
A multifaceted approach towards understanding the peculiar behavior of (α)-hydroxyiminophosphonates†
Thomas
Toupy‡
 a,
Christopher
Kune‡
b,
Kristof
Van Hecke
c,
Loïc
Quinton
*b and
Jean-Christophe M.
Monbaliu
*a
a,
Christopher
Kune‡
b,
Kristof
Van Hecke
c,
Loïc
Quinton
*b and
Jean-Christophe M.
Monbaliu
*a
aCenter for Integrated Technology and Organic Synthesis, MolSys Research Unit, University of Liège, B-4000 Liège (Sart Tilman), Belgium. E-mail: jc.monbaliu@uliege.be; Web: http://www.citos.uliege.be
bMass Spectrometry Laboratory, MolSys Research Unit, University of Liège, B-4000 Liège (Sart Tilman), Belgium
cXStruct, Department of Chemistry, Ghent University, Krijgslaan 281-S3, B-9000 Ghent, Belgium
First published on 22nd November 2021
Abstract
The peculiar isomer-selective reduction of (α)-hydroxyiminophosphonates (oxime isomers) into (α)-hydroxyaminophosphonate (hydroxylamine) derivatives is presented. A library of 16 (α)-hydroxyiminophosphonates is prepared and studied via a unique multifaceted approach involving the interplay of NMR, XRD, MS, IM-MS and computational chemistry techniques. The combination of NMR, XRD and HPLC enables the seamless separation, identification and quantification of the oxime isomers (E/Z). Tandem MS (MS/MS) enables the determination of the fragmentation patterns for both isomers. Collision energy breakdown curves highlight the order of apparition of the fragments as well as their related energy of fragmentation, demonstrating that the strength of the C–P bond in the Z isomers is much weaker than in the E isomers. Computational chemistry demonstrates that favorable protonation site is isomer-dependent with the phosphonate moiety being the favorable protonation site for the E isomers, while protonation occurs preferentially on the amino moiety for Z isomers regardless of the phosphite source. The combination of these various methods led an unprecedented level of characterization of oxime isomers, providing a better uderstanding of the isomer-dependent behavior of (α)-hydroxyiminophosphonates.
Introduction
Non-natural amino acids, such as (α)-aminophosphonic acid have been extensively used as bioisosters in the late 1970s for the preparation of peptide bond analogs. Phosphonamidate, phosphonate and phosphinate are the main moieties involved in the backbone of phosphorus analogs of peptides, commonly called phosphonopeptides (Fig. 1).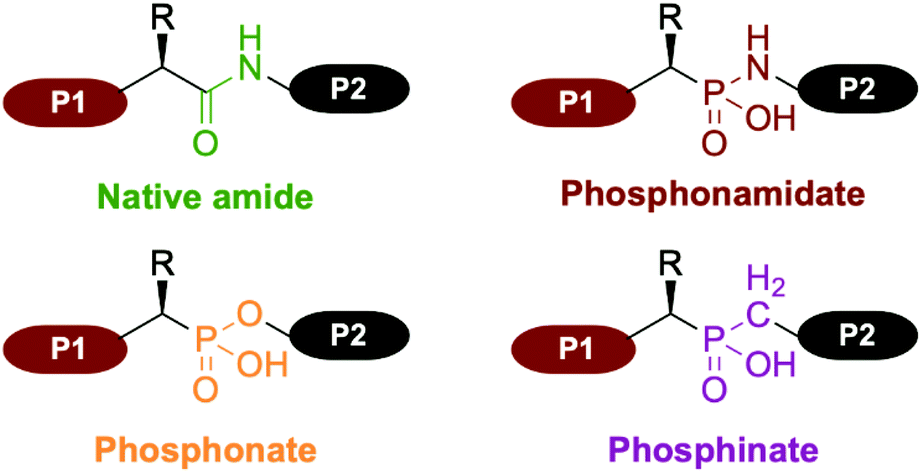 | ||
| Fig. 1 Comparison between the native amide peptide bond and the principal moieties involved in the backbone of phosphorus analogs of peptides, commonly called phosphonopeptides. | ||
Since their introduction in 1975 by Martell, they have gained significant attention as bioisosters of native peptides.1 Indeed, introduction of an aminophosphonic acid into a peptide sequence enables several structural and physico-chemical modifications. For instance, the greater lipophilicity of phosphonate compounds compared to their carboxylic analogs is a distinct advantage resulting in an increased overall biodisponibility.2 Hence, numerous examples of phosphonopeptide derivatives have been prepared for their potency as biological agents, including antibacterial agent, antithrombotic agent, inhibition of proteases (metallo-, serine, aspartic, cysteine and HIV) as well as inhibitor of Human Collagenase.3
Due to their renowned interest, numerous synthetic routes have been reported for the preparation of (α)-aminophosphonic acid, such as the three component Kabachnik–Fields reaction,4–10 the Pudovik reaction11–15 or the reduction of (α)-hydroxyiminophosphonate (aka (α)-oxime phosphonate) or (α)-hydroxyaminophosphonate16–19 (Fig. 2a). The main advantage of the latter is that it enables the preparation of the desired product as a free amine compared to the former ones. Two atom economic methods for accessing (α)-aminophosphonic acids and derivatives relies either on (a) a direct nucleophilic amination of (α)-halogenated alkylphosphonates or on (b) the reduction of an intermediate (α)-iminophosphonate derivative. It is worth mentioning that both methods proceed through the formation of an intermediate (α)-hydroxyaminophosphonate (Fig. 2b).
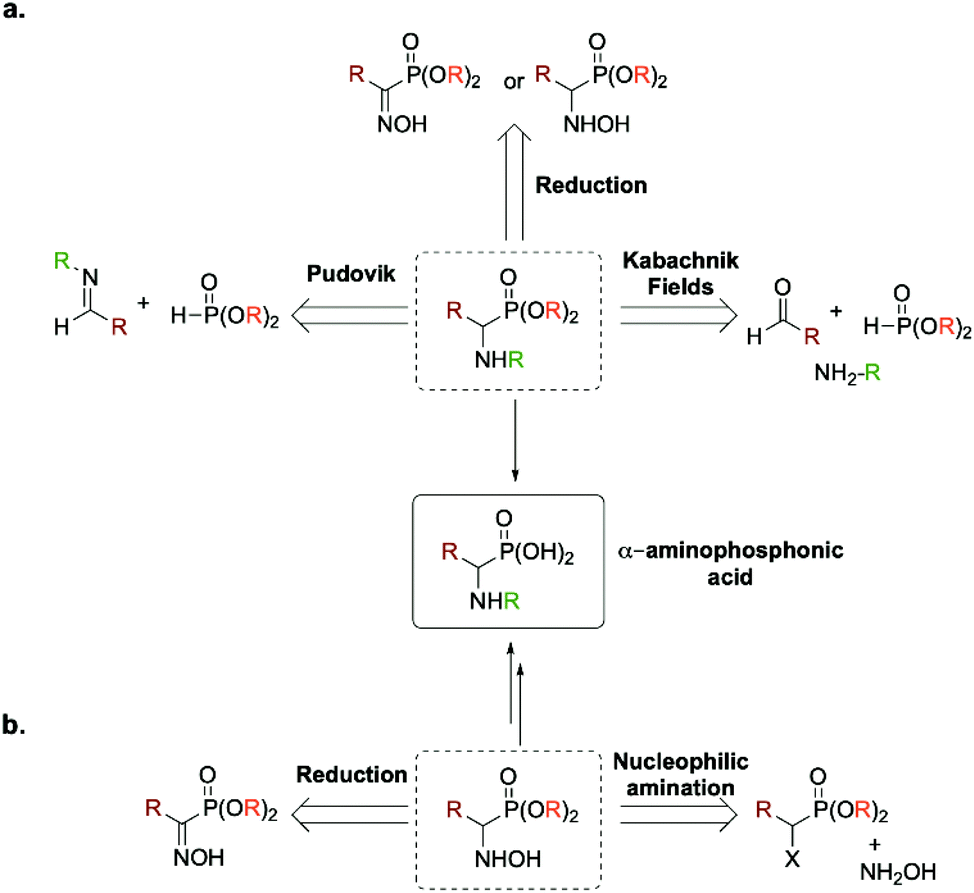 | ||
| Fig. 2 (a) Reported synthetic pathways for the preparation of (α)-aminophosphonic acids including the Pudovik reaction, the three components Kabachnik–Fields as well as the reduction of (α)-hydroxyiminophosphonates and (α)-hydroxyaminophosphonates. (b) Atom economic routes towards (α)-aminophosphonic acids relying on the reduction of (α)-hydroxyiminophosphonates or the nucleophilic amination of (α)-halogenated alkylphosphonates. | ||
Hydroxyaminophosphonate and hydroxyamino derivatives have found a wide range of synthetic utilities besides being potential intermediates en route toward (α)-aminophosphonic acids. In fact, hydroxyamino derivatives act as versatile amino source for the preparation of various nitrogen-containing molecules.20–23 Besides their common use as N-nucleophiles, they can also serve as electrophilic aminating reagents upon conversion of the hydroxyl moiety by a more potent leaving group.24–26 They have also been used as nitrogen-radical precursors in visible-light photochemistry27 as well as a more suited, non-explosive surrogate for 1-hydroxybenzotriazole (HOBT), which has been the benchmark of coupling agents in peptide chemistry.28 More recently, as antibiotic resistance has become a worldwide issue, hydroxyamino derivatives have shown promising results in the search of new antibacterial agents.29 In light of these numerous utilities, various methods have been developed for their preparation. Among those, the most used one relies on the reduction of the desired oxime (so-called hydroxyimine) derivative. This synthetic route has also been used for the preparation of their phosphono analogs, namely (α)-hydroxyaminophosphonate through reduction of the required (α)-hydroxyiminophosphonate.30 However, recent studies have reported that the yield of this pathway can be strongly influenced by the geometrical isomerism of the oxime moieties (E/Z isomerism, Fig. 3).
