex; CuI/L) was used directly to activate dormant alkyl (R–X) or macromolecular (Pn–X) halides and generating radicals (R˙/Pn˙) capable of reacting with vinyl monomers. The activation process occurs via simultaneous electron transfer process and halogen abstraction in which the CuI/L complex is oxidised to X–CuII/L and a reactive carbon-centred radical is formed. The radicals can undergo propagation until they are deactivated by a second electron transfer and halogen abstraction process in which the X–CuII/L is reduced back to CuI/L and propagating radical chain ends are oxidised back to the dormant alkyl halide chain ends (Pn–X). Control over the polymerisation was conferred by accumulation of X–CuII/L, through unavoidable radical termination reactions. This promoted deactivation of propagating radicals (Pn˙) via reformation of dormant polymer chains (Pn–X) and established the activation–deactivation equilibrium (KATRP) that governs the control over all ATRP reactions.10 It was important to perform the reactions under strict de-oxygenated conditions due to the propensity of the CuI/L complex to undergo oxidation thus removing the activating complex from the reaction system.
In recent years, advances in ATRP methodology have shown that the redox mechanism can be manipulated and controlled using external stimuli including light,11 sound,12 mechanical force13 and (bio)chemical intervention using reducing agents.14 These advances negated the need to directly use oxidatively labile CuI/L, using the external stimulus to generate it in situ from more oxidatively stable CuII/L complexes.15 The ability to control the relative [CuI/L] and [CuII/L] provides fine control over KATRP and the polymerisation as a whole. In 2011, electricity was also shown to be an excellent external stimulus leading the development of electrochemical ATRP (eATRP).16,17 The voltage and current can be readily controlled using a potentiostat or simple current generator which helps to easily alter important reaction parameters such as applied potential (Eapp) or applied current (Iapp).18 Such parameters allow the number, and relative energy of electrons involved in the reaction to be controlled.19,20 The direct use of electrons, which can be generated from sustainable/renewable energy sources, significantly improves the sustainability and reduces the carbon footprint of electrochemical reactions compared to the analogous chemically-driven processes/reactions that often require stoichiometric amounts of chemical oxidants and reductants.
In the context of Cu-mediated eATRP electrons delivered from a working electrode (WE) are used to reduce CuII/L to the active CuI/L on demand to regulate the polymer synthesis through controlling the relative [CuI/L] and [CuII/L] which allows the overall radical concentration to be accurately controlled.21 Furthermore, the use of oxidatively stable CuII/L negates the need for stringent deoxygenation since the reducing voltages/current applied throughout the polymerisation ensures a constant supply of the CuI/L activator complex. The once complex reaction set up of eATRP has been simplified over the last 10 years, to a point where eATRP can performed in a ‘plug-and-play’ configuration using commercial, standardised equipment (IKA ElectraSyn).22,23 Furthermore, it can be run in 2-electrode configuration, by virtue of the use of sacrificial counter electrodes (CE), in an undivided electrochemical cell using cheap and easy to operate current generators in simplified eATRP (seATRP) under galvanostatic (constant current) conditions.24 Both potentiostatic (constant potential) and galvanostatic eATRP have been mainly used for the controlled polymerisation of (meth)acrylates, in both organic25–27 and aqueous28–30 media.
In potentiostatic eATRP a constant potential, selected based on the redox potential of the Cu-complex used, is applied to reduce inactive CuII/L to active CuI/L resulting in generation of a current that in the presence of Pn–X rapidly decays to a steady state as the eATRP equilibrium is established. The potential is set relative to a reference electrode and the corresponding current (I) is maintained as long as there is CuII/L and Pn–X present. This configuration is attractive because it can be highly selective for a particular complex and the current output (I vs. t), can be used to qualitatively monitor the retention of CuII/L and Pn–X throughout the reaction. Integration of the I vs. t plots provides the total charge passed during a given eATRP reaction and from this a much simpler, 2-electrode configuration, through which a simple galvanostat, can be used to enable galvanostatic eATRP. Based on the total charged passed, a current profile is set; the potential output, which is reductive and measured at the cathode in this case, changes until it reaches the redox potential of the Cu-complex employed. At a given resistance, the potential output is constant as long as there is sufficient CuII/L (at the electrode surface) and Pn–X (in bulk). The galvanostatic configuration is attractive from an industrial point of view due to the lack of need for a reference electrode.
The polymerisation of acrylamides in aqueous solutions is best achieved by Cu(0)-mediated single electron transfer radical polymerisation (SET-LRP).31,32 Aqueous ATRP of acrylamides is more difficult to control due to issues such as hydrolysis and/or elimination of the ω-chain end,33 increased rates of activation (kact) leading to higher [Pn˙] and increased rates of termination and lability of the X–CuII/L bond to hydrolysis.34 These issues can be addressed by lowering reaction temperatures, adding halide salts and increasing the [CuI/L].35–37 Despite electrochemistry being an ideal way of controlling [CuI/L] there are only a few examples of eATRP being employed to synthesise polyacrylamides. Block copolymers of a primary acrylamide (acrylamide; AAm) and a secondary acrylamide (N-isopropylacrylamide; NIPAm) (AAm-b-NIPAm) have been synthesised under potentiostatic conditions.38,39 Polymers of tertiary acrylamides (dimethylacrylamide; DMAm) have also been synthesised under potentiostatic conditions.40 Using the current vs. time (I vs. t) plot generated from these reactions, the total charge passed during potentiostatic polymerisation was calculated and used to derive a current profile for galvanostatic eATRP of DMAm.
Herein, we report the potentiostatic eATRP of N-hydroxyethylacrylamide (HEAm) from which we have designed a current profile that enables the current-controlled eATRP of HEAm with good control. Moreover, the entire current-controlled investigation has been performed using a single current profile to probe the effects of temperature, degree of polymerisation (DPn), [CuII/L] and choice of monomer on the polymerisation.
Materials and methods
Copper(II) trifluoromethanesulfonate (CuII(OTf)2, Acros Organics, 98%), tris (2-pyridylmethyl)amine (TPMA, Sigma Aldrich, 98%), potassium nitrate (KNO3, Acros Organic, 99%), sodium bromide (NaBr, Alfa Aesar, >99.9%), N-hydroxyethyl acrylamide (HEAm, Sigma Aldrich, 97%) were used as received without further purification. All solutions were prepared using deionised water (15.6 MΩ, VEOLIA Elga Purelab). 2-Hydroxyethyl 2-bromoisobutyrate (HEBiB) was synthesised according to literature procedure and obtained with high spectroscopic purity.41 Potassium nitrate was used as background electrolyte. 0.05 μm MicroPolish powder (Al2O3, Buehler) was used for voltammetry electrode polishing.1H NMR
Spectra were recorded on a Bruker HD 300 spectrometer (300 MHz) using D2O (Sigma-Aldrich) as solvent. Chemical shift values (δ) are reported in ppm relative to residual solvent peaks (δ = 4.75 ppm). ACDLABS software was used to analyse the data.DMF SEC
Agilent Infinity II MDS instrument equipped with differential refractive index (DRI), viscometry (VS), dual angle light scatter (LS) and variable wavelength UV detectors. The system was equipped with 2× Agilent Polargel columns (300 × 7.5 mm) and a Polargel 5 μm guard column. The eluent is DMF with 0.01% LiBr additive. Samples were run at 1 ml min−1 at 50 °C. Poly(methyl methacrylate) standards (Agilent EasiVials) were used for calibration, and the calibration range was 600–870![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 gmol−1. Analyte samples were filtered through a Nylon membrane with 0.22 μm pore size before injection. Respectively, experimental number average molecular weights (Mn,SEC) and dispersity (Đm) values of synthesized polymers were determined by conventional calibration using Agilent GPC/SEC software.
000 gmol−1. Analyte samples were filtered through a Nylon membrane with 0.22 μm pore size before injection. Respectively, experimental number average molecular weights (Mn,SEC) and dispersity (Đm) values of synthesized polymers were determined by conventional calibration using Agilent GPC/SEC software.
