
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCore–shell Pd–P@Pt–Ni nanoparticles with enhanced activity and durability as anode electrocatalyst for methanol oxidation reaction†
Jiangbin Guo a,
Man Zhangb,
Jing Xu*a,
Jun Fanga,
Shuiyuan Luoa and
Chaolong Yang*c
a,
Man Zhangb,
Jing Xu*a,
Jun Fanga,
Shuiyuan Luoa and
Chaolong Yang*c
aCollege of Chemical Engineering and Materials Science, Quanzhou Normal University, Quanzhou 362000, P. R. China. E-mail: jingxu@qztc.edu.cn
bState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200433, P. R. China
cSchool of Materials Science and Engineering, Chongqing University of Technology, Chongqing 400054, P. R. China. E-mail: yclzjun@163.com
First published on 14th January 2022
Abstract
Pd–P@Pt–Ni core–shell nanoparticles, which consisted of a Pd–P alloy as a core and Pt–Ni thin layer as a shell, were explored as electrocatalysts for methanol oxidation reaction. The crystallographic information and the electronic properties were fully investigated by X-ray diffraction and X-ray photoelectron spectroscopy. In the methanol electrooxidation reaction, the particles showed high catalytic activity and strong resistance to the poisoning carbonaceous species in comparison with those of commercial Pt/C and the as-prepared Pt/C catalysts. The excellent durability was demonstrated by electrochemically active surface area loss and chronoamperometric measurements. These results would be due to the enhanced catalytic properties of Pt by the double synergistic effects from the core part and the nickel species in the shell part.
Introduction
Considering the limited amounts of fossil fuels and the environmental issues, there is an urgent need to develop new technology with clean and sustainable energy sources.1–3 Recently, many efforts have been made on direct methanol fuel cells (DMFCs), which are considered to be a promising candidate for energy conversion devices.4,5 In this system, methanol is utilized as the fuel molecules to be oxidized at the anodic compartment because it is abundant, renewable from wood alcohol, easily stored as well as transported, and especially has a high energy density.6 Platinum is a commonly used and most effective catalyst for methanol oxidation reaction (MOR),7 but there are some shortcomings, which should be addressed before its practical application. One is the high cost and scarcity of Pt related to the commercialization of DMFCs. In addition, Pt is easily poisoned by adsorbed intermediates such as CO, resulting in poor kinetics of methanol electrooxidation.8 Therefore, the synthesis of Pt-based electrocatalysts with high catalytic performance and low cost is extremely crucial in the field of fuel cells.It is believed that the composition, shape, and structure of Pt-based nanomaterials are key factors that determine their electrocatalytic properties.9–11 Until now, the introduction of less expensive metals, including Pd,12,13 Cu,14,15 Ni,16–19 Ru,20,21 Au,22,23 to fabricate various bi- or multi-component electrocatalysts is an effective approach to not only reduce Pt loading, but also improve the activity and durability. Another promising method is to synthesize the core–shell structure by depositing Pt as thin shells on non-Pt cores.24 In these structures, the usage of Pt is obviously reduced, while the goal of enhancing electrocatalytic properties can be achieved by the synergistic structural and electronic effects between core and shell materials.25–27 Inspired by the benefits of these two strategies, many research efforts have been dedicated to fabricating nanoparticles (NPs) with multi-composition shells like Au@CuPt,28 Pd@Pt1.8Ni,29 Au@NimPt2.30
Among the investigated anode electrocatalysts, one of the most inspiring findings is to combine Pt with a 3d transition metal Ni to prepare a series of Pt-based nanomaterials. In 2007, Stamenkovic and co-workers have reported a novel Pt3Ni catalyst with superior specific activity for oxygen reduction reaction (ORR), in which a 90-fold improvement over the commercial Pt/C catalyst.31 They found that the weak interaction between the Pt surface atoms and nonreactive oxygenated species induced by the altered Pt electronic properties makes it more active. The excellent electrocatalytic properties of PtNi system can also be observed in MOR. For instance, Zhang et al. presented a simple one-pot solvothermal method to fabricate PtNi colloidal nanocrystal clusters (CNCs), and found that the PtNi CNCs possess a higher electrooxidation activity than those of PtNi nanocrystals and Pt/C catalysts.32 By increasing the molar ratio of Pt/Ni, Chen et al. have synthesized PtNi3 CNCs with more grain boundary and interspace as the promising electrocatalyst for MOR.33 Shen and co-workers synthesized Pt–Ni cross-double dumbbell-like nanostructures exhibiting high activity and stability, owing to the high specific surface area and synergistic effect between different compositions.34 These results suggest that the core–shell structure composed of Pt–Ni shell would be promising.
