
Palladium-catalyzed direct construction of oxazoline-containing polycyclic scaffolds via tandem addition/cyclization of nitriles and arylboronic acids†
Shu-Qiang
Cui‡
a,
Na
Cheng‡
a,
Qian-Qian
Ma
a,
Zhong-Lin
Wei
*a and
Wei-Wei
Liao
 *ab
*ab
aDepartment of Organic Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: wliao@jlu.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P R China
First published on 15th November 2021
Abstract
An efficient construction of oxazoline-containing polycyclic scaffolds is presented through a Pd-catalyzed addition/cyclization of (hetero)arene tethered O-acyl cyanohydrin units and arylboronic acids. Diverse oxazoline-containing polycyclic compounds can be prepared in good to high yields under mild reaction conditions. This method can be extended to prepare multi-substituted polycyclic heterocyclic compounds via a Pd-catalyzed addition/cyclization/arylation tandem sequence using arylboronic acid and different heteroarene scaffolds such as thiophene, pyrrole and furan analogues.
Nitriles are common and fundamental chemicals, and have wide applications both in the chemical industry and academic community. The transformation of the CN triplet bond of nitriles via the nucleophilic addition of carbon nucleophiles constitutes one major category which has been extensively studied for decades.1 However, due to its relatively inert nature, harsh reaction conditions are required generally for the cleavage of the CN triple bonds of nitriles. Within this context, various stoichiometric organometallic reagents involving Mg, Zn, In etc. have been employed widely to react with nitriles to deliver imine intermediates,1,2 which are valuable and diverse organic intermediates and readily participate in diverse synthetic transformations. However, stoichiometric organometallic reagents, harsh reaction conditions and limited functional group compatibility impeded their application as a sustainable tool in organic synthesis.
Recently, transition metal-catalyzed addition of carbon-based organometallic nucleophiles to nitriles has emerged as a powerful method to assemble valuable molecular scaffolds with high efficiency and improved functional group compatibility.3 In these transformations, the key active organometallic species in situ generated from readily available starting materials such as boronic acids,4 haloarenes,5 aryl sulfonates,6 aryl acids7 and (hetero-)arenes8 reacted with nitriles via 1,2-insertion to produce valuable ketimine intermediates, which can not only deliver acyclic aryl ketone products after the hydrolysis (Fig. 1a), but also participate in assembling azaheterocyclic skeletons by means of condensation between the resulting imine functional group and the tethered carbonyl group (Fig. 1b). Nevertheless, it is still highly desirable to exploit an efficient approach for the construction of azaheterocycles from readily available starting materials under mild reaction conditions, by combining TM-catalyzed nucleophilic addition to nitriles and subsequent diverse transformations of the resultant ketimine intermediates.
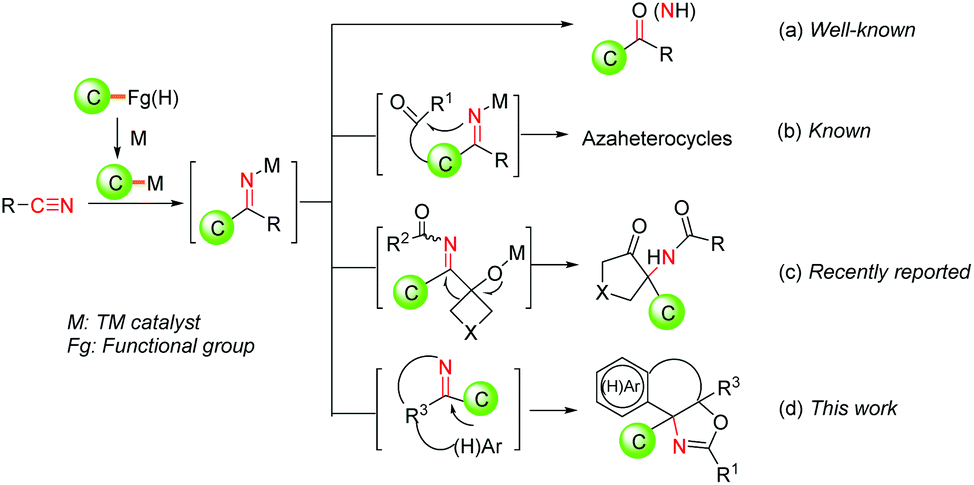 | ||
| Fig. 1 Reaction design based on TM-catalyzed addition of carbon-based organometallic nucleophiles to nitriles. | ||
O-Acyl cyanohydrins, readily prepared from aldehydes or ketones, have been utilized as diverse cyano group containing building blocks for the construction of azaheterocyclic compounds in Pd-catalyzed addition of organometallic nucleophile to nitrile/cyclization sequences.4g,9a–c,10 However, both intermolecular and intramolecular transformations rely completely on the nucleophilic reactivities of the resulting ketimine intermediates generally. Quite recently, an in situ generated ketimine intermediate of this type of reaction has been demonstrated to be involved in a Pd-triggered-iminol rearrangement, giving functionalized α-amino cyclopentanones with high efficiency (Fig. 1c).11 Given our continuous interest in the development of efficient Pd-catalyzed processes to construct diverse cyclic frameworks using nitriles,4g,9,11 we envisioned that the addition of Pd-catalyzed organometallic species in situ generated a nitrile via 1,2-insertion to produce a ketimine intermediate, followed by an intramolecular 6-endo-trig cyclization reaction with a tethered (hetero)aryl ring in which the resultant ketimine intermediate can serve as an electrophilic partner, would provide a tandem cyclization approach to construct azaheterocycles from nitriles (Fig. 1d). To the best of our knowledge, the resulting ketimine intermediates as an electrophilic partner to take part in cyclization is still under-explored. Herein, we report a novel Pd-catalyzed direct construction of oxazoline-containing polycyclic scaffolds via tandem addition/cyclization of (hetero)arene tethered O-acyl cyanohydrin units and arylboronic acids, which are important N-containing heterocyclic frameworks and incorporated in various biologically relevant compounds and chiral ligands.12,13
