
Fissisternoids A and B, two 2′,5′-quinodihydrochalcone-based meroterpenoid enantiomers with unusual carbon skeletons from Fissistigma bracteolatum†
Gui-Min
Xue
 *,
Chen-Guang
Zhao
,
Jin-Feng
Xue
,
Hui
Chen
,
Zhen-Zhu
Zhao
,
Ying-Ying
Si
,
Kun
Du
,
Yan-Le
Zhi
and
Wei-Sheng
Feng
*
*,
Chen-Guang
Zhao
,
Jin-Feng
Xue
,
Hui
Chen
,
Zhen-Zhu
Zhao
,
Ying-Ying
Si
,
Kun
Du
,
Yan-Le
Zhi
and
Wei-Sheng
Feng
*
College of Pharmacy, Henan University of Chinese Medicine, Zhengzhou 450046, China. E-mail: xueguimin123@126.com; fwsh@hactcm.edu.cn
First published on 19th November 2021
Abstract
Two pairs of meroterpenoid enantiomers, (±)-fissisternoids A (1) and B (2), along with their putative biogenetic precursor 3 were isolated from the branches and leaves of Fissistigma bracteolatum. Compound 1 represents an unprecedented meroterpenoid featuring a unique tricyclo [3,3,1,01′,5′] decane central framework, and compound 2 possesses a rare 6/6/5/4 tetracyclic carbon skeleton, both of which were derived from quinodihydrochalcone and monoterpenoid via a key [4 + 2] Diels–Alder cyclization and Prins reaction. Their structures and absolute configurations were established using spectroscopic data, X-ray diffraction, and electronic circular dichroism calculations. Compounds 1 and 2 exhibit anti-inflammatory activity (IC50: 13.2 μM, 1; 9.8 μM, 2) via suppression of inflammatory cytokines IL-1β, IL-6, and iNOS.
Introduction
The genus Fissistigma (Annonaceae) consists of more than 70 species, which are mainly distributed in tropical and subtropical regions in Asia.1 Several species, such as F. oldhamii, F. polyanthum, and F. retusum, have been used as folk medicines for curing rheumatoid arthritis and inflammation-related diseases.2 Phytochemical studies on Fissistigma were started in the 1980s, and flavonoids, alkaloids (aristolactam, phenanthrene, aporphine), sesquiterpenoids, and cyclopentenones have been reported from this genus.3–5F. bracteolatum, an evergreen shrub, is grown mainly in Vietnam and southern China.6 As a traditional medicine, its root-bark is commonly used for the treatment of bone fracture, contusion, and for stopping bleeding.6 Previous work on F. bracteolatum has revealed the presence of a series of chalcone derivatives and several phenanthrene type alkaloids.7,8 Nevertheless, four extraordinary meroterpenoids, fissistigmatins A–D, composed of a flavonoid and a sesquiterpene unit connected via a single C–C bond directly, were also isolated from F. bracteolatum.9As a part of our exploration on metabolites with unique structures from traditional medicines,10 two novel meroterpenoids fissisternoids A and B (1 and 2) and a known dihydrochalcone (3) were isolated from F. bracteolatum (Fig. 1). Compound 1 possesses an unprecedented carbon skeleton with a rare tricyclo [3,3,1,01′,5′] decane core, formed by an uncommon oxidative quinodihydrochalcone and an acyclic monoterpenoid unit. The different coupling pattern of the same units formed compound 2 that features a specific 6/6/5/4 fused tetracyclic ring skeleton. These two isolates were deduced biosynthetically via a key [4 + 2] Diels–Alder cyclization and Prins reaction. To the best of our knowledge, the coupling modes of the two units to form complex ring systems such as 1 and 2 have never been reported. Herein, the isolation, structural elucidation, plausible biosynthetic pathway, and inflammatory activity of compounds 1 and 2 are described.
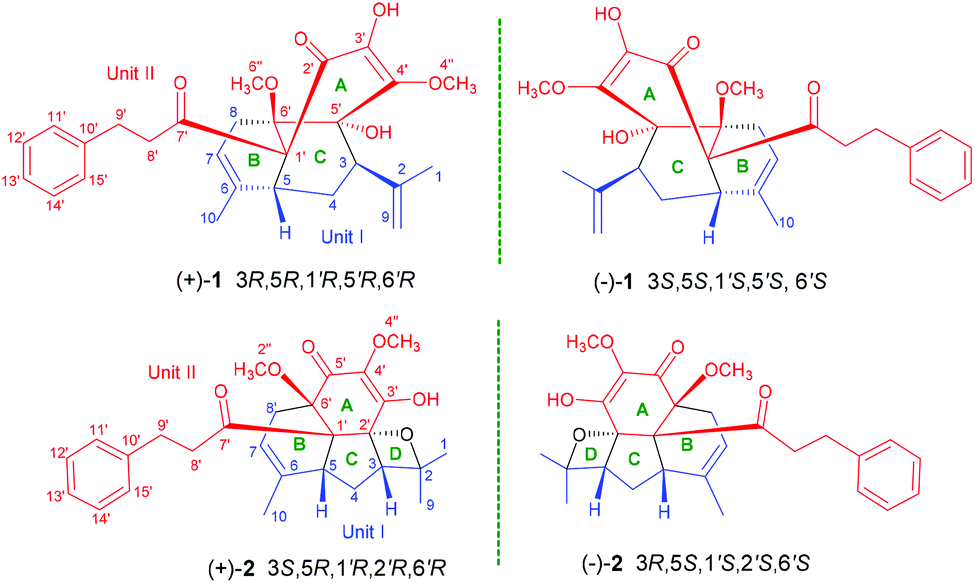 | ||
| Fig. 1 Structures of compounds (+)-1/(−)-1 and (+)-2/(−)-2. | ||
Results and discussion