Cyclic voltammetry (CV)
CV was conducted on a CH-Instruments 600 E potentiostat using a 3 mm glassy carbon disc electrode. The electrode was polished with 0.05 μm alumina powder, and then sonicated in MilliQ water for 30 seconds between each use. The counter electrode was a platinum wire coil. The reference electrode was Ag/AgCl, and the silver wire was rinsed in MilliQ water placed into a glass capillary tube fitted with a vycor frit and filled with 3 M KCl solution. Before all CVs, the reaction cell was purged with N2 for 10 minutes. The supporting electrolyte KNO3 (0.111 g, 1.1 mmol) and water (11 mL) were mixed as background electrolyte, and used to perform background CVs. Background CVs were taken before any measurements to exclude the possibility of impurities adsorbed to the glassy carbon electrode. Solutions were prepared for CV and simplified eATRP as shown below.Electrolysis reaction set-up
Potentiostatic and galvanstatic electrolyses were performed using an ElectraSyn 2.0 device. The IKA electrochemical cell consisted of a reaction vial and an electrode head to which working, counter and reference electrodes were attached. An IKA manufacture Pt-coated electrode was used as the working electrode (cathode). The sacrificial counter electrode (anode) was initially Al wire (Alfa-Aesar, length = 15 cm, diameter = 1.0 mm, annealed) manipulated to a size similar to the IKA Pt-coated electrode as reported in our previous work.22 This was replaced during the investigation by an IKA manufacture Al electrode, which was used for the majority of the experiments. An IKA manufacture Ag/AgCl (using 3 M KCl) reference electrode was included for potentiostatic reactions. For reactions performed in an ice bath, an ElectraSyn GoGo module was used enabling the reaction vial to be placed in an ice bath on a stirrer hotplate adjacent to the ElectraSyn 2.0 (Fig. S1†), which was still used to deliver the required potential or current. Stirring rates of 400 rpm or 800 rpm were used depending on the experiment.General procedure for ‘plug-and-play’ seATRP of HEAm (10 wt%) under potentiostatic conditions
For the polymerisation of HEAm using [HEAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.1]:[0.4]:[0.1]. Cu(OTf)2 (9.1 mg, 0.025 mmol) was dissolved in MilliQ water (10 mL) and TPMA (29.0 mg, 0.1 mmol) was added, resulting in a blue solution. KNO3 (0.11 g, 1.1 mmol), NaBr (2.6 mg, 0.025 mmol), and HEAm (1.1 g, 1.0 mmol) were then added to this solution. After purging with N2 for 1 minute, a CV of the catalyst was recorded to measure its standard reduction potential (Eθ = E1/2) in the reaction solution (10% v/v HEAm in H2O). HeBiB (36 μL, 1.0 mmol) was then added to the solution and CV was repeated to evaluate the redox activity and activation behaviour of CuII(OTf)2/TPMA and HEBiB respectively. The solution was then transferred to an IKA ElectraSyn reaction vial, then fitted with an IKA ElectraSyn electrode head. This was itself equipped with an IKA Pt-coated working electrode (cathode), IKA aluminium sacrificial counter electrode (anode) and an IKA manufacture Ag/AgCl (containing 3 M KCl) reference electrode. The ElectraSyn 2.0 was programmed to perform electrolysis in potentiostatic mode at the desired Eapp. Current vs. time plots were automatically collected and stored on the ElectraSyn 2.0 Android mobile app, obtained from Google Play store. Reactions were sampled periodically for reaction monitoring by 1H NMR (D2O) and SEC (DMF).
General procedure for ‘plug-and-play’ seATRP of HEAm (10 wt%) under current-controlled conditions
For the polymerisation of HEAm using [HEAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.1]:[0.4]:[0.1]. Cu(OTf)2 (9.1 mg, 0.025 mmol) was dissolved in MilliQ water (10 mL) and TPMA (29.0 mg, 0.1 mmol) was added resulting in a blue solution. KNO3 (0.11 g, 1.1 mmol), NaBr (2.6 mg, 0.025 mmol), and HEAm (1.1 g, 1.0 mmol) were then added to this solution. HeBiB (36 μL, 1.0 mmol) was then added to the solution and CV was repeated to evaluate the redox activity and activation behaviour of CuII(Otf)2/TPMA and HEBiB respectively. The solution was then transferred to an IKA ElectraSyn reaction vial, sparged with N2 for an additional 5 minutes then fitted with an IKA ElectraSyn electrode head. This was itself equipped with a IKA Pt-coated working electrode (cathode) and IKA aluminium sacrificial counter electrode (anode) only. Based on the current vs. time plots collected during potentiostatic electrolysis, the ElectraSyn 2.0 was programmed to perform electrolysis in galvanostatic mode. Current steps of −4 mA, −3.1 mA, −1.9 mA, −1.3 mA, −0.9 mA were each applied for 5 minutes, and finally −0.8 mA was applied for the total remainder reaction time. Reactions were sampled periodically for reaction monitoring by 1H NMR (D2O) and SEC (DMF).
Results and discussion
Potentiostatic seATRP of N-hydroxyethyl acrylamide
Initially, cyclic voltammetry (CV) was performed to determine standard reduction potential (Eθ ≈ E1/2 = (Epc + Epa)/2) of the CuII/TPMA complex in the reaction solution (10 wt% HEAm/H2O/KNO3). The E1/2 was found to be −0.13 V (vs. Ag/AgCl, Fig. 1A) which is similar to values reported in the literature of CuII/TPMA in aqueous solutions of other water soluble acrylamides and (meth)acrylates.28,42 The initiator, 2-hydroxyethyl-2-bromoisobutyrate (HEBiB), was then added to the reaction solution and CV was repeated to determine changes in the redox activity of the CuII/TPMA complex and the activation behaviour of HEBiB (Fig. 1B). A large current enhancement in the cathodic scan (Epc) and a significant reduction in current in the anodic scan (Epa) was observed when HEBiB was present in the reaction solution. This is consistent with our previous work and the work of others with CuII/TPMA complexes in related reaction systems.22,24 During the cathodic scan, CuII/TPMA is reduced to CuI/TPMA, which in the presence of HEBiB undergoes rapid activation of the HEBiB via one-electron transfer to form a carbon-based radical and reform CuII/TPMA, thus leading to a current enhancement. If the activation process occurs faster than the timescale of the of CV scan, there is significantly less, or no, CuI/TPMA present during the anodic scan which diminishes or removes the anodic current.43 | ||
| Fig. 1 Cyclic voltammetry of (A) 2.3 mM CuIITPMA in 10 wt% HEAm in H2O containing KNO3 (0.1 M); (B) 2.3 mM CuIITPMA in 10 wt% HEAm in H2O containing KNO3 (0.1 M) before, (red) and after, (blue) addition of HEBiB. Recorded at 20 °C at 0.1 V s−1 using a glassy carbon electrode (GCE). | ||
Potentiostatic eATRP was then performed at room temperature using [HEAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.1]:[0.4]:[0.1] at Eapp = E1/2, E1/2 − 0.06 V and E1/2 − 0.12 V. The polymerisation was performed in an undivided cell containing a Pt-coated working electrode (IKA), Al-wire counter electrode (l = 15 cm, d = 1.0 mm, annealed) and a Ag/AgCl reference (containing 3 M KCl). At Eapp = E1/2, conversion reached 77% within 2 hours. Pseudo-first order kinetics were observed, with a kappp = 1.07 × 10−4 s−1 and Mn,SEC increasing linearly with conversion, which was indicative of a controlled polymerisation (Fig. 2). This was further supported by the low dispersity obtained (Table 1, entry 1; Đm = 1.33), whilst deviations between the Mn,SEC (8400 g mol−1) and Mn,th (3700 g mol−1) suggest poor initiation efficiency or – more likely – gradual loss of the ω-Br chain end, which has been reported previously in Cu-mediated RDRP of acrylamides.33 Applying more reducing potentials (Eapp = E1/2 − 0.06/−0.12 V) resulted in slower reactions, lower conversions, bimodal SEC traces and higher dispersities, indicating that the polymerisations were not well controlled (Table 1, entries 2 and 3, Fig. S2†). The loss of control at more reducing potentials is not uncommon16,28,44 and can be attributed to higher [CuI/TPMA], which results in higher radical concentrations due to the high activity of the CuI/TPMA species.
