
The first N-ligand assisted Pd catalyzed asymmetric synthesis of 3-arylsuccinimides as novel antifungal leads†
Jixing
Lai
a,
Juan
Yang
a,
Chen
Yang
a,
René
Csuk
 b,
Baoan
Song
a and
Shengkun
Li
*a
b,
Baoan
Song
a and
Shengkun
Li
*a
aLaboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Guizhou University, Huaxi District, Guiyang 550025, China. E-mail: SKL505@outlook.com
bOrganic Chemistry, Martin-Luther-University Halle-Wittenberg, Kurt-Mothes-Str. 2, D-06120 Halle (Saale), Germany
First published on 22nd November 2021
Abstract
The first palladium/chiral nitrogenous ligand-catalyzed enantioselective addition of aryl boronic acids to various maleimides was reported. This protocol features mild conditions combined with good functional group tolerance. The resultant 3-arylsuccinimides were shown to be novel chiral antifungal leads and inspired the discovery of novel ligands to address the challenging issues of this transformation.
Introduction
Succinimide-derived scaffolds have been recognized as core characteristics in a wide variety of pharmaceutical leads. Approximately half a century ago, N-alkylated succinimides were optimized and reported as potential long-acting anticonvulsants based on phensuximide and methsuximide1 (Fig. 1). The great potential in treating petit mal epilepsy triggered enormous efforts in the synthesis and clinical evaluation of 3-aryl-succinimides,1,2 which was exemplified by the discovery of anticonvulsant or antiepileptic drug leads 1–4 targeting 5-HT7 receptors, sodium channels or calcium channels. Several naphthoquinone-containing succinimides, for example, 5 and 6, were highly cytotoxic against the human breast adenocarcinoma cell line MCF-7.3 Compound 7 acted as a novel and selective inhibitor of indoleamine 2,3-dioxygenase (IDO-1) for cancer immunotherapy and the (R)-enantiomer was found to be the active agent4 (Fig. 1). Reducing the two carbonyl groups of a 3-aryl-succinimide skeleton furnished chiral pyrrolidines showing various activities including α-2-adrenoceptor antagonists5 and HSD-1 inhibitors.6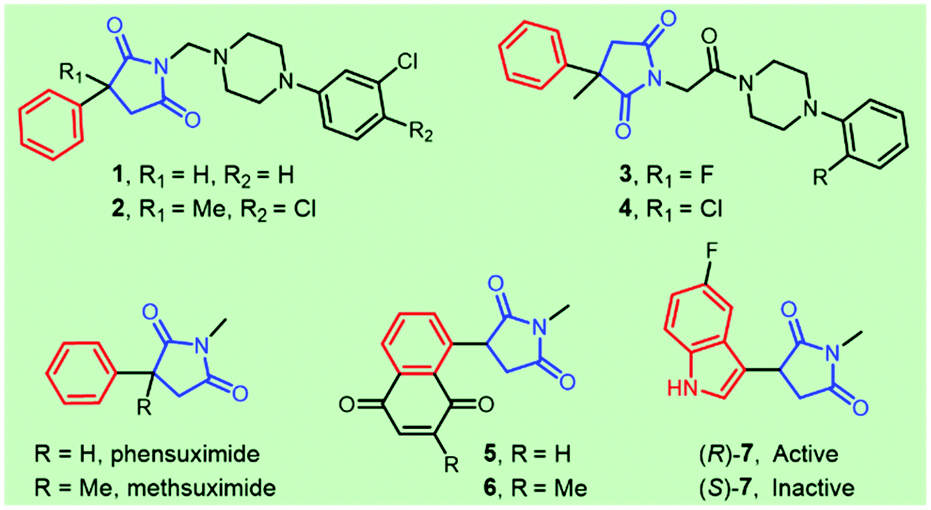 | ||
| Fig. 1 Pharmaceutically important 3-aryl-succinimides. | ||
Considerable attention from both synthetic chemists and the medicinal community has been given to accessing enantio-enriched 3-aryl-succinimides due to their pharmaceutical importance, especially for the affirmed chiral differentiation effect on bioactivity (e.g., compound 7 in Fig. 1). Therefore, transition-metal-catalyzed asymmetric additions of organoboron compounds to the electron-deficient double bonds of maleimides represent a straightforward and practical approach due to the variety, commercial availability, or synthetic tractability of both reaction partners. Intensive efforts have been made to develop rhodium catalysts for the asymmetric addition of arylboron reagents to maleimides (Fig. 2A). Significant progress has been achieved in the development of a wide range of chiral ligands, including chiral diene ligands,6,7 chiral phosphine-olefin ligands,5,8 chiral diphosphine ligands based on SYNPHOS9 and MeOBIPHEP,10 and sulfoxide–alkene hybrids.11
 | ||
| Fig. 2 Asymmetric addition of arylborons to maleimides. | ||
Although rhodium catalysis has been extensively investigated, we envisioned applying palladium catalysts as an attractive alternative for such transformation with lower costs. Only one palladium catalysis has been reported, and just three examples with the air-sensitive chiraphos have been detailed (Fig. 2B).12 We were encouraged to investigate the possibility of developing a practical palladium catalyst for the asymmetric addition of aryl boronic acids to maleimides. In addition, the protocol would avoid the inconvenient factors usually encountered in rhodium catalytic systems, e.g., multistep synthesis, resolution, cryogenic conditions, organometallic reagents or air sensitivity of the chiral ligands. To date, there have been no documented examples utilizing the catalytic combination of palladium and chiral N,N-ligands for this transformation. Herein, we document new palladium catalytic systems for the highly enantioselective addition of aryl boronic acids to maleimides with easily available and air-stable chiral heterocycle-oxazoline ligands. Enantioenriched 3-aryl-succinimides were investigated in detail for the first time and were confirmed as novel chiral antifungal leads, which intrigued the discovery of new heterocycle-oxazoline ligands to address the challenging issues of this transformation (Fig. 2B).