We have recently developed Pd–P@Pt–Ni nanoparticles as the electrocatalysts for hydrogen-evolution reaction.35 The existences of an amorphous core and a low-crystalline shell have the favorable effect on the catalytic properties. In this work, the performances of Pd–P@Pt–Ni core–shell NPs in MOR are also studied. Compared with commercial Pt/C and as-prepared Pt/C, Pd–P@Pt–Ni core–shell NPs show remarkable catalytic activity and excellent durability in MOR.
Experimental section
Chemicals and materials
Palladium acetylacetonate (Pd(acac)2, 99%), platinum acetylacetonate (Pt(acac)2, 97%), triphenylphosphine (TPP, 99%), borane tert-butylamine complex (BTB, 97%), trioctylphosphine oxide (TOPO, technical grade, 90%), oleylamine (OAm, technical grade, 70%), tetrabutylammonium bromide (TBAB, 99%) and Nafion perfluorinated ion-exchange resin (5 wt% solution in a lower aliphatic alcohol/H2O mixture that contains 15–20% water) were all purchased from Sigma-Aldrich. Acetic acid (CH3COOH, 99.7%) and methanol (CH3OH, 99%) were purchased from Wako Pure Chemical Industries Ltd. Commercial Pt/C (50% platinum on Vulcan XC-72) was purchased from Fuel Cell Store. Carbon black (Vulcan XC-72) was purchased from Moubic Inc. Polishing alumina (0.05 μm) was purchased from BAS Inc. All the chemicals were used as received without further purification.Synthesis of Pd–P@Pt–Ni NPs
30.5 mg of Pd(acac)2 (0.1 mmol), 77 mg of Ni(acac)2 (0.3 mmol), 232 mg of TPP (0.88 mmol), 322 mg of TBAB (1 mmol), 1.16 g of TOPO (3 mmol), and 6.5 mL of OAm were mixed under a nitrogen flow at 50 °C for 25 min. The formed solution was heated to 220 °C at a constant heating rate of 12 °C min−1 and kept at 220 °C for 30 min. Then, the mixture was cooled to room temperature, followed by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 8 min and washed with ethanol. Finally, the Pd–P@Ni NPs were collected and dried at room temperature for 1 h.
000 rpm for 8 min and washed with ethanol. Finally, the Pd–P@Ni NPs were collected and dried at room temperature for 1 h.
The obtained Pd–P@Ni NPs were dispersed in 17 mL OAm and then 322 mg TBAB (1 mmol), 1.16 g of TOPO (3 mmol), 51.1 mg of Pt(acac)2 (0.13 mmol) were added into the mixture, followed by deaerating (nitrogen gas) for 25 min at room temperature. The resulting solution was slowly (2–5 °C min−1) heated to 200 °C and kept at 200 °C for 30 min. After cooling to room temperature, the product was collected by centrifugation and washed with ethanol twice.