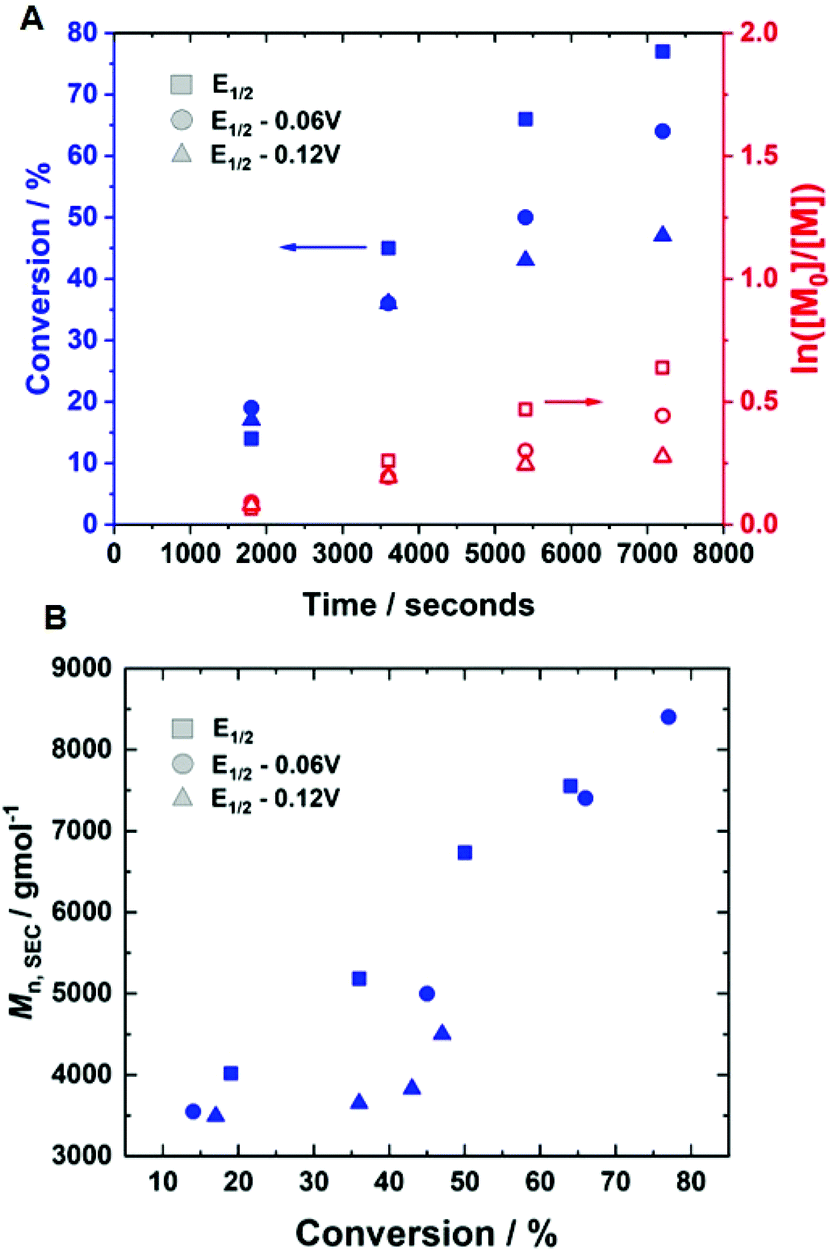 | ||
| Fig. 2 (A) Conversion vs time and first order kinetic plot; (B) Mn,SECvs. conversion for potentiostatic seATRP of 10 wt% HEAm in H2O containing NaBr (0.1 M) and KNO3 (0.1 M) at room temperature, performed at Eapp = E1/2, E1/2 − 0.06 V, E1/2 − 0.12 V. Conditions: [HEAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.1]:[0.4]:[0.1]. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | DPn,th | E app/V | I app/mA 6 steps | Conv.a |
M
n,thb/g mol−1 |
M
n,SECc/g mol−1 |
Đ
mc |
| Time = 2 h; room temperature; 400 rpm; 10% v/v HEAm; [CuII]:[TPMA]:[NaBr] = [0.1]:[0.4]:[0.1].a Determined via1H NMR of reaction samples performed in D2O.b Mn,th = [(conv./100 × DPn,th) × (115)] + 211.c From DMF SEC. |
|||||||
| 1 | 40 | −0.13 | — | 77% | 3800 | 8400 | 1.33 |
| 2 | 40 | −0.19 | — | 64% | 3100 | 7600 | 1.60 |
| 3 | 40 | −0.25 | — | 47% | 2400 | 4500 | 3.13 |
| 4 | 60 | −0.13 | — | 72% | 5200 | 11200 |
1.31 |
| 5 | 60 | — | −4.0 to −0.8 | 71% | 5100 | 9000 | 1.39 |
| 6 | 60 | — | −4.0 to −0.8 | 86% | 6100 | 11000 |
1.32 |
| 7 | 40 | — | −4.0 to −0.8 | 74% | 3600 | 8200 | 1.35 |
| 8 | 80 | — | −4.0 to −0.8 | 86% | 8100 | 14700 |
1.36 |
| 9 | 100 | — | −4.0 to −0.8 | 75% | 8800 | 17000 |
1.50 |
| 10 | 160 | — | −4.0 to −0.8 | 71% | 13300 |
16500 |
1.55 |
| 11 | 320 | — | −4.0 to −0.8 | 52% | 19300 |
20900 |
1.70 |

Current-controlled seATRP of N-hydroxyethyl acrylamide at room temperature
In order to obtain values for the applied current (Iapp) to be utilised during current-controlled seATRP, a potentiostatic reaction was repeated at room temperature using [HEAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [60]:[1]:[0.1]:[0.4]:[0.1] at Eapp = E1/2 = −0.13 V and a current vs. time (I vs. t) plot was collected. The polymerisation exhibited almost identical results to those reported above. Conversion reached 72% within 2 hours and a low dispersity was obtained (Table 1, entry 4; Đm = 1.31). The I vs. t plot displayed an exponential current decay, reaching a constant, steady state in which I ≠ 0 mA (Fig. S3†). This profile was expected for an electrochemical reduction (CuII/TPMA to CuI/TPMA), which is coupled to a chemical activation process (R–X/Pn–X by CuI/TPMA to form R˙/Pn˙ and CuII/TPMA), and has been previously reported for eATRP reactions.24,28 Integration of this gave the total charge passed (Q) during the potentiostatic reaction and enabled a 6-step current profile to be designed with Iapp gradually decreasing from −4.0 mA to −0.8 mA. This would then be applied to the subsequent current-controlled reactions.
Initially, current-controlled eATRP was performed in an undivided cell using a 2-electrode configuration consisting of a Pt-coated working electrode (IKA) and an Al-wire counter electrode (l = 15 cm, d = 1.0 mm, annealed). A current profile with regimes of Iapp = −4 mA (5 min), −3.1 mA (5 min), −1.9 mA (5 min), −1.3 mA (5 min), −0.9 mA (5 min) and −0.8 mA (95 min) resulted in 71% conversion within 2 hours at room temperature, which was similar to that achieved via potentiostatic eATRP (Table 1, entry 5; Đm = 1.39, Fig. S4†). To simplify the reaction set-up, the Al-wire counter electrode was replaced with a commercial, standardised IKA Al electrode. In this configuration, the polymerisation reached 86% conversion within 2 hours (Table 1, entry 6, Fig. S5†). The reaction exhibited pseudo-first order kinetics, and a linear increase of Mn,SEC with conversion indicative of a well-controlled eATRP (Mn,SEC = 11000 g mol−1, Đm = 1.32, Fig. 3).