Results and discussion
To test the aforementioned hypothesis, the asymmetric addition of commercially available 4-methoxyphenylboronic acid 8a to N-benzylmaleimide 9a was used as a model reaction to elucidate the parameters of the reaction (Scheme 1). Following the pioneering work on palladium/N,N-bidentate ligand catalyzed asymmetric conjugate addition of arylboronic acids reported by Stoltz's group13 and our recent successful revelation of such addition to nitroolefins,14 a variety of bench-stable and readily available heterocycle-oxazolines were screened for this transformation (Scheme 1A). We were delighted to observe that the β-carboline-oxazolines tBu-β1-CarOx L1 and tBu-β3-CarOx L2 resulted in good enantioselectivities of 80% ee and 86% ee respectively, although the isolated yields were moderate. The chiral ligand isoquinoline-oxazoline L3 increased the efficacy with a moderate decrease in enantioselectivity. These outcomes witnessed the crucial influence of the substituted types in the pyridine segment on both the efficiency and selectivity. The isomeric variants, including isoquinoline-oxazoline L4 and quinoline-oxazoline L5, were then tested to improve this reaction. Both the yield and enantioselectivity of the desired product 10a were significantly enhanced (64% yield and 91% ee) while using quinoline-oxazoline L5. Formally removing the phenyl ring from L5 led to the classic PyOx ligand L6, which did not inhibit the reactivity, but the enantioselectivity was greatly reduced. The effect of the oxazoline ring substitution on the reaction outcome demonstrated that the t-butyl group outperformed Bn and the phenyl group based on L5 (see the ESI† for more details). Other substituted PyOx ligands, such as L7, L8, and L9, were also tested and showed that C-6 substitution on the pyridine ring had a crucial role in achieving both the reactivity and selectivity. So a range of representative C6-substituted PyOx ligands (L10–L14) with different electronic or steric effects were tested. The “magic methyl effect” was observed (L9) as efforts in introducing other substituents were detrimental to this transformation (L10–L14).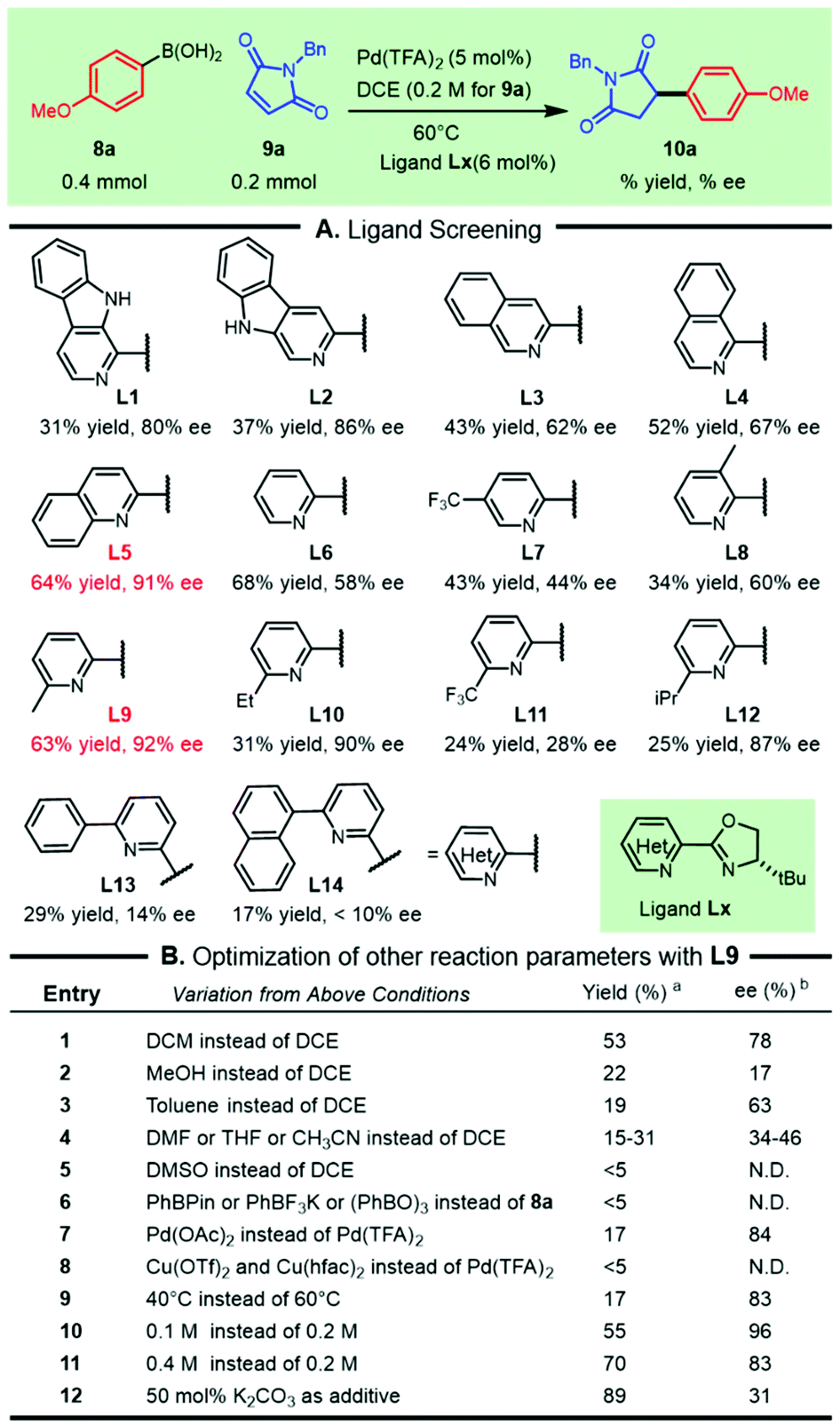 | ||
Scheme 1 Optimization of the asymmetric addition. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Isolated yield. bDetermined by HPLC on a chiral stationary phase. N.D. = not detected. Isolated yield. bDetermined by HPLC on a chiral stationary phase. N.D. = not detected. | ||
With the chiral ligand L9, we further scrutinized other reaction parameters, including solvents, metal salts, temperature, organoboronic variants and reaction concentration, which had the possibility of improving the reactivity and/or enantioselectivity (Scheme 1B). Analogously replacing DCE by DCM led to a slight decrease in both the yield and selectivity (Scheme 1B, entry 1). Other solvents, including the polar protic solvent (MeOH), polar aprotic solvents (DMF, THF, and CH3CN) and nonpolar solvent (toluene), were extremely detrimental to this transformation (Scheme 1B, entries 2–4). The selection of DMSO as the reaction media did not lead to detectable succinimide 10a (Scheme 1B, entry 5). Replacing aryl boronic acid 8a with other aryl boronic congeners (e.g., PhBPin, PhBF3K, (PhBO)3, and PhBneop) did not give a good result (Scheme 1B, entry 6). Replacing the counteranion of the Pd species with OAc− sharply dropped the yield and moderately decreased the selectivity (Scheme 1B, entry 7). Various copper salts, such as Cu(OTf)2 and Cu(hfac)2, could not act as palladium succedaneums for this reaction (Scheme 1B, entry 8). Reaction temperature variations had a significant effect on the productivity of the desired compound 10a. Although increasing the concentration or adding basic additives enhanced the yield of 10a, a decrease in enantioselectivity was observed (Scheme 1B, entry 11 and entry 12).
