
Fluoride-free synthesis of high-silica, medium-pore zeolites PST-22 and PST-30†
Donghui
Jo
 a and
Suk Bong
Hong
*b
a and
Suk Bong
Hong
*b
aPetrochemical Catalyst Research Center, Korea Research Institute of Chemical Technology, Daejeon 34114, Korea
bCenter for Ordered Nanoporous Materials Synthesis, Division of Environmental Science and Engineering, POSTECH, Pohang 37673, Korea. E-mail: sbhong@postech.ac.kr
First published on 8th November 2021
Abstract
We recently reported the synthesis of three high-silica (Si/Al ∼ 10) zeolites with new framework structures (i.e., PST-21, PST-22, and PST-30) via an excess fluoride approach which uses synthesis mixtures with HF/OSDAn+ = 2/n (n = 1 or 2). However, the use of toxic and corrosive fluoride ions should be avoided for the green manufacturing of zeolites. Here we examine the structure-directing abilities of eight diazolium-based mono- and dications in hydroxide media which could crystallize PST-21, PST-22, or PST-30 under excess fluoride conditions. Among them, 1,1′-(1,4-butanediyl)bis(2,4-dimethyl-1H-pyrazol-2-ium) (14DMP-C42+) and 1,1′-(1,4-butanediyl)bis(2,5-dimethyl-1H-pyrazol-2-ium) ions were found to direct the synthesis of PST-22 and PST-30 in the presence of Na+ ions, respectively. Powder X-ray diffraction and Rietveld analyses of as-made PST-22 reveal that while the pyrazolium moieties in 14DMP-C42+ are located near the center of t-pww cages, the tetramethylene chain runs through a 10-ring window connecting two t-pww cages. The proton forms of PST-22 and PST-30 synthesized here exhibit better performance in 1-butene skeletal isomerization than H-ferrierite, one of the most selective catalysts for this reaction.
Introduction
Zeolites are a class of stable, microporous aluminosilicates of industrial relevance that are demonstrated by their widespread use in ion exchange, separations, and catalysis.1,2 The shape-selective properties of these crystalline solids differ primarily in the size, shape, and dimensionality of their channels and cages. Thus, the synthesis of new zeolite structures has been a topic of keen interest to researchers in both academia and industry. However, although millions of energetically feasible hypothetical structures have so far been proposed,3,4 the number of distinct framework type codes (FTCs) approved by the Structure Commission of the International Zeolite Association (IZA) is only about 250.5On the other hand, since zeolite crystallization is very complex and involves a large number of chemical reactions and equilibria that are mutually dependent in nature,6,7 developing fresh synthetic strategies with a degree of rational design is a prerequisite for discovering previously unseen zeolite structures. One recent example of this is the excess fluoride approach where the molar concentration of F− ions in aluminosilicate synthesis mixtures is intentionally chosen to be considerably higher than that of a specific organic structure-directing agent (OSDA) in an attempt to alter the phase selectivity of crystallization by increasing the competition between the isomorphous substitution of Al atoms in the zeolite framework and the F− encapsulation within small cages such as double 4-rings.2,8,9 Indeed, when the diazolium-based mono- and dications in Fig. 1 were used as OSDAs under highly concentrated, excess F− conditions, we were able to find three novel zeolites, i.e., PST-21 (FTC PWO), PST-22 (PWW), and PST-30 (PTY).8,9
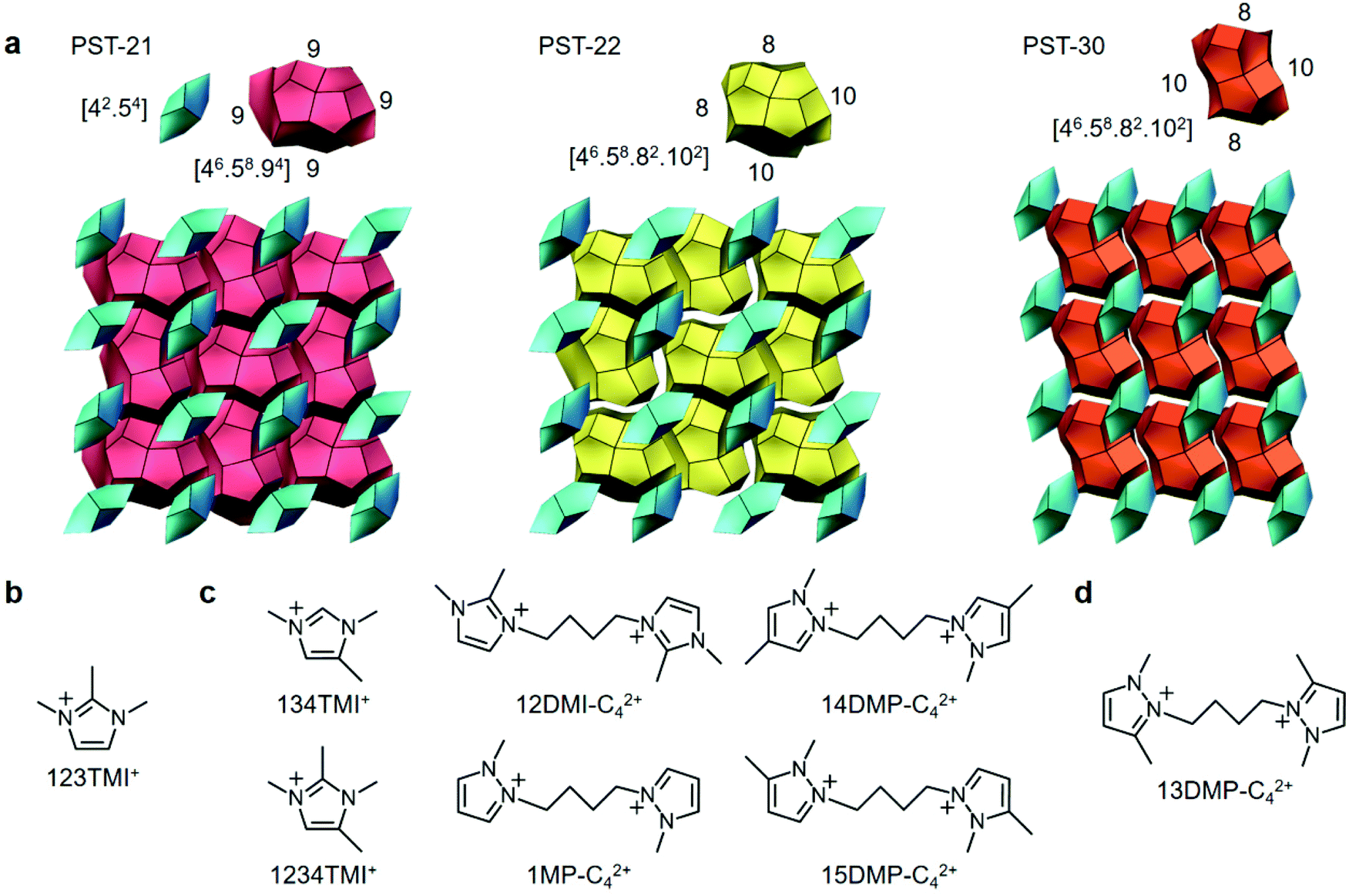 | ||
| Fig. 1 (a) The tiling structures of PST-21, PST-22, and PST-30 and (b–d) the OSDA(s) leading to the crystallization of these three zeolites under excess fluoride conditions, respectively.8,9 | ||
All three of these zeolite structures are built up of 6-hedral bre ([42·54]) cages as secondary building units in a nonjoint way and characterized by the presence of only one type of 18-hedral cavity.8,9 However, the type of their large cage is different from one another: t-pwo ([46·58·94]), t-pww ([46·58·82·102]), and t-pty ([46·58·82·102]) cages for PST-21, PST-22, and PST-30, respectively. Consequently, while PST-21 contains a two-dimensional (2D) pore system consisting of 9-ring (4.2 × 4.4 Å) channels only, PST-22 has one type of 10-ring (5.2 × 6.0 Å) channels, as well as two intersecting 8-ring (3.3 × 3.6 Å) channels. Also, PST-30 possesses a 2D pore system with intersecting 10-ring (4.1 × 6.7 Å) and 8-ring (3.1 × 4.3 Å) channels, like ferrierite (FER) (Fig. 1). To our knowledge, PST-30 is the first case where the targeted synthesis of a particular hypothetical zeolite structure has been successfully achieved by computationally searching for an appropriate OSDA.9