Fissisternoid A (1) was obtained as a faint yellow crystal (CH3OH). Its molecular formula was determined as C27H32O6 by HR-ESI-MS (m/z 475.2081 [M + Na]+, calcd for C27H32O6Na, 475.2091), indicating 12 degrees of unsaturation. The presence of absorption bonds for the hydroxyl (3401 cm−1), carbonyl (1711, 1668 cm−1), and benzene (1604, 1510 cm−1) functionalities was observed in the IR spectrum. The 1H NMR data of 1 showed typical signals for two methyl groups (δH 1.63, s, 3H; 1.80, d, J = 1.50, 3H), two methoxyl groups (δH 3.43, s, 3H; 4.18, s, 3H), a pair of exocyclic methylene protons (δH 4.80, 4.98, each, br s), an olefinic proton (δH 5.30, br s), and a mono-substituted benzene ring (δH 7.24, m, 2H; 7.16, 3H) (Table 1). The 1H and 13C NMR data of 1, interpreted with the help of the HSQC spectrum, revealed the presence of 27 carbons, which were attributed to 4 methyls (including two methoxyls δC 60.6, 53.1), 5 methylenes (including one olefinic carbon δC 116.2), 8 methines (including six olefinic carbons δC 118.7, 128.4 × 2, 128.6 × 2, 125.9), and 10 quaternary carbons (including two carbonyls δC 193.8, 205.9; two oxygenated carbons δC 81.4, 81.7; six olefinic carbons δC 132.2, 149.2, 142.6, 135.8, 142.0) (Table 1). The above observations (including three double bonds, two carbonyls, and one benzene ring) accounted for nine out of the twelve unsaturations, suggesting 1 to be tricyclic.| No. | 1 | 2 | ||
|---|---|---|---|---|
| δ H (J in Hz) | δ C | δ H (J in Hz) | δ C | |
| 1 | 1.63, s | 21.6 | 1.37, s | 25.3 |
| 2 | 142.6 | 92.4 | ||
| 3 | 2.55, dd (12.0, 5.0) | 46.7 | 2.33, m | 53.0 |
| 4a | 1.50, m | 26.9 | 1.13, ddd (12.5, 11.5, 11.5) | 30.4 |
| 4b | 1.50, m | 1.74, m | ||
| 5 | 2.59, dd (4.0, 2.5) | 41.7 | 1.99, t (9.7) | 42.4 |
| 6 | 135.8 | 135.6 | ||
| 7 | 5.30, br s | 118.7 | 5.28, d (6.5) | 118.6 |
| 8a | 2.01, m | 28.8 | 2.33, m | 29.7 |
| 8b | 3.00, m | 3.40, m | ||
| 9a | 4.80, brs | 116.2 | 1.74, s | 26.7 |
| 9b | 4.98, brs | |||
| 10 | 1.80, d (1.5) | 21.6 | 1.48, s | 20.4 |
| 1′ | 68.6 | 61.2 | ||
| 2′ | 193.8 | 88.6 | ||
| 3′ | 132.2 | 163.3 | ||
| 4′ | 149.2 | 132.6 | ||
| 5′ | 81.4 | 188.6 | ||
| 6′ | 81.7 | 78.1 | ||
| 7′ | 205.9 | 211.6 | ||
| 8′ | 2.79, m | 45.0 | 2.83, m | 42.6 |
| 9′a | 2.80, m | 30.9 | 2.93, m | 30.4 |
| 9′b | 2.91, m | 3.04, m | ||
| 10′ | 142.0 | 141.1 | ||
| 11′ | 7.24, m | 128.4 | 7.29, d (7.0) | 128.7 |
| 12′ | 7.16, m | 128.6 | 7.18, t (7.0) | 128.7 |
| 13′ | 7.16, m | 125.9 | 7.22, t (7.0) | 126.5 |
| 14′ | 7.16, m | 128.6 | 7.18, t (7.0) | 128.7 |
| 15′ | 7.24, m | 128.4 | 7.29, d (7.0) | 128.7 |
| 3′-OH | 5.63, s | 6.09, s | ||
| 4′′-OCH3 | 4.18, s | 60.6 | 3.74, s | 60.1 |
| 6′′-OCH3 | 3.43, s | 53.1 | 3.31, s | 3.31 |
In the HMBC spectrum, the correlations from CH3-1/H-9 to C-2 and C-3, from H-3 to C-2, C-4, and C-9, from H-4 to C-2, C-5, and C-6, from H-5 to C-4 and C-6, from CH3-10 to C-5 and C-7, and from H-8 to C-6 and C-7, together with the 1H–1H COSY correlations of H-3/H-4/H-5 and H-7/H-8 (Fig. 2) indicated the presence of unit I as an acyclic monoterpenoid moiety.2,11 For the remaining part unit II, the HMBC correlations of methylenes H-8′ (δH 2.79, m, 2H) and H-9′ (δH 2.80, m; 2.91, m; 2H) with carbonyl C-7′ (δC 205.9) and aromatic carbon C-10′ (δC 142.0), and H-9′ with C-11′ (δC 128.4) and C-15′ (δC 128.4), assisted with the 1H–1H COSY correlation between H-8′ and H-9′ indicated the presence of the 3-phenylpropan-1-one moiety, which is a part of the framework of dihydrochalcone.12 Dihydrochalcones have been widely reported from F. bracteolatum,7 thus, unit II was speculated as a dihydrochalcone. Apart from the acyclic monoterpenoid, two methoxyls, and 3-phenylpropan-1-one moiety, six extra carbons were observed in the 13C NMR spectrum. Thus, the remaining six carbon signals should be attributed to the other 6-membered aromatic ring (A) of dihydrochalcone, and the aromatic ring was highly oxidized with the chemical formula C6O5H2, including two olefinic, one carbonyl, one quaternary and two oxygenated carbons. For ring A, the signal of a hydroxyl (3′-OH, δH 5.63, s) was observed, although 1 was dissolved in CDCl3. The HMBC correlations from the hydroxyl 3′-OH to C-2′ (δC 193.8), C-3′ (δC 132.2), and C-4′ (δC 149.2) and from methoxyl (4′′-OCH3) at δH 4.18 to C-4′ (Fig. 2) established a 3′-hydroxy-4′-methoxy-α,β unsaturated ketone moiety. The other methoxyl was attached to C-6′ (δC 81.7) according to the HMBC correlations of CH3-6′′, H-7, and H-8 with C-6′ (Fig. 2), and the remaining chemical shift at δC 81.4 was of the oxygenated carbon C-5′ (δC 81.4) supported by the HMBC correlations of H-3and H-4 with C-5′. Thus, the fragment of ring A was deduced as shown in Fig. 2, and eventually, the dihydrochalcone for unit II was established.