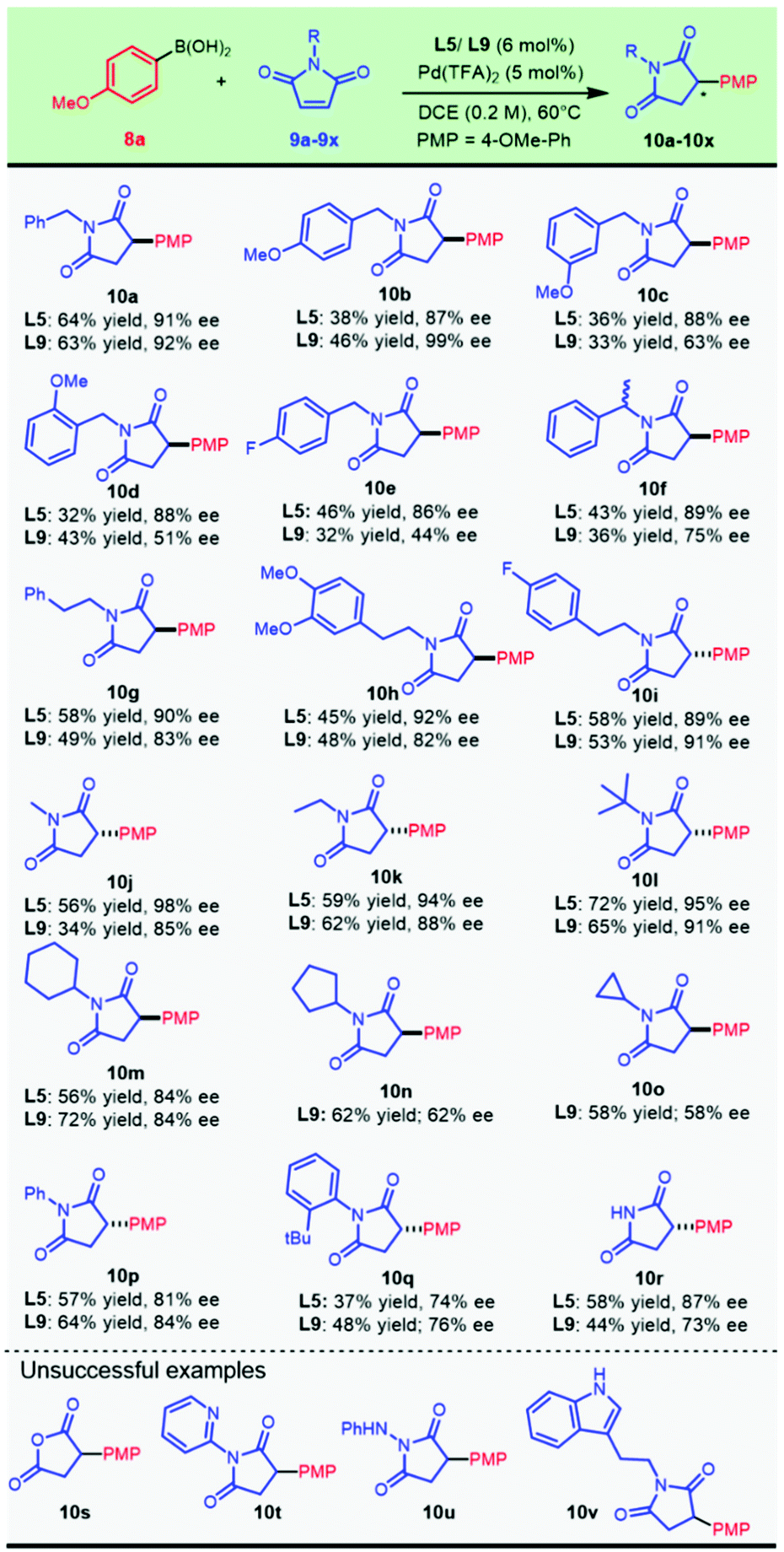
Following the condition optimization, we set out to explore the application of this chemistry for the asymmetric construction of chiral 3-arylsuccinimides with the optimal ligands L5 and L9. It was delightful and interesting to observe that 4-methoxyphenylboronic acid 8a underwent smooth enantioselective addition to a variety of N-substituted maleimides which were readily prepared from the condensation of maleic anhydride and various amines. A plethora of N-substituted succinimides, with substituted and unsubstituted benzyl, alkyl, and phenyl groups, were successfully prepared from this transformation (Scheme 2).