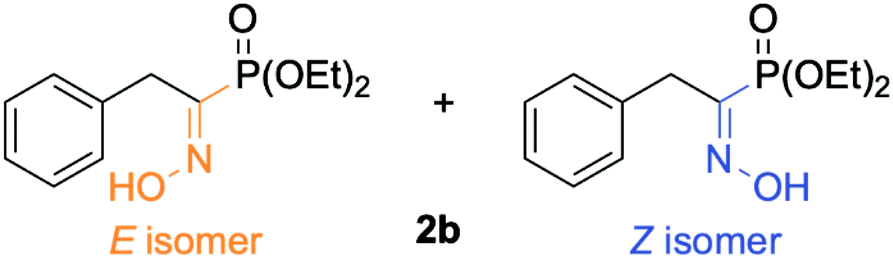 | ||
| Fig. 3 Representation of the geometrical isomerism (E/Z) of the oxime moiety for diethyl (1-(hydroxyimino)-2-phenylethyl)phosphonate 2b. | ||
For instance, Cramer et al. recently showed that the metal-assisted hydrogenation of the Z isomer of an oxime derivative was much faster than for its E counterpart.31 While studying the reduction of the oxime isomers of (S)-strepetenol A, Blechert et al. also observed a clear difference in both reactivity and selectivity.32 However, in this case, they highlighted that the E-oxime was reduced faster and under milder conditions that the Z-oxime. Nonetheless, both studies showed that under appropriate conditions and reaction time, both isomers reacted to give the desired hydroxylaminated derivative. Very few studies were conducted on the (α)-hydroxyiminophosphonates (i.e., phosphono analogs of (α)-hydroxyimino derivatives) reduction towards (α)-hydroxyamino derivatives. In 2012, Beletskaya et al. showed that under regular metal-assisted hydrogenation, only one of the two oxime isomers underwent reduction, regardless of the nature of the ligand, temperature, hydrogen pressure or reaction time. The nature of the two isomers was determined by 31P NMR due to their difference in chemical shifts. The isomer which remained untouched was attributed to the E-isomer while the Z-isomer completely reacted towards the desired product.30
From these observations, it seems of paramount importance to have a comprehensive understanding of the oxime reduction mechanism involving isomerism for improving the synthesis of hydroxyamino derivatives. This is only reachable with a complete characterization of the oxime isomers. In the 1990s and early 2000s, Eli Breuer and co-workers reported in a series of research papers, as well as in a review, on the preparation, characterization and study of (α)-hydroxyiminophosphonate derivatives.33–40 In their study, they showed that NMR was an invaluable tool for the assessment of the two isomers. In fact, in 31P NMR, the E-isomer resonates at lower field than the Z-isomer, which is backed by the expected shielding effect of the phosphorus by the lone pairs of the oxime oxygen.35 Also consistent with this shielding effect is the upfield shift observed in the 13C NMR spectra of the signal corresponding to the oxime carbon of the E-isomer relative to the Z-isomer. Moreover, a noticeable difference is observed in the coupling constant 1JPC of the oxime carbon with the phosphorus that is higher than 200 Hz for the E-isomer, while for the Z-isomer it usually ranges from 150 to 160 Hz.41 In the case of crystalline compounds, X-ray diffraction (XRD) was proven to be a valuable method for the assessment of the two isomers. During their study, they also discovered a peculiar behavior difference in the degradation of those species. Under thermal stress, the Z-isomer undergoes fragmentation into the corresponding nitrile and dimethyl hydrogen phosphonate through a 4-membered ring intermediate, while the E-isomer did not react. Though this approach proved to be useful in the study of the (α)-hydroxyiminophosphonate isomers and their properties, the main drawback is that it requires isolation of large amounts of the pure isomers, which can be extremely difficult or impossible in some instances.
Besides NMR and XRD, mass spectrometry (MS) is a characterization method widely used to determine the elemental composition and/or partial structural insight using tandem mass spectrometry (MS/MS) such as the atomic connectivity.42 MS is very useful for the analysis of low abundance analytes in complex mixtures, which can be challenging in NMR and XRD. MS can also be hyphenated with other separation methods, such as liquid chromatography (LC-MS), gas chromatography (GC-MS), capillary electrophoresis (CE-MS) and ion mobility (IM-MS). Based on the separation technique involved with MS analyser, isomeric compound can be isolated before their analysis by MS and MS/MS. Moreover, these separation techniques give access to other compound descriptors (e.g., retention time and migration time), increasing the characterization level. For instance, IM-MS has emerged as a well-suited technique to probe the shape of ions in gas-phase based on their mobility or collision cross sections (CCS).43 Separation and characterization of geometrical isomers from IM-MS analysis have been recently reported.44–47
Within the context of synthesizing (α)-aminophosphonic acid analogs of some of the 20 proteogenic amino acids through the reduction of (α)-hydroxyiminophosphonates, a systematic and thorough study of their peculiar behavior was envisioned. Herein is reported the development of a multifaceted approach to characterize (α)-hydroxyiminophosphonates involving the interplay of NMR, XRD, MS, IM-MS and computational chemistry techniques. This study encompasses 16 (α)-hydroxyiminophosphonates synthetized according to a revisited version of Viveros-Ceballos’ classical route for the preparation of (α)-aminophosphonates.19 The reported multifaceted approach leads to a high level of characterization, pushing forward the understanding of the oxime isomers behavior on the reduction mechanism towards hydroxyamino analogs.