However, the F− ion is highly toxic and corrosive, as well as a potentially environmentally unfriendly component in zeolite synthesis. This motivated us to develop the F−-free synthesis of new zeolites initially discovered via the excess fluoride approach. Here we describe our attempts to synthesize PST-22 and PST-30 using pyrazolium-based OSDAs and Na+ ions as inorganic structure-directing agents (ISDAs) in hydroxide media. While the synthesis of PST-30 includes the use of the original OSDA, we were able to synthesize PST-22 by using only one OSDA among the six azolium-based OSDAs that could direct the formation of this zeolite under excess F− conditions.8,9 Powder X-ray diffraction and Rietveld analyses of as-made PST-22 synthesized in this study demonstrate that the central tetramethylene chain in its OSDA, i.e., the 1,1′-(1,4-butanediyl)bis(2,4-dimethyl-1H-pyrazol-2-ium) ion, spans two adjacent t-pww cages through a 10-ring window. We also show that the proton forms of PST-22 and PST-30 synthesized in hydroxide media outperform H-ferrierite in the skeletal isomerization of 1-butene to isobutene, a raw material for the production of value-added chemicals such as isooctane, methacrolein, and butyl rubber.10
Experimental
Synthesis
The iodide salt of 1,2,3-trimethylimidazolium (123TMI+), 1,3,4-trimethylimidazolium (134TMI+), 1,2,3,4-tetramethylimidazolium (1234TMI+) monocations and the dibromide salt of 1,1′-(1,4-butanediyl)bis(1,2-dimethyl-1H-imidazol-2-ium) (12DMI-C42+), 1,1′-(1,4-butanediyl)bis(2-methylpyrazol-2-ium) (1MP-C42+), 1,1′-(1,4-butanediyl)bis(2,3-dimethyl-1H-pyrazol-2-ium) (15DMP-C42+), 1,1′-(1,4-butanediyl)bis(2,4-dimethyl-1H-pyrazol-2-ium) (14DMP-C42+), and 1,1′-(1,4-butanediyl)bis(2,5-dimethyl-1H-pyrazol-2-ium) (13DMP-C42+) dications used as OSDAs in this study (Fig. 1) were prepared and characterized as previously described.8,9 Other reagents used include NaOH (Aldrich, 50%), KOH (Aldrich, 45%), colloidal silica (Ludox AS-40, DuPont, 40%), and aluminum hydroxide (Al(OH)3·H2O, Aldrich).The hydroxide-mediated synthesis of zeolites was carried out using aluminosilicate gels with the chemical composition 0.25R+I (or 0.125R2+Br2)·xM2O·1.0SiO2·0.05Al2O3·30H2O, where R is the diazolium-based OSDA, M is Na+ or K+, and x is varied between 0.10 ≤ x ≤ 0.20. In a typical synthesis, aluminum hydroxide, as-prepared OSDA, and NaOH or KOH solution were mixed with some water and stirred at room temperature for 1 h. To this solution, a given amount of Ludox AS-40 was added. After being stirred at room temperature for 3 h, the final synthesis mixture was transferred into a Teflon-lined 23 mL-autoclave and heated under rotation (60 rpm) at 175 °C for 7 days. The solid products were recovered by filtration or centrifugation, washed repeatedly with water, and dried overnight at room temperature. To prepare their proton forms (H-PST-22 and H-PST-30), as-made PST-22 and PST-30 were calcined in air at 600 °C for 8 h, refluxed twice in 1.0 M NH4NO3 solutions for 6 h, and then recalcined in air at 550 °C for 2 h. For catalytic comparison, H-PST-22 and H-PST-30 were also prepared via the excess fluoride approach.8,9
Analytical methods
Powder X-ray diffraction (PXRD) patterns were recorded on a PANalytical X'Pert diffractometer (Cu Kα radiation) with an X'Celerator detector. Data were collected with a fixed divergence slit (0.50°) and Soller slits (incident and diffracted = 0.04 rad). Elemental analysis of Si and Al was performed on a Shimadzu ICPE-9000 inductively coupled plasma spectrometer. The C, H, and N contents of the samples were analyzed using a Vario EL III elemental organic analyzer. Thermogravimetric analyses (TGAs) were carried out in air on an SII EXSTAR 6000 thermal analyzer, where the weight losses related to the OSDA combustion and the coke deposits formed during the reaction were confirmed by differential thermal analyses (DTAs) using the same analyzer. The crystal morphology and average size were determined using a Hitachi S-4800 field emission scanning electron microscope (FE-SEM). N2 sorption experiments were performed on a Mirae SI nanoPorosity-XG analyzer. NH3 temperature-programmed desorption (TPD) was carried out on a fixed bed, flow-type apparatus with a VICI thermal conductivity detector. A sample of ca. 0.1 g was activated in flowing He (50 mL min−1) at 550 °C for 2 h. Then, 10% NH3 was passed over the sample at 150 °C for 0.5 h. The treated sample was subsequently purged with He at the same temperature for 1 h to remove physisorbed NH3. Finally, the TPD was performed in flowing He (30 mL min−1) from 150 to 750 °C at a temperature ramp of 10 °C min−1.All solid-state NMR measurements were carried out using a Bruker AVANCE III 500 HD spectrometer at a spinning rate of 20 kHz. 1H–13C cross polarization (CP) MAS NMR spectra were recorded at a 13C frequency of 125.77 MHz with a π/2 rad pulse length of 1.8 μs, a contact time of 2.0 ms, a recycle delay of 5.0 s, and an acquisition of ca. 1000 pulse transients. 