
Electrochemical synthesis of functionalized gem-difluoroalkenes with diverse alkyl sources via a defluorinative alkylation process†
Haoxiang
Zhang
a,
Mengze
Liang
a,
Xiao
Zhang
a,
Meng-Ke
He
b,
Chao
Yang
 a,
Lin
Guo
*a and
Wujiong
Xia
*ac
a,
Lin
Guo
*a and
Wujiong
Xia
*ac
aState Key Lab of Urban Water Resource and Environment, School of Science, Harbin Institute of Technology (Shenzhen), Shenzhen, 518055, China. E-mail: guolin@hit.edu.cn; xiawj@hit.edu.cn
bWenzhou University, Wenzhou, Zhejiang 325000, China
cSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, China
First published on 3rd November 2021
Abstract
An electrochemical defluorinative alkylation protocol of α-trifluoromethyl alkenes is described. This reaction enables the preparation of functionalized gem-difluoroalkenes with the use of diverse alkyl sources including organohalides, N-hydroxyphthalimide (NHP) esters and Katritzky salts. This method exhibits lots of synthetic advantages including mild conditions, simple operation, and convenience of amplification, and provides a new route for the synthesis of gem-difluoroalkenes.
Introduction
Gem-difluoroalkenes are privileged structural motifs present in many bioactive molecules and drug candidates. This fascinating functionality features unique properties that affect the metabolic stability, absorbability and lipophilicity of organic molecules.1 The strong electronegative nature of fluorine and the chemical reactivity of the gem-difluorovinyl moiety make gem-difluoroalkenes formidable electrophiles that act as irreversible inhibitors of specific enzymes.2 In addition, as gem-difluoroalkenes are bioisosteres of carbonyl groups (aldehydes and ketones), they can mimic the chemical reactivity of a carbonyl group, providing new opportunities in the drug discovery pipeline.3Fig. 1 shows some representative organic molecules containing the gem-difluorovinyl scaffold with various pharmacological activities, such as antiepilepsy and antitubulin.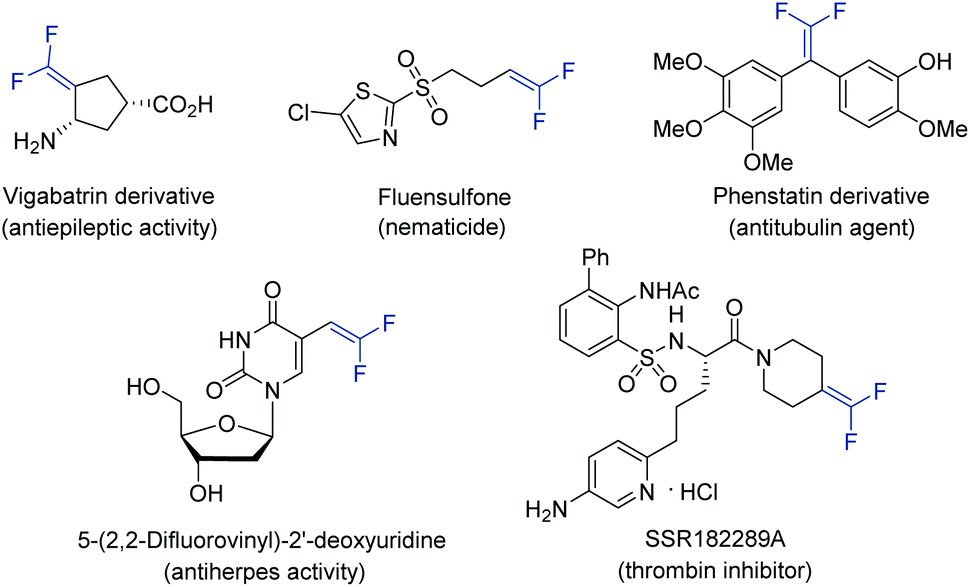 | ||
| Fig. 1 Representative applications of gem-difluoroalkenes in medicinal chemistry. | ||
During the past few decades, chemists have synthesized a series of structurally diverse gem-difluoroalkenes by a variety of simple and efficient methods. In addition to the direct conversion of carbonyl compounds into the corresponding gem-difluoroalkenes via the Wittig, Horner–Wadsworth–Emmons or Julia–Kocienski reactions,4 more common synthetic routes to these valuable compounds are through a defluorinative coupling reaction of α-trifluoromethyl alkenes owing to the ready availability of substrates as well as its high efficiency. Traditionally, gem-difluoroalkenes are prepared by a SN2′-type reaction involving highly reactive and hazardous organometallic reagents, such as organolithiums, Grignard reagents, N-lithiated amines, ester enolates, and silyl lithium reagents (Scheme 1a).5 Despite their high efficiency in certain instances, many of these protocols suffer from narrow substrate scope concerning structural diversity or functional group tolerance. In order to overcome these disadvantages, great efforts have been made to develop efficient methods for the construction of the gem-difluoroethylene motif, and two major synthetic strategies have been established. On one hand, visible light photoredox catalysis as an eco-friendly synthetic approach has provided an array of protocols for the radical defluorinative cross-coupling reaction under user-friendly conditions, with the aid of visible light-excitable metal complexes or conjugated organic molecules (Scheme 1b).6 On the other hand, considerable progress has been made in the field of transition metal (mainly nickel) catalyzed defluorinative reductive cross-coupling with organohalides and prefunctionalized backbones, affording diverse gem-difluoroalkenes bearing sensitive functional groups with high efficiency (Scheme 1c).7