Experimental section
General information
Conversion and yield were determined by high performance liquid chromatography coupled to Diode-Array Detection (HPLC/DAD) or by 1H and/or 31P NMR spectroscopy (Magritek Spinsolve 43 MHz NMR spectrometer). Structural identity was confirmed by 1H, 13C and 31P NMR spectroscopy (400 MHz Bruker Avance spectrometer) in CDCl3 or D2O (ESI†) and by X-ray diffraction (XRD) on single crystals of compounds 2b and 3b. The chemical shifts are reported in ppm relative to TMS as internal standard or to solvent residual peak. Solvents were used as received, unless otherwise stated. Acetic acid, hydroxylamine hydrochloride, pyridine, sodium bicarbonate, triethyl phosphite, trimethyl phosphite, triisopropyl phosphite, acetyl chloride, phenylacetyl chloride, isovaleryl chloride, 4-methoxyphenylacetyl chloride, 3-(methylthio)propionyl chloride, isobutyryl chloride, DL-2-methylbutyryl chloride, 3-indoleacetic acid, (phenylthiol)acetic acid, trimethylacetyl chloride, thionyl chloride, oxalyl chloride, sodium cyanoborohydride, sodium borohydride, borane pyridine complex, zinc dust, hydrogen chloride (2 N in Et2O) were obtained from commercial sources and used as received.Mass spectrometry (breakdown curve)
Mass spectrometry analysis, ion mobility measurement and collision energy breakdown curve (breakdown curve in short) analysis have been performed on a SynaptG2 HDMS instrument from Water (Manchester, UK) by monitoring the signal of the precursor ion and its fragment ions formed upon collisions with Argon (Collision Induced Dissociation, CID) with regard to the collision energy (expressed in V). This instrument is equipped with an electrospray ionization (ESI) source and a traveling wave ion mobility spectrometry (TWIMS) cell and a time-of-flight (ToF) mass analyzer. The ESI, sampling cone and voltages were respectively set on 2.5 kV, 22 V and 4 V. The ESI temperature and desolvation temperature were set to 100 and 200 °C, respectively. Precursor ions have been isolated by the quadrupole with a LM resolution of 5. CID experiments have been performed in the TRANSFER cell after ion mobility (IM) separation to further increase the isolation of the precursor ions. The ion mobility cell was filled with 2.47 mbar of N2. This pressure has been measured using independent capacitance gage (Oerlikon Q5 Leybold Vacuum, CERAVAC CTR 90) linked to a vacuum controller (Vacom, MVC-UVH Multichannel Vacuum Controller). The TWIMS wave height were set to 30 V and TWIMS waves velocities were set to 900 m s−1 for all compounds except for 16b, which required 600 m s−1 in order to optimize IMS separation. CID voltages have been scanned from 2 V to 32 V with an increment of 2 V. The ToF mass analyzer was systematically calibrated in positive from m/z 50 to m/z 2000 with a 0.1 wt% sodium iodide solution in H2O. Each mass spectrum has been acquired from m/z 50 to m/z 1200 during 2 min.Computational study
Structure optimization of neutral and protonated oxime isomers were performed at the B3LYP/6-31G(d) level of theory with the Gaussian 09 software (revision D.01).48 For each compound, all potential protonation sites have been considered. Standard enthalpy, standard entropy and standard free enthalpy of proton transfer at 298.15 K were calculated from the thermochemical calculations (298.15 K, 1 atm) to determine preferential protonation sites.Results and discussion
Preparation and characterization of a library of (α)-hydroxyiminophosphonates
The envisioned route for the preparation of the library of (α)-hydroxyiminophosphonates matches the one reported by Viveros-Ceballos. The process features two steps: (a) the preparation of (α)-ketophosphonates from the corresponding acyl chloride using an alkyl phosphite source and (b) the preparation of the desired (α)-hydroxyiminophosphonates using hydroxylamine hydrochloride. The starting point of our study was the preparation of diethyl (1-(hydroxyimino)-2-phenylethyl)phosphonate (2b)(Scheme 1).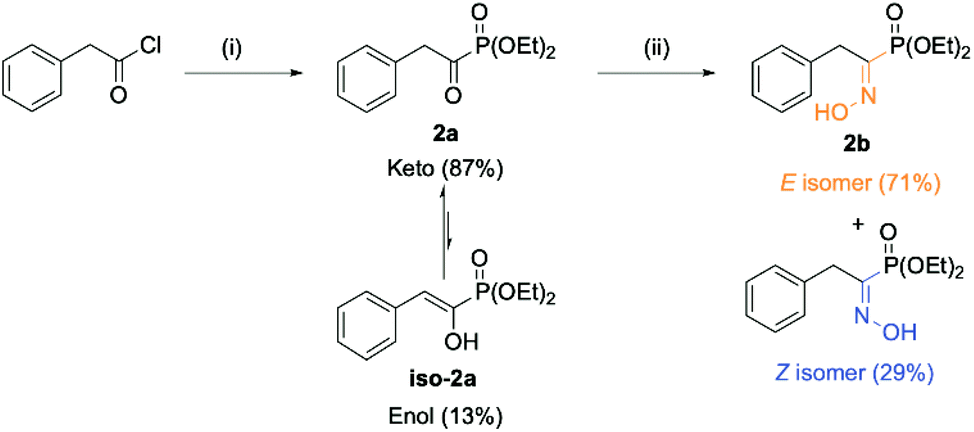 | ||
| Scheme 1 Viveros-Ceballos’ strategy towards (α)-hydroxyiminophosphonates applied for the preparation of diethyl (1-(hydroxyimino)-2-phenylethyl)phosphonate 2b highlighting the presence of two geometrical isomers of the final oxime derivative. The ratio of isomer was determined by 31P NMR. (i) P(OEt)3 in Et2O, 0 °C → RT, overnight. (ii) NH2OH·HCl and dry pyridine in absolute EtOH, RT, 12 h. | ||
The first step involved the Michaelis–Arbuzov reaction between the starting phenylacetyl chloride and triethylphosphite to yield the product as a mixture of tautomers (keto–enol) as reported by Zantour49 and Tishler,50 respectively. In this case, the product was recovered in 91% yield as an 83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17 (keto/enol) mixture as determined by 31P NMR. The second key step relied on the formation of the oxime moiety using hydroxylamine hydrochloride and pyridine as the organic base. The desired (α)-hydroxyiminophosphonate 2b was thus obtained in 66% isolated yield as a mixture of two geometrical isomers (E:71, Z:29). The isomer attribution was conducted using Eli Breuer previous (31P and 13C) NMR results.35 Because 2b is crystalline as a mixture of isomers, XRD analysis was also carried out on the mixture, which reinforced the result from the NMR attribution with a 67:33 E/Z ratio (see ESI section 2.6†). Next, the preparation of a small library of oxime derivatives was envisioned by varying the starting acyl chloride to mimic the side chains of the natural amino acids as well as the phosphite source. The results are displayed in Fig. 4 below. In the case of 15b and 16b, the starting acyl chloride were not commercially available and were prepared from their commercially available carboxylic acid derivatives (see ESI section 2.1.1†).
:17 (keto/enol) mixture as determined by 31P NMR. The second key step relied on the formation of the oxime moiety using hydroxylamine hydrochloride and pyridine as the organic base. The desired (α)-hydroxyiminophosphonate 2b was thus obtained in 66% isolated yield as a mixture of two geometrical isomers (E:71, Z:29). The isomer attribution was conducted using Eli Breuer previous (31P and 13C) NMR results.35 Because 2b is crystalline as a mixture of isomers, XRD analysis was also carried out on the mixture, which reinforced the result from the NMR attribution with a 67:33 E/Z ratio (see ESI section 2.6†). Next, the preparation of a small library of oxime derivatives was envisioned by varying the starting acyl chloride to mimic the side chains of the natural amino acids as well as the phosphite source. The results are displayed in Fig. 4 below. In the case of 15b and 16b, the starting acyl chloride were not commercially available and were prepared from their commercially available carboxylic acid derivatives (see ESI section 2.1.1†).
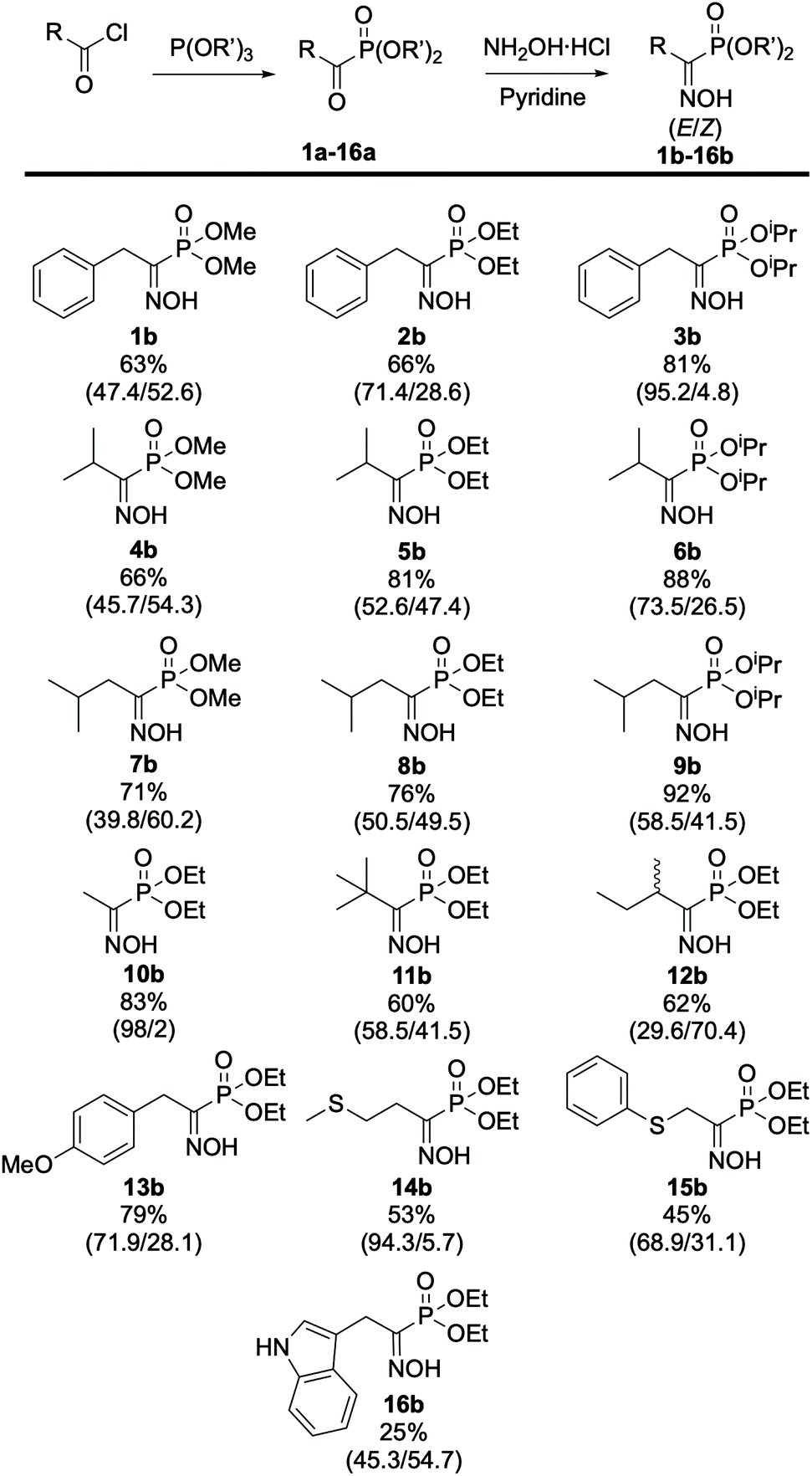 | ||
| Fig. 4 Scope of (α)-hydroxyiminophosphonates. The isolated yield and ratio of isomers E/Z (in parentheses) are indicated. All compounds were obtained according to the procedure described in Scheme 1. | ||
From these results, the effect of the phosphite source, and more specifically of the steric hindrance of the alkyl phosphite, can be highlighted on the ratio of isomers. In fact, comparing the results obtained for 2b and its phosphite derivatives, the more steric hindering triisopropryl phosphite 3b leads to a higher content of E isomer (73.5%) in the mixture compared to triethyl 2b (52.6%) and trimethyl phosphite 1b (45.7%), respectively. The same trend applies for the phosphite derivatives of 4b, 5b, 6b and 7b, 8b, 9b. Looking at the remaining compounds in the scope, which were prepared using triethyl phosphite, it appeared that the nature of the side chain also has an effect on the ratio of isomers. In most cases, the E isomer is predominant, showing that it is more favored compared its Z counterpart. In the case of 10b we determined a 98:2 (E/Z) ratio, which can be attributed to the lack of steric hindrance on the side chain, resulting in almost complete formation of the sole E isomer while for 14b the 94:6 (E/Z) ratio was determined after column purification, which resulted in loss of some of the Z isomer. During this study, HPLC/DAD also emerged as a potent method for the separation and quantification of the (α)-hydroxyiminophosphonate isomers. Indeed, the ratio of isomers obtained by HPLC/DAD matches the one obtained via31P NMR and XRD with a 1–4% margin of error. Moreover, the elution order of the oxime isomers was kept identical, regardless of the structure: for the library of 16 compounds, the Z isomer has a 0.5 min lower retention time than its E counterpart (see ESI Fig. S1†).