29Si MAS NMR spectra were recorded at a 29Si frequency of 99.377 MHz with a π/2 rad pulse length of 4 μs, a recycle delay of 150 s, and an acquisition of ca. 500 pulse transients. The 13C and 29Si shifts are referenced relative to TMS. 27Al MAS NMR spectra were recorded at an 27Al frequency of 130.351 MHz with a π/6 rad pulse length of 1.0 μs, a recycle delay of 2.0 s, and an acquisition of ca. 1000 pulse transients. The 27Al chemical shifts are referenced relative to Al(H2O)63+ solution.
The stabilization energies of OSDAs in PST-21, PST-22, and PST-30 were calculated following the method developed by Deem and co-workers.11 The framework atomic position coordinates of these zeolite structures were obtained from the IZA database,5 and the Dreiding force field was used in the Forcite module in Materials Studio 7.0 to calculate the OSDA-zeolite stabilization energies.12 The number of OSDAs per unit cell of each structure was determined so that one diazolium ring unit occupies an 18-hedral cavity surrounded by eight bre units. Detailed calculation procedures can be found in our recent work.9
Structural analysis
Synchrotron PXRD data for as-made PST-22 synthesized using 14DMP-C42+ in hydroxide media were collected on the 9B beamline at the Pohang Acceleration Laboratory (PAL; Pohang, Korea) using monochromated X-rays (λ = 1.5167 Å). The detector arm of the vertical scan diffractometer is composed of seven sets of Soller slits, flat Ge(111) crystal analyzers, anti-scatter baffles, and scintillation detectors, with each set separated by 20°. Data were obtained at room temperature in the flat plate mode, with a step size of 0.02° and an overlap of 0.5° to the next detector bank.The framework atomic positions of calcined PST-228 were taken as the starting model for the zeolite framework in Rietveld analysis using the GSAS suite of programs and EXPGUI graphical interface.13–15 The refinement on the occluded OSDAs was performed using the rigid-body method.16 During the refinement, the framework T–O and O–O distances were soft-constrained to 1.62 Å (σ = 0.005 Å) and 2.62 Å (σ = 0.005 Å), respectively. The peak shape was modeled using the pseudo-Voigt profile function.17 The isotropic atomic displacement parameters of the framework atoms have been constrained in groups for the tetrahedral Si and O atoms, respectively. The positions of OSDA molecules were derived from Fourier difference maps obtained after refining only the framework atoms against the high-angle (d-space < 2.0 Å) data. The 14DMP-C42+ cation with two pyrazolium rings was divided into two rigid-bodies (2C6N2) and two middle C atoms in the central methylene chain during the refinement, whereas the tetrahedral geometry for the two methylene C atoms was maintained by soft-constraining the bonding C–C and nonbonding (C–C)C and (C–N)C distances to 1.55 (σ = 0.01 Å), 2.64 Å (σ = 0.02 Å) and 2.54 Å (σ = 0.02 Å), respectively. The convergence was achieved by refining simultaneously all profile parameters, scale factors, lattice constants, 2θ zero-points, atomic positions, thermal displacement parameters, and occupancy factors for both framework and extra-framework atoms. Additional crystal data are available at the Cambridge Crystallographic Data Centre, deposition no. CCDC 2101479.†
Catalysis
1-Butene skeletal isomerization was performed at 400 °C with a weight hourly space velocity (WHSV) of 7.5 h−1. Prior to the catalytic experiments, the zeolite catalyst was activated under flowing N2 (50 mL min−1) at 550 °C for 2 h and kept at 400 °C, allowing time for the distribution of the reactant/carrier gas to be stabilized. Then, 1-butene (99%, RIGAS) was fed into the microreactor containing the zeolite catalyst at the same temperature. The reaction products were analyzed online using a Varian CP-3800 gas chromatograph equipped with a CP-PoraPLOT Q capillary column (25 m × 0.25 mm) and a flame ionization detector, with the first analysis being carried out after 5 min on stream. Selectivity to isobutene was calculated by dividing the isobutene yield by 1-butene conversion.Results and discussion
Table 1 presents the representative products obtained from hydroxide-mediated zeolite syntheses using a series of diazolium-based OSDAs, which were found to give PST-21, PST-22, and PST-30 under highly concentrated (H2O/SiO2 = 5.0), excess fluoride (HF/OSDAn+ = 2/n) conditions,8,9 and sodium aluminosilicate gels with the same SiO2/Al2O3 ratio (20) but different NaOH/SiO2 ratios (0.20–0.40) at 175 °C for 7 days. Here, the SiO2/Al2O3 ratio of the gel, as well as the synthesis conditions used, was the same as that employed in the typical excess fluoride approach,8,9 whereas its H2O/SiO2 ratio was increased to 30 in order to obtain the homogeneous final synthesis mixture.| Ra | NaOH/SiO2 | Productb | R | NaOH/SiO2 | Product |