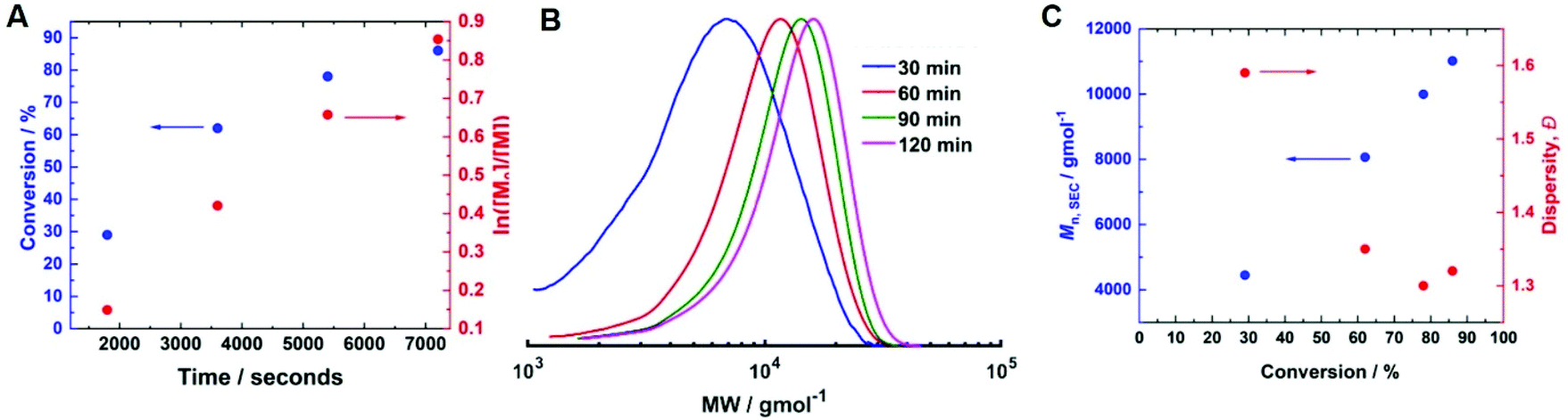 | ||
| Fig. 3 (A) Conversion v time and first order kinetic plot; (B) SEC in DMF (for the final PHEAm, Mn,SEC = 11000 g mol−1, Đm = 1.32); (C) Mn,SEC and Đmvs. conversion for current-controlled seATRP of 10 wt% HEAm in H2O containing KNO3 (0.1 M) at room temperature using a commercial IKA Al CE. Conditions: [HEAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.1]:[0.4]:[0.1]; Iapp = −4 mA (5 min), −3.1 mA (5 min), −1.9 mA (5 min), −1.3 mA (5 min), −0.9 mA (5 min) and −0.8 mA (95 min). | ||
The current-control over the reaction was then investigated in an experiment, wherein the current profile was applied and removed at regular intervals during the course of the reaction (Fig. 4). At the beginning of the reaction, Iapp = −4.0 mA. After working through the current profile, the polymerisation reached 23% conversion (tON = 30 min, kapp,1p = 6.3 × 10−5 s−1). At this point, the Iapp was removed and the reaction was left for a further 10 minutes, during which a 1% increase in conversion occurred, reaching 24% conversion (tOFF = 10 min, koff,1p = 2.2 × 10−5 s−1). When the current was switched back on (Iapp = −0.8 mA), polymerisation restarted, reaching 36% conversion (tON = 60 min, kapp,2p = 4.2 × 10−5 s−1). Again, polymerisation was halted/slowed upon removal of Iapp, before restarting again after 10 minutes, this time with no increase in conversion (tOFF = 20 min, koff,2p = 0 s−1). Conversion continued to increase up to 65% (tON = 120 min, kapp,3p = 7.0 × 10−5 s−1), before Iapp was switched off for the final time to effectively stop the polymerisation, with a final 1% increase in conversion, reaching 66% (tOFF = 30 min, koff,3p = 2.2 × 10−5 s−1), resulting in PHEAm with Mn,SEC = 9200 g mol−1 and Đm = 1.41 (Fig. S6†). The incomplete deactivation when electrolysis is removed is similar to observations made during investigations into temporal control afforded during photo-ATRP, and is related to the activity of the Cu-complex.45
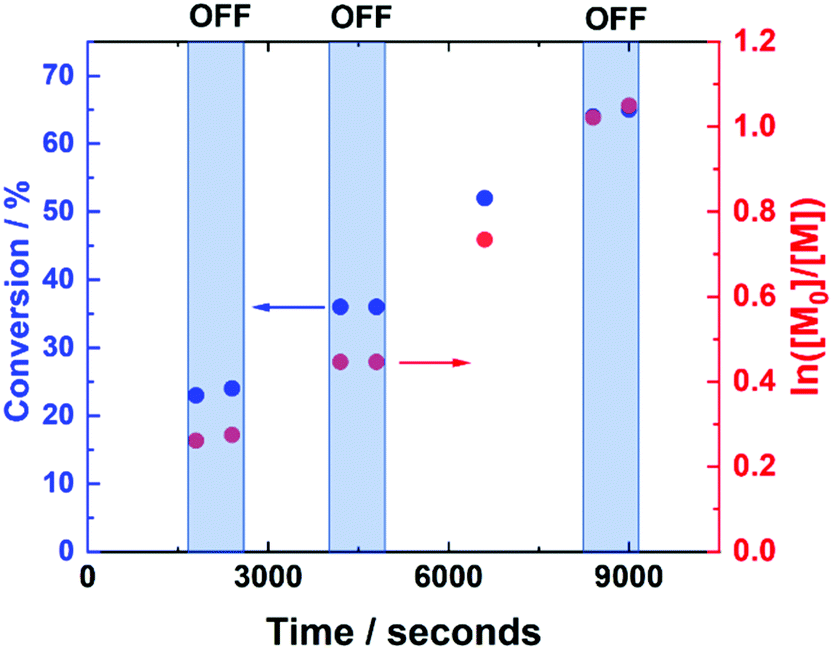 | ||
| Fig. 4 Conversion vs. time and first order kinetic plot demonstrating the temporal control exhibited during the current controlled seATRP of HEAm conducting using [HEAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.1]:[0.4]:[0.1] at room temperature. Between t = 0 and t = 30 min Iapp = −4 mA (5 min), −3.1 mA (5 min), −1.9 mA (5 min), −1.3 mA (5 min), −0.9 mA (5 min) and −0.8 mA (5 min). Thereafter, during 10 min intervals, Iapp = 0 mA and during 30 min intervals, Iapp = −0.8 mA. | ||
To determine if shorter and longer degrees for polymerisation (DPn) could be achieved using the same current profile, a series of polymerisations was performed in which the [M]/[I], i.e. DPn,th, was varied. When DPn,th was decreased to 40 the polymerisation reached 74% conversion within 2 hours yielding PHEAm with Mn,SEC = 8200 g mol−1 and Đm = 1.35 (Table 1, entry 7). Increasing DPn,th to 80 resulting in 86% conversion to PHEAm after 2 hours with control over the polymerisation comparable to DPn,th = 40 and 60 (Table 1, entry 8; Mn,SEC = 14700 g mol−1 and Đ = 1.36). Increasing DPn,th further to 100, 160 and 320 led to gradual loss of control over the polymerisation (Table 1, entries 9–11). Conversions diminished, reaching only 52% in 2 hours and dispersity values increased reaching Đ = 1.7 when DPn,th = 320. A plot of Mn,SECvs. DPn revealed that Mn,SEC increased linearly with DPn up to DPn,th = 100 but then plateaued suggesting that the polymerisations were well controlled up to DPn,th = 100 (Fig. 5A). This is supported by the SEC traces which show that the molecular weight distributions increase with DPn,th. However, the molecular weight distributions also became less symmetrical as DPn,th increased due to tailing to low molecular weight, which is most apparent when [M]/[I] = 160 and 320 (Fig. 5B). This could be attributed to loss of the ω-Br chain end which has been widely reported for acrylamides in water,33 and could be addressed by performing reactions at lower temperatures (vide infra), as has been reported for the aqueous Cu-mediated RDRP of acrylamides.32,36,37
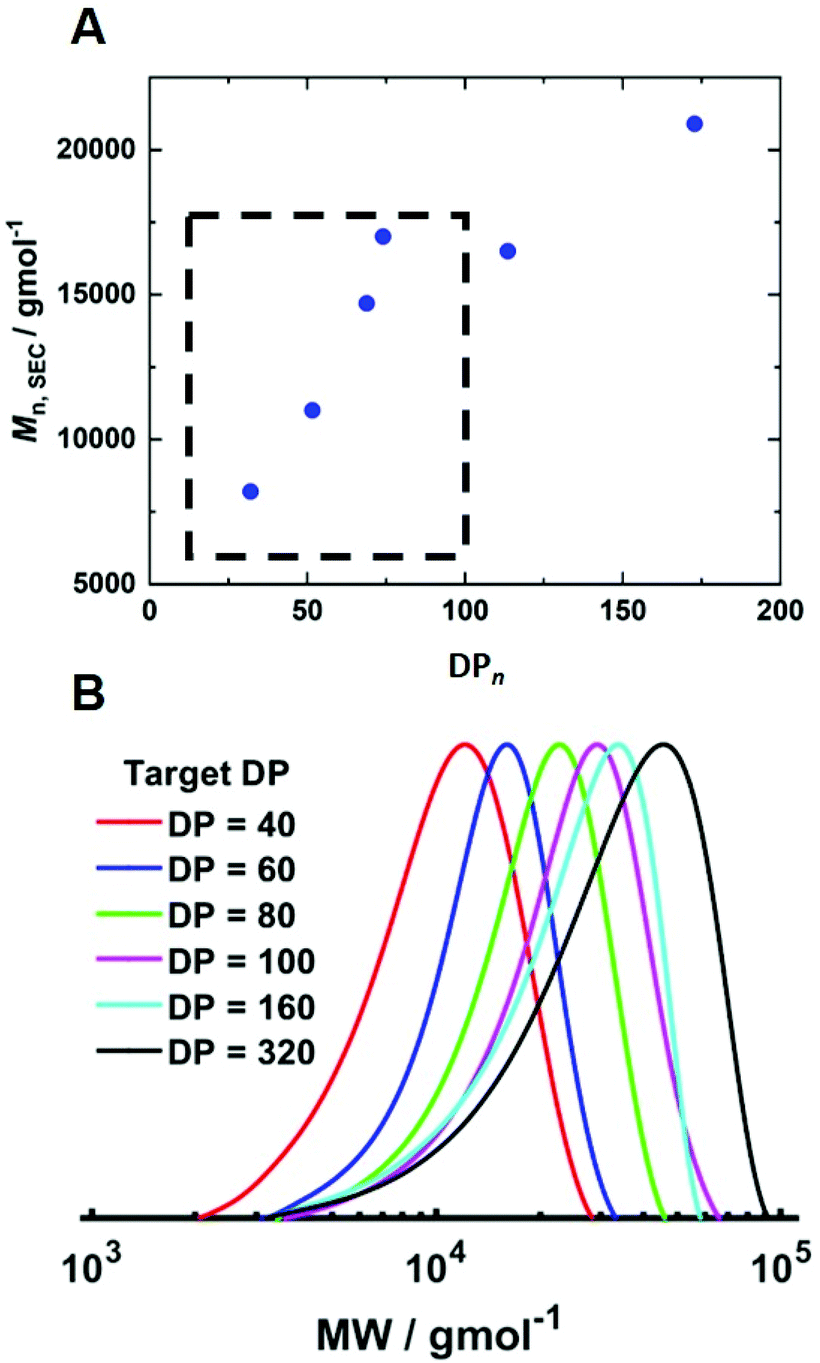 | ||
| Fig. 5 (A) Plot of Mn,SECvs. DPn and (B) SEC in DMF (Table 1, entries 6–11) for the current controlled seATRP of PHEAm synthesised using different [HEAm]/[HEBiB]. | ||
Alternatively, the loss of conversion and control in the polymerisation could also be due to disruption of the activation–deactivation equilibrium during the course of the reaction when high DPn,th were targeted. When DPn,th = 160 and 320, electrodeposition of Cu0 onto the working electrode was observed, whilst no electrodeposition was observed when DPn,th = 40–100. At the lower [I] values used when targeting higher DPn,th, more reducing potentials were required to reach and maintain Iapp (Fig. S7†). At these reducing potentials, electrodeposition of Cu0 can occur more readily which removes Cu from reaction media, thus decreasing [CuI/TPMA] and [CuII/TPMA] to the detriment of the reaction rate and control over the polymerisation.
