
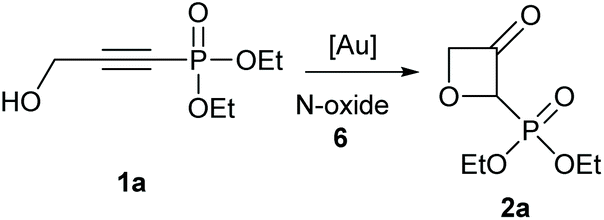
Gold-catalysed synthesis of phosphonate-substituted oxetan-3-ones – an easy access to highly strained HWE reagents†
Shaista
Tahir
a,
Jonas F.
Wunsch
a,
Matthias
Rudolph
a,
Frank
Rominger
a and
A. Stephen K.
Hashmi
 *ab
*ab
aOrganisch-Chemisches Institut, Heidelberg University, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: hashmi@hashmi.de
bChemistry Department, Faculty of Science, King Abdulaziz University, 21589 Jeddah, Saudi Arabia
First published on 12th November 2021
Abstract
A convenient approach for the attainment of strained phosphonate-substituted oxetan-3-ones, starting from easy to synthesise alkynyl phosphonates, is presented. With the aid of a pyridine-N-oxide, in a gold-catalysed step an α-oxo carbene species is generated as a key intermediate. After formal OH-insertion, synthetically useful HWE reagents can be attained. The synthetic utility of the obtained building blocks could be demonstrated, which yielded the 2-alkenyl oxetan-3-one scaffold in high yields after the HWE reaction.
Introduction
Organophosphorus compounds are important from the standpoint of pharmaceutical chemistry,1 materials science2 and organic synthesis.3 This can be attributed to the fact that a large number of naturally occurring biological molecules,4 a great number of n-type organic semiconductors5 and important organic reagents6 bear phosphorus moieties as an integral part of the structure contributing to the said properties. In organic syntheses, the most commonly employed phosphorus-based reagents include Wittig reagents7 and Horner–Wadsworth–Emmons (HWE) reagents,8 which exploit the strong oxygen phosphorous bond as driving force for deoxygenative couplings of organic substrates thus enabling the formation of new double bonds with great chemoselectivity. These type of reagents are extremely important and such reactions have been studied in much detail,9 rendering the synthesis of large natural product analogues for pharmaceutical use, relatively easier as opposed to a multistep process.10Oxeta-3-ones can be found as important substructures in many natural and pharmaceutical products. Due to the ring strain, the synthesis of strained four-membered oxetanone derivatives is an uphill task often requiring a multistep process. Even the synthesis of simple unsubstituted rings, often necessitates either the use of sensitive substrates or the use of a strong base such as LDA alongside TMEDA. The synthesis of the substrates required in the latter case, is also carried out under very tough conditions via acetal route involving protection and deprotection steps.11 An attractive alternative synthetic protocol towards these heterocycles, by employing homogeneous gold catalysis has been reported by the Zhang group.12 This method affords oxetanones in a very simple fashion from propargylic alcohols under mild conditions. While