
One-pot enantioselective construction of 3,4-dihydro-2H-1,4-oxazines over Ru/Au relay catalysis and its mechanistic serendipity†
Dongfeng
Yang‡
,
Chengyi
Wang‡
,
Yu
Wang
,
Guohua
Liu
 *,
Tanyu
Cheng
and
Rui
Liu
*
*,
Tanyu
Cheng
and
Rui
Liu
*
Joint Laboratory of International Cooperation of Resource Chemistry of Ministry of Education, Key Laboratory of Resource Chemistry of Ministry of Education, and Shanghai Key Laboratory of Rare Earth Functional Materials, Shanghai Normal University, Shanghai, China. E-mail: rliu@shnu.edu.cn; ghliu@shnu.edu.cn
First published on 17th November 2021
Abstract
The preparation of enantiomerically pure 1,4-oxazines remains a continuous challenge in synthetic chemistry because of their potential application in the total synthesis of morpholines. Herein, a one-pot asymmetric transfer hydrogenation/cyclization enantio-relay process catalyzed by Ru and Au bimetallic catalysts was developed. This enantio-relay process firstly allows the asymmetric transfer hydrogenation of alkynones to form chiral alkynols in the presence of (S,S)-mesityleneRuClTsDPEN (TsDPEN = N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine) and then the chiral alkynols could be converted into 3,4-dihydro-2H-1,4-oxazines in moderate to good yields with excellent enantioselectivity retention accompanied by AuCl(PPh3)/AgNTf2 and HCOOH. A series of sulfonamide-tethered alkynones were well tolerated in this process. Mechanistic studies indicated that the alkynols formed in the first step were initially transformed into a methylenemorpholine intermediate when AuCl(PPh3)/AgNTf2 and HCOOH were added, which can be further isomerized into chiral 3,4-dihydro-2H-1,4-oxazines with the addition of a second HCOOH. This protocol offers not only a practical method to access 3,4-dihydro-2H-1,4-oxazines from alkynones in an enantioselective version, but also enriches the arsenal of gold chemistry from a mechanistic point of view.
Introduction
Enantiomerically pure 1,4-oxazines, as important core units in a wide variety of biologically active compounds, have aroused great interest in the past several decades.1 In particular, some representative oxazines have chiral side chains at the 2-position, such as the antidepressant drug GABAB receptor antagonist1c and reboxetine1d and have been successfully used in clinical medicine as shown in Scheme 1. Unfortunately, a brief survey of previous reports shows that the main method for the preparation of chiral 1,4-oxazines is stoichiometric synthesis using chiral β-amino alcohols and analogs as starting materials.2 Therefore, developing facile and practical methods to access 1,4-oxazines bearing a chiral center at the 2-position from easily available starting materials is of great significance.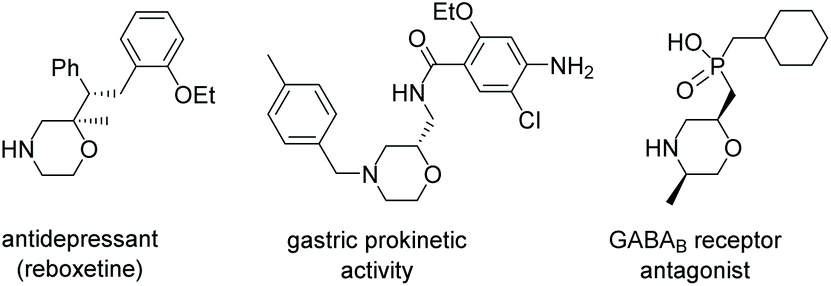 | ||
| Scheme 1 Pharmacologically active morpholine derivatives. | ||
Our synthetic strategy for the synthesis of 1,4-oxazines was inspired by the cyclization of those starting materials containing an alkyne motif because a transition metal-activated η2-coordination unit could serve as an electrophile to combine epoxide, amino and alcohol motifs, and thus give rise to the desired heterocycles.3 For example, Fang and co-workers described the synthesis of allyl vinyl ethers via the isomerization reaction of alkynyl epoxides using PtCl2 as a catalyst as shown in Scheme 2-(1).3b Shortly after that, Kang and co-workers found that AgOTf can convert alkynyl oxiranes into 1,4-oxazine derivatives, albeit with moderate yields.3c Furthermore, Wang and co-workers reported the regioselective cyclization of alkynols to access 1,4-oxazines under solvent and metal-free conditions.4 A landmark in the synthesis of 1,4-oxazines was made by Huang and co-workers, where a catalytic amount of the gold(I) complex AuCl(PPh3) was employed as the catalyst as shown in Scheme 2-(2).5 This protocol provides a convenient method to access 1,4-oxazines from the environment-friendly raw materials alkynols. As an ongoing extension of this topic, it would be highly desirable to realize the catalytic enantioselective construction of 3,4-dihydro-2H-1,4-oxazines from chiral alkynols (step II in Scheme 2-(3)).6