Synthesis of Pt NPs
39.3 mg of Pt(acac)2 (0.1 mmol) was dispersed in 6 mL OAm and then 322 mg TBAB (1 mmol), 1.16 g of TOPO (3 mmol) were added into the mixture, followed by deaerating (nitrogen gas) for 25 min at room temperature. The resulting solution was slowly (2–5 °C min−1) heated to 200 °C and kept at 200 °C for 30 min. After cooling to room temperature, the product Pt NPs were collected by centrifugation and washed with ethanol twice.Synthesis of Pd–P@Pt–Ni/C and Pt/C
Vulcan XC-72 (112 mg) in hexane (230 mL) was sonicated for 30 min, and a suspension of different nanoparticles (Pd–P@Pt–Ni NPs and Pt NPs) in hexane was added, respectively. The resultant mixtures were sonicated again for further 30 min to get the homogeneous solutions. After evaporating the hexane, the remained solid materials were dispersed in acetic acid (100 mL) and heated at 70 °C under a nitrogen gas atmosphere for 12 h. The resulting materials were separated by centrifugation, washed with ethanol three times, and dried to afford the NPs as electrocatalysts.Electrochemical measurements
The electrocatalytic activities of the samples were evaluated on an Iviumstat electrochemical analyzer (Ivium Technologies, Japan) using a three-electrode cell. The system included a Ag/AgCl (saturated KCl) electrode as the reference electrode, a pure platinum sheet as the counter electrode, and a modified glassy carbon electrode (GCE) with a surface area of 0.196 cm2 as the working electrode. All potentials were converted to values relative to a reversible hydrogen electrode (RHE) according to the formula E(RHE) = E(Ag/AgCl) + 0.199 volts + 0.05916pH volts. For preparation of working electrode, the catalyst ink was prepared by mixing catalyst (2 mg), Nafion (4.6 μL), distilled water (164 μL), and ethanol (164 μL), followed by sonication for 20 min to get a homogeneous dispersion. Then, the modified GCE was coated with 8 μL of the catalyst ink and dried naturally at room temperature. The cyclic voltammetry measurements were recorded in N2-saturated 0.1 M HClO4 between −0.06 V and 1.14 V at a scan rate of 50 mV s−1. The electrochemically active surface area (ECSA) was estimated by integrating the charge associated with the adsorption of hydrogen between −0.06 V and 0.3 V on metal nanoparticles surface. Methanol oxidation reaction (MOR) was performed with a scan rate of 50 mV s−1 in a solution containing 0.5 M H2SO4 + 1 M CH3OH at potential range between 0.2 V and 1.2 V. To investigate the long-term stability, the tests were carried out at a scan rate of 50 mV s−1 in N2-saturated 0.1 M HClO4 for 500 cycles. The chronoamperometry (CA) measurements were recorded in 0.5 M H2SO4 + 1 M CH3OH at 0.85 V for 3600 s.Results and discussion
Synthesis
According to our previous work, the Pd–P@Pt–Ni NPs were repeated and the transmission electron microscopy (TEM) images are shown in Fig. S1 (ESI†). Energy dispersive spectroscopy (EDS) in scanning transmission electron microscopy (STEM) (Fig. S2, ESI†) clearly show that the distributions of elements are not homogeneous. While Pd and P are rich in the core domain, Pt and Ni are distributed in the shell structure.For fabrication of Pd–P@Pt–Ni NPs, the synthetic condition is optimized by tuning the amounts of Pt precursor and OAm (Table S1, ESI†). According to the TEM images in Fig. S3 (ESI†), it can be seen that the undesired Pt NPs are also obtained when the amounts of Pt precursor and OAm are not appropriate. The coating of Pd–P@Ni NPs with 0.13 mmol of Pt(acac)2 in 17 mL of OAm is the best condition, and the desired core–shell NPs are produced efficiently without any by-products.