|---|---|---|---|---|---|
| a The composition of the synthesis mixture was 0.25R+I (or 0.125R2+Br2)·xNa2O·1.0SiO2·0.05Al2O3·30H2O, where R is a diazolium-based OSDA and x varies between 0.10 ≤ x ≤ 0.20. All syntheses were performed under rotation (60 rpm) at 175 °C for 7 days. b The product appearing first is the major phase, and the product obtained in a trace amount is given in parentheses. A, D, and U indicate amorphous, dense, and unknown phases, respectively. | |||||
| 123TMI+ | 0.20 | A | 0.20 | A | |
| 0.25 | A | 0.25 | A | ||
| 0.30 | Mordenite + RUB-13 | 12DMI-C42+ | 0.30 | A | |
| 0.35 | Mordenite | 0.35 | Mordenite | ||
| 0.40 | Mordenite | 0.40 | Mordenite | ||
| 134TMI+ | 0.20 | A | 0.20 | A | |
| 0.25 | A | 0.25 | A | ||
| 0.30 | A + (ferrierite) | 1MP-C42+ | 0.30 | A | |
| 0.35 | A + ferrierite | 0.35 | Mordenite + ferrierite | ||
| 0.40 | A + ferrierite | 0.40 | Mordenite + PST-22 | ||
| 1234TMI+ | 0.20 | A | 0.20 | A | |
| 0.25 | RUB-13 | 0.25 | A | ||
| 0.30 | RUB-13 | 13DMP-C42+ | 0.30 | PST-30 | |
| 0.35 | RUB-13 + Mordenite | 0.35 | RUB-13 + PST-30 | ||
| 0.40 | Mordenite + RUB-13 | 0.40 | RUB-13 + PST-30 | ||
| 0.20 | A | ||||
| 0.25 | A | ||||
| 14DMP-C42+ | 0.30 | PST-22 | |||
| 0.35 | PST-22 | ||||
| 0.40 | Mordenite + U | ||||
| 0.20 | A | ||||
| 0.25 | A | ||||
| 15DMP-C42+ | 0.30 | A + ferrierite | |||
| 0.35 | Mordenite + U | ||||
| 0.40 | Mordenite + D | ||||
The 123TMI+ monocation, one of the first reported OSDAs yielding PST-21 under excess F− conditions,8 showed no signs of formation of PST-22 and PST-30, as well as PST-21, in hydroxide media, which is also the case for 134TMI+ and 1234TMI+. As shown in Table 1, however, the 14DMP-C42+ dication directed the synthesis of PST-22 in the narrow NaOH/SiO2 ratio region (0.30–0.35), although the other three dications (i.e., 12DMI-C42+, 1MP-C42+, and 15DMP-C42+), which gave this zeolite under excess F− conditions,8 gave no pure PST-22 in hydroxide media. Table 1 also shows that 13DMP-C42+, the original OSDA for PST-30, again directed the synthesis of the same zeolite at NaOH/SiO2 = 0.25–0.30. Therefore, it is clear that while the excess fluoride approach is a useful strategy for synthesizing zeolites with novel framework topologies, such materials can be also synthesized without the aid of F− ions. However, we found that the SiO2/Al2O3 ratio range yielding pure PST-22 and PST-30 in hydroxide media is significantly narrow. When a sodium aluminosilicate gel was used with NaOH/SiO2 = 0.30 and SiO2/Al2O3 = 30 in the presence of 14DMP-C42+ as an OSDA, for example, we always obtained PST-31 (PTO), a new zeolite containing all odd-membered rings from 5 to 11,18 instead of PST-22. Also, an amorphous phase product was obtained when the SiO2/Al2O3 ratio of the synthesis mixture was slightly decreased from 20 to 15.
The replacement of NaOH with an equivalent amount of KOH under the optimized synthesis conditions for obtaining pure PST-22 or PST-30 did not give the corresponding zeolite. As shown in Table S1,† however, SUZ-4 (SZR) with Si/Al = 11 was the phase formed from a synthesis mixture with KOH/SiO2 = 0.35–0.40 in the presence of 13DMP-C42+ as an OSDA (Fig. S1†). This can be rationalized by considering the strong influence of the type of inorganic cation on the product selectivity in the OSDA-mediated synthesis of zeolites in hydroxide media. To date, tetraethylammonium and N,N,N,N′,N′,N′-hexaethylpentanediammonium ions have been the only two known OSDAs used for SUZ-4.19,20
The stabilization energy calculation results in Table 2 show that the 134TMI+ and 1234TMI+ monocations favor the formation of PST-22 compared to the 14DMP-C42+ dication because of their lower stabilization energies (−13.4 or −13.8 vs. −12.9 kJ (mol Si)−1). In addition, the stabilization energy (−12.8 kJ (mol Si)−1) of 1MP-C42+ in PST-22 was calculated to be essentially the same as that of 14DMP-C42+. As shown in Table 1, however, 134TMI+, 1234TMI+, or 1MP-C42+ is not an effective SDA for this zeolite in hydroxide media, which is also the case for 123TMI+ with a rather low stabilization energy (−13.7 kJ (mol Si)−1) in PST-21. These results reveal the limited accuracy of the current OSDA-zeolite stabilization energy calculations in explaining the experimental synthesis results, probably due to the lack of considering kinetic factors during the calculation process.