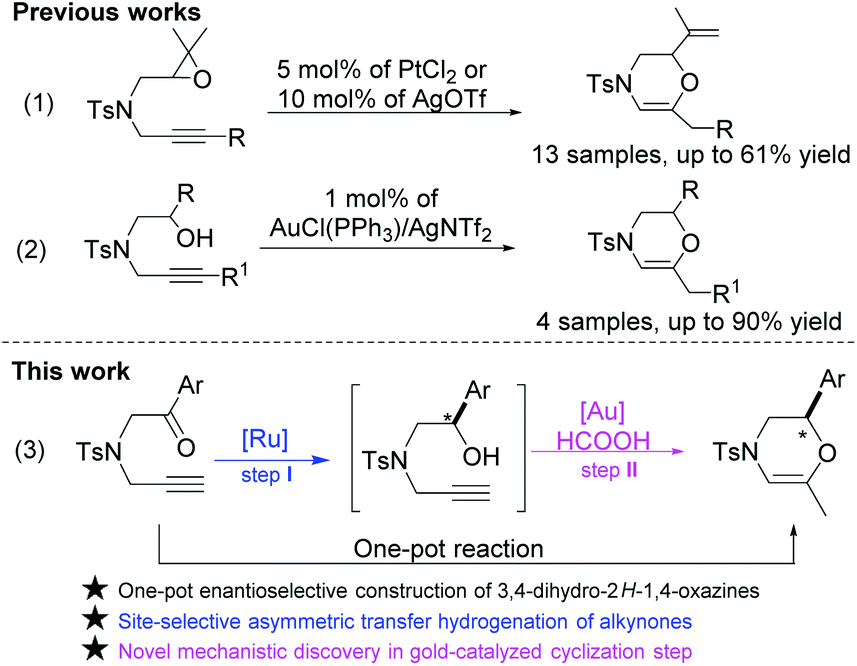 | ||
| Scheme 2 Synthesis of morpholine derivatives. | ||
Asymmetric transfer hydrogenation (ATH) as a powerful methodology has been extensively studied in the reduction of ketones, imines, and even alkynes.7 The advantages of ATH reactions include low catalyst loading, wide substrate scope, mild reaction conditions, etc.7 Generally, the ATH reaction of ketones could provide the corresponding alcohols in excellent yields and enantioselectivities as demonstrated in our previous work.8 Therefore, we consider the possibility of synthesizing the aforementioned alkynols using alkynones as starting materials through the ATH reaction (step I in Scheme 2-(3)).9 More importantly, the integration of the ATH reaction of alkynones and cyclization of alkynols10 in a one-pot reaction would also be highly interesting because of its step/cost-economical fashion.11
Herein, we report a one-pot sequential ATH/cyclization enantio-relay process of alkynones to afford chiral 3,4-dihydro-2H-1,4-oxazines. The alkynones were firstly converted into chiral alkynols using (S,S)-configurational mesityleneRuClTsDPEN as a catalyst. Then, in the presence of AuCl(PPh3)/AgNTf2 and HCOOH, the alkynols were further transformed into 3,4-dihydro-2H-1,4-oxazines in good yields with high enantioselectivity retention. Interestingly, a reaction intermediate has been isolated during the cyclization process of alkynols.
Results and discussion
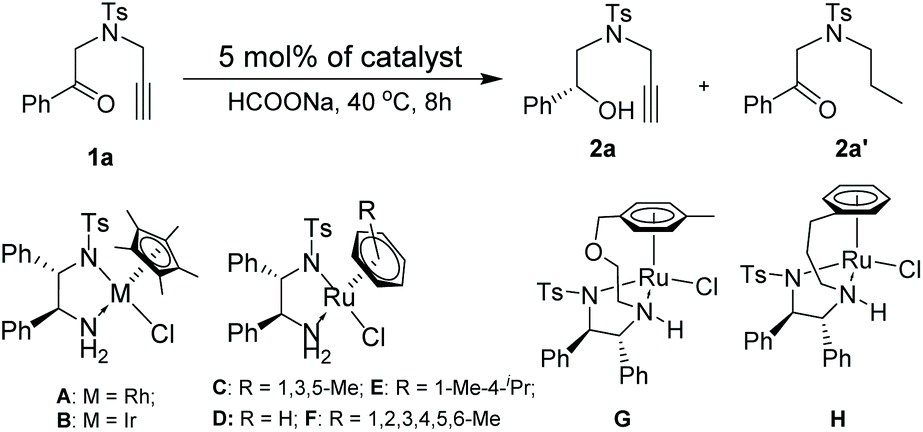
Our designed protocol consists of the ATH reaction of alkynones and cyclization of alkynols. Therefore, at the beginning of our research, we firstly focused on the selective reduction of alkynones using 4-methyl-N-(2-oxo-2-phenylethyl)-N-(prop-2-yn-1-yl)benzenesulfon-amide (1a) as a model substrate and commonly used (S,S)-1,2-diphenylethylenediamine-based complexes12 as catalysts. 1a was reacted with a toluene solution of the commonly used Cp*RhClTsDPEN (A) (Cp* = pentamethylcyclopentadiene) using HCOOH/Et3N (5/2) as the hydrogen source, affording the alkyne-selective reduced product 2a′ in 58% isolated yield (please see the ESI†). Solvent screening indicated that the carbonyl group of 1a could be solely reduced only in DCE in the presence of A and HCOOH/Et3N (5/2) (please see the ESI†). Gratifyingly, the use of HCOONa as the hydrogen source allows for a further improvement of the yield (89%) and ee value (94%) of 2a (entry 1, Table 1). Based on the above catalytic results, we then shifted our attention to the optimization of the metal source using the extensively employed chiral diamine-based complex Cp*IrClTsDPEN (B)13 and η6-arene-Ru complexes C–H (arene = substituted benzene) (entries 2–8, Table 1).8 Interestingly, the selective formation of 2a′ is not favored regardless of the type of metal complexes in the case of HCOONa as the hydrogen source. All screened transition metal complexes exclusively released the desired 2a in moderate to excellent yields with good enantioselectivities, and the best yield and ee value of 96% and 97% (entry 3, Table 1) were observed when (S,S)-mesityleneRuClTsDPEN (C) was employed. However, the ATH reaction of 1a using other hydrogen sources, for instance, iPrOH and HCOOH, did not occur under the same reaction conditions (entries 10 and 11, Table 1). A further screening of solvents also confirmed that the reaction in DCE gave the best result since the reactions in DMF and MeCN only produced 2a in moderate yields (entries 12 and 13, Table 1). Finally, the reactions performed at 35 °C and 45 °C gave inferior outcomes in terms of the yield and ee value of 2a (entries 14 and 15, Table 1).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Solvent/H-source | Yield of 2ab [%] |
ee of 2ac [%] |