 | ||
| Scheme 2 Substrate scope with respect to maleimides. Isolated yield. | ||
The chiral ligands L5 and L9 provided comparable results for compound 10a. Introducing a methoxyl group at the para-position of the N-benzyl substituent allowed this addition to progress in an almost completely enantioselective manner by virtue of ligand L9, producing 10b in up to 99% ee and 46% isolated yield. Migration of the methoxyl substituent position resulted in a significant decrease in enantioselectivity (10c and 10d) with L9, and this could be compensated by replacing the chiral ligand with L5. Gratifyingly, para-F-substituted benzyl succinimides (10e) could be obtained in good enantioselectivity by applying a Pd/L5 catalyst. A highly enantioselective synthesis of chiral α-methyl-benzyl succinimide 10f (90% ee) was achieved. Pharmaceutically important maleimides derived from phenylethylamines with both an electron-enriched characteristic (10h) and an electron-deficient property (10i) provided 92% ee and 89% ee with the Pd/L5 catalyst, respectively. Other N-alkylated maleimides, with both simple acyclic (10j, 10k, and 10l) and cyclic alkyl groups (10m, 10n, and 10o), also demonstrated good compatibility in the nucleophilic attack by aryl boronic acid 8a. The catalytic system composed of Pd(TFA)2 and chiral L5 showed substantial advantages in the asymmetric addition to N-acyclic alkylated maleimides, wherein the methyl (10j), ethyl (10k), and bulky t-butyl (10l) substrates all provided excellent ee values ranging from 90% to 98%. Maleimides alkylated with secondary cyclic substituents, including cyclohexyl (10m), cyclopentyl (10n), and cyclopropyl (10o), were all attacked efficiently by employing 6-methyl PyOx L9.
N-Phenyl maleimide could perform enantioselective arylation to afford enantio-enriched 10p in parallel outcomes with both catalytic systems. Interestingly, the chiral catalyst with L5 delivered much better results when sterically bulky N-arylated substrates (10q, 10r, and 10s) were used. It is of interest to note that N-unsubstituted maleimide was also tolerated in this transformation to produce the desired chiral succinimide 10t in an appreciable yield. Thus, a new direct and modular late-stage derivation strategy could be devised for the expeditious construction of 3-arylsuccinimides for biological explorations. Unfortunately, the bioisosteric displacement of NH by oxygen failed to proceed with the desired reaction (10u). The established optimal catalysts did not work well for transforming N-heteroarylated (10v) or N-heterocyclic alkylated (10x) substrates and an acyl hydrazine analog (10w).
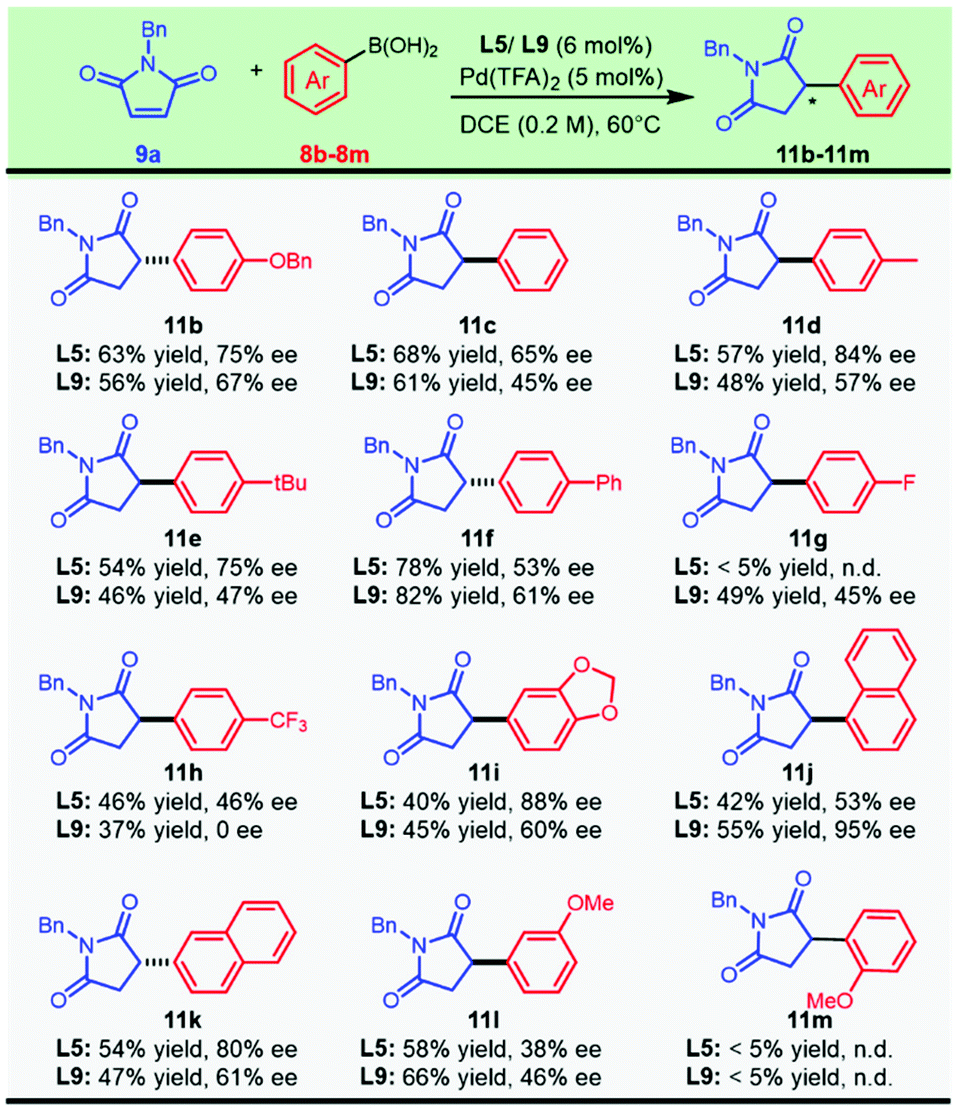
We turned our attention to executing the optimized protocols in exploring the scope with respect to aryl boronic acids (Scheme 3). Various functional groups, including ether, biphenyl, and halogen substituents, afforded the products in moderate to good yields and enantioselectivities. This asymmetric transformation exhibited a surprising dependence on the electronic effect of the aryl boronic acids. Decorating the para-position of the aryl boronic acids with electron-withdrawing groups, including fluoro- (11g) or a trifluoromethyl group (11h), resulted in a dramatically negative effect on the enantioselectivity. We speculated that the nucleophilic characteristics of the strongly electron-deficient boronic acids were slashed and caused the current system to be less reactive. Regardless of the similar electron-withdrawing para-substituent, it was noted that the corresponding aryl boronic acids had a pronounced difference in the catalytic outcome with the chiral ligand L5 or L9 (11gvs.11h). A prominent impact on enantiocontrol was noticed with the migration of the methoxyl group from the para position to the meta position, as compound 11l was synthesized with moderate enantioselectivity from both catalytic systems. Naphthyl boronic acids could also be employed, and a significant improvement in the enantioselectivity was observed when the substituted position was switched from the 2-position to the 1-position, furnishing compound 11k and its regioisomer 11j with 68% ee and 95% ee, respectively. However, this protocol was not applicable to sterically unfavored ortho-methoxyl phenylboronic acid (11m).