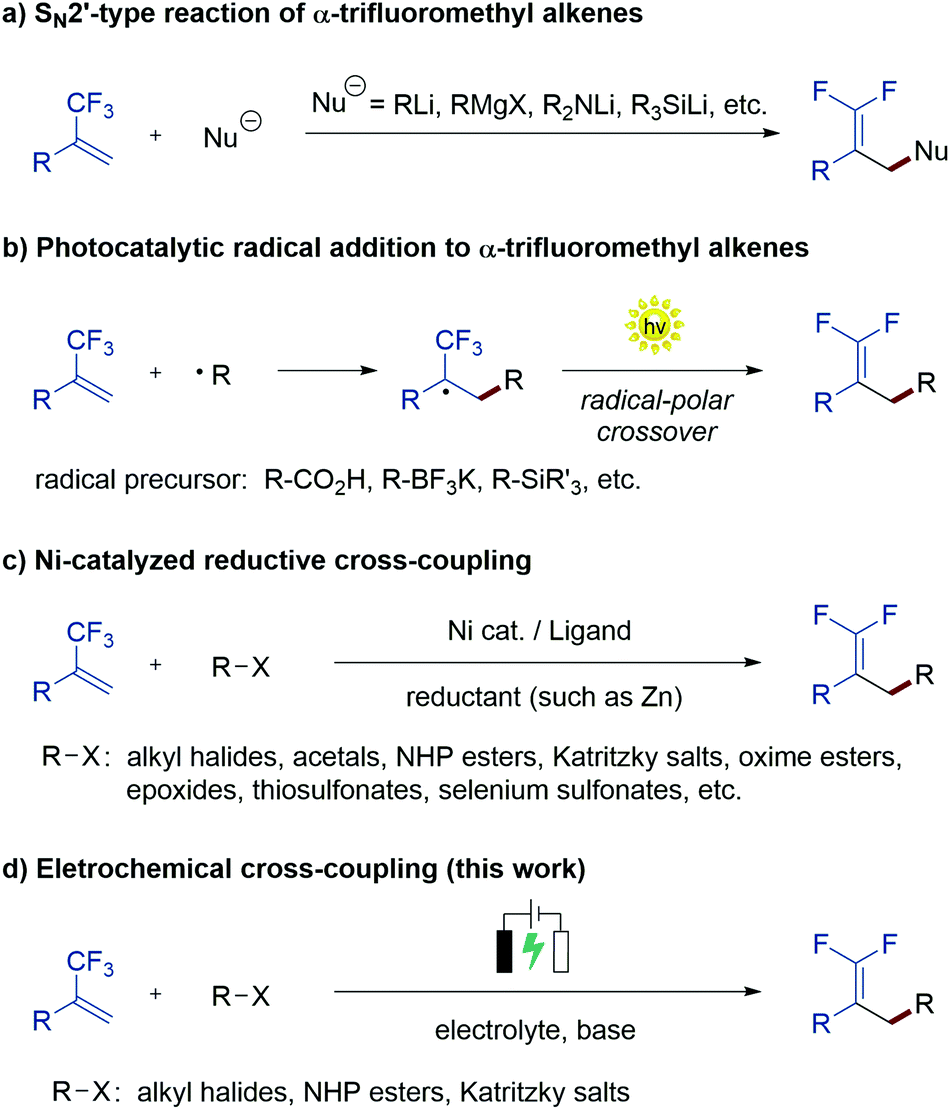 | ||
| Scheme 1 Synthesis of gem-difluoroalkenes from α-trifluoromethyl alkenes. | ||
As a widely acknowledged sustainable synthetic methodology, the reviving organic electrochemistry has recently emerged as a powerful synthetic tool that enables a vast variety of mild reductive transformations, which can be readily achieved via direct electrolysis without the requirement of catalysts or reducing agents.8 Driven by the prevalence of the gem-difluorovinyl moiety in a myriad of biologically relevant molecules, we envisioned that the exploration of a new and sustainable synthetic approach to gem-difluoroalkenes from α-trifluoromethyl alkenes under electrolysis would be highly rewarding. To date, there are very few examples on the electrochemical synthesis of gem-difluoroalkenes, although recently, Zhou and co-workers reported an electrochemical defluorinative carboxylation of α-trifluoromethyl alkenes with CO2,9a and Fu, Lu, and co-workers developed a nickel promoted electrochemical reductive cross-coupling to access functionalized gem-difluoroalkenes.9b While these works undoubtedly bring new space in this research field, these electrochemical strategies are still at their infancy in terms of diverse alkyl sources that can be introduced. As part of our ongoing interest in the electrochemical reactions,10 we reported herein a sustainable electrochemical system that allows defluorinative cross-coupling of α-trifluoromethyl alkenes to proceed smoothly (Scheme 1d). The successful coupling partners include organohalides (iodides and bromides) as well as simple prefunctionalized compounds such as N-hydroxyphthalimide (NHP) esters and Katritzky salts.
Results and discussion
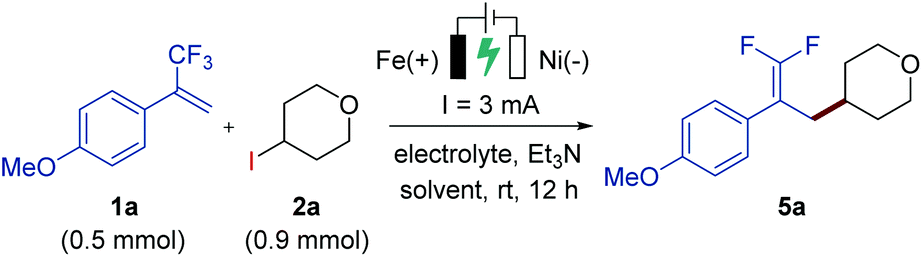
Our study began by the investigation of the reaction between trifluoromethyl-substituted alkene 1a and 4-iodotetrahydro-2H-pyran (2a) in a simple undivided cell equipped with an iron plate anode and a nickel plate cathode as the working electrodes (Table 1). After the initial evaluation of reaction parameters, we obtained a promising result when employing nBu4NPF6 as the supporting electrolyte and triethylamine as the base under 3 mA constant current electrolysis at room temperature, affording the desired gem-difluoroalkene product 5a in 32% yield (entry 1). Replacing nBu4NPF6 with nBu4NI as the supporting electrolyte under otherwise identical reaction conditions greatly improved the yield (entry 3), while the use of other electrolytes gave lower yields (entries 2 and 4) or no yield (entry 5). Further optimizations were conducted by screening a variety of reaction solvents, such as DMA, ethanol, acetonitrile, and a mixed solvent of THF/CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2.5 mL/2.5 mL), but they all provided unsatisfactory results (entries 6–9, respectively). Moreover, changing the reaction time to 6 h or 14 h reduced the reaction efficiency (entries 10 and 11). During the optimization studies, we found that the concentration of the electrolyte was critical: a low concentration of nBu4NI (0.013 M) improved the yield to 78% (with 76% isolated yield), but a high concentration (0.1 M or 0.2 M) greatly reduced the efficiency (entries 12–14). As anticipated, the control experiment revealed that electric current was critical for the reaction (entry 15).