| a Reaction conditions: 1a (0.30 mmol), catalyst (0.015 mmol), hydrogen sources (1.20 mmol), 2.0 mL of solvent, reaction time (8 h). b Isolated yield. c Determined by HPLC on a Daicel Chiralcel AD-H column. d 0.30 mmol of KOH was added. e Data were obtained at 35 °C. f Data were obtained at 45 °C. | ||||
| 1 | A | DCE/HCOONa | 89 | 94 |
| 2 | B | DCE/HCOONa | 50 | 96 |
| 3 | C | DCE/HCOONa | 96 | 97 |
| 4 | D | DCE/HCOONa | 84 | 92 |
| 5 | E | DCE/HCOONa | 60 | 96 |
| 6 | F | DCE/HCOONa | 86 | 96 |
| 7 | G | DCE/HCOONa | 86 | 91 |
| 8 | H | DCE/HCOONa | 89 | 90 |
| 9 | C | DCE/HCOOH-NEt3 (5/2) | 93 | 95 |
| 10 | C | iPrOHd | — | — |
| 11 | C | DCE/HCOOH | — | — |
| 12 | C | DMF/HCOONa | 53 | 78 |
| 13 | C | MeCN/HCOONa | 72 | 85 |
| 14 | C | DCE/HCOONa | 92 | 97e |
| 15 | C | DCE/HCOONa | 97 | 95f |
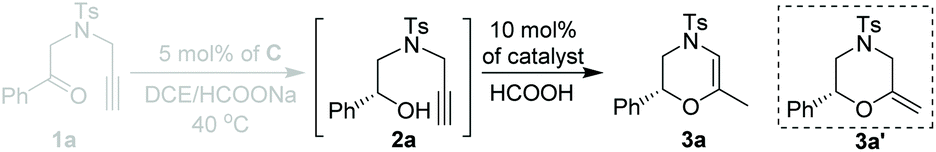
Having established the optimal ATH conditions (catalyst C, HCOONa, DCE, 40 °C) of 1a, we then tried to integrate the ATH reaction of 1a and cyclization of 2a in a one-pot reaction to prepare chiral 3,4-dihydro-2H-1,4-oxazines (3a). However, no reaction occurred when PPh3AuCl/AgNTf2 was added to a reaction mixture of 2a obtained from the ATH process (entry 1, Table 2). Inspired by Huang's report,5 it was suggested that the large excess of HCOONa might affect the activity of AuCl(PPh3)/AgNTf2. To solve this problem, 2 equiv. of HCOOH was injected prior to the addition of AuCl(PPh3)/AgNTf2. However, the further in situ NMR monitoring experiment indicated that methylenemorpholine 3a′14 was exclusively formed rather than the desired 3a, strongly suggesting the mechanistic divergence with respect to the work of Huang (entry 2, Table 2).5 Notably, the addition of a second HCOOH was able to convert 3a′ into 3a in 65% yield (entry 3, Table 2). Additional screening of metal catalysts indicated that both AuCl(PPh3)/AgOTf and PtCl2 exhibited inferior outcomes with respect to AuCl(PPh3)/AgNTf2 in terms of the yields and ee values of 3a (entries 4 and 5, Table 2).
a
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield of 3ab [%] |
ee of 3ac [%] |
| a Reaction conditions: Step I: Reaction conditions: alkynone 1a (0.30 mmol), catalyst C (9.3 mg, 0.015 mmol), HCOONa (81.6 mg, 1.20 mmol), 40 °C, DCE (2.0 mL), 8–10 h; Step II: HCOOH (0.6 mmol), AuCl(PPh3) (14.8 mg, 0.030 mmol) AgNTf2 (11.6 mg, 0.030 mmol), 40 °C, 5–7 h. b Isolated yield. c Determined by HPLC on a Daicel Chiralcel OJ-H column. d 88% of 2a was recovered. e 2 equiv. of HCOOH was added before addition of the catalyst. f 16% of 3a′ was isolated. g Reaction solution was treated with 1 equiv. of HCOOH before separation. | |||
| 1d | AuCl(PPh3)/AgNTf2 | — | — |
| 2e,f | AuCl(PPh3)/AgNTf2 | — | — |
| 3e,g | AuCl(PPh3)/AgNTf2 | 65 | 96 |
| 4e,g | Ph3PAuCl/AgOTf | 47 | 95 |
| 5e,g | PtCl2 | 34 | 95 |
Based on the best one-pot reaction conditions (step I: catalyst C, HCOONa, DCE, 40 °C and step II: HCOOH/AuCl(PPh3)/AgNTf2 + HCOOH), a series of alkynones were further investigated under the optimal reaction conditions (Table 3). Generally, all tested alkynones were capable of providing their corresponding chiral 3,4-dihydro-2H-1,4-oxazine derivatives (3) in moderate yields with high enantioselectivities (columns 6–8, Table 3). Gratifyingly, the electronic properties of the substituents connected with the aromatic ring did not have a significant impact on the enantioselectivities of the final products, since the reactions with various electron-withdrawing and -donating substituents were highly enantioselective. However, the steric hindrance of the substituents at the aromatic rings had an obvious impact on their enantioselectivities. When the substituents were at the 2-position of the aromatic ring, decreased enantioselectivities were observed, for instance, 3d and 3g (entries 3 and 6 in column 8, Table 3). Also, this ATH/cyclization enantio-relay process with naphthyl and heterocyclic aromatic rings as substrates also led to high ee values (entries 15 and 16 in column 8, Table 3). In particular, to determine their enantiospecificities, a single-step ATH reaction catalyzed by catalyst C was also performed (columns 3–5, Table 3). It was notable that the obtained chiral products 3a–3u had high enantiospecificities (% es) (column 9, Table 3). For example, in the cases of 3e, 3f, 3i, 3k, 3l, 3r, 3s and 3t, their es values were more than 100%, suggesting the high enantioselectivity retention of the present ATH/cyclization process.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R, Ar, 1 | 2a–2u | 3a–3u | % es | ||||