 | ||
| Scheme 3 Substrate scope with respect to aryl boronic acids. Isolated yield. | ||
Considering the prevalence of the phenylacetamide scaffold in pharmaceutical science, our continuing interest in the discovery of novel antifungal leads15 strengthened the preliminary structure and activity relationship of the synthetic collection of 3-arylsuccinimides (Fig. 3). To the best of our knowledge, no reports were previously disclosed on the asymmetric synthesis and antifungal profiles for this scaffold. The optimization of the antifungal coniothyriomycin16 encouraged us to more closely examine both the N-substitutions and α-aromatic variations. Based on the general elaboration of enantio-enriched 3-arylsuccinimides, we investigated the antifungal potential of these compounds. Both racemic 3-arylsuccinimides and the corresponding enantio-enriched compounds were evaluated at a concentration of 100 μM in a phenotypic antifungal screening (see the ESI† for more details). Substitution at the N-position was shown to be essential for obtaining significant antifungal activity (rac-10tvs. N-substituted 3-arylsuccinimides). N-Substitution with a long tail or a sterically bulky aliphatic substituent afforded good antifungal potency (Fig. 3A, rac-10avs. rac-10g; rac-10jvs. rac-10kvs. rac-10l).
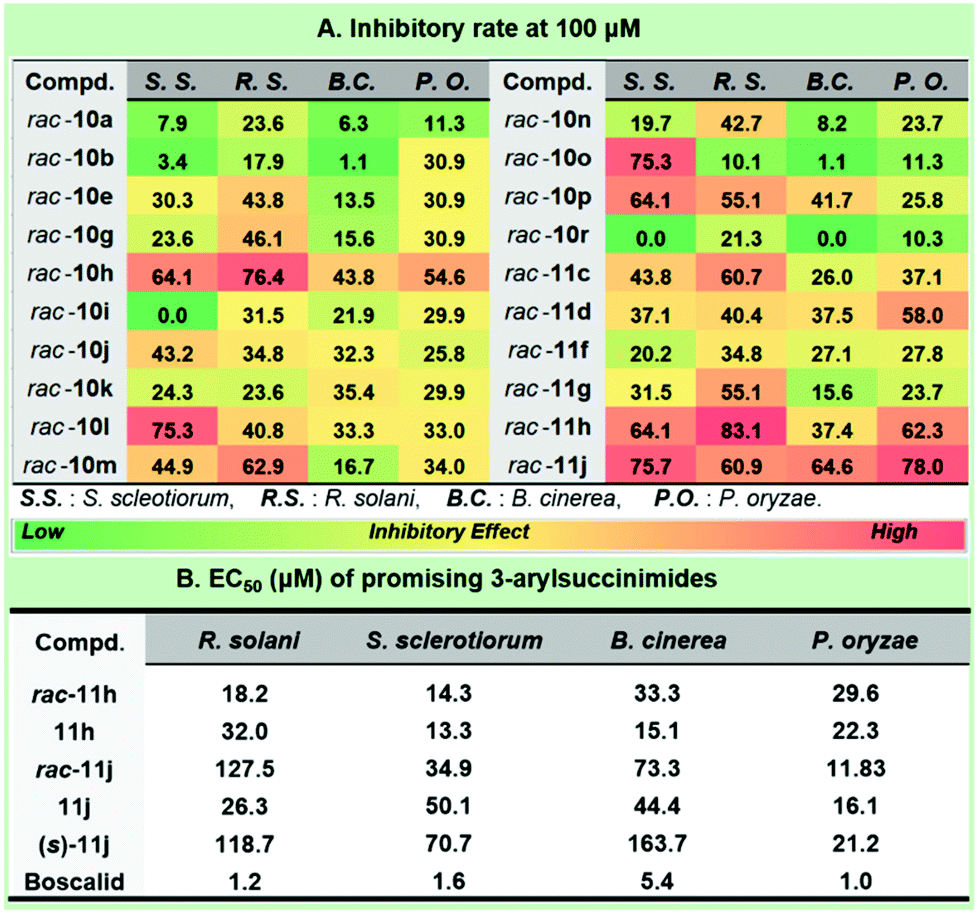 | ||
| Fig. 3 Antifungal profiles of selected 3-arylsuccinimides. | ||
Importantly, the introduction of electron-donating groups to N-phenethyl succinimide enhanced the inhibitory potential (rac-10gvs. rac-10h), while the presence of an electron-withdrawing group was detrimental (rac-10gvs. rac-10i). Subsequently, aromatic substitution modification was demonstrated to boost antifungal potency by removing the electron-rich substituent (rac-11cvs. rac-10a), and this was further enhanced by the introduction of an electron-poor group (rac-11gvs. rac-11h). Incorporating a naphthyl group (rac-11j) into the succinimide unit dramatically improved the inhibitory effect against S. sclerotiorum and P. oryzae and the inhibitory rate was >75% at a concentration of 100 μM. The chiral compounds 11h and 11j were selected to probe the effect of the configuration on the antifungal results (Fig. 3B). Interestingly, these two 3-arylsuccinimides showed quite different chiral selectivity in antifungal screening. It is worth mentioning that the R enantiomer of 11h (EC50 = 32.0 μM) was less effective against R. solani than the racemic mixture (EC50 = 18.2 μM), while a preference for the R-isomer was observed against the other three fungi. Interestingly, the antifungal potency of 11h (EC50 = 15.1 μM) was over 2-fold that of its racemate (EC50 = 33.3 μM) against B. cinerea. We typically observed that the antifungal potential of 11j against R. solani was approximately 5-fold that of the corresponding racemate. Surprisingly, racemic 3-aryl-succinimide 11j demonstrated a much better effect against P. oryzae with an EC50 value of 11.8 μM. Though the following synthesis and evaluation didn't underline the significance of maintaining the specific configuration, compound 11j possessed a reasonable antifungal enhancement compared with its (S)-enantiomer.