To push the characterization of the (α)-hydroxyiminophosphonate isomers one step further, we envisioned using complementary methods such as mass spectrometry. However, regular mass spectrometry gives us the mass-to-charge ratio (m/z) which, in this case, is not sufficient to be able to characterize geometrical isomers. Therefore, the use of tandem MS (MS/MS) was required in order to access partial structural insights such as the atomic connectivity through fragmentation. Although such insights were within reach of MS/MS analysis, isolation of the isomers was still mandatory in order to characterize them. Hence, a Synapt G2 spectrometer coupled with ion-mobility (IM) was used as a mean of separation prior to MS/MS analysis. Even though the separation of the isomers was poorly resolved, this enabled the acquisition of isomeric interference-free MS/MS spectra for the isomers of 2b. The results showed that the CID-mediated fragmentation exhibits different patterns for the two isomers studied (Fig. 5). In fact, the Z isomer was concerned with a first fragmentation occurring at the C–P bond, releasing the phosphorus copula while for the E isomer, the first fragmentation occurred through a McLafferty rearrangement resulting in the loss of one of the ethyl esters. For the remaining compounds of the scope, the IMS resolution of the instrument used was not sufficient to acquire isomeric interference-free spectra. Therefore, HPLC was used for the isomer separation prior to MS/MS analysis. Similar fragmentation pathways were observed on most of the 16 compounds studied. Predominant fragments for the isomers of each compound are reported in ESI Table S2.†
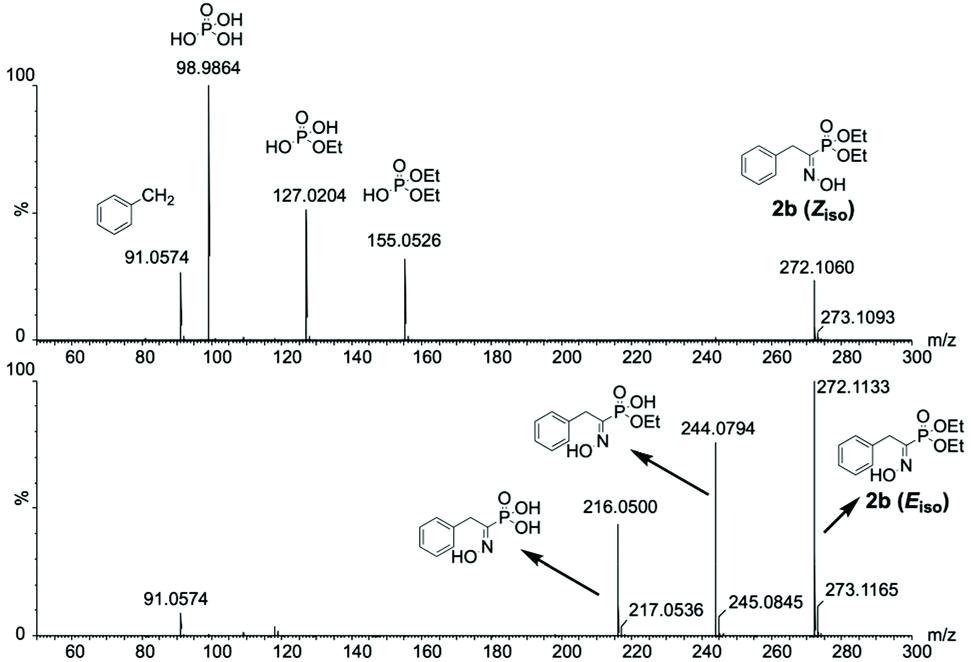 | ||
| Fig. 5 Isomeric interference-free MS/MS spectra for the isomers of 2b highlighting the difference in their CID-mediated fragmentation pattern. | ||
Reactivity assessment of (α)-hydroxyiminophosphonates towards their reduction and observations
Upon characterization of the (α)-hydroxyiminophosphonate isomers, we decided to assess their reactivity towards reduction. Various reducing agents and conditions were envisioned starting with zinc in acetic acid as described by Viveros-Ceballos.19 This enabled the seamless preparation of the (α)-aminophosphonate diester (am-2b) derivative of the natural amino acid phenylalanine in 71% isolated yield (see ESI section 2.1.5† for experimental details). Then, the preparation of the (α)-hydroxyaminophosphonate (hydroxylamine) 2c was envisioned using milder reducing agents such as borohydride derivatives (NaBH4, NaBH3CN, PyBH3). Upon optimization of the reaction conditions, NaBH3CN proved to be the most efficient reducing agent. However, no matter the borohydride derivative used, we observed the same peculiar phenomenon during the monitoring of the reaction. In fact, thanks to the thorough characterization of the oxime isomers of 2b, HPLC monitoring showed that only the Z isomer underwent reduction while the E isomer remained untouched even after a week of reaction (see ESI Fig. 1–4†). This observation was reinforced upon work up of the reaction mixture and NMR analysis of the resulting liquid/liquid extraction fractions. Then we extended the study of the reduction reaction into (α)-hydroxyaminophosphonates to the whole scope of oxime derivatives (Fig. 6). The 16 studied oxime derivatives showed the same peculiar behavior during the reduction reaction with the Z isomer properly reducing into the desired hydroxylamine derivative while the E isomer remained untouched, in all cases, as demonstrated by both HPLC and NMR.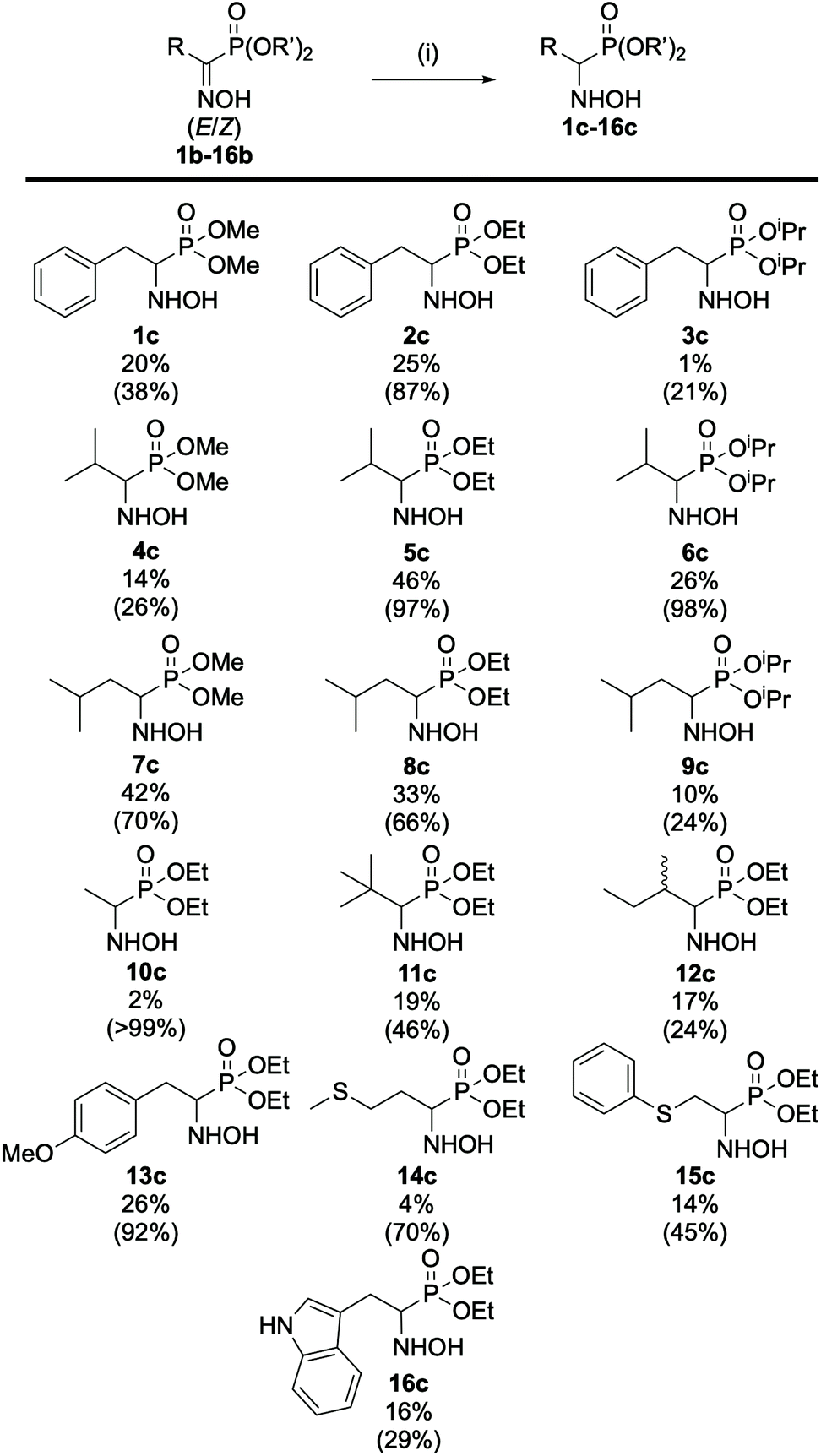 | ||
| Fig. 6 Scope of the reduction of (α)-hydroxyiminophosphonates. The isolated yield and corrected yield (in parentheses) are indicated. Corrected yield = isolated yield divided by the proportion of Z isomer in the mixture. All compounds were obtained according to the procedure described in (i). (i) NaBH3CN in EtOH, HCl in MeOH, pH 3, RT, overnight. | ||
From these observations we can highlight that the two oxime isomers have a different behavior on a multitude of facets, including HPLC, NMR, MS/MS and towards their reduction into hydroxylamine derivatives. Therefore, there is a need to better comprehend their behavior, which could extend to a better understanding of their overall reactivity.