Crystallography
The crystalline structures of Pd–P@Pt–Ni/C, the as-prepared Pt/C, commercial Pt/C and Ni powder were characterized by XRD (Fig. 1). The results indicate that the core–shell NPs have a face-centered cubic (fcc) structure, and these diffraction peaks above 30° correspond to the (111), (200), (220) and (311) planes. In addition, the peak at around 24.9° can be attributed to (002) plane of hexagonal carbon structure. It is noticeable that the diffraction peaks except for C (002) are located between those of pure Pt and Ni, revealing the formation of an alloy shell structure. Table S2 (ESI†) lists the peak position, d-spacing, and lattice parameter for (111) plane obtained from the XRD data of Pd–P@Pt–Ni/C, commercial Pt/C and Ni powder. Compared to the pure Pt, the higher angle shift of the (111) peak with a smaller d-spacing for Pd–P@Pt–Ni/C NPs would be only resulting from the insertion of Ni atoms into the Pt lattice.36 Combining the lattice parameters of Pt and Ni with the Pt/Ni ratio (3.63/1) determined by inductively coupled plasma mass spectrometry (ICP-MS), the lattice parameter of Pd–P@Pt–Ni/C NPs can be calculated as 0.3840 nm from Vegard's law,37 which is smaller than the value (0.3887 nm) based on Bragg's law.38 The result suggests that some of the nickel is in a non-alloyed form (as metal or as NiO species). Although no characteristic peaks of metallic Ni or Ni oxides are observed, their presence cannot be discarded because they may exist in a small amount or in an amorphous structure.39 It has been reported that the alloying degree of a PtNi catalyst (xa) is defined as the ratio of alloyed Ni atoms to total Ni in the material, which is estimated through eqn (1) and (2):40,41
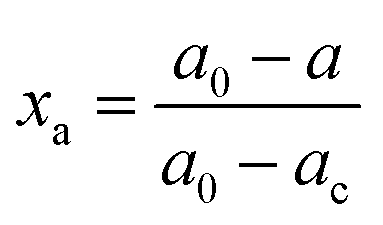 | (1) |
| ac = a0 − kxNi | (2) |
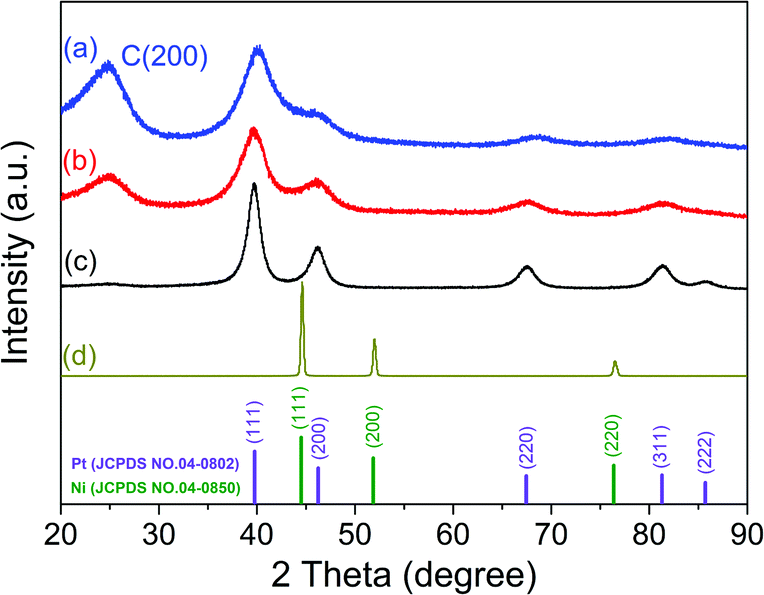 | ||
| Fig. 1 XRD patterns of (a) Pd–P@Pt–Ni/C NPs, (b) the as-prepared Pt/C, (c) commercial Pt/C and (d) commercial Ni powder. | ||
X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy were carried out to probe the electronic properties of Pd–P@Pt–Ni/C NPs (Fig. 2). As shown in Fig. 2a, the raw data of C 1s is fitted with four individual Gaussian curves, which correspond to hydrocarbons (C–H and C–C, 284.6 eV), hydroxyls or esters (C–O, 286.1 eV), carbonyls (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 287.3 eV), and carboxylic acids or esters (O–CO, 290.4 eV).43 The Pd 3d spectrum of Pd–P@Pt–Ni/C shows one doublet. The binding energies centered at 335.9 eV (Pd 3d5/2) and 341.2 eV (Pd 3d3/2) are attributed to the characteristic signature of metallic Pd (Fig. 2b). A positive shift (0.5 eV) of the Pd 3d5/2 peak relative to the pure Pd peak (335.4 eV) is observed in the core–shell NPs.44 The Ni 2p and P 2p spectra cannot be detected due to the small amounts of Ni and P. Importantly, platinum plays a critical role in methanol oxidation reaction, and its electronic property will be discussed in detail as well as compared with that of commercial Pt/C. The Pt 4f spectrum features two peaks at 71.44 eV and 74.77 eV, assigned to Pt 4f7/2 and Pt 4f5/2 of Pt0, respectively (Fig. 2c). The XPS result shows no signature for any oxidized Pt species in Pd–P@Pt–Ni/C. Furthermore, the comparison of Pt 4f7/2 