| OSDA | Zeolite host | Stabilization energy | OSDA | Zeolite host | Stabilization energy |
|---|---|---|---|---|---|
| 123TMI+ | PST-21 | −13.7 | 12DMI-C42+ | PST-22 | −12.0 |
| 134TMI+ | PST-22 | −13.4 | 1MP-C42+ | PST-22 | −12.8 |
| 1234TMI+ | PST-22 | −13.8 | 13DMP-C42+ | PST-30 | −13.7 |
| 14DMP-C42+ | PST-22 | −12.9 | |||
| 15DMP-C42+ | PST-22 | −11.8 |
The PXRD patterns of PST-22 and PST-30 synthesized using sodium aluminosilicate synthesis mixtures with NaOH/SiO2 = 0.30 in hydroxide media show that they are highly crystalline and phase-pure (Fig. 2). For convenience sake, we will refer to them as PST-22(OH) and PST-30(OH), respectively, while designating their corresponding zeolites synthesized in fluoride media as PST-22(F) and PST-30(F), respectively. The 1H–13C CP MAS NMR spectra of as-made PST-22(OH) and PST-30(OH) show that the occluded OSDAs remain intact (Fig. S2 and S3†). We also note that both of them appear as nanocrystals with ill-defined shapes (Fig. 2), unlike the cases of PST-22(F) and PST-30(F),8,9 but maintain the structural integrity after being converted to their proton form (Fig. S4†). The 27Al and 29Si MAS NMR spectra of as-made and the proton forms of PST-22(OH) and PST-30(OH) are shown in Fig. 3. Unlike that of as-made PST-30(OH), the 27Al MAS NMR spectrum of as-made PST-22(OH) exhibits two tetrahedral 27Al resonances at around 58 and 61 ppm. The appearance of an additional weak resonance at around 0 ppm in the spectra of their proton form indicates that a small portion of framework Al atoms has been removed from the zeolite framework during the initial calcination at 600 °C to remove the occluded OSDAs and the successive NH4+ ion exchange and calcination steps. This can be further supported by noticeable differences in the 29Si NMR spectra of as-made and proton forms of PST-22(OH) and PST-30(OH).
 | ||
| Fig. 2 PXRD patterns and FE-SEM images (scale bars, 1 μm) of as-made (a) PST-22 and (b) PST-30 synthesized using 14DMP-C42+ and 13DMP-C42+ as an OSDA in hydroxide media, respectively. | ||
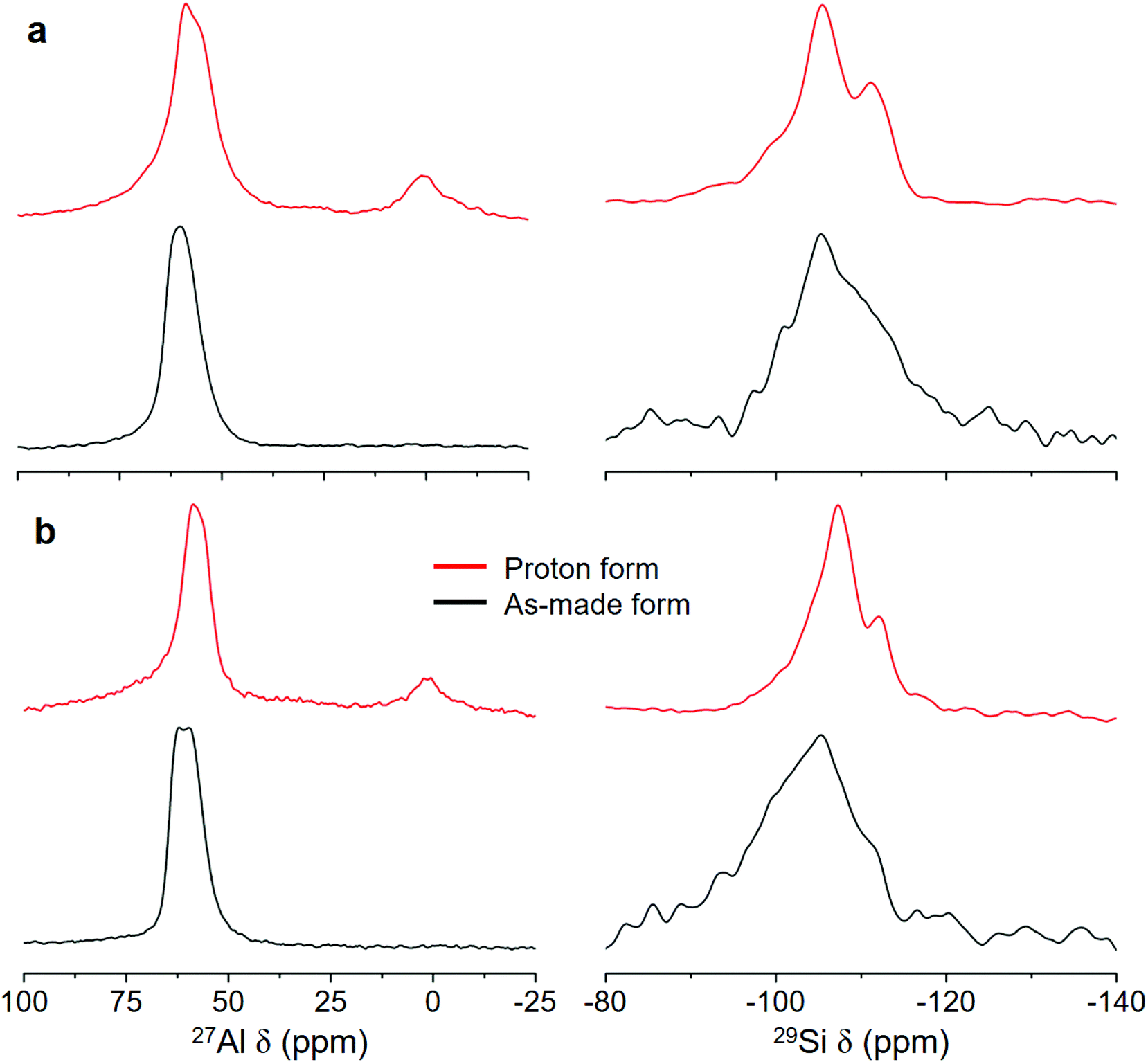 | ||
| Fig. 3 27Al (left) and 29Si (right) MAS NMR spectra of the as-made and proton forms of (a) PST-22(OH) and (b) PST-30(OH). | ||
When the tetramethylene chain of 14DMP-C42+ moves its two pyrazolium rings apart from each other at a particular distance, this OSDA could be more effective in directing the crystallization of PST-22 than imidazolium-based monocations. To check whether our hypothesis is sensible, we performed Rietveld analysis of the synchrotron PXRD data of as-made PST-22(OH) synthesized using 14DMP-C42+ as an OSDA (Fig. S5 and Tables S2–S4†). No attempts to determine the OSDA location in as-made PST-30(OH) obtained using 13DMP-C42+ were made here, because the structure of as-made PST-30(F) synthesized using the same OSDA has already been reported.9 As shown in Fig. 4, the positive difference electron density map after initial scaling, which corresponds to the refined, disordered 14DMP-C42+ molecules, fits well with the 10-ring channel composed of 18-hedral t-pww cages. Consequently, the tetramethylene linker in 14DMP-C42+ with an inversion center was found to span two adjoining t-pww cages through the 10-ring window. However, its two pyrazolium rings are located around the center of each cage (Fig. S6†), like the corresponding ring of 1234TMI+ monocations in as-made PST-22(F).8 The distance between the centers of two pyrazolium rings (i.e., the ring-to-ring distance) of 14DMP-C42+ in as-made PST-22(OH) was calculated to be 7.43 Å that is essentially identical to the distance between the centers of t-pww cages (7.49 Å). We have also noted that the crystallographically determined conformation (Fig. S6†) of 14DMP-C42+ in as-made PST-22(OH) is similar to that of the same OSDA in as-made PST-31 obtained in hydroxide media.18 Therefore, when combined with the synthesis results in Table 1, the central chain length of 14DMP-C42+ with an inversion symmetry appears to be crucial for directing the formation of PST-22. If so, the role of 13DMP-C42+ in the synthesis of PST-30(OH) can be understood in a similar way.