Collision energy breakdown curves of (α)-hydroxyiminophosphonates
To increase our comprehension of the oxime isomers behavior, advanced mass spectrometry techniques relying on collision energy breakdown curve experiments were envisioned. These analyses enable the monitoring of the parent precursor (protonated oxime isomer) as well as the fragment ions generated by collision-induced dissociation (CID) with regard to the collision energy (expressed in V). Breakdown curve experiments have been performed on the isolated (α)-hydroxyiminophosphonate isomers as well as on the mixture of both isomers for each compound from the scope. The monitored fragments were the main fragments observed (see ESI Table S2†). Resulting breakdown curves are reported in section 3 of the ESI.†Upon analysis of the main fragments and their order of appearance for the scope of isomers, we observed a few trends. The first one relies on the rupture of the C–P bond (between the carbon of the oxime and the phosphonate moiety), while the second and third ones rely on the impact of the isopropyl esters and the ethyl esters on the fragmentation pattern of the isomers, respectively. Regarding the first trend, for the studied compounds, the fragmentation of the C–P bond only occurs for the Z isomers, while it remains untouched for the E isomers except for the methyl ester derivatives 1b, 4b and 7b, where the fragmentation of the C–P bond occurs for both isomers. However, looking at the curve of appearance of the ions resulting from the C–P fragmentation for the isomers of 1b (Fig. 7) (see ESI section 3.1† for the detailed breakdown curves), clearly emphasizes that fragmentation is predominant for the Z isomer. These observations highlight that the strength of the C–P bond in the Z isomer is significantly weaker than in its E counterpart. The second trend concerns the isopropyl ester derivatives 3b, 6b and 9b, which exhibit the same first two fragmentations for both isomers with the successive loss of the two isopropyl esters. This suggests that for these compounds, the P–O isopropyl esters bonds are weaker than the C–P bond. The last trend relies on the ethyl ester derivatives. In fact, we observed that all the Z isomers of the studied ethyl ester derivatives had the same fragmentation pattern regardless of the side chain. Firstly, fragmentation of the C–P bond occurs, releasing diethyl hydrogen phosphate ((HO)P(O)(OEt)2) which then undergoes successive loss of the ethyl esters through McLafferty rearrangement to yield phosphoric acid. Whereas, for the fragmentation of the E isomers, only the successive loss of the two ethyl esters through rearrangement is observed. These observations expand to the whole scope of ethyl ester derivatives except for 14b, 15b and 16b, which have a different behavior compared to the rest of the scope. In fact, these three compounds exhibit a similar fragmentation pattern for both isomers, which can be explained by the presence of heteroatoms along the side chain, potentially influencing the nature and strength of the weakest bond.
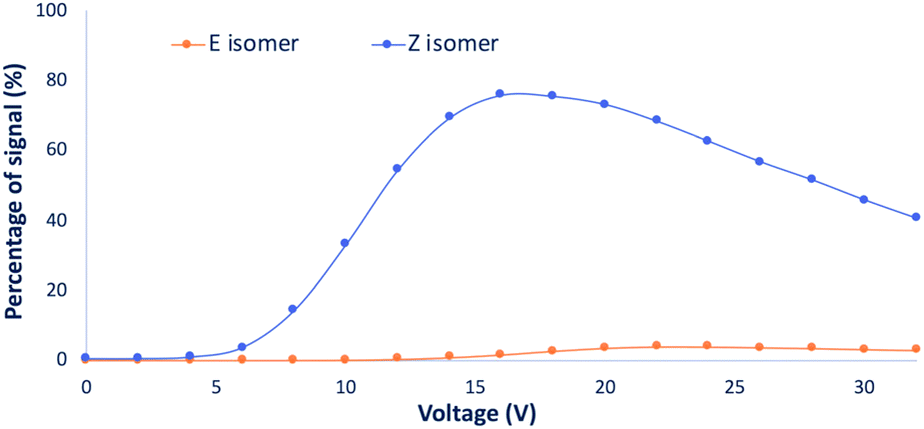 | ||
| Fig. 7 Fragment apparition curves for the ions resulting from the C–P fragmentation for both isomers of 1b. The fragment apparition curve for the Z isomer appears in blue and in orange for the E isomer. | ||
Moreover, thanks to the breakdown curve analyses, the monitoring of the parent precursor ion with respect to the collision energy (V) for the two isomers and the mixture of isomers can be used for measuring a valuable information on the strength of the weakest bond, namely, V50. In fact, the V50 corresponds to the collision energy (V), for which we observe 50% fragmentation of the parent precursor. These values highlight the difference in the strength of the weakest bond between isomers. Since CID is an ergodic process, it is important to note that the V50 values estimated from CID are directly related to the number of vibration modes. Therefore, only geometrical isomers (i.e. having the same number of vibration modes) can be compared in terms of V50. We therefore calculated ΔV50 to compare compound behavior upon fragmentation.
The V50 values for each isomers of the scope as well as the mixture of isomers are gathered in Table 1.
| Compound | V 50 (E)a (V) | V 50 (Z)a (V) | V 50 (mix)a (V) | ΔV50 (V) |
|---|---|---|---|---|
| a The V50 values have been extracted from the knee point of the sigmoid equation obtained by fitting the decreasing signal of precursor ion. Random sample consensus (RANSAC) algorithm has been applied to remove potential outliers. b Isolation of the Z isomer via HPLC failed due to its too low content in the mixture of isomers. | ||||
| 1b | 17.68 | 10.77 | 16.78 | 6.91 |
| 2b | 15.40 | 12.41 | 15.08 | 2.99 |
| 3b | 9.56 | 12.20 | 9.59 | −2.64 |
| 4b | 18.90 | 10.09 | 18.67 | 8.81 |
| 5b | 15.07 | 11.33 | 15.07 | 3.74 |
| 6b | 9.36 | 10.88 | 9.41 | −1.52 |
| 7b | 20.42 | 10.36 | 20.27 | 10.06 |
| 8b | 15.53 | 12.25 | 15.55 | 3.28 |
| 9b | 9.48 | 11.13 | 9.48 | −1.65 |
10b
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif)
|
12.76 | — | — | — |
| 11b | 16.48 | 10.79 | 16.08 | 5.69 |
| 12b | 14.67 | 10.67 | 13.81 | 4.00 |
| 13b | 16.56 | 12.06 | 16.41 | 4.50 |
| 14b | 8.72 | 8.99 | 8.74 | −0.27 |
| 15b | 11.56 | 10.13 | 11.42 | 1.43 |
| 16b | 13.57 | 11.70 | 13.17 | 1.87 |
Upon analysis of the results obtained from the V50 values for the various compounds of the scope, we observe that the nature of the parent alkyl phosphite has an impact on the ΔV50. In fact, if we look at the trimethyl phosphite (P(OMe)3) derivative 1b, a higher ΔV50 is obtained by comparison to the triethyl phosphite (P(OEt)3) derivative 2b, translating in a higher difference in the strength of the weakest bond between the two isomers. Whereas for the triisopropyl phosphite (P(OiPr)3) derivative 3b, a negative ΔV50 is observed. This trend also applies for the derivatives 4b, 5b, 6b and 7b, 8b, 9b. In the cases of 14b, 15b and 16b, which showed similar fragmentation patterns for the two isomers unlike the other triethyl phosphite derivatives, a smaller ΔV50 than for the other P(OEt)3 derivatives is observed, probably due to the presence of heteroatoms along the side chain which drives the fragmentation independently to the isomer form (i.e. same breakdown curves for both isomers). Moreover, the survival yield curves for the mixture of isomers highlight a peculiar phenomenon. In fact, for each compound of the scope, the curve for the mixture of isomers is almost a perfect match of the curve for the E isomer whereas it should consider the weighted contribution of each isomers (i.e. sum of the two sigmoid curves) (see ESI section 3†). This observation suggests that the E/Z ratio is influenced by the MS analysis.