:1, 2.5 mL/2.5 mL), but they all provided unsatisfactory results (entries 6–9, respectively). Moreover, changing the reaction time to 6 h or 14 h reduced the reaction efficiency (entries 10 and 11). During the optimization studies, we found that the concentration of the electrolyte was critical: a low concentration of nBu4NI (0.013 M) improved the yield to 78% (with 76% isolated yield), but a high concentration (0.1 M or 0.2 M) greatly reduced the efficiency (entries 12–14). As anticipated, the control experiment revealed that electric current was critical for the reaction (entry 15).
|
|
||||
|---|---|---|---|---|
| Entry | Electrolyte | Solvent | Time (h) | 5a (%) |
| a Standard conditions: iron plate anode (10 × 10 × 0.15, mm), Pt plate cathode (10 × 10 × 0.15, mm), 1a (0.5 mmol), 2a (0.9 mmol, 1.8 equiv.), Et3N (1.0 mmol, 2.0 equiv.), electrolyte (0.1 mmol, 0.02 M), anhydrous DMF (5.0 mL), constant current = 3 mA under air at room temperature for 12 h (2.7 F mol−1). b Yield determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. c nBu4NI (0.50 mmol, 0.1 M). d nBu4NI (1.0 mmol, 0.2 M). e nBu4NI (0.13 mmol, 0.013 M) in DMF (10 mL). f Yield of the isolated product. g Reaction was performed without constant current. N.R.: no reaction. | ||||
| 1 | nBu4NPF6 | DMF | 12 | 32 |
| 2 | nBu4NOAc | DMF | 12 | 17 |
| 3 | nBu4NI | DMF | 12 | 60 |
| 4 | nBu4NBF4 | DMF | 12 | 29 |
| 5 | NaIO4 | DMF | 12 | N.R. |
| 6 | nBu4NI | DMA | 12 | 37 |
| 7 | nBu4NI | EtOH | 12 | Trace |
| 8 | nBu4NI | CH3CN | 12 | Trace |
| 9 | nBu4NI | THF/CH3CN | 12 | 20 |
| 10 | nBu4NI | DMF | 6 | 26 |
| 11 | nBu4NI | DMF | 14 | 45 |
| 12c | nBu4NI | DMF | 12 | 47 |
| 13d | nBu4NI | DMF | 12 | 22 |
| 14 | nBu 4 NI | DMF | 12 | 78 (76) |
| 15g | nBu4NI | DMF | 12 | N.R. |
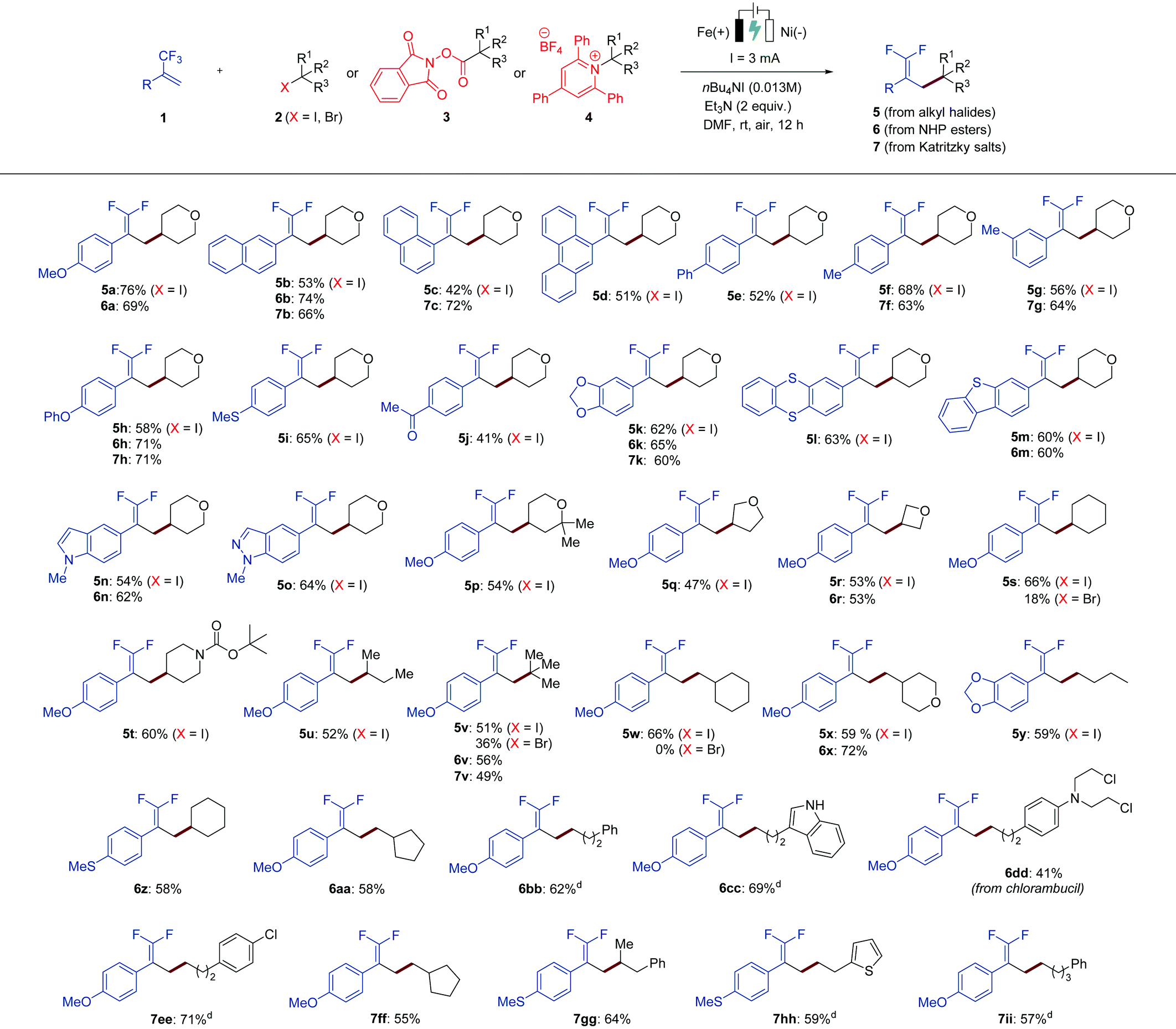
With the optimized conditions in hand, we next sought to examine the scope of this electrochemical defluorinative cross-coupling reaction. At the outset, a variety of α-trifluoromethyl alkenes were examined with 4-iodotetrahydro-2H-pyran (2a) under the standard conditions, affording the desired gem-difluoroalkene products in good to moderate yields (Table 2, 5a–5o). We found that not only the benzene ring system can be applied to this protocol, but also naphthalene and anthracene were well compatible (5b–5d). Next, different substitution patterns on the benzene ring of trifluoromethyl-substituted alkenes were studied, all of which selectively gave the desired gem-difluoro olefins in decent yields. For example, substrates with phenyl (5e), methyl (5f and 5g), ether (5h), thioether (5i), ketone (5j), and dioxole (5k) at different positions on the aromatic ring were tolerated well under the optimized conditions. It is worth noting that α-trifluoromethyl alkenes bearing heterocycles, such as thianthrene (5l), dibenzothiophene (5m), indole (5n), and indazole (5o), were also effectively transformed into the corresponding products. The scope with respect to the analogues of organohalides (2) as the coupling partner was further explored. Herein, alkyl iodides were mainly tested, as iodides exhibit a less negative redox potential in comparison with the corresponding alkyl bromides.11 Secondary alkyl iodides were found to be suitable substrates for this reaction. Cyclic iodides with different ring sizes were all converted into the corresponding gem-difluoroalkenes (5p–5t) in good to moderate yields. Iodides bearing acyclic hydrocarbons also worked well (5u). In addition, tertiary and primary aliphatic iodides could also be employed as suitable reaction partners, and again the formation of the corresponding products was observed (5v–5y). Notably, the reaction with respect to alkyl bromides as the coupling partner was further tested. Secondary and tertiary alkyl bromides were capable of proceeding the electrochemical defluorinative coupling with 1a to furnish the desired products with lower yields (5s and 5v), whereas the employment of a primary alkyl bromide gave no desired product (5w).