Due to the significant chiral effect on obtaining promising antifungal candidates, it is necessary to develop novel catalytic systems to address the challenging addition of electron-deficient aryl boronic acids to N-benzylmaleimide. New chiral heterocycle-oxazolines L15–L18 (Scheme 4) with substitution or fusion at the 6-position were conceived and prepared from readily available materials (see the ESI† for synthetic details). The newly synthesized sterically bulky 6-carbazolyl PyOx ligand L15 produced 17% yield with 18% ee, while the naphthalene-fused PyOx L16 and pyridine-fused counterpart L17 failed to afford enantio-enriched succinimide 11h. Much to our delight, the pyrrole fused pyridine oxazoline L18 inspired by the hybridization of the optimal ligands in the current methodology and our previously developed CarOx ligands significantly improved the enantioselectivity of the promising antifungal lead 11h and relevant flavanones (Scheme 4). This progress demonstrated the achievement in employing electron-withdrawing aryl boronic acids in palladium-catalyzed highly enantioselective addition to maleimides.
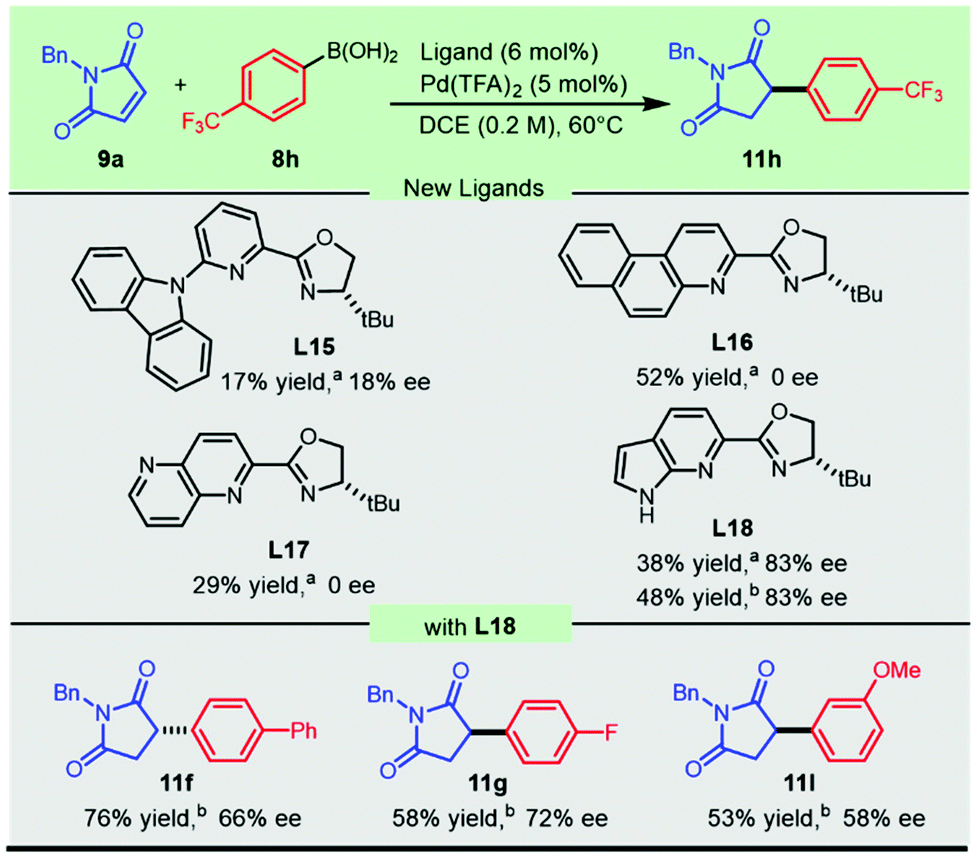 | ||
| Scheme 4 Novel heterocycle-oxazolines for the asymmetric synthesis of antifungal 11h and relevant flavanones. aHPLC yield of 11h. bIsolated yield. | ||
Conclusions
In conclusion, the current progress constituted the first application of palladium/chiral nitrogenous ligands in highly enantioselective addition of aryl boronic acids to various maleimides under rather mild conditions with good functional group tolerance. Importantly, some products exhibit good inhibitory effects for controlling agriculturally important fungi, demonstrating good potential for the discovery of novel fungicides. The new active chiral ingredients in turn inspired the acquirement of novel chiral ligands for addressing the challenging asymmetric addition of electron-deficient arylboronic acids to maleimides. Continued work will focus on synchronous and iterative optimization, involving the discovery and development of new heterocycle-oxazoline ligands to expand the substrate scope that thus far proved ineffective, and ultimately facilitate the discovery of pharmaceutically important leads.Author contributions
S. Li conceived and designed this work, J. Lai, J. Yang and C. Yang performed the experiments and provided the results, B. Song provided useful advice, S. Li and J. Lai analyzed the data and wrote the manuscript, and R. Csuk helped to check and revise the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21772094, 21977049) and the Natural Science Foundation of Jiangsu Province (BK20191306).Notes and references
- M. J. Kornet, A. M. Crider and E. O. Magarian, Potential long-acting anticonvulsants. 1. Synthesis and activity of succinimides containing an alkylating group at the 2 position, J. Med. Chem., 1977, 20, 405–409 CrossRef CAS PubMed.