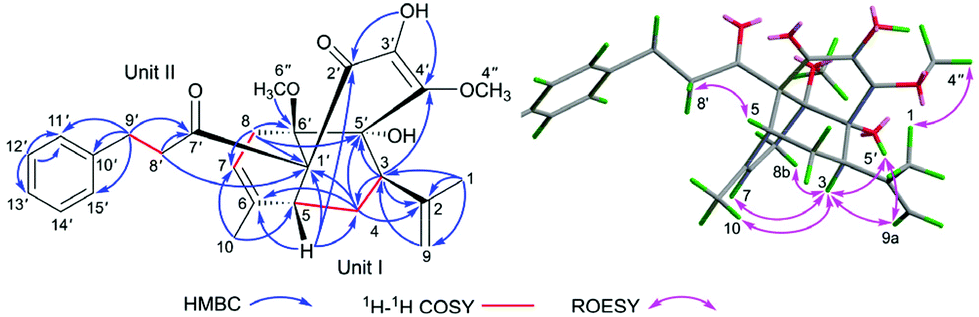 | ||
| Fig. 2 Key NMR correlations of 1. | ||
The linking mode between units I and II was supported by HMBC correlations. The correlations of H-5 (δH 2.59, dd, J = 4.0, 2.5 Hz) with C-1′ and C-2′, H-4 (δH 1.5, m, 2H) with C-1′ and H-7 (δH 5.30, s) and H-8 (δH 2.01, 3.00, each, m, 2H) with C-6′ (Fig. 2) demonstrated the respective linkages of C-5–C-1′ and C-8–C-6′, to form the six membered ring B fused with ring A. In addition, the correlations of H-3 (δH 2.55, dd, J = 12.0, 5.0 Hz) with C-4′ and C-5′ and H-4 (δH 1.5, m, 2H) with C-5′ indicated the connection between C-3 and C-5′, to establish another six-membered ring C, which satisfied the remaining one degree of unsaturation for 1.
In the ROESY spectrum, the correlations of H-3 with H-8b (δH 3.00, m), H-7, and CH3-10 and 5′-OH (Fig. 2) revealed that these protons were in a co-facial position of the cyclohexane ring C, and thus the C-6–CH-7–CH2-8 moiety, 5′-OH and H-3 were randomly assigned to the α-configuration, while 6′′-OCH3 and H-5 at both end carbons of the C-6–CH-7–CH2-8 moiety were accordingly assigned to the β-orientation. Subsequently, the ROESY correlations of H-5/H-8′ (δH 2.79, m, 2H) indicated the same orientation of H-5 and 3-phenylpropan-1-one in ring B (Fig. 2). The ROESY correlations of H-3/5′-OH, H-3/H-9a, 5′-OH/H-9a, and CH3-1/4′′-OCH3 suggested that the 3′-hydroxy-4′-methoxy-α,β unsaturated ketone moiety was on the β-orientation of ring C. The β-orientation of 3-phenylpropan-1-one related to ring B and the β-orientation of the 3′-hydroxy-4′-methoxy-α,β unsaturated ketone moiety related to ring C established the configuration of C-1′ as shown in Fig. 2. Furthermore, X-ray crystallography data were used to determine its structure (Fig. 3, CDCC 2107685†). The X-ray diffraction analysis of 1 showed the space group P-1, indicating its racemic nature, as also evidenced by a small optical value ([α]25D +2.1). Compound 1 was separated via chiral HPLC to afford the optical enantiomers (+)-1 and (−)-1 (Fig. S1†). The stereochemistry of the enantiomers of 1 was further determined by comparison of their experimental ECD spectra with those predicted by time-dependent density functional theory (TDDFT) calculations at the B3LYP/6-31+G(d,p) level. As a result, the calculated ECD curves of 3R,5R,1′R,5′R,6′R-1 and 3S,5S,1′S,5′S,6′S-1 matched well with the experimental ones of (+)-1 and (−)-1, respectively (Fig. 4), and thus their absolute configurations were determined.
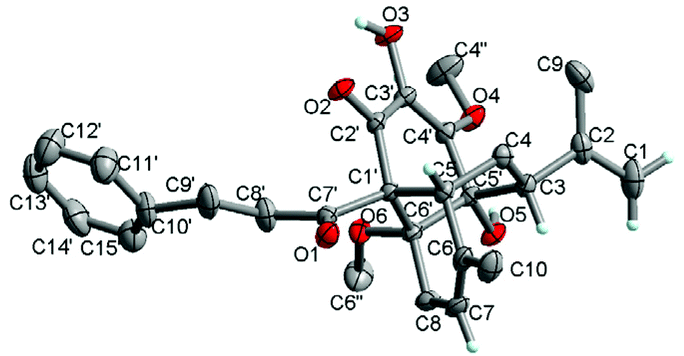 | ||
| Fig. 3 X-ray ORTEP drawing of 1. | ||
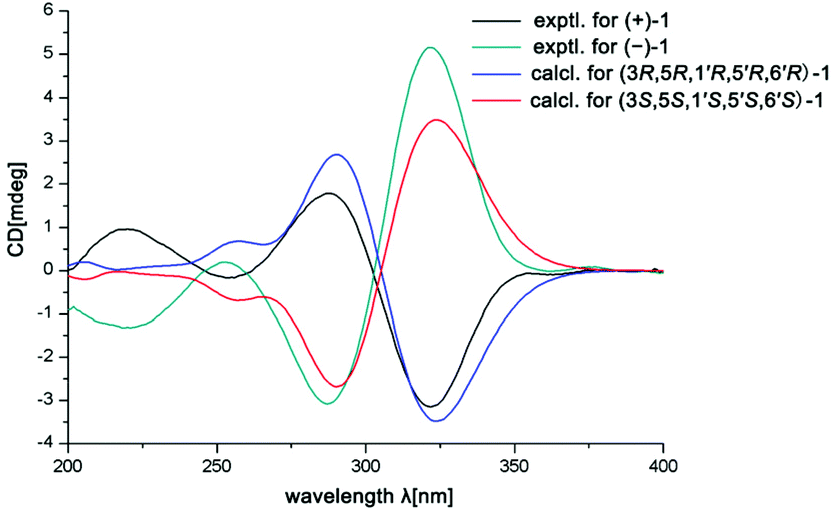 | ||
| Fig. 4 Calculated and experimental ECD spectra of (+)-1 and (−)-1. | ||
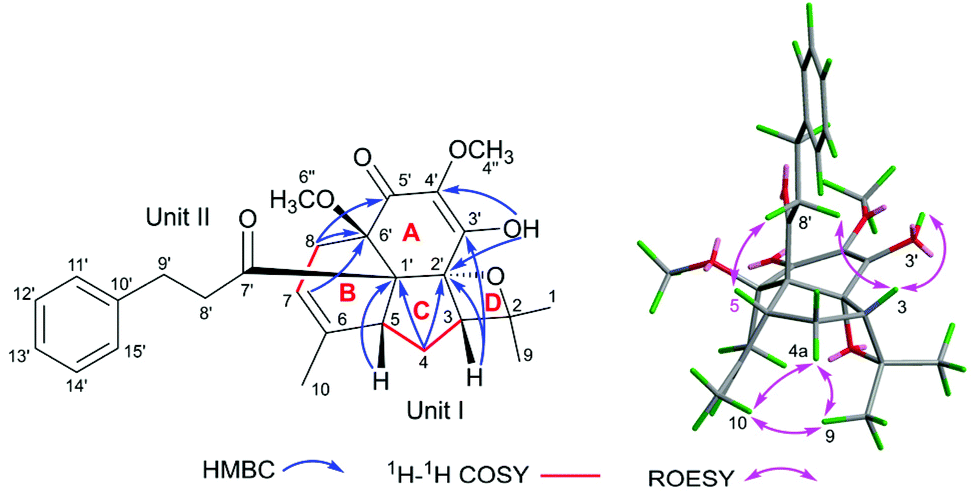
The molecular formula of fissisternoid B (2) was deduced to be C27H32O6 based on its HR-ESI-MS at m/z 475.2085 [M + Na]+ (calcd for C27H32O6Na, 475.2091), which also required 12 degrees of unsaturation. By comparison of the 1H NMR data of 2 with those of 1, the notable differences are the disappeared signals of the exocyclic terminal double bond, which was replaced with a singlet methyl group (δH 1.74, s). Besides, an additional oxygenated carbon at δH 92.4 was also observed. These data implied the exocyclic methylene in 1 to be oxygenated. The proposal was further confirmed by the HMBC correlations of CH3-1 (δH 1.37, s) and H-3 (δH 2.33, m) with C-2 (δC 92.4) and C-9 (δC 20.4), and H-4 with C-2 (Fig. 5).