Current-controlled seATRP of N-hydroxyethyl acrylamide at 0 °C
To investigate the effect of temperature on the current-controlled eATRP of HEAm, we decided to perform a series of reactions at 0 °C using the current profile established at room temperature.Initially, the current profile with regimes of Iapp = −4 mA (5 min), −3.1 mA (5 min), −1.9 mA (5 min), −1.3 mA (5 min), −0.9 mA (5 min) and −0.8 mA (215 min) was applied using [HEAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [160]:[1]:[0.1]:[0.4]:[0.1] for comparison to a room temperature reaction that was not well controlled. Disappointingly, at 0 °C the polymerisation only reached 41% conversion within 4 hours (Table 2, entry 1), and the dispersity of the PHEAm obtained was too high for a controlled polymerisation (Đm = 2.32). As well as reducing the rate of deleterious side reactions, lowering the reaction temperature was having an effect on the rate of polymerisation and also the rate of mass transport to and from the electrode interfaces46 and the activity coefficients47 of the CuII ions present. This led to higher effective resistance in the electrochemical cell and significant electrodeposition being observed, due to the highly reducing potentials required to maintain Iapp.
| Entry | [CuII] mM | [CuII]:[TPMA] |
Conv.a |
M
n,thb/g mol−1 |
M
n,SECc/g mol−1 |
Đ
mc |
|---|---|---|---|---|---|---|
| Time = 4 h; temperature = 0 °C; 800 rpm; 10% v/v HEAm; DPn,th = 160.a Determined via1H NMR of reaction samples performed in D2O.b Mn,th = [(conv./100 × DPn,th) × (115)] + 211.c From DMF SEC.d 400 rpm.e Reaction stopped after 3.25 h.f DPn,th = 40.g DPn,th = 80.h DPn,th = 320. | ||||||
| 1d | 2.30 | 1:4 |
41% | 7800 | 10700 |
2.32 |
| 2 | 2.30 | 1:4 |
56% | 10700 |
15900 |
1.49 |
| 3 | 2.30 | 1:2.7 |
66% | 12400 |
18700 |
1.47 |
| 4 | 2.30 | 1:2 |
64% | 12000 |
17500 |
1.46 |
| 5 | 2.30 | 1:1.5 |
56% | 10500 |
13200 |
1.51 |
| 6e | 1.15 | 1:2.7 |
20% | 3900 | 10000 |
2.48 |
| 7 | 4.60 | 1:2.7 |
82% | 15300 |
12300 |
1.45 |
| 8 | 6.90 | 1:2.7 |
77% | 14400 |
17700 |
1.38 |
| 9f | 6.90 | 1:2.7 |
91% | 4400 | 8000 | 1.26 |
| 10g | 6.90 | 1:2.7 |
79% | 7500 | 12500 |
1.26 |
| 11h | 6.90 | 1:2.7 |
53% | 19700 |
24700 |
1.52 |
The reaction was repeated under identical conditions, except for the stirring rate which was increased from 400 rpm and 800 rpm (Table 2, entry 2). The conversion increased to 56% after 4 hours, and the control over the polymerisation was improved (Mn,SEC = 15900 g mol−1, Mn,th = 10700 g mol−1, Đm = 1.49). Kinetic analysis of the reactions performed at 400 rpm and 800 rpm revealed the rate of the reaction increased at the higher stirring rate (kapp,400p = 2.2 × 10−5 s−1, kapp,800p = 5.6 × 10−5 s−1, Fig. 6A). Mn,SEC increased linearly with conversion at 800 rpm (Fig. 6B), which was not the case at 400 rpm, and the SEC trace of the final polymer obtained at 800 rpm was more symmetrical than the one obtained at 400 rpm (Fig. 6C). However, the dispersity obtained was still higher than expected for a polymer synthesised by RDRP and electrodeposition at the working electrode was still prevalent.
 | ||
| Fig. 6 (A) Conversion vs. time and first order kinetic plot; (B) Mn,SECvs. conversion; (C) SEC in DMF (at 400 rpm (blue), Mn,SEC = 10700 g mol−1, Đm = 2.32; at 800 rpm (red), Mn,SEC = 15900 g mol−1, Đm = 1.49) for the current-controlled seATRP of 10 wt% HEAm in H2O containing KNO3 (0.1 M) at 0 °C at 400 rpm (blue) and 800 rpm (red). Conditions: [HEAm]/[HEBiB] = [160]; Iapp = −4 mA (5 min), −3.1 mA (5 min), −1.9 mA (5 min), −1.3 mA (5 min), −0.9 mA (5 min) and −0.8 mA (215 min). | ||
In an attempt to improve the control over the polymerisation at 0 °C, the [CuII]:[TPMA] ratio and the relative [CuII/TPMA] employed was investigated. When employing a sacrificial electrode, having an excess of the ligand relative to CuII is important as the metal ions released from the electrodes (Al3+ in the case of the Al-electrodes) can compete with Cu for the ligand.48 Thus far, the reactions carried out herein have employed [CuII]:[TPMA] = [1]:[4] which is excessive. Decreasing the ratio to [CuII]:[TPMA] = [1]:[2.7] and [1]:[2] led to slightly higher conversions, reaching ∼65% conversion within 4 hours without further compromising the control over the polymerisation as Đm = 1.47 and 1.46 respectively (Table 2, entries 3 and 4, Fig. S8†). When the ratio was decreased further to [CuII]:[TPMA] = [1]:[1.5], conversion dropped back down to 56% within 4 hours and the dispersity increased slightly (Đm = 1.51, Table 2, entry 5). Based on these results, it was decided to investigate the effect of [CuII/TPMA] using [CuII]:[TPMA] = [1]:[2.7] at 800 rpm.
In the experiments performed above, in which electrodeposition was observed and as a result the control over the polymerisation was limited, the [CuII/TPMA] was 2.30 mM. Unsurprisingly, initially decreasing [CuII/TPMA] to 1.15 mM resulted in increased electrodeposition which prompted us to stop the reaction after 3.25 hours, at which point only 20% conversion had been reached (Table 2, entry 6). The lower [CuII/TPMA] had a detrimental effect on both reaction conversion and polymerisation control with a significant discrepancy between Mn,SEC (10000 g mol−1) and Mn,th (3900 g mol−1), and high dispersity (Đm = 2.48) obtained from SEC. In the literature, increasing the catalyst concentration was reported to have a positive effect on the reaction conversion, rate, and control over polymerisation during aqueous eATRP of DMAm.40 Here, when the [CuII/TPMA] was increased to 4.60 mM and 6.90 mM, electrodeposition was not observed during the course of the reactions. This is because at higher [CuII/TPMA], the potential required to maintain Iapp is less reducing so the reduction of CuI/II to Cu0 is less favourable. When [CuII/TPMA] = 4.60 mM the conversion reached 82% within 4 hours (Table 2, entry 7). However, the control over the polymerisation was only slightly improved, with Mn,SEC = 12300 g mol−1 (Mn,th = 15300 g mol−1) and Đm = 1.45. Increasing [CuII/TPMA] further to 6.90 mM resulted in 77% conversion within 4 hours (Table 2, entry 8). Kinetic analysis revealed a linear growth of Mn,SEC with conversion suggesting good control over the polymerisation (Fig. 7A). This was supported by good agreement between Mn,SEC (17700 g mol−1) and Mn,th (14400 g mol−1) and the lowest dispersity (Đm = 1.38) when targeting DPn,th = 160 (Fig. S9†).