- (a) J. Obniska and A. Zagorska, Synthesis and anticonvulsant properties of new N-[(4-arylpiperazin-1-yl)-methyl] derivatives of 3-aryl pyrrolidine-2,5-dione and 2-aza-spiro[4.4]nonane-1,3-dione, Farmaco, 2003, 58, 1227–1234 CrossRef CAS; (b) J. Obniska, I. Chlebek, K. Kaminski, A. J. Bojarski and G. Sstala, Synthesis, anticonvulsant activity and 5-HT1A/5-HT7 receptors affinity of 1-[(4-arylpiperazin-1-yl)-propyl]-succinimides, Pharmacol. Rep., 2012, 64, 326–335 CrossRef CAS; (c) K. Kaminski, J. Obniska, I. Chlebek, P. Liana and E. Pekala, Synthesis and biological properties of new N-Mannich bases derived from 3-methyl-3-phenyl- and 3,3-dimethyl-succinimides. Part V, Eur. J. Med. Chem., 2013, 66, 12–21 CrossRef CAS PubMed; (d) K. Kaminski, J. Obniska, I. Chlebek, B. Wiklik and S. Rzepka, Design, synthesis and anticonvulsant properties of new N-Mannich bases derived from 3-phenylpyrrolidine-2,5-diones, Bioorg. Med. Chem., 2013, 21, 6821–6830 CrossRef CAS PubMed; (e) J. Obniska, A. Rapacz, S. Rybka, B. Powroznik, E. Pekala, B. Filipek, P. Zmudzki and K. Kaminski, Design, synthesis and biological activity of new amides derived from 3-methyl-3-phenyl-2,5-dioxo-pyrrolidin-1-yl-acetic acid, Eur. J. Med. Chem., 2015, 102, 14–25 CrossRef CAS; (f) J. Obniska, I. Chlebek, K. Kaminski and J. Karolak-Wojciechowska, Synthesis and anticonvulsant properties of new N-Mannich bases derived from 3,3-diphenyl- and 3-ethyl-3-methyl-pyrrolidine-2,5-diones. Part III, Arch. Pharm., 2013, 346, 71–82 CrossRef CAS; (g) M. Gora, A. Czopek, A. Rapacz, A. Gebska, K. Wojcik-Pszczola, E. Pekala and K. Kaminski, Synthesis, anticonvulsant, and antinociceptive activity of new 3-(2-chlorophenyl)- and 3-(3-chlorophenyl)-2,5-dioxo-pyrrolidin-1-yl-acetamides, Molecules, 2021, 26, 1564 CrossRef CAS PubMed.
- S. H. Han, S. Kim, U. De, N. K. Mishra, J. Park, S. Sharma, J. H. Kwak, S. Han, H. S. Kim and I. S. Kim, Synthesis of Succinimide-Containing Chromones, Naphthoquinones, and Xanthones under Rh(III) Catalysis: Evaluation of Anticancer Activity, J. Org. Chem., 2016, 81, 12416–12425 CrossRef CAS.
- S. Crosignani, P. Bingham, P. Bottemanne, H. Cannelle, S. Cauwenberghs, M. Cordonnier, D. Dalvie, F. Deroose, J. L. Feng, B. Gomes, S. Greasley, S. E. Kaiser, M. Kraus, M. Negrerie, K. Maegley, N. Miller, B. W. Murray, M. Schneider, J. Soloweij, A. E. Stewart, J. Tumang, V. R. Torti, B. Van Den Eynde and M. Wythes, Discovery of a Novel and Selective Indoleamine 2,3-Dioxygenase (IDO-1) Inhibitor 3-(5-Fluoro-1H-indol-3-yl)pyrrolidine-2,5-dione (EOS200271/PF-06840003) and Its Characterization as a Potential Clinical Candidate, J. Med. Chem., 2017, 60, 9617–9629 CrossRef CAS.
- (a) R. Shintani, W.-L. Duan, T. Nagano, A. Okada and T. Hayashi, Chiral phosphine-olefin bidentate ligands in asymmetric catalysis: Rhodium-catalyzed asymmetric 1,4-addition of aryl boronic acids to maleimides, Angew. Chem., Int. Ed., 2005, 44, 4611–4614 CrossRef CAS; (b) W.-L. Duan, H. Iwamura, R. Shintani and T. Hayashi, Chiral Phosphine-Olefin Ligands in the Rhodium-Catalyzed Asymmetric 1,4-Addition Reactions, J. Am. Chem. Soc., 2007, 129, 2130–2138 CrossRef CAS PubMed.
- B. Gopula, S.-H. Yang, T.-S. Kuo, J.-C. Hsieh, P.-Y. Wu, J. P. Henschke and H.-L. Wu, Direct Synthesis of Chiral 3-Arylsuccinimides by Rhodium-Catalyzed Enantioselective Conjugate Addition of Arylboronic Acids to Maleimides, Chem. – Eur. J., 2015, 21, 11050–11055 CrossRef CAS.