| a Reaction conditions: iron plate anode (10 × 10 × 0.15, mm), Pt plate cathode (10 × 10 × 0.15, mm), 1 (0.5 mmol), 2 (0.9 mmol, 1.8 equiv.), nBu4NI (0.13 mmol, 0.013 M), Et3N (1.0 mmol, 2.0 equiv.), anhydrous DMF (10.0 mL), constant current = 3 mA under air at room temperature for 12 h. b Reaction conditions: iron plate anode (10 × 10 × 0.15, mm), Pt plate cathode (10 × 10 × 0.15, mm), 1 (0.50 mmol), 3 or 4 (0.90 mmol, 1.8 equiv.), nBu4NI (0.13 mmol, 0.013 M), Et3N (1.0 mmol, 2.0 equiv.), anhydrous DMF (10.0 mL), constant current = 6 mA under air at room temperature for 12 h. c Yield of the isolated product. d Reaction time: 10 h. |
|---|
|
To further demonstrate the compatibility and applicability of our protocol, we expanded the substrate scope to carboxylic acid derivatives instead of organohalides, because carboxylic acids are among the most prevalent organic molecules found in nature. They can be simply activated to form redox-active N-hydroxyphthalimide esters (NHP esters, 3), which is known in many other synthetic transformations,7g,12 then generate alkyl radicals via facile single-electron reduction, and undergo the designed electrochemical defluorinative reaction with trifluoromethyl-substituted alkenes (1). As shown in Table 2, using the NHP ester of tetrahydro-2H-pyran-4-carboxylic acid as the model substrate, several trifluoromethyl alkenes were evaluated under the optimized conditions. These results revealed that trifluoromethyl alkenes bearing different functional groups and heterocyclic functionalities could be successfully converted into the desired products (6a, 6b, 6h, 6k, 6m and 6n), affording similar yields in comparison tothe reactions using alkyl halides. Moreover, it could be applied to a variety of secondary (6r and 6z), tertiary (6v), and primary (6x and 6aa–6cc) aliphatic redox-active esters. Both cyclic and acyclic esters were suitable substrates in this transformation. Notably, carboxylic acid-containing medicinal compounds bearing multiple reactive functional groups, such as chlorambucil that has antineoplastic properties, provided the desired coupling product 6dd in 41% yield, which greatly highlights our methodology.
On the basis of our existing work, we envisaged that under the appropriate conditions, an electrochemical cross-coupling of trifluoromethyl alkenes with alkyl radicals generated from amine derivatives via a deaminative manner would facilitate the production of gem-difluoroalkenes. Such a sustainable synthetic pathway is highly attractive as amines, especially primary amines, are readily available in nature. Inspired by the works of Watson, Wang, and others,7h,13 we began to investigate the reaction of trifluoromethyl-substituted alkenes (1) with Katritzky salts 4 by electrolysis (Table 2). To our delight, all α-trifluoromethyl alkenes bearing various functional groups turned out to be suitable substrates for this electrochemical defluorinative cross-coupling under slightly amended reaction conditions, providing the desired gem-difluoroalkene products (7a–7b, 7f–7h, 7k) in acceptable yields. Additionally, diverse aliphatic Katritzky salts, including primary, secondary, and tertiary alkyl-substituted ones, were reacted with α-trifluoromethyl alkenes to furnish 7v and 7ee-7ii with ease. All these results in Table 2 have well demonstrated the broad applicability of our electrochemical method, which is suitable not only for a wide variety of α-trifluoromethyl alkenes, but also for diverse alkyl radical precursors including organohalides, N-hydroxyphthalimide (NHP) esters, and Katritzky salts.