between Pd–P@Pt–Ni/C and commercial Pt/C is illustrated in Fig. 2d, and they are identified as 71.44 and 71.54 eV, respectively. Considering the standard data of Pt0 (71.2 eV) in either bulk Pt or platinized carbon electrodes,45 the binding energy of commercial Pt/C NPs is shifted to higher value due to a contribution from metal–support interaction or small cluster-size effects.46,47 However, it is observed that the Pt binding energy in as-synthesized Pd–P@Pt–Ni/C NPs is negatively shifted by 0.1 eV relative to commercial Pt/C. It is found that these results are different (the absence of oxidized Pt species) or even opposite (the negative shift of the Pt 4f7/2 peak) with our previous work,48 which may be attributed to the effects of nickel on platinum. On one hand, the presence of Ni could prevent the coexisting surface Pt from air-oxidation due to the larger oxygen affinity of Ni as compared to Pt.26 On the other hand, the shell part consists of Pt and Ni with different ionization energies (Pt: 9.02 eV; Ni: 7.63 eV),49 resulting in the electron donation of Ni to Pt. The change in the electronic properties of the metals can improve the electrocatalytic performance.
O, 287.3 eV), and carboxylic acids or esters (O–CO, 290.4 eV).43 The Pd 3d spectrum of Pd–P@Pt–Ni/C shows one doublet. The binding energies centered at 335.9 eV (Pd 3d5/2) and 341.2 eV (Pd 3d3/2) are attributed to the characteristic signature of metallic Pd (Fig. 2b). A positive shift (0.5 eV) of the Pd 3d5/2 peak relative to the pure Pd peak (335.4 eV) is observed in the core–shell NPs.44 The Ni 2p and P 2p spectra cannot be detected due to the small amounts of Ni and P. Importantly, platinum plays a critical role in methanol oxidation reaction, and its electronic property will be discussed in detail as well as compared with that of commercial Pt/C. The Pt 4f spectrum features two peaks at 71.44 eV and 74.77 eV, assigned to Pt 4f7/2 and Pt 4f5/2 of Pt0, respectively (Fig. 2c). The XPS result shows no signature for any oxidized Pt species in Pd–P@Pt–Ni/C. Furthermore, the comparison of Pt 4f7/2 between Pd–P@Pt–Ni/C and commercial Pt/C is illustrated in Fig. 2d, and they are identified as 71.44 and 71.54 eV, respectively. Considering the standard data of Pt0 (71.2 eV) in either bulk Pt or platinized carbon electrodes,45 the binding energy of commercial Pt/C NPs is shifted to higher value due to a contribution from metal–support interaction or small cluster-size effects.46,47 However, it is observed that the Pt binding energy in as-synthesized Pd–P@Pt–Ni/C NPs is negatively shifted by 0.1 eV relative to commercial Pt/C. It is found that these results are different (the absence of oxidized Pt species) or even opposite (the negative shift of the Pt 4f7/2 peak) with our previous work,48 which may be attributed to the effects of nickel on platinum. On one hand, the presence of Ni could prevent the coexisting surface Pt from air-oxidation due to the larger oxygen affinity of Ni as compared to Pt.26 On the other hand, the shell part consists of Pt and Ni with different ionization energies (Pt: 9.02 eV; Ni: 7.63 eV),49 resulting in the electron donation of Ni to Pt. The change in the electronic properties of the metals can improve the electrocatalytic performance.