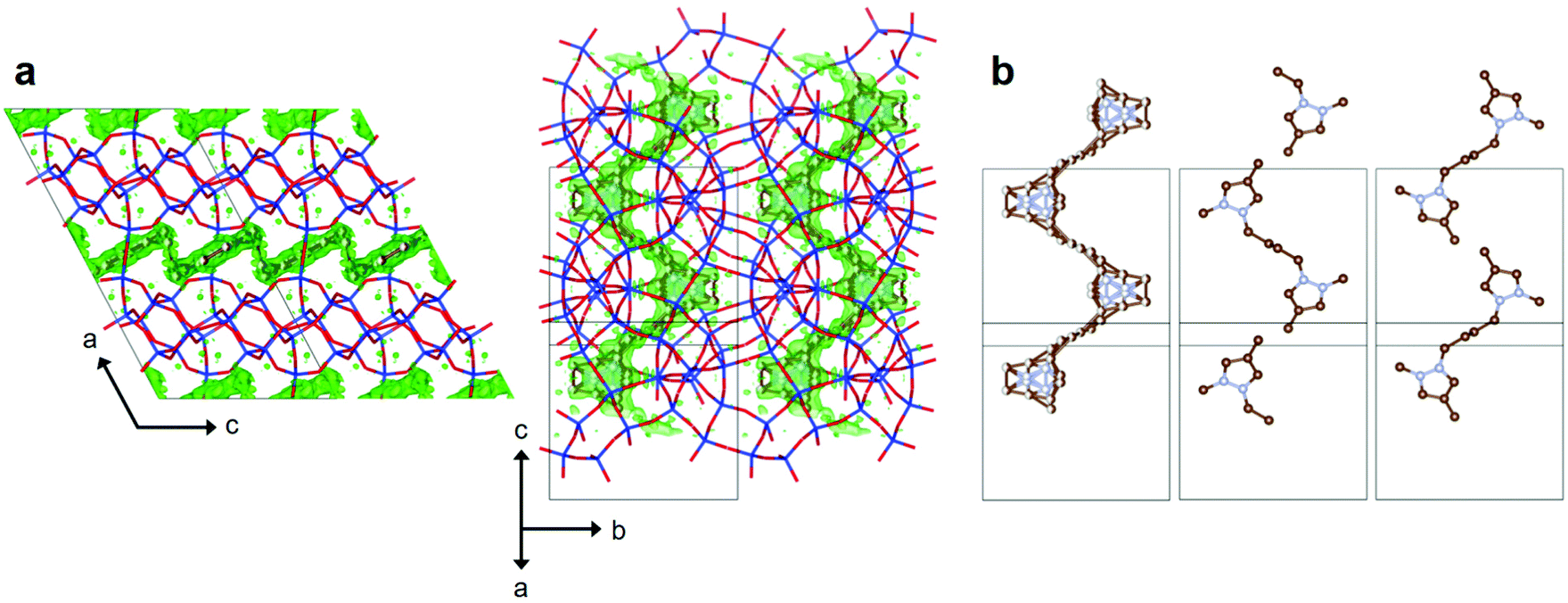 | ||
| Fig. 4 (a) Two views of the refined structure of as-made PST-22(OH) with occluded 14DMP-C42+. Si, blue; O, red; C, brown; and N, pale blue. The regions marked in translucent green indicate the positive difference electron density maps visualized using the whole data set, after refining the framework atoms only against the high-angle (d-space < 2.0 Å) data. (b) Conformations of intrazeolitic 14DMP-C42+ ions: disordered (left) and ordered (middle and right) conformations in two different ways. | ||
The N2 adsorption data reveal that the micropore volume of H-PST-22(OH) is 0.15 cm3 g−1 (Fig. S7†), which is very similar to the value (0.16 cm3 g−1) of H-PST-22(F).8 The same trend was observed for H-PST-30(OH) and H-PST-30(F). Also, all these four zeolites were determined to have essentially the same bulk Si/Al ratio (11), with no noticeable difference in the Al content compared to their respective synthesis mixtures. However, their NH3 TPD profiles in Fig. 5 show that the total area of NH3 desorption or the density of acid sites, especially for the area of the high-temperature desorption peak with a maximum of around 450 °C, is significantly lower for H-PST-22(OH) and H-PST-30(OH) than for H-PST-22(F) and H-PST-30(F). This could be due to the high extent of dealumination in the former two zeolites, because the formation of framework defects in zeolites is more favorable in hydroxide media than in fluoride media. When compared with the 27Al MAS NMR spectra of H-PST-22(F) and H-PST-30(F),8,9 in fact, the relative intensity of the octahedral 27Al resonance around 0 ppm is stronger for H-PST-22(OH) and H-PST-30(OH) (Fig. 3). We also found non-negligible differences in their 27Al and 29Si NMR line shapes compared to H-PST-22(F) and H-PST-30(F), which is also the case of the as-made forms of these two sets of zeolites.8,9 This led us to speculate that the Al distributions in PST-22 and PST-30 may differ according to the synthesis method (i.e., hydroxide route vs. fluoride route), partially explaining notable differences in the density of strong acid sites (Fig. 5).