In order to demonstrate the practicability of the developed electrochemical defluorinative cross-coupling, a gram-scale experiment of 1a and 2a has been performed on a 4.5 mmol scale, and the corresponding gem-difluoroalkene product 5a was isolated in 67% yield (Scheme 2a). To further evaluate this electrochemical process, a series of cyclic voltammetry (CV) experiments were carried out. The Ep values of 4-iodotetrahydro-2H-pyran, NHP ester and Katritzky salt were measured and determined to be −2.58 V, −1.35 V, and −1.26 V, respectively (see the ESI† for details), which indicated that all of these substrates are likely to undergo cathodic reduction to produce the corresponding aliphatic carbon-centered radicals. Moreover, a divided-cell experiment was later carried out in order to gain more insight into the reaction mechanism. Substrates 1a and 2a as well as triethylamine were placed into the cathode chamber with an iron plate anode and a nickel plate cathode as the working electrodes. As expected, the desired product 5a was detected in the cathode chamber with 71% isolated yield (Scheme 2b), suggesting that the electrochemical defluorinative reaction occurred around the nickel plate cathode, and the iron plate anode was used as a sacrificial anode.
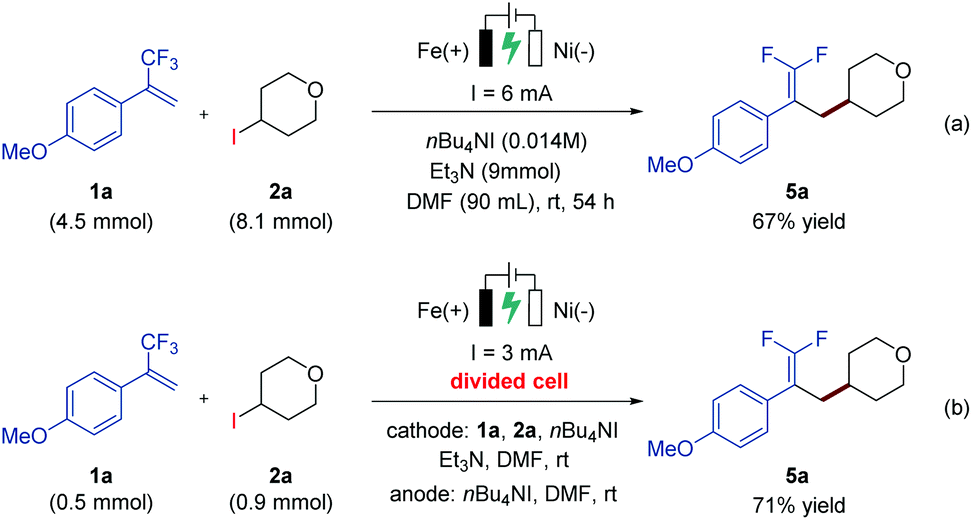 | ||
| Scheme 2 Gram-scale synthesis and divided-cell experiment. | ||
On the basis of the above experimental results and previous literature, a plausible mechanism for this electrochemical defluorinative cross-coupling reaction was proposed, as depicted in Scheme 3. Substrate 2 (including organohalides, NHP esters, and Katritzky salts) undergoes a single electron transfer (SET) process by cathodic electrolysis to provide a crucial aliphatic carbon-centered radical intermediate A, which further reacts with α-trifluoromethyl alkene 1 to form an α-CF3 carbon radical B. Cathodic reduction of this radical furnishes α-CF3 carbanion C. Finally, a β-fluoride elimination process of intermediate C affords the gem-difluoroalkene 5 as the desired product.
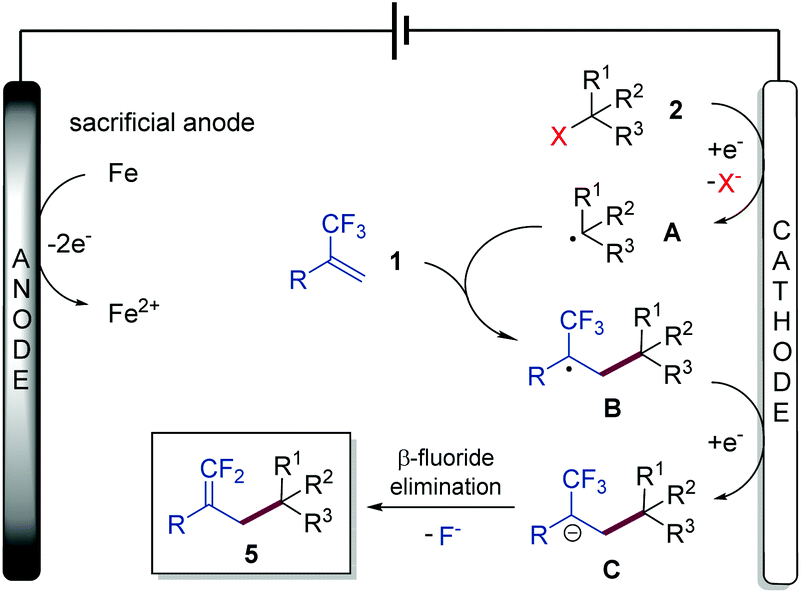 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
We have developed a highly efficient, green, and concise system for the electrochemical synthesis of diverse gem-difluoroalkenes. This protocol is applicable not only to a wide variety of α-trifluoromethyl alkenes, but also to different kinds of alkyl radical precursors such as organohalides, N-hydroxyphthalimide (NHP) esters, and Katritzky salts. This method has many significant advantages including mild conditions, high reaction efficiency, low cost, and convenience of purification and amplification. We believe that this sustainable, convenient and environmentally friendly system will find wide application in high value-added transformations, and provide novel inspiration for late-stage functionalization of pharmaceutical intermediates. Further development of new electrochemical reactions is underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (no. 21672047 and 22101066), the State Key Laboratory of Urban Water Resource and Environment (no. 2018DX02), the Science and Technology Plan of Shenzhen (JCYJ20210324133001004), and the Natural Science Foundation of Guangdong (no. 2020A1515010564). W. X. is grateful for the Talent Plan of the Pearl River in Guangdong, a starting up fund from the Shenzhen Government and the financial support from the Guangdong Province Covid-19 Pandemic Control Research Fund (no. 2020KZDZX1218). The project was also supported by the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University.Notes and references
- (a) Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, Wiley-Blackwell, West Sussex, 2009 Search PubMed; (b) X. Zhang and S. Cao, Recent advances in the synthesis and C-F functionalization of gem-difluoroalkenes, Tetrahedron Lett., 2017, 58, 375–392 CrossRef CAS; (c) C. Ni and J. Hu, Recent Advances in the Synthetic Application of Difluorocarbene, Synthesis, 2014, 842–863 Search PubMed; (d) Y. Pan, J. Qiu and R. B. Silverman, Design, Synthesis, and Biological Activity of a Difluoro-Substituted, Conformationally Rigid Vigabatrin Analogue as a Potent γ-Aminobutyric Acid Aminotransferase Inhibitor, J. Med. Chem., 2003, 46, 5292–5293 CrossRef CAS PubMed; (e) J.-M. Altenburger, G. Y. Lassalle, M. Matrougui, D. Galtier, J.-C. Jetha, Z. Bocsker, C. N. Berry, C. Lunven, J. Lorrain, J.-P. Herault, P. Schaeffer, S. E. O'Connor and J.-M. Herbert, SSR182289A, a selective and potent orally active thrombin inhibitor, Bioorg. Med. Chem., 2004, 12, 1713–1730 CrossRef CAS.