Initially, a reaction of N-methylindole 1a bearing an O-(p-nitro)benzoyl substituted cyanohydrin unit and phenylboronic acid 2a was chosen as a model for evaluating the feasibility of this Pd-catalyzed tandem process (Table 1). In the presence of Pd(OAc)2 (10 mol%)/2,2′-bipyridine (12 mol%), the effect of solvent was investigated first. A range of screened solvents can give the desired product 3a, but the yields varied considerably (entries 1–6). Nevertheless, to our delight, a high yield of the desired product 3a can be obtained in DMA (entry 7). The ligands were examined next. Other 2,2′-bipyridine-derived ligands such as 4,4′-dimethyl-2,2′-bipyridine (L1) and 5,5′-dimethyl-2,2′-bipyridine (L2) afforded similar results to that of 2,2′-bipyridine, while 1,10-phenanthroline gave the desired product in moderate yield (entries 8–10). The reaction can also be completed in 12 hours (entry 11). With this promising result in hand, we estimated the influence of additives. It turned out that bases such as Cs2CO3 and KF have an obvious inhibition effect on this reaction, while acids provided product 3a in a slightly increased yield (entries 12–14). It was noteworthy that further reducing the loading of Pd(OAc)2/2,2′-bipyridine and the amount of phenylboronic acid 2a had negligible effects on this transformation, which provided product 3a in comparable yield (entry 15). Notably, no N-acylated carbazol-1-amine was observed which was the major product in our previous work.9a,14 Both the catalyst and ligand are essential for this transformation, and the absence of either the Pd(II) catalyst or ligand completely shut down the reaction (for details see the ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Ligand | Additive | t (h) | Yieldb (%) |
a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), Pd(OAc)2 (10 mol%), and ligand (12 mol%) in solvent (c = 0.2 M) at 80 °C.
b Isolated yields.
c Additive (1.0 equiv.).
d Additive (0.2 equiv.).
e Pd(OAc)2 (5 mol%) and bpy (6 mol%).
f ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2a (0.4 mmol). bpy: 2,2′-bipyridine; L1: 4,4′-dimethyl-2,2′-bipyridine; L2: 5,5′-dimethyl-2,2′-bipyridine; phen: 1,10-phenanthroline; NMA: N-methylacetamide; NMP: N-methylpyrrolidone; and DMA: N,N-dimethylacetamide. 2a (0.4 mmol). bpy: 2,2′-bipyridine; L1: 4,4′-dimethyl-2,2′-bipyridine; L2: 5,5′-dimethyl-2,2′-bipyridine; phen: 1,10-phenanthroline; NMA: N-methylacetamide; NMP: N-methylpyrrolidone; and DMA: N,N-dimethylacetamide.
|
|||||
| 1 | Toluene | bpy | — | 24 | 15 |
| 2 | THF | bpy | — | 24 | 47 |
| 3 | 1,4-Dioxane | bpy | — | 24 | 47 |
| 4 | NMA | bpy | — | 24 | 44 |
| 5 | DMF | bpy | — | 24 | 64 |
| 6 | NMP | bpy | — | 24 | 85 |
| 7 | DMA | bpy | — | 24 | 90 |
| 8 | DMA | L1 | — | 24 | 89 |
| 9 | DMA | L2 | — | 24 | 90 |
| 10 | DMA | Phen | — | 24 | 76 |
| 11 | DMA | bpy | — | 12 | 90 |
| 12c | DMA | bpy | CsF | 24 | 15 |
| 13d | DMA | bpy | TFA | 12 | 92 |
| 14d | DMA | bpy | HOAc | 12 | 92 |
| 15d,e,f | DMA | bpy | HOAc | 24 | 91 |
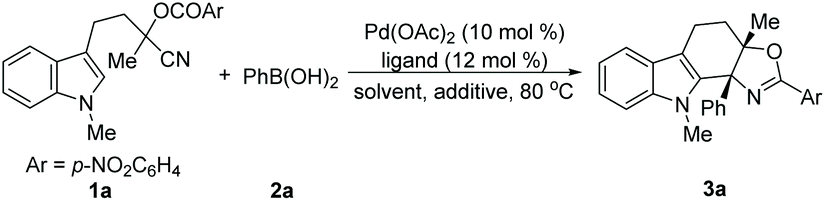
With the optimized conditions in hand, we then evaluated the scope of the reaction by employing indole derivatives bearing different O-acyl substituted cyanohydrin units and 2a (Table 2). At the outset, indoles with different N-substituents were examined. It was found that all reactions worked well, giving rise to the corresponding products (3a–3c) in moderate to good yields, even with the electron-deficient indole core (3d), which was unachievable in the Pd-catalyzed intramolecular addition of an organometallic nucleophile to the nitrile/cyclization sequence.9a Then, the pattern and electronic effect of the substituents on N-methylindoles were examined, and all reactions proceeded well and gave rise to the corresponding C(2) cyclization products (3e–3h) in good to excellent yields. Additionally, the reactions showed good functional group compatibility towards the substituents on cyanohydrin units (R3 and R4) (3i–3q), except for N-methylindole bearing the O-cinnamoyl substituted cyanohydrin unit, which did not give the desired product 3r. The structure of the oxazoline-containing polycyclic product 3a was confirmed by X-ray crystal structural analysis.15 Besides, this protocol is highly scalable, and the 3.0 mmol scale synthesis of 3a was performed, which delivered the desired product in 80% yield (1.05 g).