substituents at the former propargylic position can easily be introduced, the substituent directly attached to the alkyne was restricted to either a hydrogen atom or a carbonyl moiety. Inspired by the work on specially activated alkynes, reported in the past decades,13,14 we were curious if due to the electron-withdrawing ability of a phosphonate moiety, P-substituted alkynes could also serve as precursors for oxetanones via a gold-catalysed approach. The addressed oxetanones as targets would be extremely valuable as the attached HWE unit could serve as a suitable synthetic handle for many downstream transformations. The ability for further derivatizations at the former alkyne position should essentially contribute to the implementation of this protocol for the synthesis of medicinally active pharmaceutical molecules and or natural product analogues15–25 as the success of phosphonate-substituted heterocycles as HWE reagents to yield products of vital significance has already been demonstrated.11
Experimental
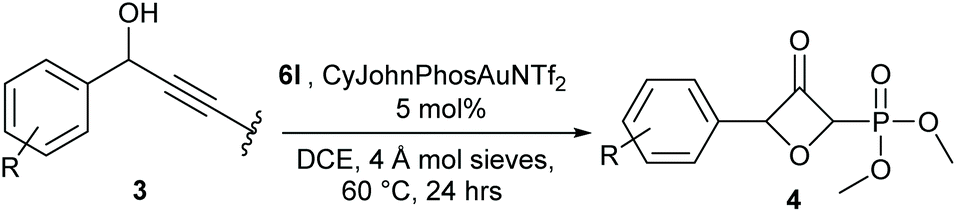
The synthesis of various alkynyl phosphonates was carried out, according to reported procedures (see ESI† for experimental details). The simplest substrate 1a was first selected for the screening of the gold-catalysed reaction (Table 1). The reaction was optimized by using different gold catalysts and pyridine-N-oxides, under varied reaction conditions. With simple pyridine N-oxide only a poor yield was obtained with the common IPr ligand (entry 1) and no yield at all was obtained with AuCl3 (entry 2). Among the tested Phosphine-based ligands (entries 3–5) CyJohnPhosAuNTf2 was the best candidate (21%, entry 4). With this ligand, we next evaluated the effect of different N-oxides and different reaction temperatures (entries 6–19). The temperature screening at three different temperatures showed an optimum at 60 °C, lower or higher temperatures were less efficient (entries 4, 7 and 9). Among the screened N-oxides mostly electron-deficient N-oxides were the best candidates (entries 16–19) with 2-trifluoromethylpyridine (6l) being by far the best candidate.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | N-Oxide | Solvent | T [°C] | Yieldb [%] |
| a Generalised reaction conditions: 1a (0.3 mmol, 1.0 equiv), 6l (2.0 equiv;a1.2 equiv.), catalyst (5 mol%; *2.5 mol%), solvent (2.0 mL, 0.1 M), 24 h. b NMR yield of product 2a. c n.d.: not detected. d Isolated yield of 54%, accompanied by 42% hydration side-product. | |||||
| 1 | IPrAuNTf2 | Pyridine (6a) | DCM | 30 | 11 |
| 2 | AuCl3 | (6a) | DCM | 30 | n.dc |
| 3 | BrettPhosAuNTf2 | (6a) | DCM | 30 | 5 |
| 4 | CyJohnPhosAuNTf2 | (6a) | DCM | 30 | 21 |
| 5 | PPh3AuAuNTf2 | (6a) | DCM | 30 | 5 |
| 6 | CyJohnPhosAuNTf2 | (6a) | DCE | 60 | 25 |
| 7 | CyJohnPhosAuNTf2 | 8-Methylquinoline (6b) | DCM | 30 | 16 |
| 8 | CyJohnPhosAuNTf2 | (6b) | DCE | 60 | 49 |