| 2 | % yieldb | % eec | 3 | % yieldb | % eec | |||
| a Reaction conditions: Step I: Reaction conditions: alkynone 1a (0.30 mmol), catalyst A (0.015 mmol), HCOONa (1.20 mmol), 40 °C, DCE (2.0 mL), 8–10 h; Step II: HCOOH (0.6 mmol), AuCl(PPh3) (14.8 mg, 0.030 mmol), AgNTf2 (11.6 mg, 0.030 mmol), 40 °C, 5–7 h. b Isolated yield. c Determined by chiral HPLC. | ||||||||
| 1 | Me, 4-FPh, 1b | 2b | 93 | 94 | 3b | 60 | 94 | 100 |
| 2 | Me, 3-FPh, 1c | 2c | 92 | 96 | 3c | 64 | 95 | 99 |
| 3 | Me, 2-FPh, 1d | 2d | 88 | 85 | 3d | 61 | 85 | 100 |
| 4 | Me, 4-ClPh, 1e | 2e | 96 | 95 | 3e | 62 | 98 | >100 |
| 5 | Me, 3-ClPh, 1f | 2f | 90 | 95 | 3f | 56 | 97 | >100 |
| 6 | Me, 2-ClPh, 1g | 2g | 93 | 81 | 3g | 53 | 79 | 98 |
| 7 | Me, 4-BrPh, 1h | 2h | 95 | 95 | 3h | 61 | 93 | 98 |
| 8 | Me, 3-BrPh, 1i | 2i | 93 | 94 | 3i | 60 | 98 | >100 |
| 9 | Me, 4-CF3Ph, 1j | 2j | 90 | 94 | 3j | 67 | 91 | 97 |
| 10 | Me, 4-CNPh, 1k | 2k | 92 | 92 | 3k | 70 | 96 | >100 |
| 11 | Me, 4-MePh, 1l | 2l | 96 | 96 | 3l | 69 | 97 | >100 |
| 12 | Me, 4-OMePh, 1m | 2m | 97 | 98 | 3m | 68 | 96 | 98 |
| 13 | Me, 3-OMePh, 1n | 2n | 91 | 95 | 3n | 59 | 95 | 100 |
| 14 | Me, biphenyl, 1o | 2o | 93 | 95 | 3o | 63 | 94 | 99 |
| 15 | Me, 2-naphthyl, 1p | 2p | 95 | 96 | 3p | 70 | 96 | 100 |
| 16 | Me, 3-thienyl, 1q | 2q | 91 | 98 | 3q | 73 | 96 | 98 |
| 17 | F, Ph, 1r | 2r | 94 | 94 | 3r | 63 | 94 | >100 |
| 18 | Cl, Ph, 1s | 2s | 92 | 93 | 3s | 70 | 94 | >100 |
| 19 | Br, Ph, 1t | 2t | 90 | 94 | 3t | 75 | 95 | >100 |
| 20 | NO2, Ph, 1u | 2u | 87 | 97 | 3u | 61 | 96 | 99 |
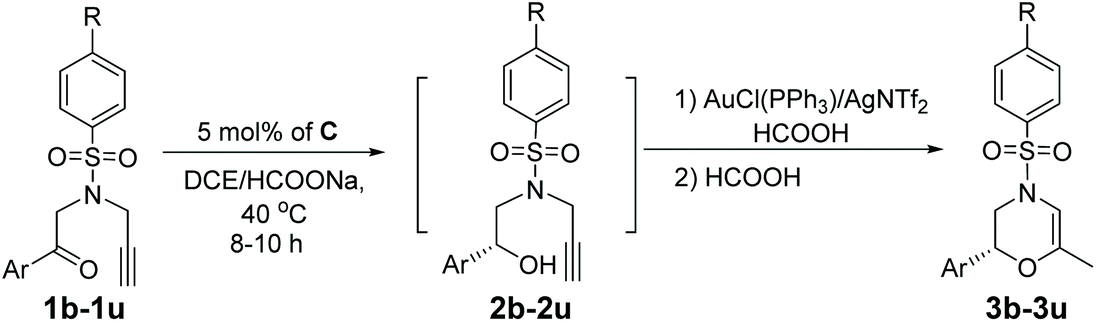
Based on our catalytic results and previous report,5,10 a plausible mechanism for the cyclization step was proposed using 2a as the model (Scheme 3) (for the proposed ATH mechanism of 1a, please see the ESI†). The cationic gold species firstly coordinate with the alkyne side of the alkynol to provide the gold η2-complex I. The following intramolecular cyclization of I provides the vinyl gold carbene II that undergoes the deprotonation process to afford III. The following protonation of intermediate III gave rise to methylenemorpholine 3a′ that has been successfully isolated as shown in Fig. 1.15 The peaks at 4.42 ppm and 94.2 ppm demonstrated that an exocyclic methylene unit was involved in 3a′ (blue dot). Finally, the isomerization of 3a′ promoted by HCOOH afforded the corresponding 3a with excellent enantioselectivity retention.
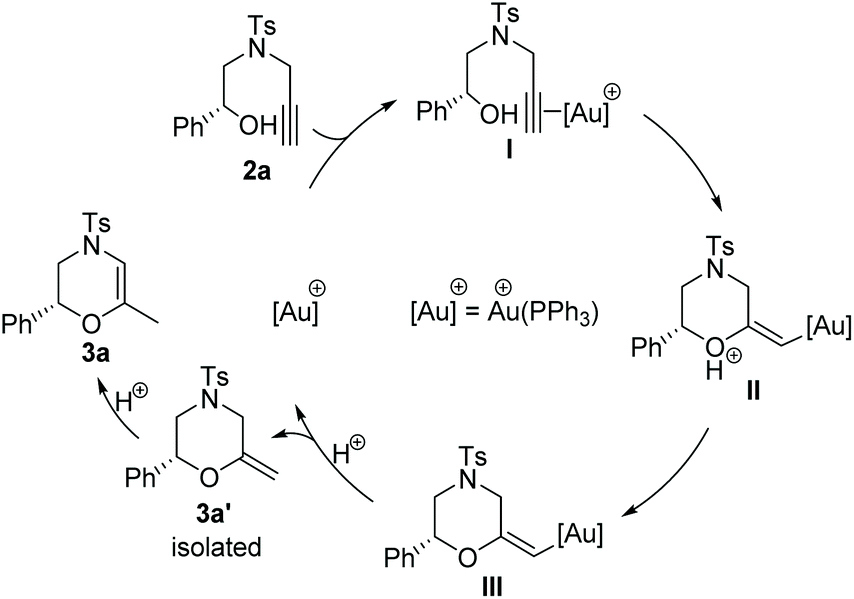 | ||
| Scheme 3 Proposed mechanism for the cyclization step of alkynols. | ||
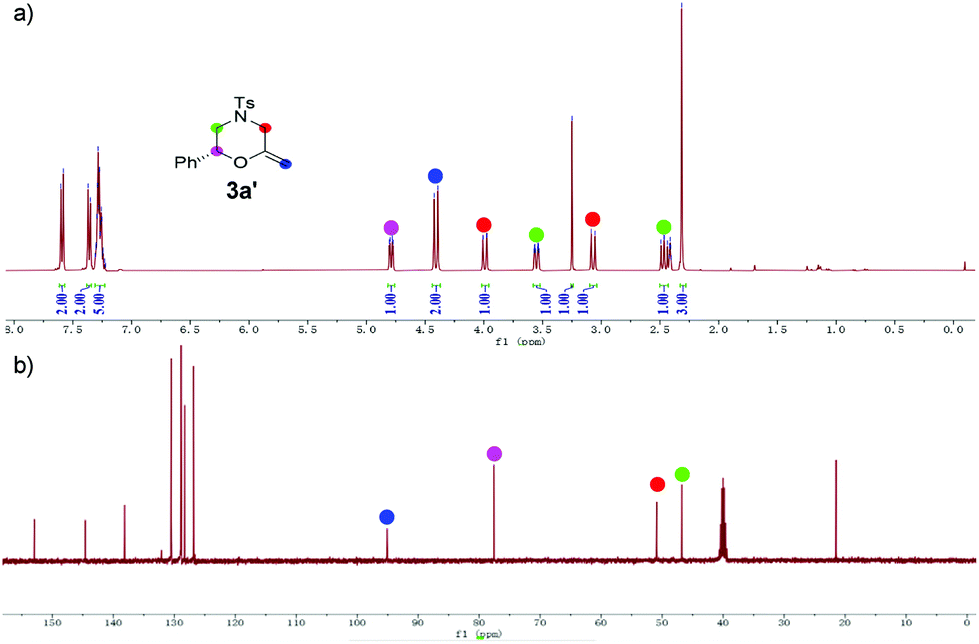 | ||
| Fig. 1 (a) 1HNMR spectra of 3a′; (b) 13C{1H}NMR spectra of 3a′. | ||
Besides the general feasibility, this ATH/cyclization enantio-relay process could be used as an efficient synthetic protocol for the gram-scale preparation. Taking the ATH/cyclization enantio-relay process of 1t as a representative, the desirable targeting product of (R)-4-((4-bromophenyl)sulfonyl)-6-methyl-2-phenyl-3,4-dihydro-2H-1,4-oxazine (3t) can be steadily obtained in the gram-scale synthesis without an obvious loss of yield (59%) and enantioselectivity (94% ee), as shown in Scheme 4. More importantly, as an important chiral core of morpholines, the obtained 3,4-dihydro-2H-1,4-oxazines could be conveniently transformed into the corresponding morpholine derivatives. In this case, this representative gram-scale 3t was able to react with H2O under acidic conditions, producing (2S,6R)-4-((4-bromophenyl)sulfonyl)-2-methyl-6-phenylmor pholin-2-ol (4) (CCDC: 2016445†) in 91% yield with 98% ee and 94/6 dr. This observation suggests a practical approach to access the morpholine derivatives with dual chiral centers.