a
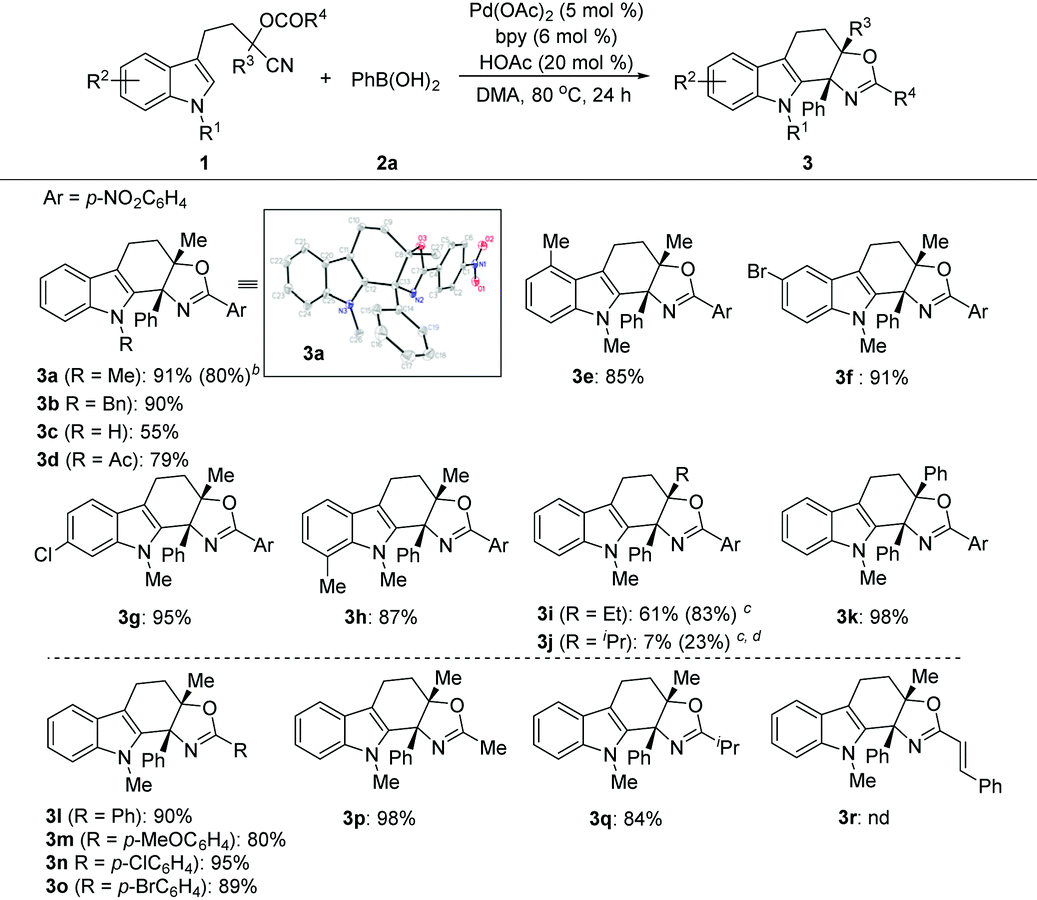
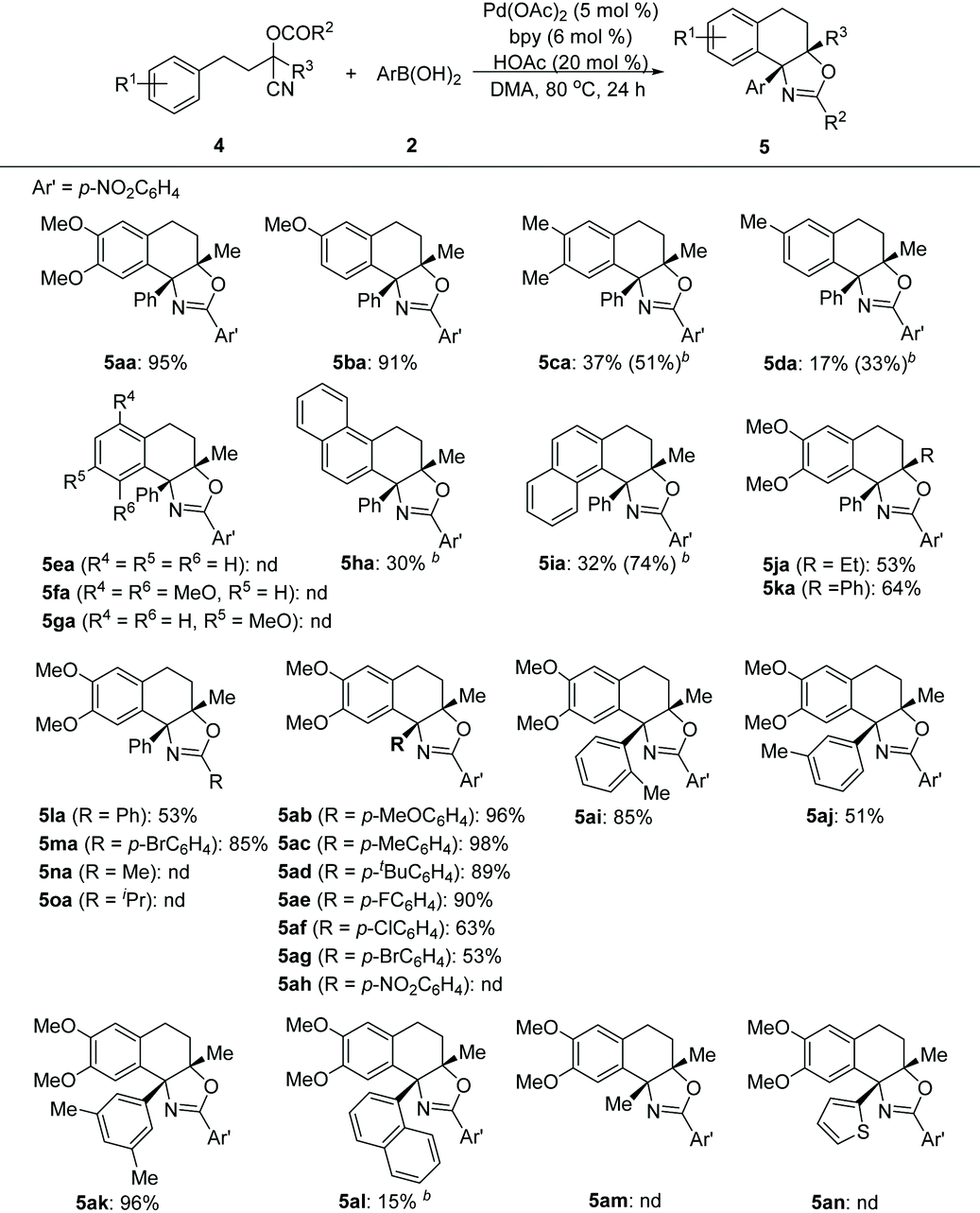
Next, the feasibility of this Pd-catalyzed tandem cyclization of arene tethered O-acyl cyanohydrin units was evaluated (Table 3). It turned out that this tandem transformation was susceptible to the electronic nature and pattern of the substituent on the aromatic ring system of arenes 4. The reactions between 3-methoxyl or 3,4-dimethoxyl substituted arenes with phenylboronic acid 2a proceeded smoothly under the optimized reaction conditions, and delivered the desired products (5aa–5ba) in high yields, while methyl substituted analogues provided inferior results (5ca–5da). Unsubstituted, 4-monsubstituted and 2,5-disubstituted substrates with electron-rich groups did not afford any products (5ea–5ga), which indicated that the reaction may involve a Friedel–Crafts-type pathway. Although both α- and β-naphthyl based substrates can furnish the desired products under the slightly modified reaction conditions (5ha–5ia), the β-naphthyl analogue provided a superior yield of the product (5ia). The substituents on the cyanohydrin units (R2 and R3) of arenes were also examined. The R3 groups such as Et and Ph, and R2 groups such as Ph and p-bromophenyl were well tolerated (5ja–5ka and 5la–5ma), while O-acetyl or O-isobutyryl substrates (R2 = Me or R2 = iPr) cannot give the desired products (5na or 5oa). Subsequently, the scope of substrates with respect to arylboronic acids was investigated. Both electron-donating (5ab–5ad) and electron-withdrawing substituents (5ae–5ag) at the para-position of the benzene ring of arylboronic acids were well tolerated, giving the desired products in good to