| 9 | CyJohnPhosAuNTf2 | (6b) | DCE | 80 | 18 |
| 10 | CyJohnPhosAuNTf2 | 8-Isopropylquinoline (6c) | DCE | 60 | 46 |
| 11 | CyJohnPhosAuNTf2 | 4-Nitroquinoline (6d) | DCE | 60 | 15 |
| 12 | CyJohnPhosAuNTf2 | 4-Methylpyridine (6e) | DCE | 60 | 11 |
| 13 | CyJohnPhosAuNTf2 | 4-Acetylpyridine (6f) | DCE | 60 | 9 |
| 14 | CyJohnPhosAuNTf2 | Methyl-5-bromopyridine-3-carboxylate (6g) | DCE | 60 | 37 |
| 15 | CyJohnPhosAuNTf2 | 2-Bromopyridine (6h) | DCE | 60 | 26 |
| 16 | CyJohnPhosAuNTf2 | 2,5-Dibromopyrdine (6i) | DCE | 60 | 55 |
| 17 | CyJohnPhosAuNTf2 | 3,5-Dibromopyridine (6j) | DCE | 60 | 51 |
| 18 | CyJohnPhosAuNTf2 | 3,5-Dichloropyridine (6k) | DCE | 60 | 52 |
| 19 | CyJohnPhosAuNTf2 | 2-Trifluoromethylpyridine (6l) | DCE | 60 | 80, (54%)d |
| 20 | CyJohnPhosAuNTf2* | (6l) | DCE | 60 | 47 |
| 21 | CyJohnPhosAuNTf2 | (6l)α | DCE | 60 | 47 |
Results and discussion
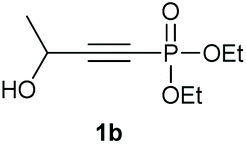
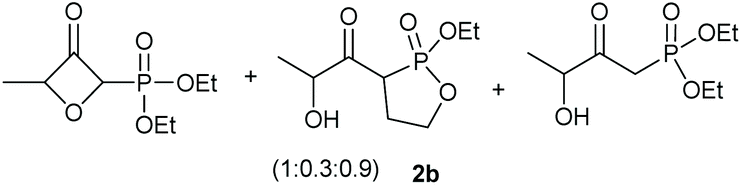
Under the optimised conditions, the scope of the reaction was tested for various alkyl and aryl substituted substrates. At first, diethyl phosphonate-substituted substrates 1b–1d were tested for the transformation towards the desired oxetanones (Table 2). But unfortunately, these substrates showed a selectivity problem. Even though the desired products were formed as the major products, as evident by the given ratios in Table 2, the synthetic value for these starting materials was strongly limited. This drawback is based on the unsuccessful separation of side products, possessing the same polarity. In addition to the desired pathway, a competing C–H insertion of the methyl group of the phosphonate26–28 (Scheme 1) and a simple hydration reaction in the presence of the gold catalyst29 were observed and only combined yields can be reported. While these were good for open-chained systems (entries 1 and 2), substrate 1d containing a cyclooctyl moiety, only delivered a combined yield of 25% (entry 3). Concerning the ratio of the obtained products it becomes obvious, that tertiary alcohols are leading to higher ratios of the desired OH insertion products which we attribute to the Thorpe-Ingold effect.30,31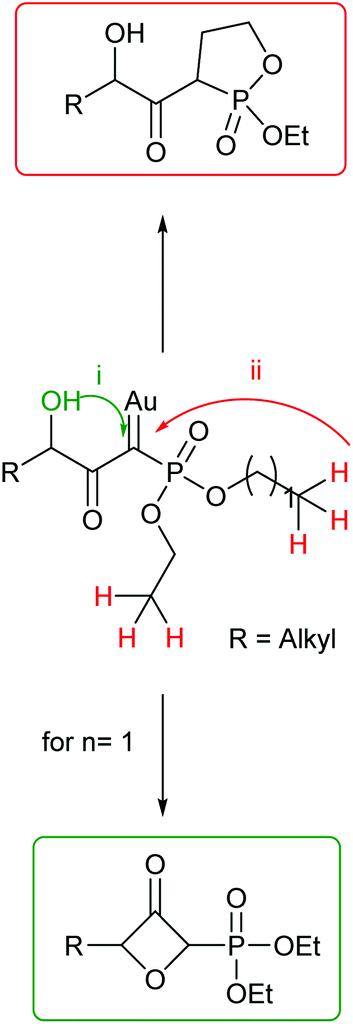 | ||
| Scheme 1 Competing CH-insertion for ethyl phosphonates. | ||
| Entry | Substrate | Mixture | Overall yield |
|---|---|---|---|
| 1 |
|
|
81% |
| 2 |
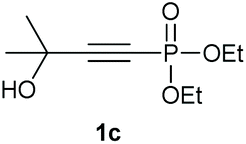
|
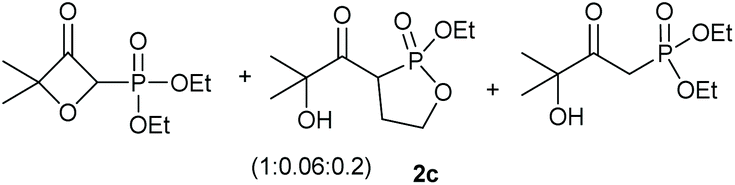
|
95% |
| 3 |
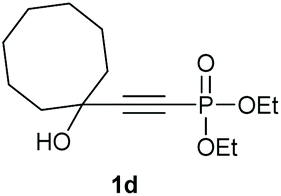
|
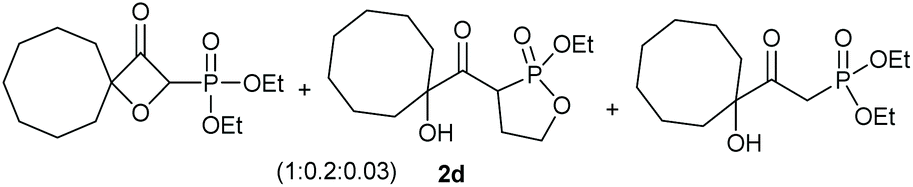
|
25% |
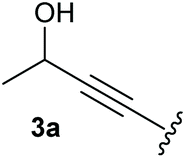
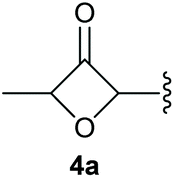
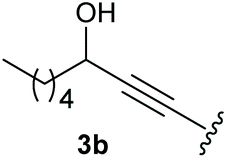
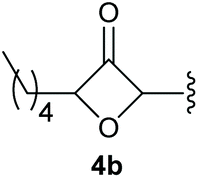
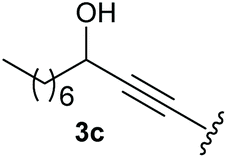
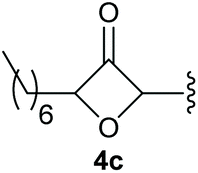
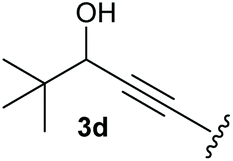
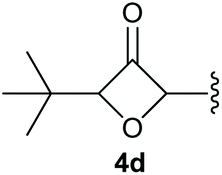