- (a) M. Bobek, I. Kavai and E. De Clercq, Synthesis and biological activity of 5-(2,2-difluorovinyl)-2′-deoxyuridine, J. Med. Chem., 1987, 30, 1494–1497 CrossRef CAS PubMed; (b) Y.-C. C. Cheng, B. A. Domin, R. A. Sharma and M. Bobek, Antiviral Action and Cellular Toxicity of Four Thymidine Analogues: 5-Ethyl-, 5-Vinyl-, 5-Propyl-, and 5-Allyl-2′-Deoxyuridine, Antimicrob. Agents Chemother., 1976, 10, 119–122 CrossRef CAS PubMed; (c) J. Reefschlager, D. Barwolff and P. Langen, Acta Virol., 1984, 28, 282–286 CAS.
- (a) G. Magueur, B. Crousse, M. Ourévitch, D. Bonnet-Delpon and J.-P. Bégué, Fluoro-artemisinins: When a gem-difluoroethylene replaces a carbonyl group, J. Fluorine Chem., 2006, 127, 637–642 CrossRef CAS; (b) N. A. Meanwell, Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS; (c) C. Leriche, X. He, C.-W. T. Chang and H.-W. Liu, Reversal of the Apparent Regiospecificity of NAD(P)H-Dependent Hydride Transfer: The Properties of the Difluoromethylene Group, A Carbonyl Mimic, J. Am. Chem. Soc., 2003, 125, 6348–6349 CrossRef CAS.
- (a) J. Zheng, J. Cai, J.-H. Lin, Y. Guo and J.-C. Xiao, Synthesis and decarboxylative Wittig reaction of difluoromethylene phosphobetaine, Chem. Commun., 2013, 49, 7513–7515 RSC; (b) I. Nowak and M. Robins, New Methodology for the Deoxygenative Difluoromethylenation of Aldehydes and Ketones; Unexpected Formation of Tetrafluorocyclopropanes, Org. Lett., 2005, 7, 721–724 CrossRef CAS PubMed; (c) D. J. Burton, Z.-Y. Yang and W. Qiu, Fluorinated Ylides and Related Compounds, Chem. Rev., 1996, 96, 1641–1643 CrossRef CAS PubMed; (d) S. R. Piettre and L. Cabanas, Reinvestigation of the Wadsworth-Emmons reaction involving lithium difluoromethylenephosphonate, Tetrahedron Lett., 1996, 37, 5881–5884 CrossRef CAS; (e) Y. Zhao, W. Huang, L. Zhu and J. Hu, Difluoromethyl 2-Pyridyl Sulfone: A New gem-Difluoroolefination Reagent for Aldehydes and Ketones, Org. Lett., 2010, 12, 1444–1447 CrossRef CAS PubMed; (f) B. Gao, Y. Zhao and J. Hu, Difluoromethyl 2-pyridyl sulfone: a versatile carbonyl gem-difluoroolefination reagent, Org. Chem. Front., 2015, 2, 163–168 RSC.
- (a) H. M. Park, T. Uegaki, T. Konno, T. Ishihara and H. Yamanaka, Chemistry of fluorinated enamines. Novel reaction of trifluoromethylated enamine with grignard reagents, Tetrahedron Lett., 1999, 40, 2985–2988 CrossRef CAS; (b) H. Yamanaka, T. Uegaki, T. Ishihara, T. Kubota and J. T. Gupton, Chemistry of fluorinated vinamidinium salts: novel reactions of β-trifluoromethyl vinamidinium salt with Grignard reagents, J. Fluorine Chem., 1999, 97, 101–108 CrossRef CAS; (c) T. Kitazume and T. Ohnogi, Synthesis of α-Trifluoromethylated Carboxylic Acid Esters, Synthesis, 1988, 614–615 CrossRef CAS; (d) J. Ichikawa, H. Fukui and Y. Ishibashi, 1-Trifluoromethylvinylsilane as a CF2=C−−CH2+ Synthon: Synthesis of Functionalized 1,1-Difluoro-1-alkenes via Isolable 2,2-Difluorovinylsilanes, J. Org. Chem., 2003, 68, 7800–7805 CrossRef CAS; (e) K. Fuchibe, M. Takahashi and J. Ichikawa, Substitution of Two Fluorine Atoms in a Trifluoromethyl Group: Regioselective Synthesis of 3-Fluoropyrazoles, Angew. Chem., Int. Ed., 2012, 51, 12059–12062 CrossRef CAS; (f) M. Fujita, M. Obayashi and T. Hiyama, Fluoride ion-catalyzed generation and carbonyl addition of α-halo carbanions derived from α-halo organosilicon compounds, Tetrahedron, 1988, 44, 4135–4145 CrossRef CAS; (g) P. Gao, C. Yuan, Y. Zhao and Z. Shi, Copper-Catalyzed Asymmetric Defluoroborylation of 1-(Trifluoromethyl)Alkenes, Chem, 2018, 4, 2201–2211 CrossRef CAS.