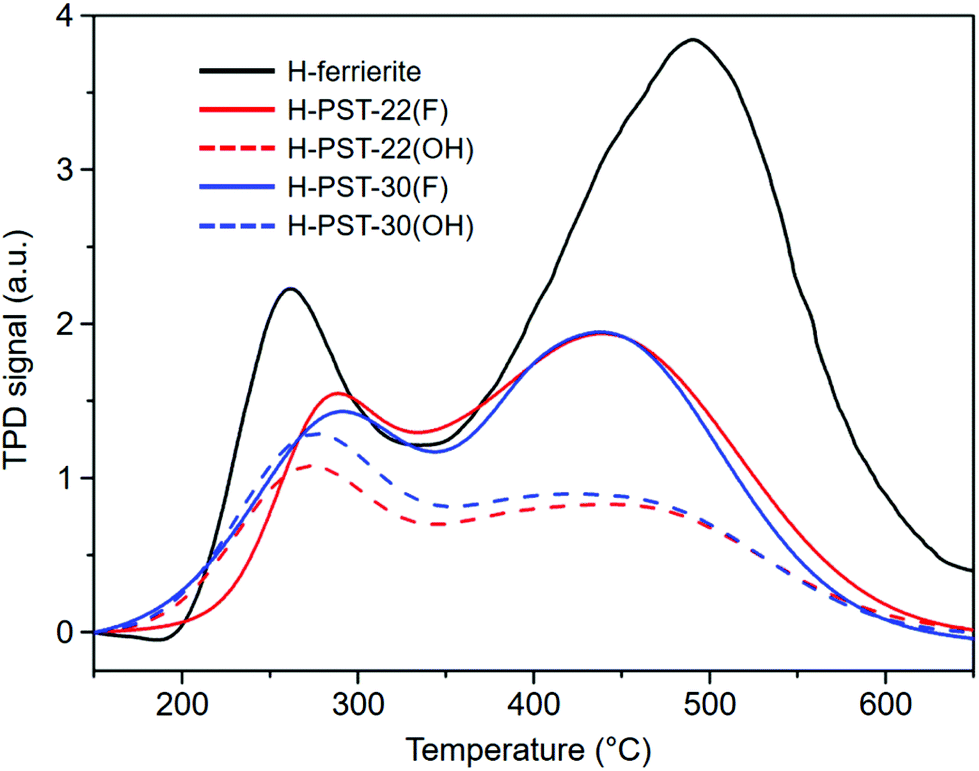 | ||
| Fig. 5 NH3 TPD profiles of H-ferrierite, H-PST-22(F), H-PST-22(OH), H-PST-30(F), and H-PST-30(OH). The bulk Si/Al ratios of H-ferrierite and the other four catalysts are 8.9 and 11, respectively. | ||
Fig. 6 shows 1-butene conversion and selectivity to and yield in isobutene as a function of time on stream (TOS) in the skeletal isomerization of 1-butene over H-PST-22(F), H-PST-22(OH), H-PST-30(F), and H-PST-30(OH), as well as over H-ferrierite with Si/Al = 8.9, one of the best catalysts for this reaction,10 measured at 400 °C and 7.5 h−1 WHSV. The former four catalysts, except H-PST-30(F), are characterized by considerably higher isobutene yields than H-ferrierite over the period of TOS studied. This can be attributed to the lower density and strength of acid sites, as well as to the unique pore structures of PST-22 and PST-30. Despite the similarity in acidity, however, the isobutene yield of H-PST-30(F) is much lower than the yield of H-PST-22(OH) and is similar to that of H-ferrierite. Thermal analysis indicates that the organic contents (7–10 wt%) accumulated in all five zeolites after 12 h on stream at 400 °C are not significantly different from one another. If such is the case, the poor isomerization activity of H-PST-30(F) would then originate mainly from its unique crystallite size and morphology. While H-PST-30(F) appears as heavily overlapped plate-like crystallites of approximately 1.0 μm width and 0.1 μm thickness,9 PST-30(OH) consists of highly agglomerated, nano-sized (< 200 nm) spheres, like PST-22(OH) (Fig. 2).