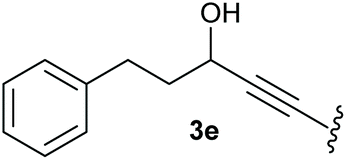
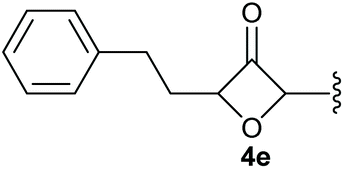
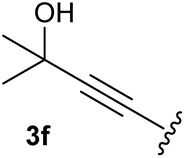
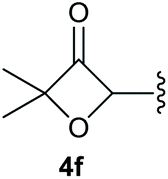
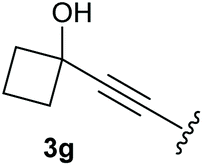
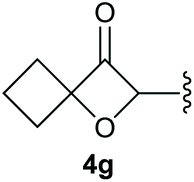
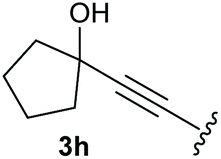
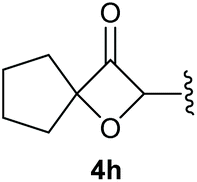
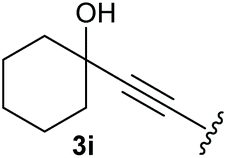
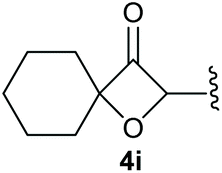
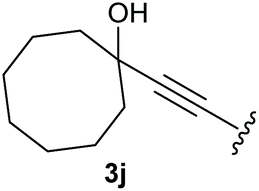
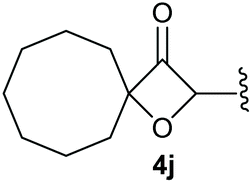
To supress the potential side reaction stemming from the insertion of the phosphonate side chain, we switched to dimethyl phosphonate-substituted substrates for further scope evaluation. Indeed, for none of the converted substrates the insertion of the methyl group next to the P-atom was observed, which can be explained by the less favourable formation of a four–membered ring. Also, to supress the side hydration reaction (which also occurred for the test substrate 1a in a preparative scale – Table 1, entry 18 yield in brackets), 4 Å pore-sized molecular sieve were added to ensure dry reaction conditions. All starting materials delivered the corresponding oxetanones in moderate to good yields depending on the substituent (Table 3). In case of secondary alkynols, the yields decreased with an increase in the aliphatic chain length and the n-pentyl-substituted substrate 3b showed a better yield (entry 2) compared to the n-heptyl substituted substrate 3c (entry 3). This effect can be rationalised by side products derived from the CH-insertion of the propargylic substituent which led to a mixture of the desired oxetanone 4b or 4c and CH inserted end products in a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively. A 1-phenyl-substituted hexynol 3e showed the best yield with 88% noteworthy no competing aryl CH insertion was monitored (entry 5). A t-butyl-substituted alkynol 3d delivered only a low yield of 39%, which might be caused by the bulkiness of this moiety (entry 4). For tertiary alkynols, the best yield was observed for dimethyl-substituted alkynol 3f, 82% (entry 6) and yields were comparatively lower for cycloalkyl-substituted alkynols. In this case an increase in the aliphatic ring size of the cycloalkyl substituent, resulted in decreased yields, with cyclooctyl-substituted substrate 3j showing no conversion at all (entry 10). It should be mentioned that the corresponding ethyl phosphonate 1d (Table 2, entry 3) was converted under the same conditions, which indicates that ethyl phosphonates seem to be more reactive substrates. A cyclobutyl-substituted substrate 3g delivered product 4g consisting of two spirofused 4-membered rings in a yield of 42% (Table 3, entry 7). The corresponding cyclopentyl 3h and cyclohexyl substituted substrates 3i (Table 3, entries 8 and 9, respectively) delivered higher yields, which can be explained by the reduced strain of the resulting products.