 | ||
| Fig. 5 Key NMR correlations of 2. | ||
Detailed analysis of 2D NMR data for 2 suggested that its structure should be also established from the identical units of monoterpenoid (I) and quinodihydrochalcone (II) to those of 1. The same connectivities between 1 and 2via C-5–C-1′ and C-8–C-6′ for ring B were established by the key HMBC correlations from H-5 and H-4 to C-1′ and H-8 and H-7 to C-6′ (Fig. 5). The HMBC correlations of H-3 with C-2′ and C-3′, H-4 with C-2′, OH-3′ (δH 6.09, s) with C-2′, and C-3′, and C-4′, indicated the linkage of C-3–C-2′ in 2 to form ring C, which was distinct from that of 1 (C-3–C-5′). Additionally, the HMBC correlation of H-8 with carbonyl C-5′ (δC 188.6) (Fig. 5) indicated the nearby side of the carbonyl with H-8, and thus it was also suggested that it was carbonyl C-2′, but not C-5′ that participated in the Prins reaction to establish ring C. The C-2–O–C-2′ connectivity for ring D was demonstrated by the remaining one degree of unsaturation and the oxygenated carbon in the downfield region at C-2 (δC 92.4). Thus, the linking mode of 2 between units I and II was changed as shown in Fig. 5.
In the ROESY spectrum, the observed cross-peaks of H-5 and H-3 with H-8′ (δH 2.83, m, 2H) and H-3 with OH-3′ indicated that the protons of H-3 and H-5, the 3-phenylpropan-1-one group, and the chain of C-3′–OH were co-faced and assigned to be the β-side of ring C (Fig. 5). Furthermore, the ROESY correlations of CH3-9/CH3-10, CH3-9/H-4a, and CH3-10/H-4a revealed that the fractions of C-6–CH-7–CH2-8 (ring B) and C-2–O (ring D) were on the same face and on the other side of ring C, and thus 6′′-OCH3 should be situated at the β-side of ring B. Eventually, the structure of 2 should be deduced as shown in Fig. 5.
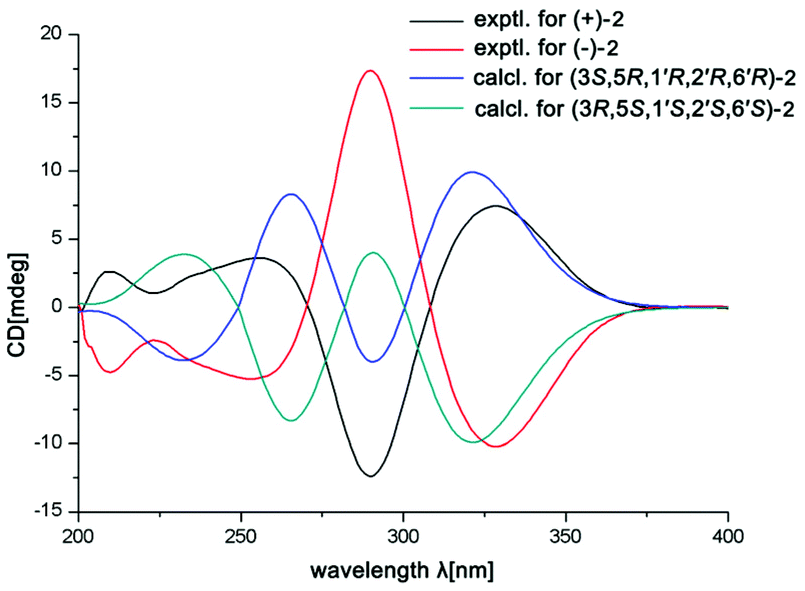
The small optical rotation value clearly suggested that compound 2 was also a pair of enantiomers. The subsequent chiral HPLC resolution of 2 yielded the anticipated enantiomers, (+)-2 and (−)-2 (Fig. S1†), which were opposite in terms of their CD curves. The experimental ECD spectra of (+)-2 and (−)-2 were also mirror images and agreed well with the calculated ECD spectra (Fig. 6). Finally, the absolute configurations of (+)-fissisternoid B and (−)-fissisternoid B were assigned as 3S,5R,1′R,2′R,6′R and 3R,5S,1′S,2′S,6′S, respectively.