Isomer specific in-source fragmentation and/or difference in protonic affinity likely weighs a lot in the appearance of such phenomenon. The former hypothesis is easily demonstrated by looking at the full MS spectra without collision activation for both isomers. In fact, in the case of 2b, a higher content of fragmentation for the full MS spectrum of the Z isomer compared to the E isomer (Fig. 8) is observed, emphasizing that the Z isomer suffers more of in-source fragmentation than the E isomer.
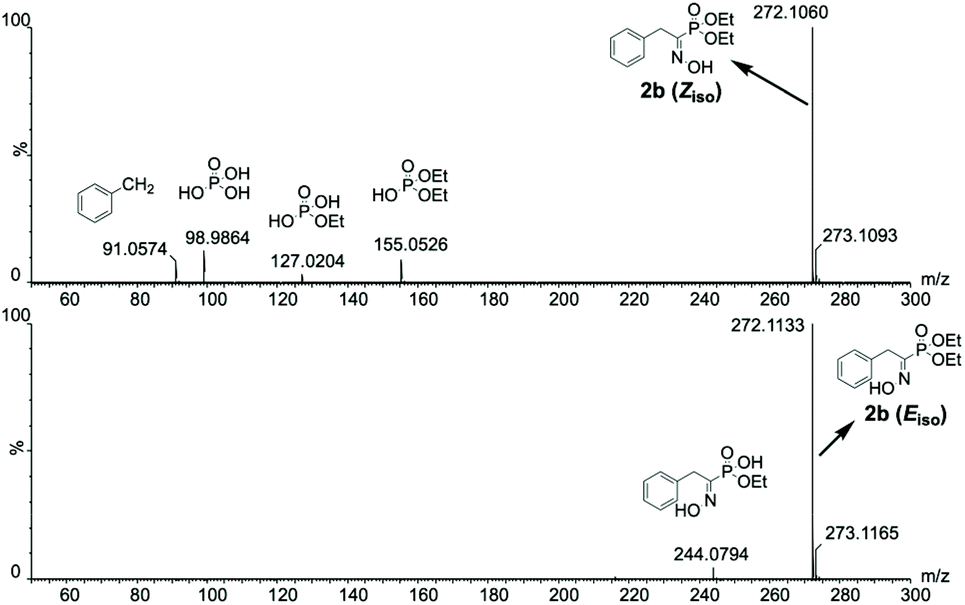 | ||
| Fig. 8 Isomeric interference-free full MS spectra without collision activation for the isomers of 2b highlighting a higher content of in-source fragmentation for the Z isomer. | ||
In order to verify the latter hypothesis, favorable protonation site calculations were conducted on 9 couple of oxime isomers (1b–9b) by DFT at the B3LYP/6-31G(d) level of theory. Calculations showed that each isomer exhibits two stable protonation sites: one in the amine moiety (–N–H+) and the other on the phosphonate moiety (–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–H+). Standard enthalpy, entropy and free enthalpy variations at 298.15 K (
O–H+). Standard enthalpy, entropy and free enthalpy variations at 298.15 K (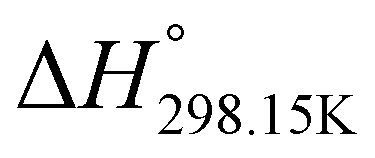 ,
, 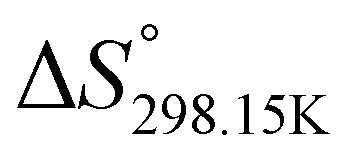 ,
, 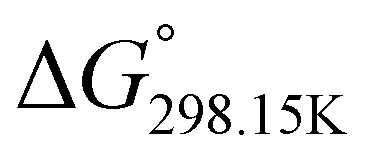 , respectively) of the proton transfer reaction (–N–H+ ⇌ –PO–H+) are reported in Table 2. A negative value of
, respectively) of the proton transfer reaction (–N–H+ ⇌ –PO–H+) are reported in Table 2. A negative value of 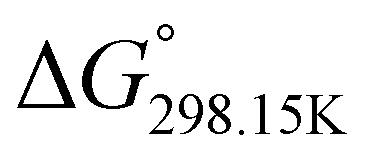 indicates that –PO–H+ is more stable than –N–H+. Inversely, a positive value of
indicates that –PO–H+ is more stable than –N–H+. Inversely, a positive value of 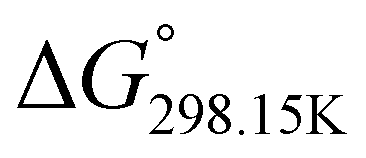 indicates that –N–H+ is more stable than –PO–H+. The calculated
indicates that –N–H+ is more stable than –PO–H+. The calculated 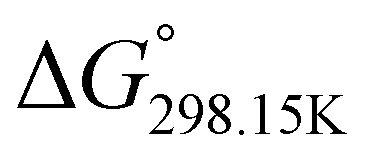 values support that the phosphonate moiety is the more favorable protonation site for E isomers while protonation occurs preferentially on the amino moiety for Z isomers regardless of the phosphite source. Since the favorable protonation site are isomer-dependent, one can consider that the protonic affinity is also dependent on the isomeric form. Based on the DFT calculations, this difference in the protonation sites may explain the difference observed for the breakdown curves of the two isomers as well as their difference in V50, except for compounds 14b, 15b and 16b which display similar behavior and V50 values for both isomers. This might be explained by the presence of heteroatoms along the side chain, potentially changing the nature of the protonation site/protonation behavior.
values support that the phosphonate moiety is the more favorable protonation site for E isomers while protonation occurs preferentially on the amino moiety for Z isomers regardless of the phosphite source. Since the favorable protonation site are isomer-dependent, one can consider that the protonic affinity is also dependent on the isomeric form. Based on the DFT calculations, this difference in the protonation sites may explain the difference observed for the breakdown curves of the two isomers as well as their difference in V50, except for compounds 14b, 15b and 16b which display similar behavior and V50 values for both isomers. This might be explained by the presence of heteroatoms along the side chain, potentially changing the nature of the protonation site/protonation behavior.