high yields, except for the nitro group (5ah). Notably, ortho-, meta- or di-substituted analogues can also serve as suitable substrates to afford products (5ai–5ak) in comparable yields. Additionally, α-naphthyl boronic acid can also deliver the corresponding product (5al), albeit with a low yield under the optimized reaction conditions. However, hetero-aromatic boronic and alkyl boronic acids failed to afford any desired products (5am–5an).
a
In addition, this method was also applicable for the cyclization of other heteroarene analogues. For example, a 5-methyl substituted thiophene analogue delivered the desired product 7a in good yield under the standard reaction conditions (Scheme 1a). Notably, 5-unsubstituted thiophene with the tethered cyanohydrin moiety at the C-3 position afforded product 7b in 44% yield incorporating the additional phenyl group at the 5-position, which indicated that the sequential Pd-catalyzed arylation of heteroarene with aryl boronic acid could be compatible under these reaction conditions.16 To our delight, an improved yield (64%) of this novel tandem sequence can be obtained under the modified reaction conditions (Scheme 1b). Moreover, this Pd-catalyzed tandem addition/cyclization/arylation approach can also be extended to pyrrole and furan analogues, which react with phenylboronic acid and afford the desired multi-substituted polycyclic heterocyclic products (7c–7d) in reasonable yields, respectively (Scheme 1b and c). However, Pd-catalyzed addition/cyclization sequences of (hetero)arene (1l or 4a) tethered O-acyl cyanohydrin units and N-methyl-indole 8 have been proven unsuccessful, and no desired products can be obtained (Scheme 2).