:1, respectively. A 1-phenyl-substituted hexynol 3e showed the best yield with 88% noteworthy no competing aryl CH insertion was monitored (entry 5). A t-butyl-substituted alkynol 3d delivered only a low yield of 39%, which might be caused by the bulkiness of this moiety (entry 4). For tertiary alkynols, the best yield was observed for dimethyl-substituted alkynol 3f, 82% (entry 6) and yields were comparatively lower for cycloalkyl-substituted alkynols. In this case an increase in the aliphatic ring size of the cycloalkyl substituent, resulted in decreased yields, with cyclooctyl-substituted substrate 3j showing no conversion at all (entry 10). It should be mentioned that the corresponding ethyl phosphonate 1d (Table 2, entry 3) was converted under the same conditions, which indicates that ethyl phosphonates seem to be more reactive substrates. A cyclobutyl-substituted substrate 3g delivered product 4g consisting of two spirofused 4-membered rings in a yield of 42% (Table 3, entry 7). The corresponding cyclopentyl 3h and cyclohexyl substituted substrates 3i (Table 3, entries 8 and 9, respectively) delivered higher yields, which can be explained by the reduced strain of the resulting products.

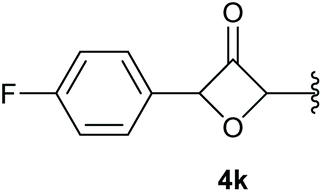
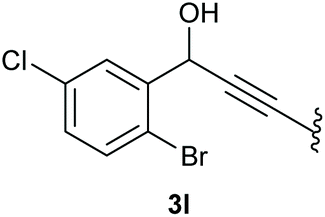
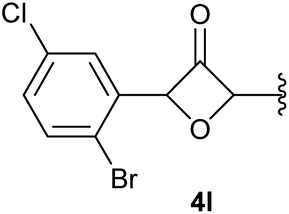
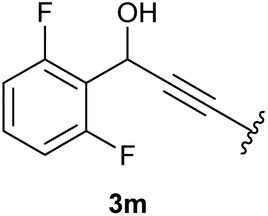
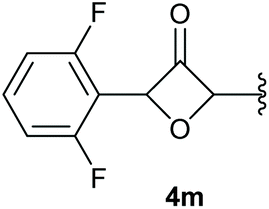
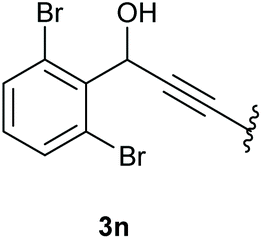
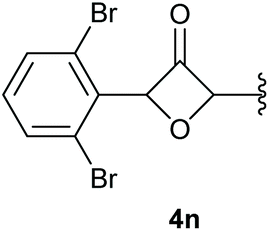
In case of aryl-substituted substrates, owing to a possible C–H functionalization pathway caused by the aromatic residue, the reaction resulted in a mixture of both the desired product and the side product in a ratio of 1.3:1 (4k and 4l) for unblocked test substrates. Respectively, blocking the ortho positions of the phenyl substituent, gave the oxetanones exclusively (Table 4). Aryl systems with fluorine substituents showed better yields in this case, with 4-fluoro-substituted aryl system 3k giving a yield of 70% (entry 1), followed by 2,6-difluorosubstituted aryl system, 3m with a yield of 66% (entry 3). For electron-deficient aryl systems, the yields were relatively lower, 2-bromo-4-chloro substituted phenyl system 3l resulted in a yield of 63% (Table 4, entry 2) and 2,6-dibromo-substituted system 3n showed a yield of 58% (Table 4, entry 4).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Overall yield |
| Reaction conditions: substrate (0.3 mmol, 1.0 equiv.), 2-trifluoromethylpyridine N-oxide (2.0 equiv), CyJohnPhosAuNTf2 (5 mol%), DCE (2.0 mL, 0.1 M).a Mixture of the desired oxetanone (excess) and the C–H functionalized products.b Selective towards O–H insertion to yield the desired oxetanone, no CH insertion side-product observed. | |||
| 1 |

|
|
70%a |
| 2 |
|
|
63%a |
| 3 |
|
|
66%b |
| 4 |
|
|
58%b |
To demonstrate the applicability of the phosphonate-substituted oxetanones as HWE reagents, substrate 4e was selected and allowed to react with benzaldehyde in the presence of lithiumdiisopropyl amide, for 30 minutes at 0 °C.11 The desired alkenyl-substituted oxetanone 5a was obtained in 95% yield, with an E/Z ratio of 1:5 (Scheme 2). A separate reaction was also tested for the in situ conversion of the gold-catalysed product to the corresponding 2-alkenyl-3-oxetanone. In this case THF was used as a solvent to avoid the deprotonation of the protic solvent in the presence of LDA, however the yield for this one pot process was comparably lower.