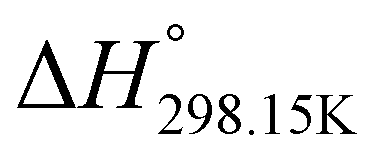 ,
, 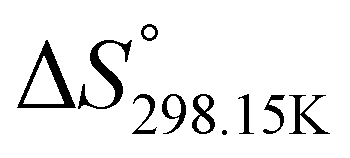 , and
, and 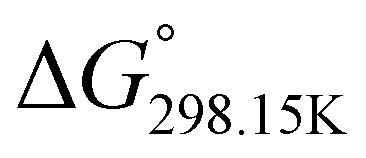 , respectively) for the proton transfer reaction from the amino moiety (reactant) to the phosphonate moiety (product) of investigated oxime isomers, –N–H+ ⇌ –PO–H+. Values are given in kJ mol−1 for ΔH° and ΔG° and in J mol−1 K−1 for ΔS°
, respectively) for the proton transfer reaction from the amino moiety (reactant) to the phosphonate moiety (product) of investigated oxime isomers, –N–H+ ⇌ –PO–H+. Values are given in kJ mol−1 for ΔH° and ΔG° and in J mol−1 K−1 for ΔS°
| Compound | Isomer | –N–H+ ⇌ –PO–H+ |
||
|---|---|---|---|---|
|
|
|
|
||
| 1b | E | −10.21 | −7.78 | −7.89 |
| Z | 70.58 | 11.78 | 67.07 | |
| 2b | E | −14.27 | −5.07 | −12.75 |
| Z | 53.71 | −11.94 | 57.27 | |
| 3b | E | −18.58 | 6.11 | −20.40 |
| Z | 8.02 | −5.79 | 9.74 | |
| 4b | E | −20.86 | −14.69 | −16.48 |
| Z | 6.95 | 4.84 | 5.51 | |
| 5b | E | −14.44 | −7.35 | −12.25 |
| Z | 71.98 | 22.29 | 65.34 | |
| 6b | E | −24.96 | −7.70 | −22.67 |
| Z | 69.26 | 47.58 | 55.07 | |
| 7b | E | −17.37 | −2.71 | −16.56 |
| Z | 81.18 | 23.05 | 74.30 | |
| 8b | E | −22.61 | 4.63 | −23.99 |
| Z | 74.88 | 21.52 | 68.47 | |
| 9b | E | −36.08 | −8.05 | −33.68 |
| Z | 64.52 | 13.59 | 60.46 | |
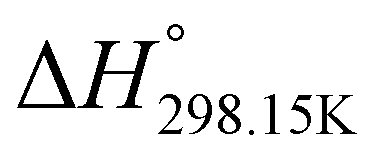
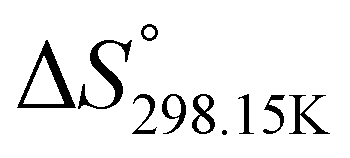
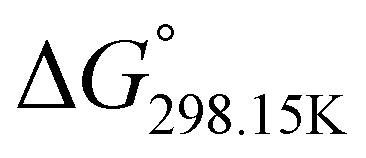
Although MS methods were unable to successfully quantify the isomeric ratio of the investigated oximes (due to overestimation of the E/Z ratio), numerous valuable structural information can be obtained. Indeed, tandem MS (MS/MS) gives access to structural insights such as the atomic connectivity through fragmentation. However, in some cases where the fragmentation pattern is the same for the two isomers, atomic connectivity is not sufficient on its own to characterize these species. Therefore, collision energy breakdown curves can be used as a powerful addition, highlighting the order of apparition of the fragments as well as their related energy of fragmentation. This allows the determination of isomer specific bond strength.
Conclusions
Within the context of synthesizing (α)-aminophosphonic acid analogs of some of the 20 proteogenic amino acids, this work discloses a study of the peculiar behavior of (α)-hydroxyiminophosphonates (oxime isomers) towards their reduction into (α)-hydroxyaminophosphonates (hydroxylamine) derivatives. A library of 16 (α)-hydroxyiminophosphonates was prepared according to Viveros-Ceballos’ classical route. A unique multifaceted approach involving the interplay of NMR, XRD, MS, IM-MS and computational chemistry techniques has been developed to enhance the compound characterization at the isomer level. The combination of NMR, XRD and HPLC analyses enabled the seamless separation, identification and quantification of the two oxime isomers for the 16 studied compounds. MS provided numerous valuable structural information, pushing further the characterization of such isomers. Tandem MS (MS/MS) of isomers isolated by HPLC and/or IMS enabled the determination of the fragmentation patterns for both isomers, while collision energy breakdown curves highlighted the precursor ion yield with regard to the collision energy, as well as the order of apparition of the fragments. This allowed the estimation of isomer specific bond strengths, demonstrating that the strength of the C–P bond in the Z isomer was much weaker than in its E counterpart. Moreover, computational chemistry demonstrated that favorable protonation site is isomer-dependent, which reflects the different fragmentation behaviors of oxime isomers. The combination of these various methods led an unprecedented level of characterization of oxime isomers and a more refined understanding of their complex behavior in reaction.Author contributions
T. T. designed the chemistry, performed the experiments, analyzed the results and wrote the manuscript. C. K. analyzed the results and wrote the manuscript. K. V. H. performed the X-ray diffraction analysis. L. Q. revised the manuscript. J.-C. M. M. designed the chemistry, supervised the project and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Liège (Welcome Grant WG-13/03, J. C. M. M.) and the F.R.S.-FNRS (Incentive grant for scientific research MIS F453020F, J. C. M. M.). Computational have been provided by the “Consortium des Équipements de Calcul Intensif” (CÉCI), funded by the “Fonds de la Recherche Scientifique de Belgique” (F.R.S.-FNRS) under Grant No. 2.5020.11. T. T. is a FRIA PhD fellow. K. V. H. thanks the Research Foundation – Flanders (FWO) (projects AUGE/11/029 and G099319N) for funding.Notes and references
- M. Hariharan, R. J. Motekaitis and A. E. Martell, Synthesis of dipeptides of aminophosphonic acids, J. Org. Chem., 1975, 40, 470–473 CrossRef CAS PubMed.
- S. J. Hecker and M. D. Erion, Prodrugs of Phosphates and Phosphonates, J. Med. Chem., 2008, 51, 2328–2345 CrossRef CAS PubMed.
- V. Kukhar and H. Hudson, Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity, John Wiley & Sons, 2000 Search PubMed.
- P. R. Varga and G. Keglevich, Synthesis of α-Aminophosphonates and Related Derivatives; The Last Decade of the Kabachnik-Fields Reaction, Molecules, 2021, 26, 2511 CrossRef CAS PubMed.
- G. Keglevich and A. Szekrenyi, Eco-Friendly Accomplishment of the Extended Kabachnik - Fields Reaction; a Solvent- and Catalyst-Free Microwave-Assisted Synthesis of α-Aminophosphonates and α-Aminophosphine Oxides, Lett. Org. Chem., 2008, 5, 616–622 CrossRef CAS.
- B. C. Ranu and A. Hajra, A simple and green procedure for the synthesis of α-aminophosphonate by a one-pot three-component condensation of carbonyl compound, amine and diethyl phosphite without solvent and catalyst, Green Chem., 2002, 4, 551–554 RSC.
- X.-J. Mu, M.-Y. Lei, J.-P. Zou and W. Zhang, Microwave-assisted solvent-free and catalyst-free Kabachnik-Fields reactions for α-amino phosphonates, Tetrahedron Lett., 2006, 47, 1125–1127 CrossRef CAS.
- M. Zahouily, A. Elmakssoudi, A. Mezdar, A. Rayadh and S. Sebti, Uncatalysed Preparation of α-Amino Phosphonates under Solvent Free Conditions, J. Chem. Res., 2005, 2005, 324–327 CrossRef.
- I. Essid and S. Touil, Efficient and Green One-Pot Multi-Component Synthesis of α-Aminophosphonates Catalyzed by Zinc Triflate, Curr. Org. Synth., 2017, 14, 272–278 CrossRef CAS.
- M. M. Kabachnik, E. V. Zobnina and I. P. Beletskaya, Catalyst-Free Microwave-Assisted Synthesis of α-Aminophosphonates in a Three-Component System: R1C(O)R2-(EtO)2P(O)H-RNH2, Synlett, 2005, 2005, 1393–1396 CrossRef.
- A. N. Pudovik and I. V. Konovalova, Addition Reactions of Esters of Phosphorus(III) Acids with Unsaturated Systems, Synthesis, 1979, 1979, 81–96 CrossRef.
- M. Ordóñez, J. L. Viveros–Ceballos and I. R. Estudillo, Amino Acid - New Insights and Roles in Plant and Animal, Stereoselective Synthesis of α-Aminophosphonic Acids through Pudovik and Kabachnik-Fields Reaction, Toshiki Asao and Md. Asaduzzaman, IntechOpen, 2017.
- K. R. Naidu, K. R. K. Kumar Reddy, C. V. Kumar, C. N. Raju and D. D. Sharma, A facile catalyst-free Pudovik reaction for the synthesis of α-amino phosphonates, S. Afr. J. Chem., 2009, 62, 185–188 CAS.
- O. I. Kolodiazhnyi, S. Sheiko and E. V. Grishkun, C3-symmetric trialkyl phosphites as starting compounds of asymmetric synthesis, Heteroat. Chem., 2000, 11, 138–143 CrossRef CAS.
- F. Palacios, T. K. Olszewski and J. Vicario, Diastereoselective hydrophosphonylation of imines using (R,R)-TADDOL phosphite. Asymmetric synthesis of α-aminophosphonic acid derivatives, Org. Biomol. Chem., 2010, 8, 4255–4258 RSC.
- A. S. Demir, C. Tanyeli, O. Şeşenoǧlu, S. Demiç and O. O. Evin, A simple synthesis of 1-aminophosphonic acids from 1-hydroxyiminophosphonates with NaBH4 in the presence of transition metal compounds, Tetrahedron Lett., 1996, 37, 407–410 CrossRef CAS.
- R. Rogers and M. Stern, An Improved Synthesis of the Phosphonic Acid Analog of Tryptophan, Synlett, 1992, 708 CrossRef CAS.
- J. L. Viveros-Ceballos, F. J. Sayago, C. Cativiela and M. Ordóñez, First Practical and Efficient Synthesis of 3-Phosphorylated β-Carboline Derivatives Using the Pictet-Spengler Reaction, Eur. J. Org. Chem., 2015, 2015, 1084–1091 CrossRef CAS.