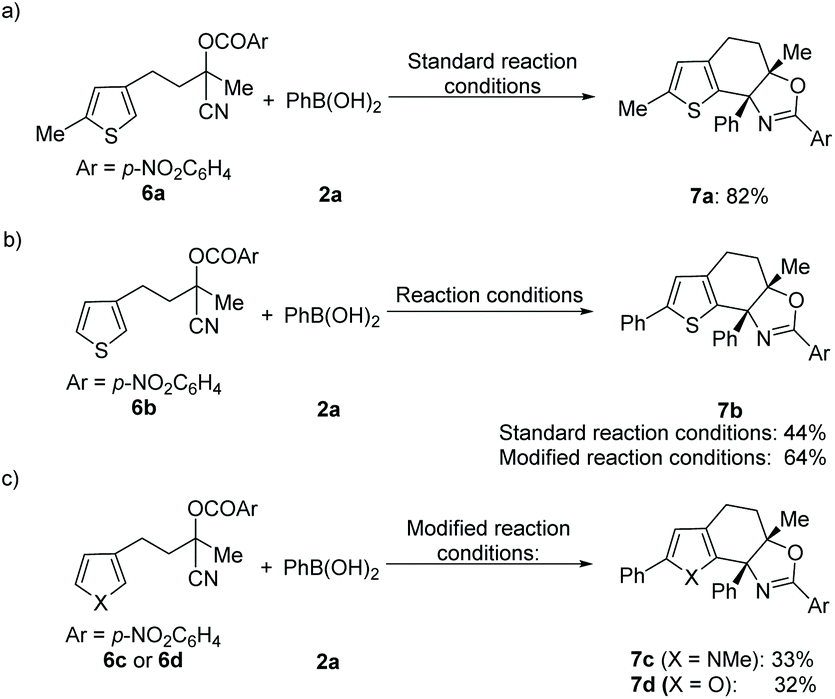 | ||
| Scheme 1 Standard reaction conditions: 6 (0.2 mmol), 2a (0.4 mmol), Pd(OAc)2 (5 mol%), bpy (6 mol%), HOAc (20 mol%), and DMA, 80 °C, 24 h. Modified reaction conditions: 6 (0.2 mmol), 2a (0.6 mmol), Pd(OAc)2 (5 mol%), bpy (6 mol%), HOAc (20 mol%), and DMA, 100 °C, 24 h under 1 atm of O2. | ||
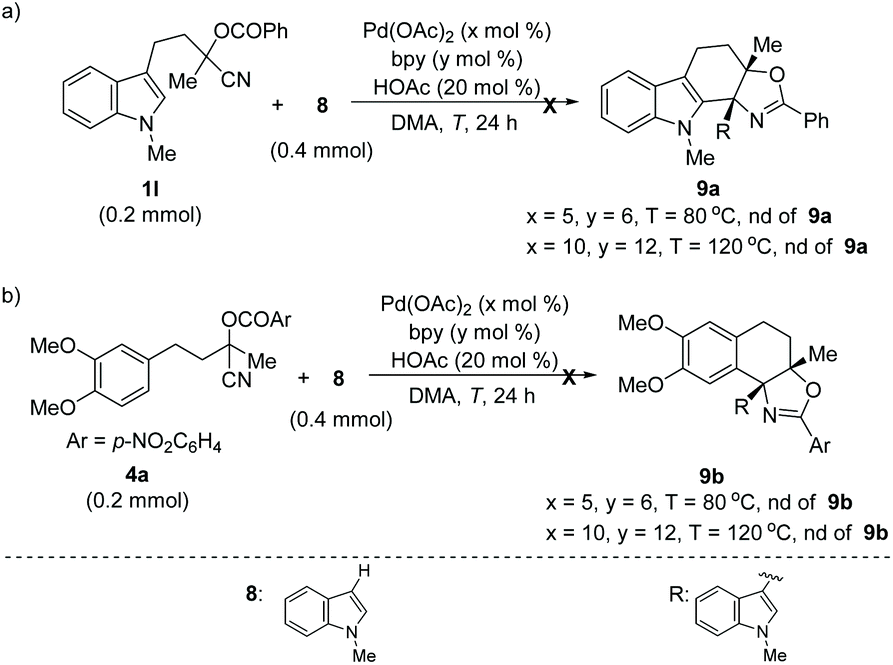 | ||
| Scheme 2 Unsuccessful Pd-catalyzed C–H addition to nitrile/cyclization sequences. | ||
On the basis of these results and other previous works,4,9 the proposed mechanism is illustrated in Scheme 3. The initial transmetalation between arylboronic acid and the Pd(II) catalyst A generates the arylpalladium species B. Then coordination of the nitrile with species B, followed by a carbopalladation of the cyano group, would result in the ketimine Pd(II) complex D, which reacts with an adjacent carbonyl group to give the species E. The intramolecular 6-endo-trig cyclization reaction of the intermediate E with the tethered (hetero)aryl ring forms intermediate F which affords the product and regenerates the Pd(II) catalyst.