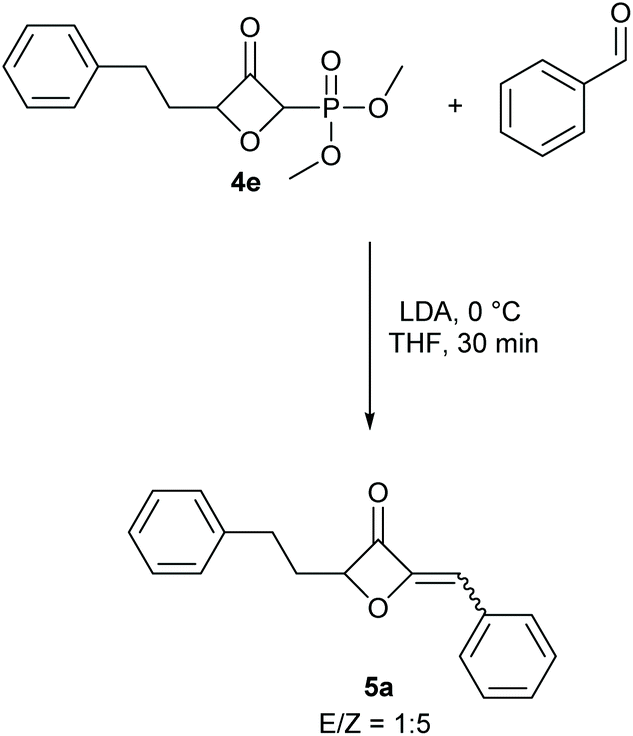 | ||
| Scheme 2 Example for the application of the phosphonate-substituted oxeta-3-ones in a HWE reaction. | ||
For the HWE reaction it is thought that the reaction proceeds through a betaine intermediate.9 Therefore, either betaine formation or elimination from the betaine intermediate can determine the selectivity depending on the employed reaction conditions. For steric reasons the former favours a trans product and the later a cis product. We modelled the corresponding betaine intermediates and alkene products by density functional theory calculations in the orca program package32 to gauge this effect. However, the energy differences for the two betaines and alkene products are small due to little steric repulsion. Betaine formation favours the E-product by 0.8 kcal mol−1 and elimination favours the Z-product by 2.2 kcal mol−1. We trace the only mediocre E:Z selectivity back to these small energy differences. But we want to mention that different sterically more demanding aldehydes will likely lead to higher selectivity.
Conclusions
In conclusion, we have developed a gold-catalysed method for the synthesis of phosphonate-substituted oxetan-3-ones. These can be used as HWE reagents, upon reaction with aldehydes to get otherwise difficult to obtain alkenes. If the starting materials are selected properly, CH-insertions as side reactions can be prevented and the target molecules can be obtained in good efficiency. Due to the easy accessibility of these highly strained HWE reagents and the possibility to further transfer these substrates this methodology might become attractive as new tool for natural product synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S.T. acknowledges the financial support by the DAAD PhD-program. J.F.W. thanks the support from the Hector Fellow Academy. The authors acknowledge the support by the state of Baden-Württemberg through bwHPC. The authors also thank Dr. Christoph Hendrich for proofreading the Manuscript.References
- J. Hassan, Use of organophosphorus compounds for the therapeutic and prophylactic treatment of infections, U.S. Patent 6680308, 2004 Search PubMed.
- P. H. Mutin, G. Guerrero and A. Vioux, Hybrid materials from organophosphorus coupling molecules, J. Mater. Chem., 2005, 15, 3761–3768 RSC.