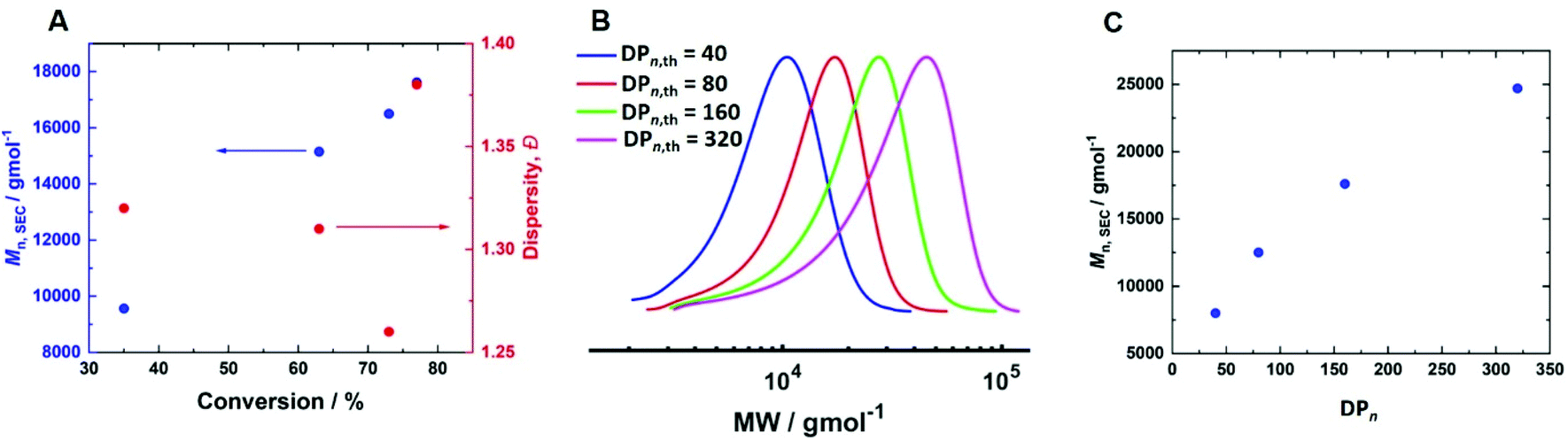 | ||
| Fig. 7 (A) Mn,SEC and Đmvs. conversion for the current-controlled seATRP of 10 wt% HEAm in H2O containing KNO3 (0.1 M) at 0 °C using [HEAm]/[HEBiB] = 160; conditions; [CuII] = 6.9 mM, [CuII]:[TPMA] = [1]:[2.7]. (B) SEC in DMF (Table 2, entries 8–11); (C) Mn,SECvs. DPn for the current controlled seATRP of 10 wt% HEAm in H2O containing KNO3 (0.1 M) at 0 °C as a function of [HEAm]/[HEBiB]; for A–C; Iapp = −4 mA (5 min), −3.1 mA (5 min), −1.9 mA (5 min), −1.3 mA (5 min), −0.9 mA (5 min) and −0.8 mA (215 min). | ||
To determine if the increased [CuII/TPMA] could be applied to target shorter and longer chain lengths, reactions in which the DPn,th was varied were performed at 0 °C, 800 rpm and [CuII/TPMA] = 6.90 mM (Fig. 7B). When DPn,th was decreased to 40, the polymerisation reached 91% conversion within 4 hours, yielding PHEAm with Mn,SEC = 8000 g mol−1 (Mn,th = 4400 g mol−1) and Đm = 1.26, which is an improvement on the analogous reaction performed at room temperature (77% in 2 hours, Mn,SEC = 8200 g mol−1, Mn,th = 3600 g mol−1, Đm = 1.35). Increasing DPn,th to 80 resulting in 79% conversion to PHEAm after 4 hours with control over the polymerisation retained (Mn,SEC = 12500 g mol−1, Mn,th = 7400 g mol−1, Đm = 1.26). When the DPn,th was increased from 160 to 320 the control over the polymerisation was compromised, as previously observed in the room temperature reactions. The reaction conversion reached 53% in 4 hours, yielding PHEAm with Mn,SEC = 24700 g mol−1 (Mn,th = 19600 g mol−1) and Đm = 1.52. Although the dispersity value is larger than we would expect for a true RDRP reaction, a plot of Mn,SECvs. DPn revealed a linear correlation between Mn,SEC and DPn,th, which represents an improvement on the analogous room temperature reactions (Fig. 7C).
The current-control over the reaction at 0 °C was then investigated using [HEAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.1]:[0.4]:[0.1] (Fig. 8A). The temporal control observed was comparable to that obtained at room temperature. At the beginning of the reaction, Iapp = −4.0 mA and the polymerisation reached 51% conversion (tON = 60 min, kapp,1p = 2.0 × 10−4 s−1, Fig. S11†). When Iapp was removed and the reaction was left for a further 15 minutes, the rate of reaction decreased significantly but not completely. The current was switched back on (Iapp = −0.8 mA) to restart the polymerisation, reaching 79% conversion (tON = 120 min, kapp,2p = 2.2 × 10−4 s−1). Again, the rate of reaction was significantly reduced upon removal of Iapp and restarted again after 15 minutes. Conversion continued to increase up to 90% (tON = 130 min, kapp,3p = 1.9 × 10−4 s−1) before Iapp was switched off and the polymerisation was stopped. The molecular weight was shown to increase during the periods when current was applied, whilst there was little or no change when the current was removed (Fig. 8B). The final polymer reached 90% conversion with Mn,SEC = 7200 g mol−1 and Đm = 1.26.