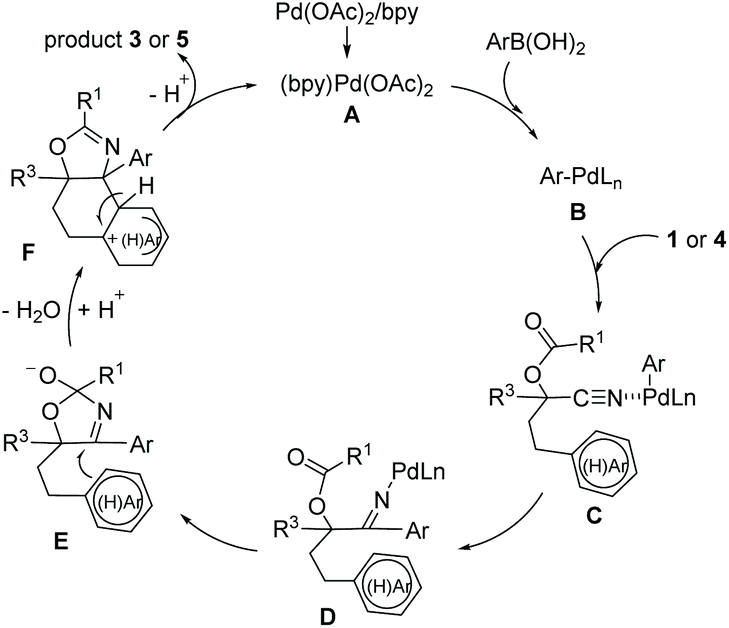 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In summary, we have developed an efficient protocol for the construction of oxazoline-containing polycyclic scaffolds via a Pd-catalyzed tandem addition/cyclization of (hetero)arene tethered O-acyl cyanohydrin units and arylboronic acids. Various functionalized oxazoline-containing polycyclic compounds can be prepared in good to high yields under mild conditions. Furthermore, Pd-catalyzed addition/cyclization/arylation tandem sequences have also been demonstrated by employing thiophene, pyrrole and furan analogues, which afforded the desired multi-substituted polycyclic heterocyclic products with reasonable yields. Further studies on the application of this synthetic method are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The NSFC (no. 21772063) and the Open Project of State Key Laboratory for Supramolecular Structure and Materials (sklssm2021010) are acknowledged.Notes and references
- (a) A. J. Fatiadi, in Preparation and Synthetic Applications of Cyano Compounds, ed. S. Patai and Z. Rappaport, Wiley, New York, 1983, pp. 1057–1303 Search PubMed; (b) R. C. Larock, Comprehensive Organic Transformations, Wiley-VCH, New York, 1989, p. 686 Search PubMed; (c) S. I. Murahashi, S. Kanemasa, L. R. Subramanian, A. Schmidt, M. North, J. Podlech, S. J. Collier, P. Langer, W. T. Wang and L. L. Lin, in Three Carbon-Heteroatom Bonds: Nitriles, Isocyanides, and Derivatives, ed. S. Murahashi, Thieme, Germany, 2004, vol. 19, pp. 41–52 Search PubMed.
- (a) B. J. Wakefield, Organomagnesium in organic synthesis, Academic Press, London, 1995, pp. 101–107 Search PubMed; (b) H. S. Prakash Rao, S. Rafi and K. Padmavathy, The Blaise reaction, Tetrahedron, 2008, 64, 8037–8043 CrossRef CAS; (c) S. H. Kim, H. S. Lee, K. H. Kim, S. H. Kim and J. N. Kim, Recent advances in allylindium reagents in organic synthesis, Tetrahedron, 2010, 66, 7065–7076 CrossRef CAS; (d) M. S. M. Pearson-Long, F. Boeda and P. Bertus, Double Addition of Organometallics to Nitriles: Toward an Access to Tertiary Carbinamines, Adv. Synth. Catal., 2017, 359, 179–201 CrossRef CAS.
- Review: (a) J.-C. Hsieh and H.-L. Su, Synthesis of N-Heterocycles via Transition-Metal-Catalyzed Tandem Addition/Cyclization of a Nitrile, Synthesis, 2020, 52, 819–833 CrossRef CAS; (b) S. Q. Cui and W. W. Liao, Recent Advances on Transition Metal-Catalyzed C-H Addition to Nitrile, Synthesis, 2021 DOI:10.1055/s-0040-1719826.
- Selected examples: (a) B. Zhao and X. Lu, Palladium(II)-catalyzed addition of arylboronic acid to nitriles, Tetrahedron Lett., 2006, 47, 6765–6768 CrossRef CAS; (b) J. Sävmarker, J. Rydfjord, J. Gising, L. R. Odell and M. Larhed, Direct Palladium(II)-Catalyzed Synthesis of Arylamidines from Aryltrifluoroborates, Org. Lett., 2012, 14, 2394–2397 CrossRef; (c) A. Yu, J. Li, M. Cui and Y. Wu, Addition of Arylboronic Acids to Nitriles in Aqueous Media Catalyzed by a 2,2′-Bipyridine-Cyclopalladated Ferrocenylimine Complex, Synlett, 2007, 3063–3067 CAS; (d) L. Qi, K. Hu, S. Yu, J. Zhu, T. Cheng, X. Wang, J. Chen and H. Wu, Tandem Addition/Cyclization for Access to Isoquinolines and Isoquinolones via Catalytic Carbopalladation of Nitriles, Org. Lett., 2017, 19, 218–221 CrossRef CAS; (e) K. Hu, Q. Zhen, J. Gong, T. Cheng, L. Qi, Y. Shao and J. Chen, Palladium-Catalyzed Three-Component Tandem Process: One-Pot Assembly of Quinazolines, Org. Lett., 2018, 20, 3083–3087 CrossRef CAS; (f) H. Yu, L. Xiao, X. Yang and L. Shao, Controllable access to multi-substituted imidazoles via palladium(II)-catalyzed C–C coupling and C–N condensation cascade reactions, Chem. Commun., 2017, 53, 9745–9748 RSC; (g) H. Song, N. Cheng, L.-Q. She, Y. Wu and W.-W. Liao, Efficient synthesis of spirooxindolyl oxazol-2(5H)-ones via palladium(II)-catalyzed addition of arylboronic acids to nitriles, RSC Adv., 2019, 9, 29424–29428 RSC.