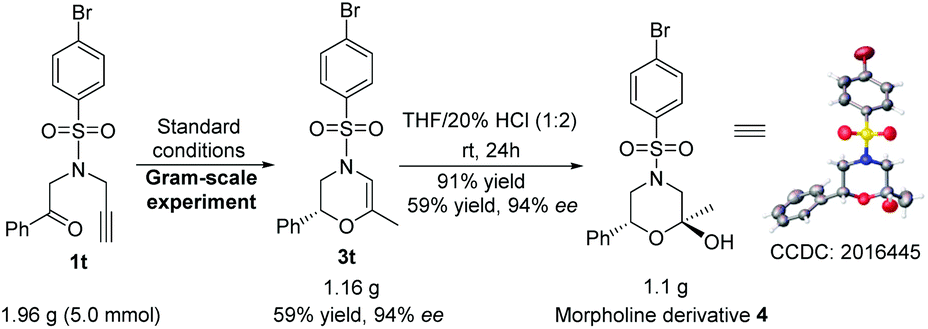 | ||
| Scheme 4 Gram-scale synthesis of 3t and 4 (CCDC: 2016445,† thermal ellipsoids are shown at 50% probability). | ||
Conclusions
In conclusion, using a one-pot ATH/cyclization enantio-relay strategy, we have realized the efficient enantioselective synthesis of 3,4-dihydro-2H-1,4-oxazines. A series of alkynones were proved to be good substrates for the presented sequential transformations, resulting in the corresponding chiral 3,4-dihydro-2H-1,4-oxazines in moderate yields and excellent enantioselectivities. Mechanistic studies suggested that the reaction intermediate methylenemorpholine firstly formed during the Au(I)-catalyzed cyclization process of alkynols in the presence of C/HCOOH/HCOONa. This approach not only integrates the advantages of both asymmetric transfer hydrogenation and 5-exo-dig cyclization of alkynols but also offers a practical methodology for the synthesis of optically pure morpholine derivatives.Experimental
Procedure for the synthesis of 2
Step I (ATH reaction of alkynone 1): Catalyst C (9.3 mg, 0.015 mmol) and HCOONa (81.6 mg, 1.20 mmol) were placed in a Schlenk tube. After filling the Schlenk tube with Ar, a solution of alkynone 1 (0.3 mmol) in DCE (2 mL) was injected. Then the resulting solution was stirred at 40 °C for 8–10 h. Once the consumption of 1 was complete (monitored by TLC), the reaction solution was washed with H2O (5 mL × 3) and brine (5 mL), and dried with Na2SO4. The crude residue was purified by flash column chromatography over silica gel.Procedure for the synthesis of 3
Step II (synthesis of 3,4-dihydro-2H-1,4-oxa zines 3): Catalyst C (9.3 mg, 0.015 mmol) and HCOONa (81.6 mg, 1.20 mmol) were placed in a Schlenk tube. After filling the Schlenk tube with Ar, a solution of alkynone 1 (0.3 mmol) in DCE (2 mL) was injected. Then the resulting solution was stirred at 40 °C for 8–10 h. Once the consumption of 1 was complete (monitored by TLC), HCOOH (22 μL, 0.6 mmol) was added. After 10 min, a solid mixture of AuCl(PPh3) (14.8 mg, 0.030 mmol) and AgNTf2 (11.6 mg, 0.030 mmol) was added. The resulting solution was maintained at 40 °C until the full conversion of alkynols. Finally, the reaction was quenched with HCOOH (11 μL, 0.3 mmol) for 15 min. The reaction solution was washed with H2O (5 mL × 3) and brine (5 mL), and dried with Na2SO4. The crude residue was purified by flash column chromatography over silica gel.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (22001170, 22071154 and 21872095), the Shanghai Sciences and Technologies Development Fund (20070502600) and the Shanghai Frontiers Science Research Base of Biomimetic Catalysis for financial support.Notes and references
- (a) S. Kato, T. Morie, T. Kon, N. Yoshida, T. Karasawa and J. Matsumoto, Novel benzamides as selective and potent gastrokinetic agents. 2. Synthesis and structure-activity relationships of 4-amino-5-chloro-2-ethoxy-N-[[4-(4-fluorobenzyl)-2-morpholinyl]methyl]benzamide citrate (AS-4370) and related compounds, J. Med. Chem., 1991, 34, 616 CrossRef CAS PubMed; (b) H. Berzewski, M. Van Moffaert and C. A. Gagiano, Efficacy and tolerability of reboxetine compared with imipramine in a double-blind study in patients suffering from major depressive episodes, Eur. Neuropsychopharmacol., 1997, 7, S37 CrossRef CAS PubMed; (c) J. Ong, D. I. B. Kerr, H. Bittiger, P. C. Waldmeier, P. A. Baumann, N. G. Cooke, S. J. Mickel and W. Froestl, Morpholin-2-yl-phosphinic acids are potent GABAB receptor antagonists in rat brain, Eur. J. Pharmacol., 1998, 362, 27 CrossRef CAS; (d) E. H. Wong, M. S. Sonders, S. G. Amara, P. M. Tinholt, M. F. Piercey, W. P. Hoffmann, D. K. Hyslop, S. Franklin, R. D. Porsolt, A. Bonsignori, N. Carfagna