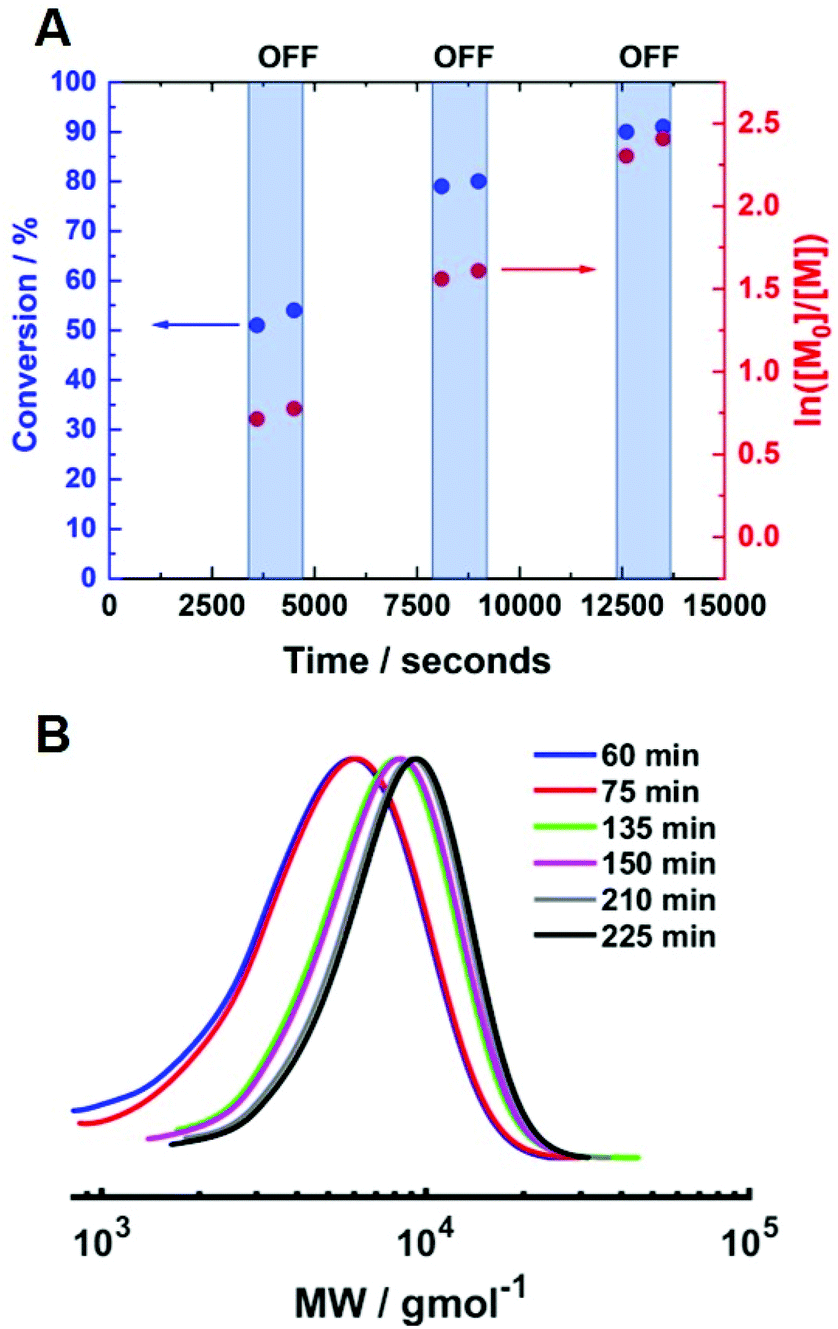 | ||
| Fig. 8 (A) Conversion vs. time plot and first order kinetic plot demonstrating the temporal control exhibited during the current controlled seATRP of HEAm conducting using [HEAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.1]:[0.4]:[0.1] at 0 °C. Between t = 0 and t = 30 min Iapp = −4 mA (5 min), −3.1 mA (5 min), −1.9 mA (5 min), −1.3 mA (5 min), −0.9 mA (5 min) and −0.8 mA (35 min). Thereafter, during 15 min intervals, Iapp = 0 mA and during 60 min intervals, Iapp = −0.8 mA. (B) SEC in DMF of samples taken at each time point during the temporal control experiment. For the final polymer, Mn,SEC = 7200 g mol−1 and Đm = 1.26. | ||
Scope of the reaction
To investigate the scope of the reaction conditions developed, we applied our current profile to a series of polymerisations using commercially available primary, secondary and tertiary acrylamides using [M]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.3]:[0.8]:[0.1]. Repeating the polymerisation of secondary acrylamide HEAm resulted in 91% in 4 hours, Mn,SEC = 8000 g mol−1 (Mn,th = 4400 g mol−1), and Đm = 1.25 (Table 3, entry 1; Fig. S10†). Secondary acrylamide NIPAm gave a comparable outcome with conversion reaching 96% in 4 hours. SEC analysis furnished a symmetrical molecular weight distribution with relatively good agreement between Mn,SEC = 7300 g mol−1 and Mn,th = 4500 g mol−1, and low dispersity (Đm = 1.31, Table 3, entry 2; Fig. S11†). Tertiary acrylamides N-acryloylmorpholine (NAM) and DMAm did not polymerise well using our current profile under these reaction conditions. Conversions after 4 hours were limited to 29% for NAM, 49% for DMAm and control over the polymerisations was poor (Đm ≥ 1.60, Table 3, entry 3–4; Fig. S12 and S13)†. The limited conversion indicates that the current profile, established for the polymerisation of secondary acrylamide HEAm was not appropriate for the tertiary acrylamide monomers. Furthermore, the loss of the ω-chain end during the aqueous Cu-mediated polymerisation of acrylamides is more prevalent with tertiary acrylamides than acrylamides, which could also contribute to loss of control during the polymerisation.32,33 The monomer acrylamide, which contains a primary amide group was then subject to our current profile reaching 86% conversion within 4 hours. Aqueous SEC analysis revealed a highly symmetrical molecular weight distribution, with excellent agreement between the Mn,SEC = 2200 g mol−1 and Mn,th = 2500 g mol−1 and low dispersity (Đ = 1.27, Table 3, entry 5; Fig. S14†).
| Entry | Monomer | Conv.a |
M
n,thb/g mol−1 |
M
n,SECc/g mol−1 |
Đ
mc |
|---|---|---|---|---|---|
| Time = 4 h; temperature = 0 °C; 800 rpm; 10% v/v monomer; [M]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.3]:[0.8]:[0.1].a Determined via1H NMR of reaction samples performed in D2O.b Mn,th = [(conv./100 × DPn,th) × (MWmonomer)] + 211.c From DMF SEC. |
|||||
| 1 | HEAm | 91% | 4400 | 8000 | 1.25 |
| 2 | NIPAm | 96% | 4600 | 7300 | 1.31 |
| 3 | NAM | 29% | 1800 | 2000 | 1.60 |
| 4 | DMAm | 49% | 2200 | 1500 | 2.02 |
| 5 | AAm | 83% | 2600 | 2200 | 1.27 |
Finally, a chain extension reaction was attempted to exemplify retention of the ω-Br chain end during the current-controlled eATRP reactions. On the balance of the high conversion and low dispersity obtained previously, homopolymerisation of NIPAm was initially repeated using [NIPAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.3]:[0.8]:[0.1]. Conversion reached 89% after 3 hours yielding PNIPAm with Mn,SEC = 8500 g mol−1 and Đm = 1.23.
Electrolysis was stopped and a second aliquot of NIPAm (DPn,th = 40) in water was added to the reaction mixture. The current profile with regimes of Iapp = −4 mA (5 min), −3.1 mA (5 min), −1.9 mA (5 min), −1.3 mA (5 min), −0.9 mA (5 min) and −0.8 mA (155 min) was applied to the reaction solution, resulting in 13% conversion within 3 hours at 0 °C. Despite the conversion being low, chain extension was evident via a shift in the mono-modal, symmetrical molecular weight distribution to higher molecular weight with Mn,SEC of chain extended PNIPAm increasing to 9400 g mol−1 and low dispersity (Đ = 1.26) being retained (Fig. 9). Thus, although chain extension is possible using our current profile, there is scope for improvement to enable higher conversions and application to block copolymerisation. This could be achieved by first performing the reactions under potentiostatic conditions to obtain a more bespoke current profile for the target polymerisation.
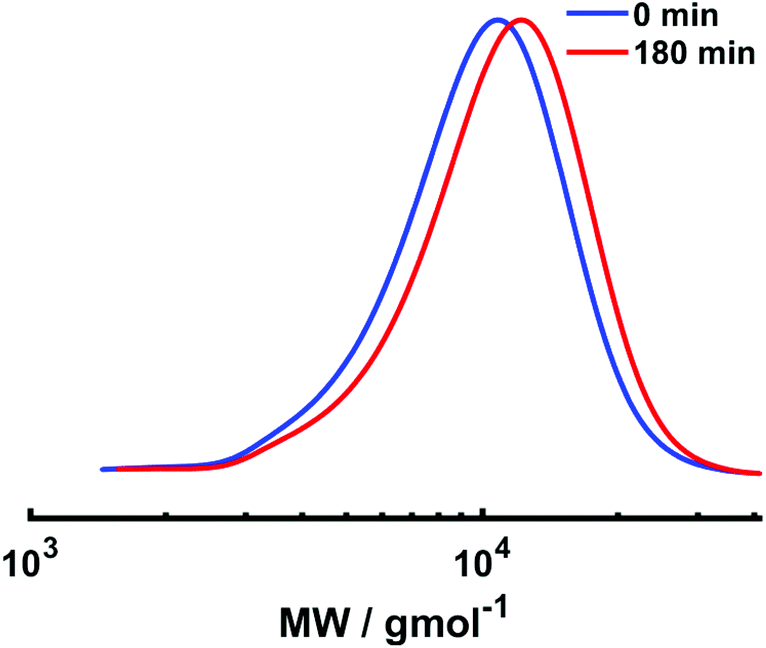 | ||
| Fig. 9 SEC in DMF for the current-controlled seATRP homopolymerisation of NIPAm (red, [NIPAm]:[HEBiB]:[CuII]:[TPMA]:[NaBr] = [40]:[1]:[0.3]:[0.8]:[0.1], Mn,SEC = 8500 g mol−1 and Đ = 1.23) and subsequent in situ chain extension using NIPAm (blue, [NIPAm]:[PNIPAm-Br] = [20]:[1], Mn,SEC = 9400 g mol−1 and Đm = 1.26). | ||
Conclusions
Current-controlled eATRP of acrylamides in water has been simplified with respect to the reaction hardware by using a 2-electrode, ‘plug-and-play’, undivided electrochemical cell enabled by the IKA ElectraSyn 2.0 device. Furthermore, the experimental reaction conditions have been simplified using an I vs. t plot from a model reaction – the aqueous eATRP of HEAm under potentiostatic conditions – to design a stepwise current profile (Iappvs. time) for current-controlled polymerisation of HEAm. The current profile was initially employed to explore the current-controlled eATRP of HEAm at room temperature. Switching the Iapp on and off at regular intervals demonstrated that conversion of monomer to polymer occurred in the presence of Iapp, with little or no conversion observed in the absence of Iapp. It was possible to target DPn,th = 20–100, whilst retaining good control over the polymerisations (Đm ≤ 1.50). However, it was not possible to target DPn,th > 100 and retain control of the polymerisation. To alleviate this, the reaction temperature was reduced to 0 °C to minimise deleterious reactions that can occur at the ω-chain end; the stirring rate was increased from 400 rpm to 800 rpm, to improve mass transfer and limit electrodeposition of Cu0 on the working electrode that was found to occur at the lower temperature and stirring rates; and the concentration of CuII/TPMA was increased to enhance the rate of reaction and improve control over the polymerisation. Under these conditions, the polymerisations were again shown to be under the control of Iapp whilst a plot of Mn,SECvs. DPn exhibited a linear increase up to DPn,th = 320. The control over the polymerisation was improved across the targeted DPn,th range, with lower Đm values and more symmetrical SEC traces obtained at 0 °C compared to room temperature. Finally, the current profile was shown to enable the polymerisation of other secondary (NIPAm) and primary (AAm) acrylamides with very good control with retention of the ω-Br chain end in PNIPAm, prepared using our current profile, verified by a short in situ chain extension.Thus, we have shown that it is possible to apply a single current profile, derived from a well-controlled potentiostatic eATRP reaction, to perform simplified, current-controlled eATRP of primary and secondary acrylamides. However, it should be noted that tertiary acrylamides (DMAm and NAM) suffered from low conversion and poor control when subjected to our current profile, indicating that it is not universal for the acrylamide monomer family. For the best results, bespoke current profiles should be obtained.