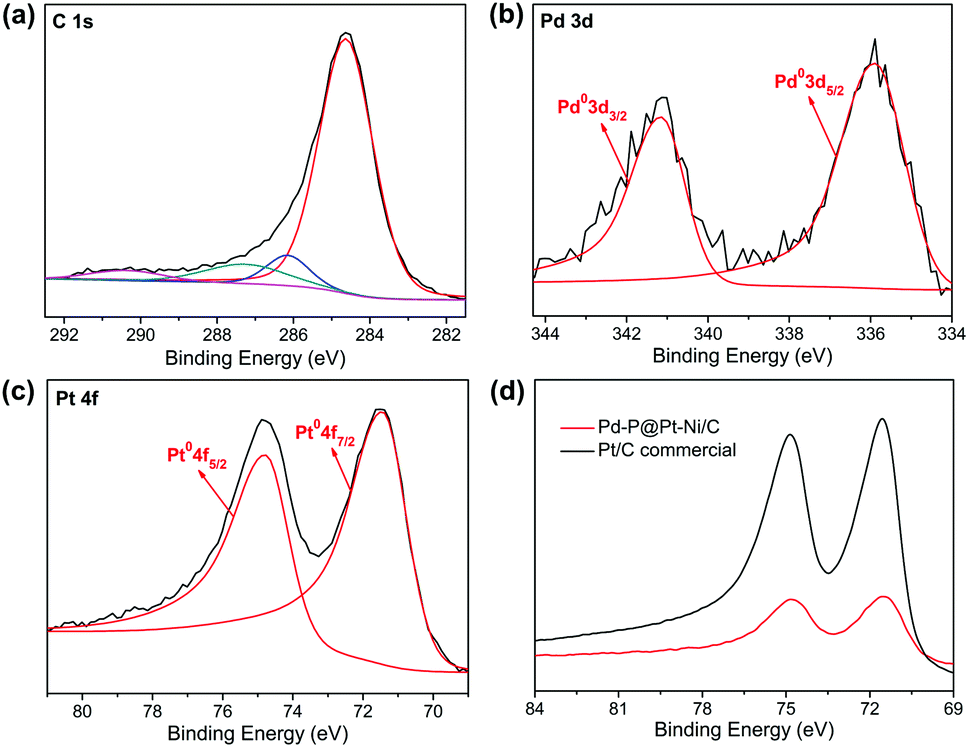 | ||
| Fig. 2 XPS spectra of Pd–P@Pt–Ni/C in (a) C 1s, (b) Pd 3d and (c) Pt 4f regions; (d) representative XPS spectra of Pt 4f in Pd–P@Pt–Ni/C NPs and commercial Pt/C. | ||
Electrocatalytic activity and durability
Determination of the electrochemically active surface area (ECSA) of Pd–P@Pt–Ni/C core–shell NPs was conducted by the cyclic voltammetry (CV), together with commercial Pt/C and as-prepared Pt/C. The CV curves of the samples were recorded in N2-saturated 0.1 M HClO4 solution at a scan rate of 50 mV s−1 (Fig. 3). The ECSA can be obtained according to the equation ECSA = Q/(0.21 × WPt), where WPt represents the amount of Pt loading (mg cm−2) on the electrode, Q is the charge by integrating the hydrogen adsorption/desorption region after double-layer correction (mC), and 0.21 is the charge required for monolayer adsorption of hydrogen on a Pt surface (mC cm−2).50,51 As a result, the ECSA is calculated as 20.9 m2 gPt−1 for Pd–P@Pt–Ni/C, 33.8 m2 gPt−1 for commercial Pt/C, and 67.4 m2 gPt−1 for the as-prepared Pt/C. The as-prepared Pt/C NPs show the highest ECSA probably due to the smaller size of nanoparticles and good dispersibility on the activated carbon (Fig. S4, ESI†).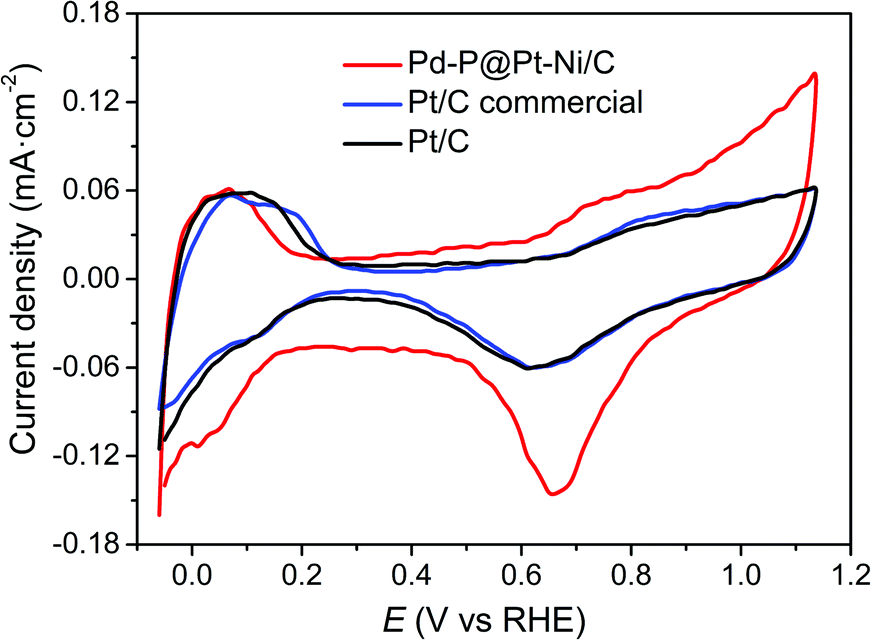 | ||
| Fig. 3 Cyclic voltammetry profiles of Pd–P@Pt–Ni/C NPs, commercial Pt/C and the as-prepared Pt/C in N2-saturated 0.1 M HClO4 solution. The sweep rate is 50 mV s−1. | ||
Based on many literature,34,52–56 the electrocatalytic properties of Pt-based catalysts were studied in 1 M CH3OH. In this work, the electrocatalytic activities of Pd–P@Pt–Ni/C, commercial Pt/C, and as-prepared Pt/C for methanol oxidation reaction are also investigated in 0.5 M H2SO4 + 1 M CH3OH solution at a sweep rate of 50 mV s−1, as shown in Fig. 4. All of the CV curves exhibit a methanol oxidation