and R. A. McArthur, Reboxetine: a pharmacologically potent, selective, and specific norepinephrine reuptake inhibitor, Biol. Psychiatry, 2000, 47, 818 CrossRef CAS; (e) M. J. Cases-Thomas, J. J. Masters, M. W. Walter, G. Campbell, L. Haughton, P. T. Gallagher, D. R. Dobson, V. Mancuso, B. Bonnier, T. Giard, T. Defrance, M. Vanmarsenille, A. Ledgard, C. White, S. Ouwerkerk-Mahadevan, F. J. Brunelle, N. A. Dezutter, C. A. Herbots, J. Y. Lienard, J. Findlay, L. Hayhurst, J. Boot, L. K. Thompson and S. Hemrick-Luecke, Discovery of novel and selective tertiary alcohol containing inhibitors of the norepinephrine transporter, Bioorg. Med. Chem. Lett., 2006, 16, 2022 CrossRef CAS PubMed; (f) W. Xu, D. L. Gray, S. A. Glase and N. S. Barta, Design and synthesis of reboxetine analogs morpholine derivatives as selective norepinephrine reuptake inhibitors, Bioorg. Med. Chem. Lett., 2008, 18, 5550 CrossRef CAS PubMed.
- (a) E. Bouron, G. Goussard, C. Marchand, M. Bonin, X. Panneconcke, J.-C. Quirion and H.-P. Husson, Stereoselective synthesis of 2,6-disubstituted morpholines from chiral non-racemic lactams, Tetrahedron Lett., 1999, 40, 7227 CrossRef CAS; (b) B. A. Lanman and A. G. Myers, Efficient, Stereoselective synthesis of trans-2,5-disubstituted morpholines, Org. Lett., 2004, 6, 1045 CrossRef CAS PubMed; (c) V. Lupi, D. Albanese, D. Landini, D. Scaletti and M. Penso, Synthesis of 2,6-disubstituted morpholines through regioselective oxiranes ring opening by tosylamide under PTC conditions, Tetrahedron, 2004, 60, 11709 CrossRef CAS; (d) R. Pedrosa, C. Andrés, P. Mendiguchía and J. Nieto, Diastereoselective synthesis of enantiopure morpholines by electrophilic selenium-induced 6-exo cyclizations on chiral 3-allyl-2-hydroxymethylperhydro-1,3-benzoxazine derivatives, J. Org. Chem., 2006, 71, 8854 CrossRef CAS PubMed; (e) D. Albanese, M. Salsa, D. Landini, V. Lupi and M. Penso, Concise synthesis of 2,6-disubstituted morpholines by cyclization of epoxy alcohols, Eur. J. Org. Chem., 2007, 2107 CrossRef CAS; (f) M. Breuning, M. Winnacker and M. Steiner, Efficient one-pot synthesis of enantiomerically pure 2-(hydroxymethyl)morpholines, Eur. J. Org. Chem., 2007, 2100 CrossRef CAS; (g) M. Penso, V. Lupi, D. Albanese, F. Foschi, D. Landini and A. Tagliabue, A straightforward synthesis of enantiopure 2,6-disubstituted morpholines by a regioselective o-protection/activation protocol, Synlett, 2008, 2451 CrossRef CAS; (h) M. L. Leathen, B. R. Rosen and J. P. Wolfe, New strategy for the synthesis of substituted morpholines, J. Org. Chem., 2009, 74, 5107 CrossRef CAS; (i) J. Nonnenmacher, F. Grellepois and C. Portella, Synthesis of enantiopure 2-aryl(alkyl)-2-trifluoromethyl-substituted morpholines and oxazepanes, Eur. J. Org. Chem., 2009, 3726 CrossRef CAS; (j) J. Bornholdt, J. Felding and J. L. Kristensen, Synthesis of enantiopure 3-substituted morpholines, J. Org. Chem., 2010, 75, 7454 CrossRef CAS PubMed; (k) B. Ritzen, S. Hoekman, E. D. Verdasco, F. L. van Delft and P. J. T. Rutjes, Enantioselective chemoenzymatic synthesis of cis- and trans-2,5-disubstituted morpholines, J. Org. Chem., 2010, 75, 3461 CrossRef CAS; (l) L. Bagnoli, C. Scarponi, M. G. Rossi, L. Testaferri and M. Tiecco, Synthesis of enantiopure 1,4-dioxanes, morpholines, and piperazines from the reaction of chiral 1,2-diols, amino alcohols, and diamines with vinyl selenones, Chem. – Eur. J., 2011, 17, 993 CrossRef CAS PubMed; (m) B. Alcaide, P. Almendros, R. Carrascosa, L. Casarrubios and E. Soriano, A versatile synthesis of β-lactam-fused oxacycles through the palladium-catalyzed chemo-, regio-, and diastereoselective cyclization of allenic diols, Chem. – Eur. J., 2015, 21, 2200 CrossRef CAS; (n) F. Foschi, D. Albanese, I. Pecnikaj, A. Tagliabue and M. Penso, Regioselective O-sulfonylation of N,N-bis(2-hydroxyalkyl)tosylamides as a synthetic key step to enantiopure morpholines, Org. Lett., 2017, 19, 70 CrossRef CAS.