 | ||
| Fig. 6 Calculated and experimental ECD spectra of (+)-2 and (−)-2. | ||
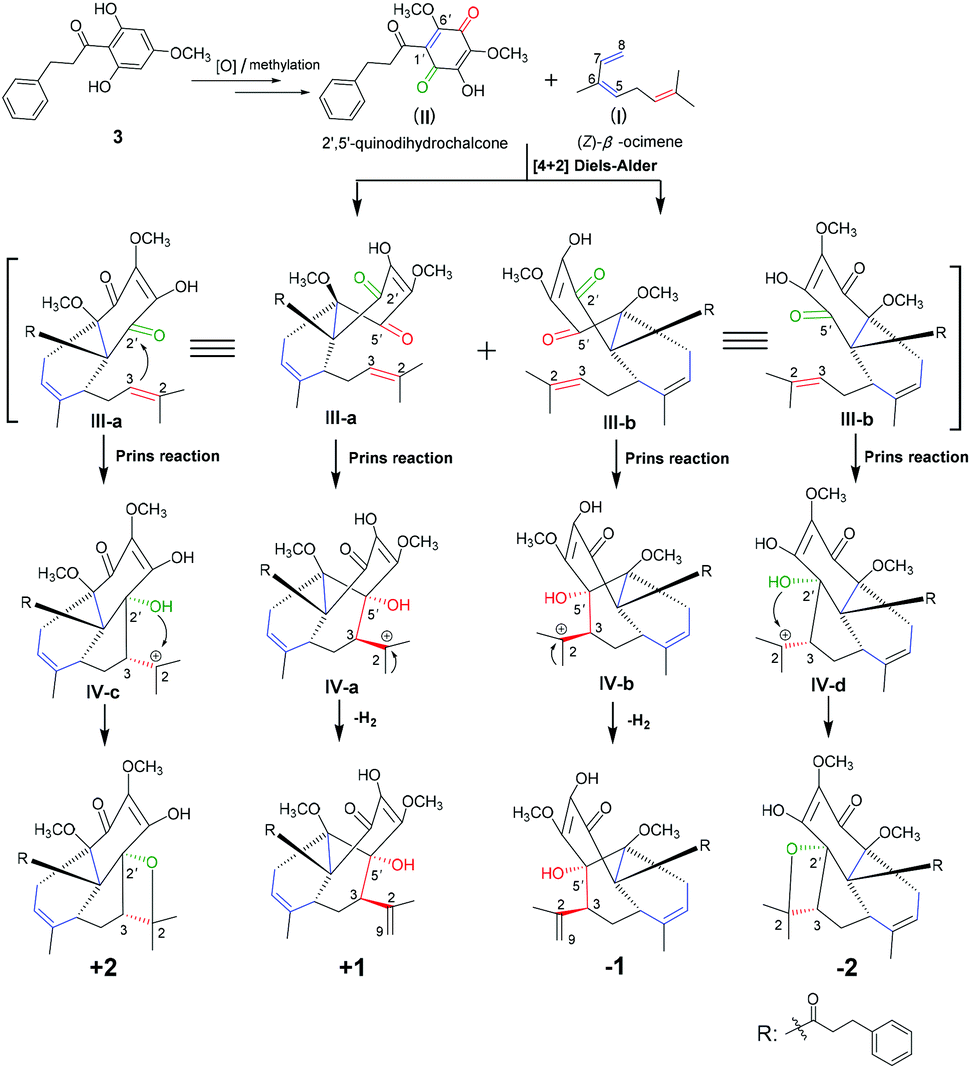
Compounds 1 and 2 represent a pair of unprecedented meroterpenoids with unusual 6/6/6 and 6/6/5/4 polycyclic core systems, respectively. The meroterpenoids were both formed from an acyclic monoterpenoid (unit I) and a 2′,5′-quinodihydrochalcone (unit II) moiety. The acyclic monoterpenoid moiety, (Z)-β-ocimene, as a diene for Diels–Alder cycloaddition, was identified from genus Fissistigma.2 The highly oxygenated 2′,5′-quinodihydrochalcones have rarely been reported from the same genus,3 and its precursor of 2,6-dihydroxy-4-methoxydihydrochalcone133 was isolated by us. Thus, the biosynthetic pathways of compounds 1 and 2 should be proposed as shown in Scheme 1. Initially, precursor 3 would undergo oxidation and methylation processes to generate the intermediate unit II. The [4 + 2] Diels–Alder reaction between II (Δ1′(6′)) and (Z)-β-ocimene (Δ5(6), 7(8)) established the connectivity of C-5–C-1′ and C-8–C-6′ for ring B of III-a and b, and thus 6′′-OCH3 and the 3-phenylpropan-1-one moiety of both 1 and 2 should be at the same side according to the Diels–Alder reaction mode.14 Moreover, the Prins reaction of the remaining alkene (Δ2(3)) for (Z)-β-ocimene with the carbonyls of C-2′ and C-5′ for 2′,5′-quinodihydrochalcone establishes the connectivity of C-3–C-2′ (IV-c and d) and C-3–C-5′ (IV-a and b), respectively.15 On one hand, the 2′-hydroxyl group in intermediates IV-c and d attacks the carbocation C-2 to construct the oxetane moiety for the D ring to form compound 2. On the other hand, IV-a and b would undergo the dehydrogenation reaction to form the Δ2(9)-double bond for compound 1.
 | ||
| Scheme 1 Plausible biosynthetic pathways of 1 and 2. | ||
The racemic mixtures 1 and 2 were evaluated for their inhibitory effects on NO production in LPS-activated RAW 264.7 cells. MTT results revealed that the cell viabilities were over 90% under the treatment of 1 and 2 at concentrations up to 30 μM (Table S5†). Furthermore, compounds 1 and 2 displayed NO production inhibitory activity with smaller IC50 values of 13.2 and 9.8 μM, respectively, compared with that of the positive control NG-monomethyl-L-arginine (L-NMMA, IC50 26.8 μM) (Table S5†). Since inflammatory cytokines such as IL-1β, TNF-α, IL-6, and iNOS play important roles in inflammation,16 we detected the release of these inflammatory cytokines in LPS-induced RAW264.7 cells through qRT-PCR analysis. Expectedly, compounds 1 and 2 dose-dependently (7.5, 15, 30 μM) inhibited the release levels of IL-1β, TNF-α, and IL-6 in mRNA expression, and exhibited a weak inhibitory effect on TNF-α compared with the LPS group (Fig. 7). These studies revealed that these enantiomers exhibited anti-inflammatory activity via the suppression of excessive inflammatory cytokines in LPS-activated RAW264.7 cells.