- (a) R. C. Larock, Q. Tian and A. A. Pletnev, Carbocycle Synthesis via Carbopalladation of Nitriles, J. Am. Chem. Soc., 1999, 121, 3238–3239 CrossRef CAS; (b) J.-C. Hsieh, Y.-C. Chen, A.-Y. Cheng and H.-C. Tseng, Nickel-Catalyzed Intermolecular Insertion of Aryl Iodides to Nitriles: A Novel Method to Synthesize Arylketones, Org. Lett., 2012, 14, 1282–1285 CrossRef CAS PubMed; (c) Y. Jaiswal, Y. Kumar, J. Pal, R. Subramanian and A. Kumar, Rapid synthesis of polysubstituted phenanthridines from simple aliphatic/aromatic nitriles and iodo arenes via Pd(II) catalyzed domino C–C/C–C/C–N bond formation, Chem. Commun., 2018, 54, 7207–7210 RSC.
- (a) J. Liu, X. Zhou, H. Rao, F. Xiao, C.-J. Li and G.-J. Deng, Direct Synthesis of Aryl Ketones by Palladium-Catalyzed Desulfinative Addition of Sodium Sulfinates to Nitriles, Chem. – Eur. J., 2011, 17, 7996–7999 CrossRef CAS; (b) M. Behrends, J. Sävmarker, P. J. R. Sjöberg and M. Larhed, Microwave-Assisted Palladium(II)-Catalyzed Synthesis of Aryl Ketones from Aryl Sulfinates and Direct ESI-MS Studies Thereof, ACS Catal., 2011, 1, 1455–1459 CrossRef CAS; (c) T. Miao and G.-W. Wang, Synthesis of ketones by palladium-catalysed desulfitative reaction of arylsulfinic acids with nitriles, Chem. Commun., 2011, 47, 9501–9503 RSC.
- (a) J. Lindh, P. J. R. Sjöberg and M. Larhed, Synthesis of Aryl Ketones by Palladium(II)-Catalyzed Decarboxylative Addition of Benzoic Acids to Nitriles, Angew. Chem., Int. Ed., 2010, 49, 7733–7737 CrossRef CAS PubMed; (b) J. Rydfjord, F. Svensson, A. Trejos, P. J. R. Sjöberg, C. Sköld, J. Sävmarker, L. R. Odell and M. Larhed, Decarboxylative Palladium(II)-Catalyzed Synthesis of Aryl Amidines from Aryl Carboxylic Acids: Development and Mechanistic Investigation, Chem. – Eur. J., 2013, 19, 13803–13810 CrossRef CAS PubMed.
- Selected examples: (a) C. Zhou and R. C. Larock, Synthesis of Aryl Ketones by the Pd-Catalyzed C–H Activation of Arenes and Intermolecular Carbopalladation of Nitriles, J. Am. Chem. Soc., 2004, 126, 2302–2303 CrossRef CAS; (b) C. Zhou and R. C. Larock, Synthesis of Aryl Ketones or Ketimines by Palladium-Catalyzed Arene C–H Addition to Nitriles, J. Org. Chem., 2006, 71, 3551–3558 CrossRef CAS PubMed; (c) H. Takaya, M. Ito and S.-I. Murahashi, Rhenium-Catalyzed Addition of Carbonyl Compounds to the Carbon–Nitrogen Triple Bonds of Nitriles: α-C–H Activation of Carbonyl Compounds, J. Am. Chem. Soc., 2009, 131, 10824–10825 CrossRef CAS PubMed; (d) T.-S. Jiang and G.-W. Wang, Synthesis of 3-Acylindoles by Palladium-Catalyzed Acylation of Free (N–H) Indoles with Nitriles, Org. Lett., 2013, 15, 788–791 CrossRef CAS PubMed; (e) Y. Ma, J. You and F. Song, Facile Access to 3-Acylindoles through Palladium-Catalyzed Addition of Indoles to Nitriles: The One-Pot Synthesis of Indenoindolones, Chem. – Eur. J., 2013, 19, 1189–1193 CrossRef CAS PubMed; (f) T.-S. Jiang and G.-W. Wang, Synthesis of 2-Acylthiophenes by Palladium-Catalyzed Addition of Thiophenes to Nitriles, Adv. Synth. Catal., 2014, 356, 369–373 CrossRef CAS; (g) B. Zhou, Y. Hu and C. Wang, Manganese-Catalyzed Direct Nucleophilic C(sp2)□H Addition to Aldehydes and Nitriles, Angew. Chem., Int. Ed., 2015, 54, 13659–13663 CrossRef CAS PubMed; (h) W. Xiong, Z. Chen, Y. Shao, R. Li, K. Hu and J. Chen, The synthesis of fluorescent benzofuro[2,3-c]pyridines via palladium-catalyzed heteroaromatic C–H addition and sequential tandem cyclization, Org. Chem. Front., 2020, 7, 756–762 RSC.