- (a) T. Xiao, L. Li and L. Zhou, Synthesis of Functionalized gem-Difluoroalkenes via a Photocatalytic Decarboxylative/Defluorinative Reaction, J. Org. Chem., 2016, 81, 7908–7916 CrossRef CAS; (b) L. Li, T. Xiao, H. Chen and L. Zhou, Visible-Light-Mediated Two-Fold Unsymmetrical C(sp3)@H Functionalization and Double C-F Substitution, Chem. – Eur. J., 2017, 23, 2249–2254 CrossRef CAS; (c) H. Chen, T. Xiao and D. Anand, Synthesis of Fluorinated Benzo[a]quinolizidines via Visible Light-induced Tandem Substitution of Two Fluorine Atoms in a0020CF3 Group, Adv. Synth. Catal., 2017, 359, 3642–3647 CrossRef CAS; (d) S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Photoredox Generation of Carbon-Centered Radicals Enables the Construction of 1,1-Difluoroalkene Carbonyl Mimics, Angew. Chem., Int. Ed., 2017, 56, 15073–15077 CrossRef CAS; (e) L.-H. Wu, J.-K. Cheng, L. Shen, Z.-L. Shen and T.-P. Loh, Visible Light-Mediated Trifluoromethylation of Fluorinated Alkenes via C-F Bond Cleavage, Adv. Synth. Catal., 2018, 360, 3894–3899 CrossRef CAS; (f) P.-J. Xia, Z.-P. Ye, Y.-Z. Hu, D. Song, H.-Y. Xiang, X.-Q. Chen and H. Yang, Photocatalytic, Phosphoranyl Radical-Mediated N-O Cleavage of Strained Cycloketone Oximes, Org. Lett., 2019, 21, 2658–2662 CrossRef CAS PubMed; (g) Y. He, D. Anand, Z. Sun and L. Zhou, Visible-Light-Promoted Redox Neutral γ,γ-Difluoroallylation of Cycloketone Oxime Ethers with Trifluoromethyl Alkenes via C-C and C-F Bond Cleavage, Org. Lett., 2019, 21, 3769–3773 CrossRef CAS PubMed; (h) R. J. Wiles, J. P. Phelan and G. A. Molander, Metal-free defluorinative arylation of trifluoromethyl alkenes via photoredox catalysis, Chem. Commun., 2019, 55, 7599–7602 RSC; (i) Y.-Q. Guo, R. Wang, H. Song, Y. Liu and Q. Wang, Visible-Light-Induced Deoxygenation/Defluorination Protocol for Synthesis of γ,γ-Difluoroallylic Ketones, Org. Lett., 2020, 22, 709–713 CrossRef CAS; (j) D. Anand, Z. Sun and L. Zhou, Visible-Light-Mediated β–C-H gem-Difluoroallylation of Aldehydes and Cyclic Ketones through C-F Bond Cleavage of 1–Trifluoromethyl Alkenes, Org. Lett., 2020, 22, 2371–2375 CrossRef CAS PubMed; (k) Y. Chen, N. Ni, D. Cheng and X. Xu, The coupling of alkylboronic acids with a-(trifluoromethyl)styrenes by lewis base/photoredox dual catalysis, Tetrahedron Lett., 2020, 61, 152425 CrossRef CAS; (l) V. I. Supranovich, V. V. Levin, V. A. Kokorekin and A. D. Dilmana, Generation of Alkyl Radicals from Thiols via Zinc Thiolates: Application for the Synthesis of gem-Difluorostyrenes, Adv. Synth. Catal., 2021, 363, 2888–2892 CrossRef CAS; (m) W. Xu, H. Jiang, J. Leng, H.-W. Ong and J. Wu, Visible-Light-Induced Selective Defluoroborylation of Polyfluoroarenes, gem-Difluoroalkenes, and Trifluoromethylalkenes, Angew. Chem., Int. Ed., 2020, 59, 4009–4016 CrossRef CAS PubMed.
- (a) T. Ichitsuka, T. Fujita and J. Ichikawa, Nickel-Catalyzed Allylic C(sp3)-F Bond Activation of Trifluoromethyl Groups via β–Fluorine Elimination: Synthesis of Difluoro-1,4-dienes, ACS Catal., 2015, 5, 5947–5950 CrossRef CAS; (b) Y. Liu, Y. Zhou, Y. Zhao and J. Qu, Synthesis of gem-Difluoroallylboronates via FeCl2–Catalyzed Boration/β-Fluorine Elimination of Trifluoromethyl Alkenes, Org. Lett., 2017, 19, 946–949 CrossRef CAS; (c) Y. Lan, F. Yang and C. Wang, Synthesis of gem-Difluoroalkenes via Nickel-Catalyzed Allylic Defluorinative Reductive Cross-Coupling, ACS Catal., 2018, 8, 9245–9251 CrossRef CAS; (d) W. Dai, Y. Lin, Y. Wan and S. Cao, Cu-Catalyzed tertiary alkylation of α-(trifluoromethyl)styrenes with tertiary alkylmagnesium reagents, Org. Chem. Front., 2018, 5, 55–58 RSC; (e) D. Ding, Y. Lan, Z. Lin and C. Wang, Synthesis of gem-Difluoroalkenes by Merging Ni-Catalyzed C-F and C-C Bond Activation in Cross-Electrophile Coupling, Org. Lett., 2019, 21, 2723–2730 CrossRef CAS PubMed; (f) Z. Lin, Y. Lan and C. Wang, Synthesis of gem-Difluoroalkenes via Nickel-Catalyzed Reductive C-F and C-O Bond Cleavage, ACS Catal., 2019, 9, 775–780 CrossRef CAS; (g) X. Lu, X.-X. Wang, T.-J. Gong, J.-J. Pi, S.-J. He and Y. Fu, Nickel-catalyzed allylic defluorinative alkylation of trifluoromethyl alkenes with reductive decarboxylation of redox-active esters, Chem. Sci., 2019, 10, 809–814 RSC; (h) Y. Jin, J. Wu, Z. Lin, Y. Lan and C. Wang, Merger of C-F and C-N Bond Cleavage in Cross-Electrophile Coupling for the Synthesis of gem-Difluoroalkenes, Org. Lett., 2020, 22, 5347–5352 CrossRef CAS PubMed; (i) C. Yao, S. Wang, J. Norton and M. Hammond, Catalyzing the Hydrodefluorination of CF3–Substituted Alkenes by PhSiH3. H• Transfer from a Nickel Hydride, J. Am. Chem. Soc., 2020, 142, 4793–4799 CrossRef CAS; (j) C. Zhang, Z. Lin, Y. Zhu and C. Wang, Chromium-Catalyzed Allylic Defluorinative Ketyl Olefin Coupling, J. Am. Chem. Soc., 2021, 143, 11602–11610 CrossRef CAS PubMed; (k) W.-J. Yue, C. S. Day and R. Martin, Site-Selective Defluorinative sp3 C–H Alkylation of Secondary Amides, J. Am. Chem. Soc., 2021, 143, 6395–6400 CrossRef CAS; (l) B.-J. Xiong, T. Wang, H. Sun, Y. Li, S. Kramer, G.-J. Cheng and Z. Lian, Nickel-Catalyzed Cross-Electrophile Coupling Reactions for the Synthesis of gem-Difluorovinyl Arenes, ACS Catal., 2020, 10, 13616–13623 CrossRef CAS.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) Y. Jiang, K. Xu and C. Zeng, Use of Electrochemistry in the Synthesis of Heterocyclic Structures, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (c) I. Chiarotto, L. Mattiello and M. Feroci, The Electrogenerated Cyanomethyl Anion: An Old Base Still Smart, Acc. Chem. Res., 2019, 52, 3297–3308 CrossRef CAS; (d) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Powering the Future: How Can Electrochemistry Make a Difference in Organic Synthesis, Chem, 2020, 6, 2484–2496 CrossRef CAS; (e) Y. Yu, J. S. Zhong and K. Xu, Recent Advances in the Electrochemical Synthesis and Functionalization of Indole Derivatives, Adv. Synth. Catal., 2020, 11, 2102–2119 CrossRef.