- O. I. Kolodiazhnyi, Methods of preparation of C-substituted phosphorus ylides and their application in organic synthesis, Russ. Chem. Rev., 1997, 66, 225 CrossRef.
- C.-M. Chen, Q.-Z. Ye, Z. Zhu, B. L. Wanner and C. T. Walsh, Molecular biology of carbon-phosphorus bond cleavage. Cloning and sequencing of the phn (psiD) genes involved in alkylphosphonate uptake and CP lyase activity in Escherichia coli B, J. Biol. Chem., 1990, 265, 4461–4471 CrossRef CAS.
- Y. Dienes, M. Eggenstein, T. Kárpáti, T. C. Sutherland, L. Nyulászi and T. Baumgartner, Phosphorus-Based Heteropentacenes: Efficiently Tunable Materials for Organic n-Type Semiconductors, Chem. – Eur. J., 2008, 14, 9878–9889 CrossRef CAS.
- S. Shah and J. D. Protasiewicz, ‘Phospha-variations’ on the themes of Staudinger and Wittig: phosphorus analogs of Wittig reagents, Coord. Chem. Rev., 2000, 210, 181–201 CrossRef CAS.
- D. J. St. Cyr and B. A. Arndtsen, A new use of wittig-type reagents as 1, 3-dipolar cycloaddition precursors and in pyrrole synthesis, J. Am. Chem. Soc., 2007, 129, 12366–12367 CrossRef.
- K. Tago and H. Kogen, Bis (2, 2, 2-trifluoroethyl) bromophosphonoacetate, a novel HWE reagent for the preparation of (E)-α-bromoacrylates: A general and stereoselective method for the synthesis of trisubstituted alkenes, Org. Lett., 2000, 2, 1975–1978 CrossRef CAS.
- J. A. Bisceglia and L. R. Orelli, Recent progress in the Horner-Wadsworth-Emmons reaction, Curr. Org. Chem., 2015, 19, 744–775 CrossRef CAS.
- A. Bisceglia and L. R. Orelli, Recent applications of the Horner-Wadsworth-Emmons reaction to the synthesis of natural products, Curr. Org. Chem., 2012, 16, 2206–2230 CrossRef.
- T. Maegawa, K. Otake, K. Hirosawa, A. Goto and H. Fujioka, Method for the Efficient Synthesis of Highly-Substituted Oxetan-and Azetidin-, Dihydrofuran-and Pyrrolidin-3-Ones and Its Application to the Synthesis of (±)-Pseudodeflectusin, Org. Lett., 2012, 14, 4798–4801 CrossRef CAS.
- L. Ye, W. He and L. Zhang, Gold-catalyzed one-step practical synthesis of oxetan-3-ones from readily available propargylic alcohols, J. Am. Chem. Soc., 2010, 132, 8550–8551 CrossRef CAS.
- D. Campeau, D. F. León Rayo, A. Mansour, K. Muratov and F. Gagosz, Gold-Catalyzed Reactions of Specially Activated Alkynes, Allenes, and Alkenes, Chem. Rev., 2021, 121(14), 8756–8867 CrossRef CAS PubMed.
- L.-W. Ye, X.-Q. Zhu, R. L. Sahani, Y. Xu, P.-C. Qian and R.-S. Liu, Nitrene transfer and carbene transfer in gold catalysis, Chem. Rev., 2021, 121(14), 9039–9112 CrossRef CAS.
- G. C. Geary, A. Nortcliffe, C. A. Pearce, D. Hamza, G. Jones and C. J. Moody, Densely functionalised spirocyclic oxetane-piperidine scaffolds for drug discovery, Bioorg. Med. Chem., 2018, 26, 791–797 CrossRef CAS.
- D. Romo, H. W. Yang and W. D. Schmitz, Concise, stereoselective syntheses and novel transformations of beta-lactones: Applications to natural product synthesis, Abstracts of Papers of the American Chemical Society, Amer Chemical Soc, 1155 16th ST, NW, Washington, DC 20036, 2004, vol. 213 Search PubMed.
- S. Roesner, J. D. Beadle, L. K. Tam, I. Wilkening, G. J. Clarkson, P. Raubo and M. Shipman, Development of oxetane modified building blocks for peptide synthesis, Org. Biomol. Chem., 2020, 18, 5400–5405 RSC.
- H. Huang, W. Yang, Z. Chen, Z. Lai and J. Sun, A mild catalytic synthesis of 2-oxazolines via oxetane ring-opening: rapid access to a diverse family of natural products, Chem. Sci., 2019, 10, 9586–9590 RSC.