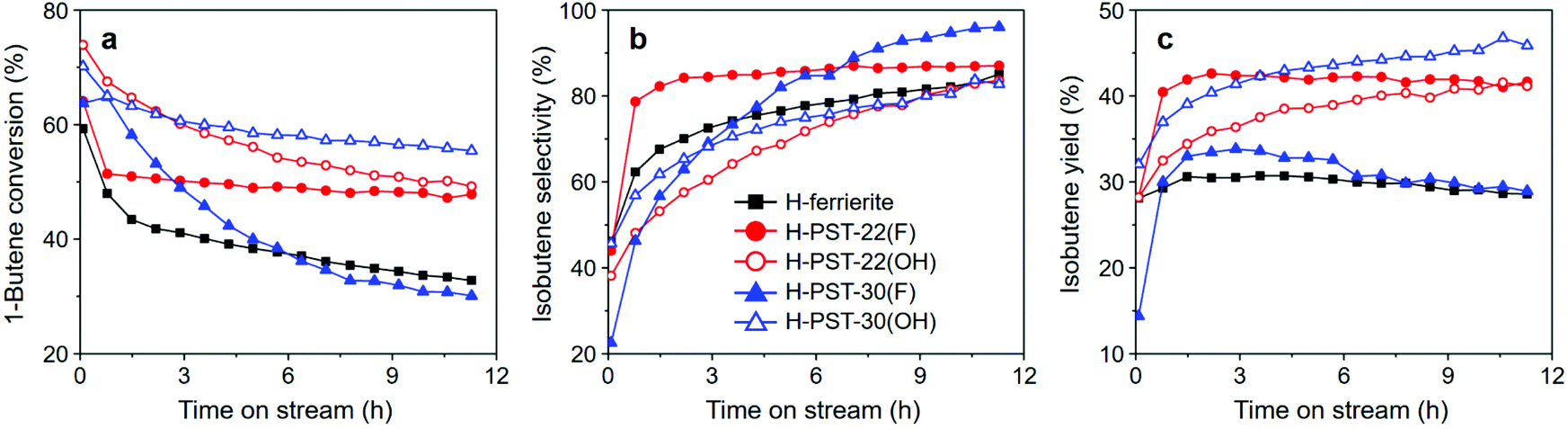 | ||
| Fig. 6 (a) 1-Butene conversion and (b) selectivity to and (c) yield in isobutene at 400 °C and 7.5 h−1 WHSV in the skeletal isomerization of 1-butene over H-ferrierite, H-PST-22(F), H-PST-22(OH), H-PST-30(F), and H-PST-30(OH). The bulk Si/Al ratios of H-ferrierite and the other four catalysts are 8.9 and 11, respectively. | ||
Conclusions
In summary, we have successfully synthesized high-silica, medium-pore zeolites PST-22 and PST-30 in hydroxide media, using 14DMP-C42+ and 13DMP-C42+ as OSDAs, respectively, and Na+ ions as ISDAs, which had been originally discovered under highly concentrated (H2O/SiO2 = 5.0), excess fluoride (HF/OSDAn+ = 2/n) conditions. 13DMP-C42+ was found to produce PST-30 in both fluoride and hydroxide media. Among the six azolium-based mono- and dications known to direct the formation of PST-22 under excess F− conditions, however, 14DMP-C42+ is the only OSDA yielding the corresponding zeolite in hydroxide media, although this dication was calculated to have a lower OSDA-zeolite stabilization energy than monocationic OSDAs such as 134TMI+ and 1234TMI+. Structural analysis of as-made PST-22 reveals a nice geometric match between the 14DMP-C42+ guest with a central tetramethylene chain and the PST-22 host. We also found that H-PST-22 and H-PST-30 prepared here are promising catalysts for the skeletal isomerization of 1-butene to isobutene.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Creative Research Initiative Program (2021R1A3A3088711) through the National Research Foundation of Korea. We thank H. J. Choi (POSTECH) for technical assistance and PAL (Pohang, Korea) for synchrotron PXRD measurements at the 9B beamline (D. Ahn). PAL is supported by MSIP and POSTECH.Notes and references
- J.-P. Gilson and W. Vermeiren, Impact of Zeolites on the Petroleum and Petrochemical Industry, Top. Catal., 2009, 52, 1131–1161 CrossRef.
- J. Shin, D. Jo and S. B. Hong, Rediscovery of the Importance of Inorganic Synthesis Parameters in the Search for New Zeolites, Acc. Chem. Res., 2019, 52, 1419–1427 CrossRef CAS.
- M. D. Foster and M. M. J. Treacy, Atlas of Prospective Zeolite Structures, http://www.hypotheticalzeolites.net, accessed September 16, 2021.
- Y. Li, X. Li, J. Liu, F. Duan and J. Yu, In silico prediction and screening of modular crystal structures via a high-throughput genomic approach, Nat. Commun., 2015, 6, 8328 CrossRef CAS PubMed.
- Ch. Baerlocher and L. B. McCusker, Database of Zeolite Structures, http://www.iza-structure.org/databases/, accessed September 16, 2021.
- C. S. Cundy and P. A. Cox, The hydrothermal synthesis of zeolites: Precursors, intermediates and reaction mechanism, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- J. Grand, H. Awala and S. Mintova, Mechanism of zeolites crystal growth: new findings and open questions, CrystEngComm, 2016, 18, 650–664 RSC.
- D. Jo, G. T. Park, J. Shin and S. B. Hong, A Zeolite Family Nonjointly Built from the 1,3-Stellated Cubic Building Unit, Angew. Chem., Int. Ed., 2018, 57, 2199–2203 CrossRef CAS PubMed.
- D. Jo and S. B. Hong, Targeted Synthesis of a Zeolite with Pre-established Framework Topology, Angew. Chem., Int. Ed., 2019, 58, 13845–13848 CrossRef CAS.
- S. van Donk, J. H. Bitter and K. P. de Jong, Deactivation of solid acid catalysts for butene skeletal isomerisation: on the beneficial and harmful effects of carbonaceous deposits, Appl. Catal., A, 2001, 212, 97–116 CrossRef CAS.
- R. Pophale, F. Daeyaert and M. W. Deem, Computational prediction of chemically synthesizable organic structure directing agents for zeolites, J. Mater. Chem. A, 2013, 1, 6750–6760 RSC.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, DREIDING: A Generic Force Field for Molecular Simulations, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- H. M. Rietveld, A profile refinement method for nuclear and magnetic structures, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR 86-748, 2004.
- B. H. Toby, EXPGUI, a graphical user interface for GSAS, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- C. H. Lake and B. H. Toby, Rigid body refinements in GSAS/EXPGUI, Powder Diffr., 2001, 26, S13–S21 CrossRef.
- R. A. Young and D. B. Wiles, Profile shape functions in Rietveld refinements, J. Appl. Crystallogr., 1982, 15, 430–438 CrossRef CAS.
- D. Jo, Y. Zhang, J. H. Lee, A. Mayoral, J. Shin, N. Y. Kang, Y.-K. Park and S. B. Hong, An Aluminosilicate Zeolite Containing Rings of Tetrahedral Atoms with All Odd Numbers from Five to Eleven, Angew. Chem., Int. Ed., 2021, 60, 5936–5940 CrossRef CAS.
- S. L. Lawton, J. M. Bennett, J. L. Schlenker and M. K. Rubin, Synthesis and proposed framework topology of zeolite SUZ-4, J. Chem. Soc., Chem. Commun., 1993, 894–896 RSC.
- W. C. Paik, C.-H. Shin and S. B. Hong, Synthesis of zeolites P1 and SUZ-4 through a synergy of organic N,N,N,N′,N′,N′-hexaethylpentanediammonium and inorganic cations, Chem. Commun., 2000, 1609–1610 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2101479. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qi01213d |
| This journal is © the Partner Organisations 2022 |