- (a) R. Shintani, K. Ueyama, I. Yamada and T. Hayashi, Chiral Norbornadienes as Efficient Ligands for the Rhodium-Catalyzed Asymmetric 1,4-Addition of Arylboronic Acids to Fumaric and Maleic Compounds, Org. Lett., 2004, 6, 3425–3427 CrossRef CAS PubMed; (b) X. Dou, Y. Lu and T. Hayashi, Base-Free Conditions for Rhodium-Catalyzed Asymmetric Arylation To Produce Stereochemically Labile α-Aryl Ketones, Angew. Chem., Int. Ed., 2016, 55, 6739–6743 CrossRef CAS; (c) Y. Luo and A. J. Carnell, Chemoenzymatic Synthesis and Application of Bicyclo[2.2.2]octadiene Ligands: Increased Efficiency in Rhodium-Catalyzed Asymmetric Conjugate Additions by Electronic Tuning, Angew. Chem., Int. Ed., 2010, 49, 2750–2754 CrossRef CAS.
- (a) E. Piras, F. Lang, H. Ruegger, D. Stein, M. Worle and H. Grutzmacher, Chiral phosphane alkenes (PALs): simple synthesis, applications in catalysis, and functional hemilability, Chem. – Eur. J., 2006, 12, 5849–5858 CrossRef CAS; (b) J. Csizmadiova, M. Meciarova, E. Rakovsky, B. Horvath and R. Sebesta, [5]Ferrocenophanene-Phosphane Ligands for Enantioselective Rh-Catalyzed Conjugate Additions, Eur. J. Org. Chem., 2011, 6110–6116 CrossRef CAS; (c) S. Vlahovic, N. Schaedel, S. Tussetschlaeger and S. Laschat, Tropanes as scaffolds for phosphorus-olefin ligands and their application in asymmetric catalysis, Eur. J. Org. Chem., 2013, 1580–1590 CrossRef CAS; (d) M. Ogasawara, Y.-Y. Tseng, S. Arae, T. Morita, T. Nakaya, W.-Y. Wu, T. Takahashi and K. Kamikawa, Phosphine-Olefin Ligands Based on a Planar-Chiral (π-Arene)chromium Scaffold: Design, Synthesis, and Application in Asymmetric Catalysis, J. Am. Chem. Soc., 2014, 136, 9377–9384 CrossRef CAS PubMed.
- (a) F. Berhal, O. Esseiva, C.-H. Martin, H. Tone, J.-P. Genet, T. Ayad and V. Ratovelomanana-Vidal, (R)-3,5-diCF3-SYNPHOS and (R)-p-CF3-SYNPHOS, Electron-Poor Diphosphines for Efficient Room Temperature Rh-Catalyzed Asymmetric Conjugate Addition of Arylboronic Acids, Org. Lett., 2011, 13, 2806–2809 CrossRef CAS PubMed; (b) F. Berhal, Z. Wu, J.-P. Genet, T. Ayad and V. Ratovelomanana-Vidal, Rh-catalyzed asymmetric 1,4-addition of arylboronic acids to α,β-unsaturated ketones with DIFLUORPHOS and SYNPHOS analogues, J. Org. Chem., 2011, 76, 6320–6326 CrossRef CAS.
- (a) F. Le Boucher d'Herouville, A. Millet, M. Scalone and V. Michelet, Room-temperature Rh-catalyzed asymmetric 1,4-addition of arylboronic acids to maleimides and enones in the presence of CF3-substituted MeOBIPHEP analogues, J. Org. Chem., 2011, 76, 6925–6930 CrossRef; (b) T. Korenaga, A. Ko and K. Shimada, Low-Temperature Rh-Catalyzed Asymmetric 1,4-Addition of Arylboronic Acids to α,β-Unsaturated Carbonyl Compounds, J. Org. Chem., 2013, 78, 9975–9980 CrossRef CAS.
- T. Thaler, L.-N. Guo, A. K. Steib, M. Raducan, K. Karaghiosoff, P. Mayer and P. Knochel, Sulfoxide-Alkene Hybrids: A New Class of Chiral Ligands for the Hayashi-Miyaura Reaction, Org. Lett., 2011, 13, 3182–3185 CrossRef CAS PubMed.
- T. Nishikata, Y. Yamamoto and N. Miyaura, Asymmetric 1,4-addition of arylboronic acids to α,β-unsaturated N-acylamides catalyzed by dicationic palladium(II)-(S,S)-chiraphos complex, Chem. Lett., 2007, 36, 1442–1443 CrossRef CAS.
- K. Kikushima, J. C. Holder, M. Gatti and B. M. Stoltz, Palladium-Catalyzed Asymmetric Conjugate Addition of Arylboronic Acids to Five-, Six-, and Seven-Membered β-Substituted Cyclic Enones: Enantioselective Construction of All-Carbon Quaternary Stereocenters, J. Am. Chem. Soc., 2011, 133, 6902–6905 CrossRef CAS PubMed.
- (a) W. Li, G. Wang, J. Lai and S. Li, Multifunctional isoquinoline-oxazoline ligands of chemical and biological importance, Chem. Commun., 2019, 55, 5902–5905 RSC; (b) J. Lai, W. Li, S. Wei and S. Li, Natural carbolines inspired the discovery of chiral CarOx ligands for asymmetric synthesis and antifungal leads, Org. Chem. Front., 2020, 7, 2263–2268 RSC.
- (a) S. Li, D. Li, T. Xiao, S. Zhang, Z. Song and H. Ma, Design, synthesis, fungicidal activity, and unexpected docking model of the first chiral boscalid analogues containing oxazolines, J. Agric. Food Chem., 2016, 64, 8927–8934 CrossRef CAS PubMed; (b) L. Zhang, W. Li, T. Xiao, Z. Song, R. Csuk and S. Li, Design and Discovery of Novel Chiral Antifungal Amides with 2-(2-Oxazolinyl)aniline as a Promising Pharmacophore, J. Agric. Food Chem., 2018, 66, 8957–8965 CrossRef CAS PubMed.
- K. Krohn, B. Elsasser, S. Antus, K. Konya and E. Ammermann, Synthesis and structure-activity relationship of antifungal coniothyriomycin analogues, J. Antibiot., 2003, 56, 296–305 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis and characteristic data for the synthesized new compounds and antifungal data. See DOI: 10.1039/d1qo01510a |
| This journal is © the Partner Organisations 2022 |