peak in the forward scan as well as another peak in the backward scan, which is commonly attributed to the reactivation of oxidized platinum.57 With regard to specific activity (normalizing mass activity with the ECSA value), the current density of Pd–P@Pt–Ni/C (1.56 mA cm−2) is much higher than those of commercial Pt/C (1.16 mA cm−2) and the as-prepared Pt/C (1.18 mA cm−2). Except for anodic peak, the oxidation current at a given potential is a significant factor for comparing the relative activity.58 For example, the current density of Pd–P@Pt–Ni/C core–shell NPs is 0.47 mA cm−2 at 0.7 V, which is ∼2 and ∼1.6 times higher than those of commercial Pt/C and the as-prepared Pt/C, respectively. The result implies that the core–shell NPs are more effective electrocatalysts. Moreover, it is worth noting that the potential of anodic peak is negatively shifted by ca. 0.14 V and 0.08 V compared with commercial Pt/C and the as-prepared Pt/C, which suggests that the oxidation of CH3OH to CO2 is much easier on the surface of Pd–P@Pt–Ni/C NPs.59 In addition, many publications have reported that the peak current ratio of the forward scan (If) to the backward scan (Ib) can be used to evaluate the tolerance of the catalyst to carbonaceous species accumulation in the direct methanol fuel cell.60,61 During the forward scan, the current density increases sharply due to the oxidation of adsorbed methanol to CO2.60 While in the reverse scan, the anodic peak is associated with the removal of the incompletely oxidized carbonaceous species formed in the forward scan.61 Thus, a higher If/Ib ratio indicates the more methanol molecules go through the complete oxidation process with less accumulation of residual carbon species. From Fig. 4, it can be seen that If/Ib of Pd–P@Pt–Ni/C core–shell catalyst is 1.00, which is significantly higher than those of