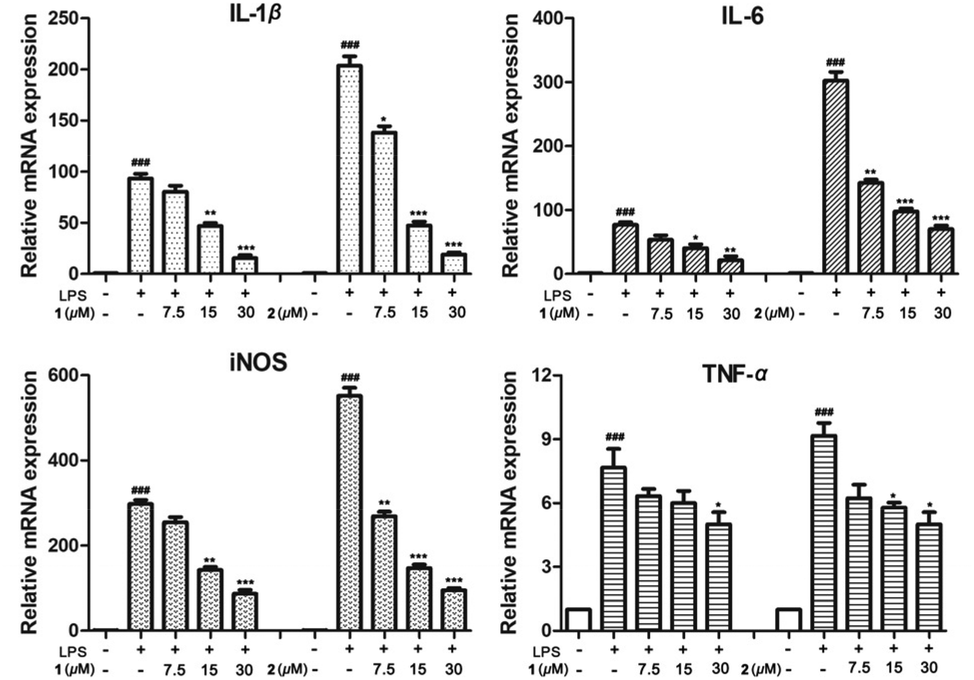 | ||
| Fig. 7 Inhibitory effects of 1 and 2 on the LPS-induced release of pro-inflammatory cytokines (IL-1β, IL-6, iNOS, and TNF-α) by qRT-PCR analysis. ###p < 0.001, compared with the control group. *p < 0.05, *p < 0.01, ***p < 0.001, compared with the LPS-treated group. | ||
Conclusions
In conclusion, fissisternoids A (1) and B (2), the first two unprecedented examples with polycyclic carbon skeletons presumably biosynthesized from 2′,5′-quinodihydrochalcone and an acyclic monoterpenoid, along with their possible precursor 3, were isolated from the traditional Chinese medicine F. bracteolatum. In our search for natural products to be a potential lead as a therapeutic agent for inflammatory diseases,17 racemic mixtures 1 and 2 exhibited anti-inflammatory activity via inhibiting the release of IL-1β, IL-6 and iNOS at mRNA levels. The structural novelty and complexity together with their bioactivity might attract the interest of pharmacologists and synthetic chemists.Experimental section
General experimental procedures
Optical rotations were measured using a JASCO P-1020 polarimeter in MeOH. UV spectra were recorded on a UV-2450 visible spectrophotometer (Shimadzu, Tokyo, Japan). IR spectra were acquired using a Nicolet iS10 microscope spectrometer in MeOH (Thermo Scientific, USA). All of the nuclear magnetic resonance (NMR) spectra were recorded using a Bruker AVIII-500 NMR instrument (1H: 500 MHz, 13C: 125 MHz) (Bruker, Karlsruhe, Germany), and tetramethylsilane (TMS) was used as an internal standard. High-resolution electrospray ionization mass spectra (HRESIMS) were recorded using an AB Sciex Triple-TOF 6600 (AB SCIEX, Framingham, MA, USA). Preparative HPLC was carried out on an LC-120 system (Separation, Beijing, China) equipped with a YMC-pack RP-C18 column (250 mm × 20 mm i.d., 5 μm, YMC, Tokyo, Japan) with a flow rate of 3 mL min−1, detected using a binary channel UV detector at 210 and 310 nm. Analytical HPLC was performed with an Agilent 1260 Series instrument equipped with a DAD detector using a Shim-pack VP-ODS column (4.6 × 250 mm; Agilent Technologies, Palo Alto, CA, USA). MCI (Mitsubishi, Tokyo, Japan) and RP-C18 silica (40–63 μm, Fuji, Tokyo, Japan) were used for column chromatography (CC). All solvents used were of analytical grade (TJshield Fine Chemicals Co., Ltd, Tianjin, China).Plant material
The branches and leaves of F. bracteolatum were collected from the Yunnan province of China in May 2019. The plant material was identified by Prof. Cheng-Ming Dong (Henan University of Chinese Medicine). A voucher specimen (no. 201905) was deposited in the Department of Natural Product Chemistry, School of Pharmacy, Henan University of Chinese Medicine.Extraction and isolation
The branches and leaves of F. bracteolatum (10.0 kg) were chopped and refluxed three times with 95% EtOH. The EtOH extract was concentrated under reduced pressure to obtain the extract (800 g), which then was dissolved in water (4.0 L) and successively partitioned with petroleum ether (PE, 4.0 L × 4), CH2Cl2 (4.0 L × 4), EtOAc (4.0 L × 4), and n-butanol to afford FrA–FrD.The CH2Cl2 extract (FrB, 180 g) was subjected to D101 macroporous resin CC using EtOH/H2O in step gradients (v/v 30%, 60%, 90% and 100%) as an eluent. The 60% MeOH fraction (50.2 g) was chromatographed by silica gel CC eluted with CH2Cl2–MeOH (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 → 0:100) to afford 7 fractions FrB1–FrB7. FrB1 (11.3 g) was chromatographed by MCI CC using a gradient elution of MeOH–H2O (v/v 20, 40, 60, 80, 100%) to yield six subfractions (FrB1a–FrB1e). FrB1d (4.3 g) was subjected to Sephadex LH-20 eluted with MeOH to afford FrB1d1–FrB1d9. FrB1d2 (0.5 g) was further subjected to reversed-phase C18 CC (40–63 μm, 40 mm × 250 mm) eluted with MeOH/H2O (v/v, 50–100%) to afford FrB1d2a–FrB1d2l. FrB1d2g (60.2 mg) was separated by pre-HPLC (10 mm × 250 mm, 5 μm, YMC, Tokyo, Japan) using 45–100% MeCN/H2O (v/v, 50 min) as the mobile phase to yield compound 1 (4.8 mg, tR 38.8 min). FrB1d2l (43.2 mg) was subjected to pre-HPLC (v/v, 40–100% MeCN/H2O) to yield compound 2 (10.2 mg, tR 33.1 min).