- (a) T.-T. Wang, L. Zhao, Y.-J. Zhang and W.-W. Liao, Pd-Catalyzed Intramolecular Cyclization via Direct C–H Addition to Nitriles: Skeletal Diverse Synthesis of Fused Polycyclic Indoles, Org. Lett., 2016, 18, 5002–5005 CrossRef CAS; (b) L. Zhao and W.-W. Liao, Org. Chem. Front., 2018, 5, 801 RSC; (c) D. Zhang, H. Song, N. Cheng and W.-W. Liao, Synthesis of 2,4,5-Trisubstituted Oxazoles via Pd-Catalyzed C–H Addition to Nitriles/Cyclization Sequences, Org. Lett., 2019, 21, 2745–2749 CrossRef CAS PubMed; (d) T.-T. Wang, D. Zhang and W.-W. Liao, Versatile synthesis of functionalized β- and γ-carbolines via Pd-catalyzed C–H addition to nitriles/cyclization sequences, Chem. Commun., 2018, 54, 2048–2051 RSC.
- L. Dai, S. Yu, Y. Shao, R. Li, Z. Chen, N. Lv and J. Chen, Palladium-catalyzed C–H activation of simple arenes and cascade reaction with nitriles: access to 2,4,5-trisubstituted oxazoles, Chem. Commun., 2021, 57, 1376–1379 RSC.
- N. Cheng, S.-Q. Cui, Q.-Q. Ma, Z.-L. Wei and W.-W. Liao, α-Iminol Rearrangement Triggered by Pd-Catalyzed C–H Addition to Nitriles Sequences: Synthesis of Functionalized α-Amino Cyclopentanones, Org. Lett., 2021, 23, 1021–1025 CrossRef CAS PubMed.
- (a) J. A. Frump, Oxazolines. Their preparation, reactions, and applications, Chem. Rev., 1971, 71, 483–505 CrossRef CAS. Selected examples: (b) H. R. Onishi, B. A. Pelak, L. S. Gerckens, L. L. Silver, F. M. Kahan, M.-H. Chen, A. A. Patchett, S. M. Galloway, S. A. Hyland, M. S. Anderson and C. R. H. Raetz, Antibacterial Agents That Inhibit Lipid A Biosynthesis, Science, 1996, 274, 980–982 CrossRef CAS PubMed; (c) D. Clark and D. A. Travis, An enantioselective synthesis of insecticidal 4-alkynyloxazolines, Bioorg. Med. Chem., 2001, 9, 2857–2862 CrossRef CAS; (d) R. J. Bergeron, M. G. Xin, W. R. Weimar, R. E. Smith and J. Wiegand, Significance of Asymmetric Sites in Choosing Siderophores as Deferration Agents, J. Med. Chem., 2001, 44, 2469–2478 CrossRef CAS; (e) Q. Li, K. W. Woods, A. Claiborne, S. L. Gwaltney, K. J. Barr, G. Liu, L. Gehrke, R. B. Credo, Y. H. Hui, J. Lee, R. B. Warner, P. Kovar, M. A. Nukkala, N. A. Zielinski, S. K. Tahir, M. Fitzgerald, K. H. Kim, K. Marsh, D. Frost, S.-C. Ng, S. Rosenberg and H. L. Sham, Synthesis and Biological Evaluation of 2-Indolyloxazolines as a New Class of Tubulin Polymerization Inhibitors. Discovery of A-289099 as an Orally Active Antitumor Agent, Bioorg. Med. Chem. Lett., 2002, 12, 465–469 CrossRef CAS PubMed; (f) M. R. Prinsep, R. E. Moore, I. A. Levine and G. M. L. Patterson, Westiellamide, a Bistratamide-Related Cyclic Peptide from the Blue-Green Alga Westiellopsis prolifica, J. Nat. Prod., 1992, 55, 140–142 CrossRef CAS; (g) S.-L. You and J. W. Kelly, Highly Efficient Biomimetic Total Synthesis and Structural Verification of Bistratamides E and J from Lissoclinum bistratum, Chem. – Eur. J., 2004, 10, 71–75 CrossRef CAS PubMed.
- R. Connon, B. Roche, B. V. Rokade and P. J. Guiry, Further Developments and Applications of Oxazoline-Containing Ligands in Asymmetric Catalysis, Chem. Rev., 2021, 121, 6373–6521 CrossRef CAS PubMed.
- In this transformation, the intermolecular reaction between indole-based substrates 1a and 2a was performed prior to the intramolecular cyclization of 1a, see ref. 9a.
- CCDC 2103937 (compound 3a), see the ESI† for details.
- S.-D. Yang, C.-L. Sun, Z. Fang, B.-H. Li, Y.-Z. Li and Z.-J. Shi, Palladium-Catalyzed Direct Arylation of (Hetero)Arenes with Aryl Boronic Acids, Angew. Chem., Int. Ed., 2008, 47, 1473–1476 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2103937. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo01260f |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2022 |