- J. L. Viveros-Ceballos, M. Ordóñez, F. J. Sayago, A. I. Jiménez and C. Cativiela, First Synthesis of (R)- and (S,)-1,2,3,4-Tetrahydroisoquinoline-3-phosphonic Acid (TicP) Using a Pictet-Spengler Reaction, Eur. J. Org. Chem., 2016, 2016, 2711–2719 CrossRef CAS.
- P. Patel and S. Chang, N-Substituted Hydroxylamines as Synthetically Versatile Amino Sources in the Iridium-Catalyzed Mild C-H Amidation Reaction, Org. Lett., 2014, 16, 3328–3331 CrossRef CAS PubMed.
- E. J. Yoo, S. Ma, T.-S. Mei, K. S. L. Chan and J.-Q. Yu, Pd-Catalyzed Intermolecular C-H Amination with Alkylamines, J. Am. Chem. Soc., 2011, 133, 7652–7655 CrossRef CAS.
- N. Matsuda, K. Hirano, T. Satoh and M. Miura, Copper-Catalyzed Direct Amination of Electron-Deficient Arenes with Hydrodryxlamines, Org. Lett., 2011, 13, 2860–2863 CrossRef CAS.
- L. Fan, J. Hao, J. Yu, X. Ma, J. Liu and X. Luan, Hydroxylamines As Bifunctional Single-Nitrogen Sources for the Rapid Assembly of Diverse Tricyclic Indole Scaffolds, J. Am. Chem. Soc., 2020, 142, 6698–6707 CrossRef CAS PubMed.
- E. Erdik, Electrophilic α-amination of carbonyl compounds, Tetrahedron, 2004, 60, 8747–8782 CrossRef CAS.
- M.-L. Louillat and F. W. Patureau, Oxidative C-H amination reactions, Chem. Soc. Rev., 2014, 43, 901–910 RSC.
- Y. Park, Y. Kim and S. Chang, Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS.
- J. Davies, S. P. Morcillo, J. J. Douglas and D. Leonori, Hydroxylamine Derivatives as Nitrogen-Radical Precursors in Visible-Light Photochemistry, Chem. – Eur. J., 2018, 24, 12154–12163 CrossRef CAS.
- A. El-Faham and F. Albericio, Synthesis and Application of N-Hydroxylamine Derivatives as Potential Replacements for HOBt, Eur. J. Org. Chem., 2009, 2009, 1499–1501 CrossRef.
- L. Miret-Casals, A. Baelo, E. Julián, J. Astola, A. Lobo-Ruiz, F. Albericio and E. Torrents, Hydroxylamine Derivatives as a New Paradigm in the Search of Antibacterial Agents, ACS Omega, 2018, 3, 17057–17069 CrossRef CAS.
- N. S. Goulioukina, I. A. Shergold, G. N. Bondarenko, M. M. Ilyin, V. A. Davankov and I. P. Beletskaya, Palladium-Catalyzed Asymmetric Hydrogenation of N-Hydroxy-α-imino Phosphonates Using Brønsted Acid as Activator: The First Catalytic Enantioselective Approach to Chiral N-Hydroxy-α-amino Phosphonates, Adv. Synth. Catal., 2012, 354, 2727–2733 CrossRef CAS.
- J. Mas-Roselló, C. J. Cope, E. Tan, B. Pinson, A. Robinson, T. Smejkal and N. Cramer, Iridium-Catalyzed Acid-Assisted Hydrogenation of Oximes to Hydroxylamines, Angew. Chem., Int. Ed., 2021, 60, 15524–15532 CrossRef.
- H. Dollt, P. Hammann and S. Blechert, Synthesis of (+)-(S)-Streptenol A and Biomimetic Synthesis of (2R,4S)- and (2S,4S)-2-(Pent-3-enyl)piperidin-4-ol, Helv. Chim. Acta, 1999, 82, 1111–1121 CrossRef CAS.
- E. Breuer, R. Karaman, H. Leader and A. Goldblum, Phosphorylation of alcohols through the acid-catalysed fragmentation of α-oxyiminophosphonates, J. Chem. Soc., Chem. Commun., 1987, 671–672 RSC.
- E. Breuer, R. Karaman, D. Gibson, H. Leader and A. Goldblum, α-Hydroxyiminophosphonic acids. New types of phosphorylating agents through monomeric metaphosphate, J. Chem. Soc., Chem. Commun., 1988, 504–506 RSC.
- E. Breuer, R. Karaman, A. Goldblum, D. Gibson, H. Leader, B. V. L. Potter and J. H. Cummins, α-Oxyiminophosphonates: chemical and physical properties. Reactions, theoretical calculations, and X-ray crystal structures of (E) and (Z)-dimethyl α-hydroxyiminobenzylphosphonates, J. Chem. Soc., Perkin Trans. 1, 1988, 3047–3057 RSC.
- E. Breuer, M. Safadi, M. Chorev, A. Vineze and P. Bel, Synthesis and characterization of amino protected peptides derived from amino-α-hydroxyiminophosphonates, Tetrahedron, 1991, 47, 1257–1264 CrossRef CAS.
- E. Breuer and M. Mahajna, Synthesis and reactions of 2,2,2-trihaloethyl α-hydroxyiminobenzylphosphonates. The influence of the ester group on the chemistry of phosphonates, Heteroat. Chem., 1992, 3, 251–260 CrossRef CAS.
- E. Breuer, Fragmentations and Rearrangements of α-Hydroxyiminoalkylphosphonates, Curr. Org. Chem., 2006, 10, 2357–2370 CrossRef CAS.
- J. Katzhendler, R. Karaman, D. Gibson, E. Breuer and H. Leader, Fragmentation of methyl hydrogen α-hydroxyiminophosphonates to monomeric methyl metaphosphate: stereochemistry and mechanism, J. Chem. Soc., Perkin Trans. 2, 1989, 589–594 RSC.
- M. Mahajna and E. Breuer, 2-Cyanoethyl and p-nitrophenethyl α-hydroxyimino- and α-methoxyimino-phosphonates: potential precursors of metaphosphate anion, J. Chem. Soc., Perkin Trans. 1, 1994, 1847–1849 RSC.
- J. Vicario, C. Alonso, J. M. de los Santos and F. Palacios, α-Hydroxyiminophosphonate, -phosphinate and -phosphonium Derivatives, Curr. Org. Synth., 2010, 7, 628–649 CrossRef CAS.
- F. W. McLafferty, Tandem Mass Spectrometry, Science, 1981, 214, 280–287 CrossRef CAS PubMed.
- C. M. Nichols, J. N. Dodds, B. S. Rose, J. A. Picache, C. B. Morris, S. G. Codreanu, J. C. May, S. D. Sherrod and J. A. McLean, Untargeted Molecular Discovery in Primary Metabolism: Collision Cross Section as a Molecular Descriptor in Ion Mobility-Mass Spectrometry, Anal. Chem., 2018, 90, 14484–14492 CrossRef CAS PubMed.
- I. Czerwinska, A. Kulesza, C. Choi, F. Chirot, A.-L. Simon, J. Far, C. Kune, E. de Pauw and P. Dugourd, Supramolecular influence on cis-trans isomerization probed by ion mobility spectrometry, Phys. Chem. Chem. Phys., 2016, 18, 32331–32336 RSC.
- A. J. Creese and H. J. Cooper, Separation of cis and trans Isomers of Polyproline by FAIMS Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2016, 27, 2071–2074 CrossRef CAS PubMed.
- S. Poyer, C. M. Choi, C. Deo, N. Bogliotti, J. Xie, P. Dugourd, F. Chirot and J.-Y. Salpin, Kinetic study of azobenzene E/Z isomerization using ion mobility-mass spectrometry and liquid chromatography-UV detection, Analyst, 2020, 145, 4012–4020 RSC.
- X. Zheng, R. S. Renslow, M. M. Makola, I. K. Webb, L. Deng, D. G. Thomas, N. Govind, Y. M. Ibrahim, M. M. Kabanda, I. A. Dubery, H. M. Heyman, R. D. Smith, N. E. Madala and E. S. Baker, Structural Elucidation of cis/trans Dicaffeoylquinic Acid Photoisomerization Using Ion Mobility Spectrometry-Mass Spectrometry, J. Phys. Chem. Lett., 2017, 8, 1381–1388 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Z. Hassen, A. Ben Akacha and H. Zantour, Synthese de Quelques α-Cetophosphonates Comportant des Hydrogenes Mobiles en Position α: Caracteristiques Spectroscopiques IR, RMN 1H, 13C, 31P et Reactivites vis-a-vis des Amines et des Derivees de L'hydrazine, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 2241–2253 CrossRef CAS.
- C. C. Tam, K. L. Mattocks and M. Tishler, Enol-keto tautomerism of alpha-ketophosphonates, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 3301–3304 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental procedures, off-line analysis with structural assignments (NMR, X-ray diffraction and Mass Spectrometry, breakdown curves) and computations. CCDC 2115990–2115991. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo01564h |
| ‡ Equal contributions. |
| This journal is © the Partner Organisations 2022 |