- (a) X.-T. Gao, Z. Zhang, X. Wang, J.-S. Tian, S.-L. Xie, F. Zhou and J. Zhou, Direct electrochemical defluorinative carboxylation of α-CF3 alkenes with carbon dioxide, Chem. Sci., 2020, 11, 10414–10420 RSC; (b) Z. Chang, J.-X. Wang, X. Lu and Y. Fu, Synthesis of gem Difluoroalkenes through Nickel Promoted Electrochemical Reductive Cross Coupling, Chin. J. Org. Chem., 2021 DOI:10.6023/cjoc202108006.
- (a) B. Huang, Y. Li, C. Yang and W. Xia, Electrochemical 1,4-reduction of α,β-unsaturated ketones with methanol and ammonium chloride as hydrogen sources, Chem. Commun., 2019, 55, 6731–6734 RSC; (b) B. Huang, C. Yang, J. Zhou and W. Xia, Electrochemically generated N-iodoaminium species as key intermediates for selective methyl sulphonylimination of tertiary amines, Chem. Commun., 2020, 56, 5010–5013 RSC; (c) B. Huang, L. Guo and W. Xia, A facile and versatile electro-reductive system for hydrodefunctionalization under ambient conditions, Green Chem., 2021, 23, 2095–2103 RSC; (d) X. Zhang, C. Yang, H. Gao, L. Wang, L. Guo and W. Xia, Reductive Arylation of Aliphatic and Aromatic Aldehydes with Cyanoarenes by Electrolysis for the Synthesis of Alcohols, Org. Lett., 2021, 23, 3472–3476 CrossRef CAS.
- (a) S. Ozaki, H. Matsushita and H. Ohmori, Indirect electroreductive addition of alkyl radicals to activated olefins using a nickel(II) complex as an electron-transfer catalyst, J. Chem. Soc., Perkin Trans. 1, 1993, 649–651 RSC; (b) Y.-W. Lu, J.-Y. Nédélec, J.-C. Folest and J. Périchon, Electrochemical coupling of activated olefins and alkyl dihalides: formation of cyclic compounds, J. Org. Chem., 1990, 55, 2503–2507 CrossRef CAS; (c) J. A. Cleary, M. S. Mubarak, K. L. Vieira, M. R. Anderson and D. G. Peters, Electrochemical reduction of alkyl halides at vitreous carbon cathodes in dimethylformamide, J. Electroanal. Chem., 1986, 198, 107–124 CrossRef CAS; (d) W. Zhang and S. Lin, Electroreductive Carbofunctionalization of Alkenes with Alkyl Bromides via a Radical-Polar Crossover Mechanism, J. Am. Chem. Soc., 2020, 142, 20661–20670 CrossRef CAS.
- (a) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, Photosensitized decarboxylative Michael addition through N-(acyloxy)phthalimides via an electron-transfer mechanism, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS; (b) G. Pratsch, G. L. Lackner and L. E. Overman, Constructing Quaternary Carbons from N-(Acyloxy)phthalimide Precursors of Tertiary Radicals Using Visible-Light Photocatalysis, J. Org. Chem., 2015, 80, 6025–6036 CrossRef CAS PubMed; (c) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents, Science, 2016, 352, 801–805 CrossRef CAS; (d) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides, J. Am. Chem. Soc., 2016, 138(15), 5016–5019 CrossRef CAS; (e) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Photoinduced decarboxylative borylation of carboxylic acids, Science, 2017, 357, 283–286 CrossRef CAS; (f) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Decarboxylative alkenylation, Nature, 2017, 545, 213–218 CrossRef CAS.
- (a) N. F. Eweiss, A. R. Katritzky, P. L. Nie and C. A. Ramsden, The Conversion of Amines into Iodides, Synthesis, 1977, 634–635 CrossRef CAS; (b) C. H. Basch, J. Liao, J. Xu, J. J. Piane and M. P. Watson, Harnessing Alkyl Amines as Electrophiles for Nickel-Catalyzed Cross Couplings via C–N Bond Activation, J. Am. Chem. Soc., 2017, 139, 5313–5316 CrossRef CAS; (c) F. J. R. Klauck, M. J. James and F. Glorius, Deaminative Strategy for the Visible-Light-Mediated Generation of Alkyl Radicals, Angew. Chem., Int. Ed., 2017, 56, 12336–12339 CrossRef CAS; (d) S.-Z. Sun, C. Romano and R. Martin, Site-Selective Catalytic Deaminative Alkylation of Unactivated Olefins, J. Am. Chem. Soc., 2019, 141, 16197–16201 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qo01460a |
| This journal is © the Partner Organisations 2022 |