:0 → 0:100) to afford 7 fractions FrB1–FrB7. FrB1 (11.3 g) was chromatographed by MCI CC using a gradient elution of MeOH–H2O (v/v 20, 40, 60, 80, 100%) to yield six subfractions (FrB1a–FrB1e). FrB1d (4.3 g) was subjected to Sephadex LH-20 eluted with MeOH to afford FrB1d1–FrB1d9. FrB1d2 (0.5 g) was further subjected to reversed-phase C18 CC (40–63 μm, 40 mm × 250 mm) eluted with MeOH/H2O (v/v, 50–100%) to afford FrB1d2a–FrB1d2l. FrB1d2g (60.2 mg) was separated by pre-HPLC (10 mm × 250 mm, 5 μm, YMC, Tokyo, Japan) using 45–100% MeCN/H2O (v/v, 50 min) as the mobile phase to yield compound 1 (4.8 mg, tR 38.8 min). FrB1d2l (43.2 mg) was subjected to pre-HPLC (v/v, 40–100% MeCN/H2O) to yield compound 2 (10.2 mg, tR 33.1 min).
Fissisternoids A (±1): faint yellow crystal (CH3OH); [α]25D +2.1 (c 0.1, MeOH), UV (MeOH) λmax (logε) 204 (4.01), 298 (3.78) nm; IR (MeOH) νmax 3401, 2926, 1711, 1668, 1604, 1509, 1379, 1251, 1102, 1034, 830 cm−1. 1H and 13C NMR data (Table 1); HRESIMS m/z 475.2081 [M + Na]+ (calcd for C27H32O6Na, 475.2091).
(+)-1: [α]25D +68.5 (c 0.1, MeOH), ECD (MeOH) λmax (Δε): 220.0 (+1.0), 254.0 (−0.2), 288.0 (+1.8), 322.0 (−3.2) nm; (−)-1: [α]25D −70.3 (c 0.1, MeOH), ECD (MeOH) λmax (Δε): 218.0 (−1.3), 253.0 (+0.2), 287.0 (−3.1), 322.0 ( + 5.2) nm.
Fissisternoids B (±2): faint yellow oil; [α]25D +1.5 (c 0.1, MeOH), UV (MeOH) λmax (logε) 276 (3.21) nm; IR (MeOH) νmax 1683, 1635, 1496, 1455, 1438, 1386, 1329, 1303, 1278, 1187, 1121, 1085, 1043, 930, 827, 768, 701; 1H and 13C NMR data (Table 1); HRESIMS m/z 475.2085 [M + Na]+ (calcd for C27H32O6Na, 475.2091).
(+)-2: [α]25D +45.0 (c 0.1, MeOH), ECD (MeOH) λmax (Δε): 256 (+3.6), 290 (−12.4), 328.7 (+7.4) nm; (−)-2: [α]25D −46.5 (c 0.1, MeOH), ECD (MeOH) λmax (Δε): 255 (−5.2), 290 (+17.4), 329 (−10.2) nm.
Quantum chemical ECD calculations
The relative configurations of 1 and 2 were established initially according to their ROESY NMR spectra. Their conformers were first identified using the Spartan software using the MMFF94S force field. Then, on the basis of the Boltzmann distribution, the selected conformers were further optimized at the B3LYP/6-31+G* level in MeOH, and TDDFT ECD calculations were performed using the Gaussian 09 program package at the B3LYP/6-31+G(d,p) level with a CPCM model. The ECD curves of different conformers were simulated using SpecDis and weighted according to the Boltzmann distribution after UV correction.Measurement of NO release
The MTT assay was first used to evaluate RAW264.7 cell viability as previously described.17 Briefly, the cells were plated in 96-well plates (5 × 103 cells per well) for 18 h and then incubated with compounds 1 and 2 at various concentrations with or without LPS (1.0 μg mL−1). Eighteen hours later, the prepared MTT solution (20 μL, 5 mg mL−1) was added, and the cells were incubated for another 4 h. After the formazan that formed was fully dissolved in DMSO (150 μL per well), the absorbance was read at 570 nm on a microplate reader. The viability of the RAW264.7 cells for the control group (with DMSO only) is defined as 100%.NO production was determined by the level of accumulated nitrite in cell culture supernatants using Griess reagent from Beyotime Institute of Biotechnology (Jiangsu, China). Briefly, RAW264.7 cells (8 × 105 cells per mL) were incubated in 96-well plates and pretreated with test samples for 1 h, followed by incubation with 1 μg mL−1 LPS stimulation for 18 h. Then, the culture supernatant was mixed with an equal volume of the Griess reagent. The absorbance of the mixture was read at 540 nm using a microplate reader. Nitrite concentrations were calculated using sodium nitrate as a standard. NG-Monomethyl-L-arginine (L-NMMA) was used as a positive control in the experiments.
Quantitative real-time PCR
The methods for the total RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR) have been described previously.18 Total RNA was extracted from spleens or cultured cells using TRIzol according to the manufacturer's instructions (Invitrogen Corp.). Subsequently, RNA was reverse transcribed into cDNA using HiScript II Reverse Transcriptase (Vazyme, Nanjing, China). Real-time quantitative PCR was performed using real-time reverse transcription (RT)-PCR (Applied Biosystems, CA, USA) based on general fluorescence detection by SYBR Green. For mRNA transcript analysis, gene-specific values were normalized to the Actb gene.Statistical analysis
Statistical analysis of the data was performed using the Prism Demo Prism 4.0 software. Statistical analysis data were expressed as mean ± SD. Values were analyzed by one-way analysis of variance (ANOVA) using the SPSS version 12.0 software. P < 0.05 was considered statistically significant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was financially supported by the National Natural Science Foundation of China (82003606), the Scientific and Technological Key Project in Henan Province (212102311093), the Key Scientific Research Projects of Universities of Henan Province (21A360002), and the Scientific Research Nursery Project (MP2020-29).Notes and references
- Z. Qi, Y. H. Fu, Y. Q. Zhang, S. Y. Wu and G. Y. Chen, Fissitungfines A and B, two novel aporphine related alkaloids from Fissistigma tungfangense, Tetrahedron Lett., 2016, 57, 4162–4164 CrossRef.
- C. Gény, A. A. Samra, P. Retailleau, B. I. Iorga, H. Nedev, K. Awang, F. Roussi, M. Litaudon and V. Dumontet, (+)- and (-)-Ecarlottones, uncommon chalconoids from Fissistigma latifolium with pro-apoptotic activity, J. Nat. Prod., 2017, 80, 179–3185 CrossRef.