- D. W. Klosowski, J. C. Hethcox, D. H. Paull, C. Fang, J. R. Donald, C. R. Shugrue, A. D. Pansick and S. F. Martin, Enantioselective halolactonization reactions using binol-derived bifunctional catalysts: Methodology, diversification, and applications, J. Org. Chem., 2018, 83, 5954–5968 CrossRef CAS PubMed.
- I. Mineeva, Asymmetric syntheses of the lactone core of tetrahydrolipstatin and tetrahydroesterastin and of the oriental hornet Vespa Orientalis pheromone, Russ. J. Org. Chem., 2015, 51, 842–848 CrossRef CAS.
- H. Tan, X. Chen, Z. Liu and D. Z. Wang, Total synthesis of angelone enabled by a remarkable biomimetic sequence, Tetrahedron, 2012, 68, 3952–3955 CrossRef CAS.
- R. J. Duffy, K. A. Morris, R. Vallakati, W. Zhang and D. Romo, Asymmetric Synthesis, Structure, and Reactivity of Unexpectedly Stable Spiroepoxy-β-Lactones Including Facile Conversion to Tetronic Acids: Application to (+)-Maculalactone A, J. Org. Chem., 2009, 74, 4772–4781 CrossRef CAS.
- B. M. Trost, J. D. Chisholm, S. T. Wrobleski and M. Jung, Ruthenium-catalyzed alkene-alkyne coupling: Synthesis of the proposed structure of amphidinolide A, J. Am. Chem. Soc., 2002, 124, 12420–12421 CrossRef CAS PubMed.
- U. Eichelberger, I. Neundorf, L. Hennig, M. Findeisen, S. Giesa, D. Müller and P. Welzel, Synthesis of analogues of the 2-O-alkyl glycerate part of the moenomycins, Tetrahedron, 2002, 58, 545–559 CrossRef CAS.
- T. Fujisawa, T. Ito, K. Fujimoto, M. Shimizu, H. Wynberg and E. Staring, Facile synthesis of (S)-β-hydroxy-β-trichloromethylated aromatic ketones by the regioselective ring cleavage of chiral β-trichloromethyl-β-propiolactone under the Friedel-Crafts conditions, Tetrahedron Lett., 1997, 38, 1593–1596 CrossRef CAS.
- M. R. Fructos, P. de Frémont, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Alkane carbon− hydrogen bond functionalization with (NHC) MCl precatalysts (M=Cu, Au; NHC=N-Heterocyclic Carbene), Organometallics, 2006, 25, 2237–2241 CrossRef CAS.
- Y. Liu, Z. Yu, Z. Luo, J. Z. Zhang, L. Liu and F. Xia, Mechanistic Investigation of Aromatic C (sp2)–H and Alkyl C (sp3)–H Bond Insertion by Gold Carbenes, J. Phys. Chem. A, 2016, 120, 1925–1932 CrossRef CAS.
- Z. Zheng, Y. Wang, X. Ma, Y. Li and L. Zhang, Non–Diazo C− H Insertion Approach to Cyclobutanones through Oxidative Gold Catalysis, Angew. Chem., Int. Ed., 2020, 59, 17398–17402 CrossRef CAS PubMed.
- L. Xie, R. Yuan, R. Wang, Z. Peng, J. Xiang and W. He, Gold(I)–Catalyzed Hydration of Alkynylphosphonates: Efficient Access to β–Ketophosphonates, Eur. J. Org. Chem., 2014, 2668–2671 CrossRef CAS.
- Y. Wang, Z. Zheng and L. Zhang, Intramolecular Insertions into Unactivated C (sp3)–H Bonds by Oxidatively Generated β-Diketone-α-Gold Carbenes: Synthesis of Cyclopentanones, J. Am. Chem. Soc., 2015, 137, 5316–5319 CrossRef CAS PubMed.
- M. Wieteck, Y. Tokimizu, M. Rudolph, F. Rominger, H. Ohno, N. Fujii and A. S. K. Hashmi, Dual Gold Catalysis: Synthesis of Polycyclic Compounds via C- H Insertion of Gold Vinylidenes, Chem. – Eur. J., 2014, 20, 16331–16336 CrossRef CAS.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci, 2018, 8, e1327 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2107149 and 2107150. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo01214b |
| This journal is © the Partner Organisations 2022 |