Data availablity
Additional data is presented in the ESI file.† The raw/processed data from which this data was prepared is available upon request.Author contributions
Mahir Mohammed: Investigation; methodology; formal analysis; validation; visualization; writing – original draft. Bryn Jones: Methodology; formal Analysis; supervision; writing – review and editing. Paul Wilson: Conceptualization; funding acquisition; investigation; methodology; project administration; resources; supervision; writing – review and editing.Conflicts of interest
No conflicts of interest to report.Acknowledgements
The authors would like to thank the Polymer Characterization Research Technology Platforms for maintenance, access and use of SEC facilities. This research was funded in whole or in part by the Royal Society and Tata companies for the award of a University Research Fellowship (URF\R1\180274) and the EPSRC for funding a DTP studentship (M. M. EP/R513374/1). For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.References
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- S. Yurteri, I. Cianga and Y. Yagci, Macromol. Chem. Phys., 2003, 204, 1771–1783 CrossRef CAS.
- K. Matyjaszewski, P. J. Miller, J. Pyun, G. Kickelbick and S. Diamanti, Macromolecules, 1999, 32, 6526–6535 CrossRef CAS.
- J. Pyun, T. Kowalewski and K. Matyjaszewski, Macromol. Rapid Commun., 2003, 24, 1043–1059 CrossRef CAS.
- K. L. Beers, S. G. Gaynor, K. Matyjaszewski, S. S. Sheiko and M. Möller, Macromolecules, 1998, 31, 9413–9415 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS PubMed.
- W. Tang, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 1598–1604 CrossRef CAS PubMed.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58–62 CrossRef CAS PubMed.
- Z. Wang, Z. Wang, X. Pan, L. Fu, S. Lathwal, M. Olszewski, J. Yan, A. E. Enciso, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 275–280 CrossRef CAS.
- H. Y. Cho and C. W. Bielawski, Angew. Chem., Int. Ed., 2020, 59, 13929–13935 CrossRef CAS PubMed.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- X. Pan, M. Fantin, F. Yuan and K. Matyjaszewski, Chem. Soc. Rev., 2018, 47, 5457–5490 RSC.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. D. Magenau, A. Sobkowiak and K. Matyjaszewski, Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS.
- A. J. D. Magenau, N. Bortolamei, E. Frick, S. Park, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 4346–4353 CrossRef CAS.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed.
- C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC.
- F. Lorandi, M. Fantin, A. A. Isse and A. Gennaro, Curr. Opin. Electrochem., 2018, 8, 1–7 CrossRef CAS.
- B. Zhao, M. Mohammed, B. A. Jones and P. Wilson, Chem. Commun., 2021, 57, 3897–3900 RSC.
- B. Zhao, F. Pashley-Johnson, B. A. Jones and P. Wilson, Chem. Sci., 2022, 13, 5741–5749 RSC.
- S. Park, P. Chmielarz, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2015, 54, 2388–2392 CrossRef CAS PubMed.
- P. Chmielarz, A. Sobkowiak and K. Matyjaszewski, Polymer, 2015, 77, 266–271 CrossRef CAS.
- F. Lorandi, M. Fantin, A. A. Isse and A. Gennaro, Polym. Chem., 2016, 7, 5357–5365 RSC.
- P. Chmielarz and A. Sobkowiak, Polimery, 2021, 61, 585–590 CrossRef.
- N. Bortolamei, A. A. Isse, A. J. D. Magenau, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 11391–11394 CrossRef CAS PubMed.
- M. Fantin, A. A. Isse, A. Venzo, A. Gennaro and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 7216–7219 CrossRef CAS PubMed.
- M. Fantin, F. Lorandi, A. A. Isse and A. Gennaro, Macromol. Rapid Commun., 2016, 37, 1318–1322 CrossRef CAS PubMed.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- F. Alsubaie, A. Anastasaki, P. Wilson and D. M. Haddleton, Polym. Chem., 2015, 6, 406–417 RSC.
- J. T. Rademacher, M. Baum, M. E. Pallack, W. J. Brittain and W. J. Simonsick, Macromolecules, 2000, 33, 284–288 CrossRef CAS.
- M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2015, 48, 6862–6875 CrossRef CAS.
- N. V. Tsarevsky, T. Pintauer and K. Matyjaszewski, Macromolecules, 2004, 37, 9768–9778 CrossRef CAS.
- P.-E. Millard, N. C. Mougin, A. Böker and A. H. E. Müller, Controlled/Living Radical Polymerization: Progress in ATRP, American Chemical Society, 2009, vol. 1023, ch. 9, pp. 127–137 Search PubMed.
- A. Anastasaki, A. J. Haddleton, Q. Zhang, A. Simula, M. Droesbeke, P. Wilson and D. M. Haddleton, Macromol. Rapid Commun., 2014, 35, 965–970 CrossRef CAS PubMed.
- P. Chmielarz, P. Krys, S. Park and K. Matyjaszewski, Polymer, 2015, 71, 143–147 CrossRef CAS.
- P. Chmielarz, S. Park, A. Simakova and K. Matyjaszewski, Polymer, 2015, 60, 302–307 CrossRef CAS.
- F. De Bon, S. Marenzi, A. A. Isse, C. Durante and A. Gennaro, ChemElectroChem, 2020, 7, 1378–1388 CrossRef CAS.
- S. E. Edwards, S. Flynn, J. J. Hobson, P. Chambon, H. Cauldbeck and S. P. Rannard, RSC Adv., 2020, 10, 30463–30475 RSC.
- T. J. Zerk, M. Martinez and P. V. Bernhardt, Inorg. Chem., 2016, 55, 9848–9857 CrossRef CAS PubMed.
- C. A. Bell, P. V. Bernhardt and M. J. Monteiro, J. Am. Chem. Soc., 2011, 133, 11944–11947 CrossRef CAS PubMed.
- F. De Bon, M. Fantin, A. A. Isse and A. Gennaro, Polym. Chem., 2018, 9, 646–655 RSC.
- S. Dadashi-Silab, I.-H. Lee, A. Anastasaki, F. Lorandi, B. Narupai, N. D. Dolinski, M. L. Allegrezza, M. Fantin, D. Konkolewicz, C. J. Hawker and K. Matyjaszewski, Macromolecules, 2020, 53, 5280–5288 CrossRef CAS.
- J. R. Black, S. John, E. D. Young and A. Kavner, Geochim. Cosmochim. Acta, 2010, 74, 5187–5201 CrossRef CAS.
- R. Schmidt and J. Gaida, ChemElectroChem, 2018, 5, 2176–2180 CrossRef CAS.
- J. Luo, C. Durante, A. Gennaro and A. A. Isse, Electrochim. Acta, 2021, 388, 138589 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00412g |
| This journal is © The Royal Society of Chemistry 2022 |