- Y. H. Lan, Y. L. Leu, Y. T. Peng, T. D. Thang, C. C. Lin and B. Y. Bao, The first bis-retrochalcone from Fissistigma latifolium, Planta Med., 2011, 77, 2019–2022 CrossRef CAS PubMed.
- H. N. Ngoc, L. Mair, D. T. Nghiem, L. K. Thien, J. M. Gostner, H. Stuppner and M. Ganzera, Phenolic compounds from the stems of Fissistigma polyanthoides and their anti-oxidant activities, Fitoterapia, 2019, 137, 104252–104257 CrossRef CAS PubMed.
- Y. C. Chia, F. R. Chang, C. M. Teng and Y. C. Wu, Aristolactams and dioxoaporphines from Fissistigma balansae and Fissistigma oldhamii, J. Nat. Prod., 2000, 63, 1160–1163 CrossRef CAS PubMed.
- Y. C. Wu, M. Sureshbabu, Y. C. Fang, Y. H. Wu, Y. H. Lan, F. R. Chang, Y. W. Chang and T. L. Hwang, Potent inhibition of human neutrophil activations by bractelactone, a novel chalcone from Fissistigma bracteolatum, Toxicol. Appl. Pharmacol., 2013, 266, 399–407 CrossRef CAS.
- T. P. Lien, A. Porzel, J. Schmidt, T. V. Sung and G. Adam, Chalconoids from Fissistigma bracteolatum, Phytochemistry, 2000, 53, 991–995 CrossRef CAS.
- Y. H. Lan, Y. C. Chia, F. R. Chang, T. L. Hwang, C. C. Liaw and Y. C. Wu, Potential anti-inflammatory activities of bractelactone and other compounds isolated form Fissistigma bracteolatum, Helv. Chim. Acta, 2005, 88, 905–909 CrossRef CAS.
- A. Porzel, T. P. Lien, J. Schmidt, S. Drosihn, C. Wagner, K. Merzweiler, T. V. Sung and G. Adam, Fissistigmatins A-D: novel type natural products with flavonoid-sesquiterpene hybrid structure from Fissistigma bracteolatum, Tetrahedron, 2000, 56, 865–872 CrossRef CAS.
- (a) G. M. Xue, C. Han, C. Chen, L. N. Li, X. B. Wang, M. H. Yang, Y. C. Gu, J. G. Luo and L. Y. Kong, Artemisians A−D, diseco-guaianolide involved heterodimeric [4+2] adducts from Artemisia argyi, Org. Lett., 2017, 19, 5410–5413 CrossRef CAS PubMed; (b) G. M. Xue, D. R. Zhu, C. Han, X. B. Wang, J. G. Luo and L. Y. Kong, Artemisianins A-D, new stereoisomers of seco-guaianolide involved heterodimeric [4+2] adducts from Artemisia argyi induce apoptosis via enhancement of endoplasmic reticulum stress, Bioorg. Chem., 2019, 84, 295–301 CrossRef CAS PubMed.
- L. F. Barahona and G. L. Rorrer, Isolation of halogenated monoterpenes from bioreactor-cultured microplantlets of the macrophytic red algae Ochtodes secundiramea and Portieria hornemannii, J. Nat. Prod., 2003, 66, 743–751 CrossRef CAS.
- (a) A. Benosman, J. M. Oger, P. Richomme, J. Bruneton, C. Roussakis, K. Ito, K. Ichino and A. H. A. Hadi, New terpenylated dihydrochalcone derivatives isolated from Mitrella kentii, J. Nat. Prod., 1997, 60, 921–924 CrossRef CAS; (b) F. Simard, C. Gauthier, É. Chiasson, S. Lavoie, V. Mshvildadze, J. Legault and A. Pichette, Antibacterial balsacones J-M, hydroxycinnamoylated dihydrochalcones from Populus balsamifera Buds, J. Nat. Prod., 2015, 78, 1147–1153 CrossRef CAS.
- B. Burke and M. Nair, Phenylpropene, benzoic acid and flavonoid derivatives from fruits of jamaican Piper species, Phytochemistry, 1986, 25, 1427–1430 CrossRef CAS.
- (a) H. Oikawa and T. Tokiwanoa, Enzymatic catalysis of the Diels–Alder reaction in the biosynthesis of natural products, Nat. Prod. Rep., 2004, 21, 321–352 RSC; (b) B. Y. Hu, S. X. Wang, Y. M. Yan, J. W. Liu, D. P. Qin and Y. X. Cheng, Spiromyrrhenes A-D: unprecedented diterpene–sesquiterpene heterodimers as intermolecular [4+2] cycloaddition products from Resina Commiphora that inhibit tumor stemness in esophageal cancer, Org. Chem. Front., 2020, 7, 2710–2718 RSC.
- W. Y. Zhang, J. X. Zhao, L. Sheng, Y. Y. Fan, J. Y. Li, K. Gao and J. M. Yue, Mangelonoids A and B, two pairs of macrocyclic diterpenoid enantiomers from Croton mangelong, Org. Lett., 2018, 20, 4040–4043 CrossRef CAS.
- J. Sung, C. T. Ho and Y. Wang, Preventive mechanism of bioactive dietary foods on obesity-related inflammation and diseases, Food Funct., 2018, 9, 6081–6095 RSC.
- (a) G. M. Xue, X. Q. Li, C. Chen, K. Chen, X. B. Wang, Y. C. Gu, J. G. Luo and L. Y. Kong, Highly oxidized guaianolide sesquiterpenoids with potential antiinflammatory activity from Chrysanthemum indicum, J. Nat. Prod., 2018, 81, 378–386 CrossRef CAS; (b) G. M. Xue, D. R. Zhu, T. Y. Zhu, X. B. Wang, J. G. Luo and L. Y. Kong, Lactone ring-opening seco-guaianolide involved heterodimers linked via an ester bond from Artemisia argyi, with NO inhibitory activity, Fitoterapia, 2019, 132, 94–100 CrossRef CAS PubMed.
- R. J. Li, C. Y. Gao, C. Guo, M. M. Zhou, J. Luo and L. Y. Kong, The anti-inflammatory activities of two major withanolides from Physalis minima via acting on NF-κB, STAT3, and HO-1 in LPS-stimulated RAW264.7 Cells, Inflammation, 2017, 40, 401–413 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data of compound 1, and 1D NMR, 2D NMR, HRESIMS, UV, and IR spectra of 1 and 2. CCDC 2107685. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo01404h |
| This journal is © the Partner Organisations 2022 |