- (a) J. E. Baldwin, Rules for ring closure, J. Chem. Soc., Chem. Commun., 1976, 734 RSC; (b) Z. Wang, X. Lin, R. L. Luck, G. Gibbons and S. Fang, Synthesis of cyclic allyl vinyl ethers using Pt(II)-catalyzed isomerization of oxo-alkynes, Tetrahedron, 2009, 65, 2643 CrossRef CAS; (c) M. Maheswara, Y. Y. Lee and E. J. Kang, AgOTf-catalyzed cycloisomerization of alkynyl oxiranes for dihydro-1,4-oxazine synthesis, Synlett, 2012, 2481 CAS; (d) F. Cambeiro, S. Lopez, J. A. Varela and C. Saa, Vinyl dihydropyrans and dihydrooxazines: cyclizations of catalytic ruthenium carbenes derived from alkynals and alkynones, Angew. Chem., Int. Ed., 2014, 53, 5959 CrossRef CAS; (e) A. L. S. Kumari, A. S. Reddy and K. C. K. Swamy, Transition metal-free cascade cyclization of epoxy-ynamides: To go for 1,3-oxazines or 1,4-oxazines?, Org. Lett., 2016, 18, 5752 CrossRef; (f) M. Anitha, M. Shankar and K. C. K. Swamy, Reactivity of epoxy-ynamides with metal halides: nucleophile (Br/Cl/OH)-assisted tandem intramolecular 5-exo-dig or 6-endo-dig cyclisation and AgF2-promoted oxidation, Org. Chem. Front., 2019, 6, 1133 RSC.
- J. K. Vandavasi, W.-P. Hu, H.-Y. Chen, G. C. Senadi, C.-Y. Chen and J.-J. Wang, A new approach to 1,4-oxazines and 1,4-oxazepines via base-promoted exo mode cyclization of alkynyl alcohols: mechanism and DFT studies, Org. Lett., 2012, 14, 3134 CrossRef CAS PubMed.
- L.-F. Yao, Y. Wang and K.-W. Huang, Synthesis of morpholine or piperazine derivatives through gold-catalyzed cyclization reactions of alkynylamines or alkynylalcohols, Org. Chem. Front., 2015, 2, 721 RSC.
- J. Barluenga, A. Fernández, A. Satrústegui, A. Diéguez, F. Rodríguez and F. J. Fañanás, Tandem intramolecular hydroalkoxylation–hydroarylation reactions: synthesis of enantiopure benzofused cyclic ethers from the chiral pool, Chem. – Eur. J., 2008, 14, 4153 CrossRef.
- (a) F. Glorius, Asymmetric hydrogenation of aromatic compounds, Org. Biomol. Chem., 2005, 3, 4171 RSC; (b) S. Gladiali and E. Alberico, Asymmetric transfer hydrogenation: chiral ligands and applications, Chem. Soc. Rev., 2006, 35, 226 RSC; (c) R. H. Morris, Asymmetric hydrogenation, transfer hydrogenation and hydrosilylation of ketones catalyzed by iron complexes, Chem. Soc. Rev., 2009, 38, 2282 RSC; (d) M. Yoon, R. Srirambalaji and K. Kim, Homochiral metal–organic frameworks for asymmetric heterogeneous catalysis, Chem. Rev., 2012, 112, 1196 CrossRef CAS; (e) C. Zheng and S.-L. You, Transfer hydrogenation with Hantzsch esters and related organic hydride donors, Chem. Soc. Rev., 2012, 41, 2498 RSC; (f) D. Wang and D. Astruc, The Golden Age of Transfer Hydrogenation, Chem. Rev., 2015, 115, 6621 CrossRef CAS; (g) C. S. G. Seo and R. H. Morris, Catalytic homogeneous asymmetric hydrogenation: successes and opportunities, Organometallics, 2019, 38, 47 CrossRef CAS; (h) S. E. Sloane, A. Reyes, Z. P. Vang, L. Li, K. T. Behlow and J. R. Clark, Copper-Catalyzed Formal Transfer Hydrogenation/Deuteration of Aryl Alkynes, Org. Lett., 2020, 22, 9139 CrossRef CAS PubMed; (i) N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, Asymmetric transfer hydrogenation of imines, J. Am. Chem. Soc., 1996, 118, 4916 CrossRef CAS; (j) S. Hoffmann, A. M. Seayad and B. List, A powerful brønsted acid catalyst for the organocatalytic asymmetric transfer hydrogenation of imines, Angew. Chem., Int. Ed., 2005, 44, 7424 CrossRef CAS PubMed; (k) N. Fleury-Bregeot, V. de la Fuente, S. Castillon and C. Claver, Highlights of transition metal-catalyzed asymmetric hydrogenation of imines, ChemCatChem, 2010, 2, 1346 CrossRef CAS; (l) W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Amine(imine)diphosphine iron catalysts for asymmetric transfer hydrogenation of ketones and imines, Science, 2013, 342, 1080 CrossRef CAS PubMed; (m) Y.-Y. Li, S.-L. Yu, W.-Y. Shen and J.-X. Gao, Iron-, Cobalt-, and Nickel-catalyzed asymmetric transfer hydrogenation and asymmetric hydrogenation of ketones, Acc. Chem. Res., 2015, 48, 2587 CrossRef CAS.
- (a) T. Cheng, Q. Ye, Q. Zhao and G. Liu, Dynamic kinetic resolution of phthalides via asymmetric transfer hydrogenation: a strategy constructs 1,3-distereocentered 3-(2-hydroxy-2-arylethyl)isobenzofuran-1(3H)-one, Org. Lett., 2015, 17, 4972 CrossRef CAS; (b) L. Wu, R. Jin, L. Li, X. Hu, T. Cheng and G. Liu, A michael addition-asymmetric transfer hydrogenation one-pot enantioselective tandem process for syntheses of chiral γ-secondary amino alcohols, Org. Lett., 2017, 19, 3047 CrossRef CAS; (c) D. Zhang, T. Cheng, Q. Zhao, J. Xu and G. Liu, Highly enantioselective one-pot synthesis of chiral β-hydroxy sulfones via asymmetric transfer hydrogenation in an aqueous medium, Org. Lett., 2014, 16, 5764 CrossRef CAS.
- (a) W. Fu, M. Nie, A. Wang, Z. Cao and W. Tang, Highly enantioselective nickel-catalyzed intramolecular reductive cyclization of alkynones, Angew. Chem., Int. Ed., 2015, 54, 2520 CrossRef CAS PubMed; (b) J. Chen, X. Han and X. Lu, Palladium(II)-catalyzed asymmetric tandem cyclization of 2-aminoaryl alkynones: an approach to chiral 1,2,3,4-tetrahydro-β-carbolines, Org. Lett., 2018, 20, 7470 CrossRef CAS.
- (a) B. Alcaide, P. Almendros and J. M. Alonso, Gold-catalyzed cyclizations of alkynol-based compounds: synthesis of natural products and derivatives, Molecules, 2011, 16, 7815 CrossRef CAS; (b) R. Dorel and A. M. Echavarren, Gold(I)-catalyzed activation of alkynes for the construction of molecular complexity, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed; (c) B. V. S. Reddy, P. N. Nair, A. Antony and N. Srivastava, Recent advances in prins spirocyclization, Eur. J. Org. Chem., 2017, 5484 CrossRef.
- (a) B. M. Trost, Atom economy—a challenge for organic synthesis: homogeneous catalysis leads the way, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS; (b) R. A. Sheldon, Atom efficiency and catalysis in organic synthesis, Pure Appl. Chem., 2000, 72, 1233 CAS; (c) C. M. R. Volla, I. Atodiresei and M. Rueping, Catalytic C–C bond-forming multi-component cascade or domino reactions: pushing the boundaries of complexity in asymmetric organocatalysis, Chem. Rev., 2014, 114, 2390 CrossRef CAS.
- (a) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, Asymmetric transfer hydrogenation of aromatic ketones catalyzed by chiral ruthenium(II) complexes, J. Am. Chem. Soc., 1995, 117, 7562 CrossRef CAS; (b) R. Noyori and S. Hashiguchi, Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS; (c) A. M. Hayes, D. J. Morris, G. J. Clarkson and M. Wills, A class of ruthenium(II) catalyst for asymmetric transfer hydrogenations of ketones, J. Am. Chem. Soc., 2005, 127, 7318 CrossRef CAS; (d) X. Wu, J. Liu, D. Di Tommaso, J. A. Iggo, C. R. A. Catlow, J. Bacsa and J. Xiao, A multilateral mechanistic study into asymmetric transfer hydrogenation in water, Chem. – Eur. J., 2008, 14, 7699 CrossRef CAS; (e) J. E. D. Martins, G. J. Clarkson and M. Wills, Ru(II) complexes of N-alkylated TsDPEN ligands in asymmetric transfer hydrogenation of ketones and imines, Org. Lett., 2009, 11, 847 CrossRef CAS PubMed; (f) Z. Fang and M. Wills, Asymmetric Transfer Hydrogenation of Functionalized Acetylenic Ketones, J. Org. Chem., 2013, 78, 8594 CrossRef CAS PubMed.
- (a) G. Liu, J. Wang, T. Huang, X. Liang, Y. Zhang and H. Li, Mesoporous silica-supported iridium catalysts for asymmetric hydrogenation reactions, J. Mater. Chem., 2010, 20, 1970 RSC; (b) B. Deng, W. Xiao, C. Li, F. Zhou, X. Xia, T. Cheng and G. Liu, Imidazolium-based organoiridium-functionalized periodic mesoporous organosilica boosts enantioselective reduction of α-cyanoacetophenones, α-nitroacetophenones, and β-ketoesters, J. Catal., 2014, 320, 70 CrossRef CAS.
- 3a′ is highly sensitive to acid and even SiO2; it should be quickly isolated by flash chromatography over neutral aluminum oxide using an ice-cooled eluent. The NMR spectra should be performed immediately after separation under the protection of Ar. (3a′: a colorless oil, Rf = 0.3 (PE/TBME 8/1). 1H NMR (400 MHz, DMSO-d6) δ 7.59 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 7.31–7.23 (m, 5H), 4.79 (dd, J = 10.2, 2.9 Hz, 1H), 4.41 (d, J = 12.3 Hz, 2H), 3.99 (dd, J = 13.0, 1.6 Hz, 1H), 3.55 (ddd, J = 12.0, 3.0, 1.6 Hz, 1H), 3.25 (s, 1H), 3.07 (d, J = 13.0 Hz, 1H), 2.46 (dd, J = 12.0, 10.2 Hz, 1H), 2.32 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 152.9, 144.6, 138.1, 130.5, 128.9, 128.8, 128.3, 126.8, 96.3, 76.7, 49.7, 45.4, 21.5). Notably, we failed to determine the ee of 3a’ due to its high instability in iPrOH.
- Although there is no direct evidence, the control experiments suggested that the in situ formed ruthenium hydride intermediate might be beneficial for the protonation process of III (please see the ESI†).
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2016445. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo01482j |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2022 |