commercial Pt/C (0.58) and the as-prepared Pt/C (0.61). The relatively larger If/Ib ratio of Pd–P@Pt–Ni/C core–shell catalyst indicates a stronger resistance to the poisoning of carbonaceous species. In addition, the electrocatalytic activity of Pd–P@Pt–Ni/C at high concentration of methanol is also investigated in 0.5 M H2SO4 + 10 M CH3OH solution (Fig. S5, ESI†). The result suggests that the core–shell NPs exhibit the lower specific activity as well as the higher peak potential, which may be attributed to the saturation of Pt active sites and poisoning the electrode surface with adsorbed intermediates.62
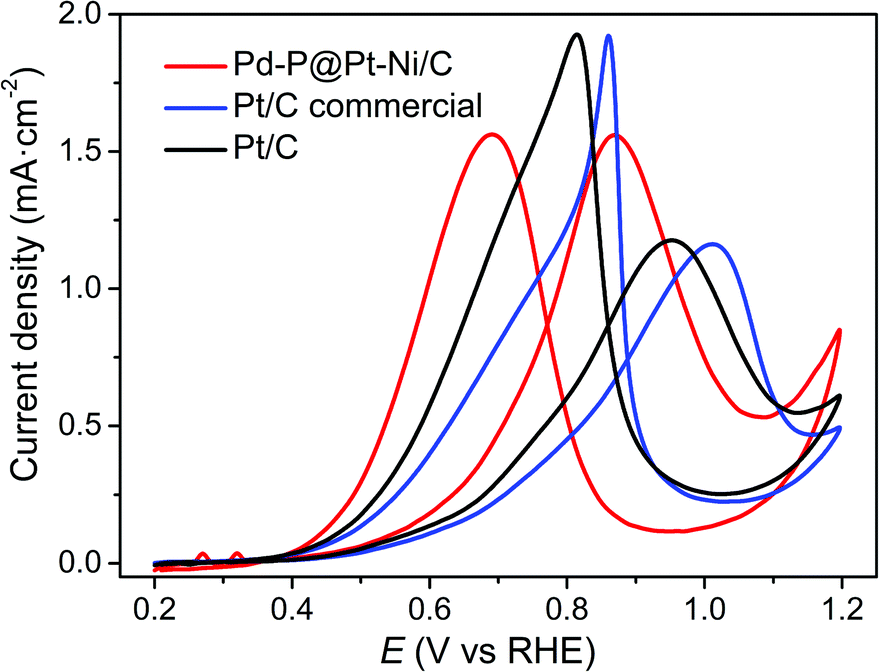 | ||
| Fig. 4 Cyclic voltammetry profiles of Pd–P@Pt–Ni/C NPs, commercial Pt/C and the as-prepared Pt/C in 0.5 M H2SO4 + 1 M CH3OH solution. The sweep rate is 50 mV s−1. | ||
Next, the attenuation of ECSA after long-term cyclic voltammetry was examined to see the stability of electrocatalysts.63 Fig. 5a–c shows the CV curves of Pd–P@Pt–Ni/C, commercial Pt/C, and the as-prepared Pt/C with increasing cycle number up to 500 cycles in N2-saturated 0.1 M HClO4 at 50 mV s−1, and the ECSA losses are summarized in Fig. 5d. The significant shrinkage is observed in the CV curves of commercial Pt/C and as-prepared Pt/C with more than 10% of ECSA loss in both cases. In contrast, there is no obvious change in the hydrogen adsorption/desorption region between −0.06 V and 0.3 V for Pd–P@Pt–Ni/C, and the 98% of ECSA is remained after 500 cycles. After the stability test, the core–shell NPs retain spherical morphology and the less aggregation of NPs is observed (Fig. S6(a) and (b), ESI†). Moreover, there are no obvious changes in the crystalline structure and electronic property of Pt (Fig. S6(c) and (d), ESI†). The results imply that the Pd–P@Pt–Ni core–shell NPs are stable during methanol oxidation reaction.
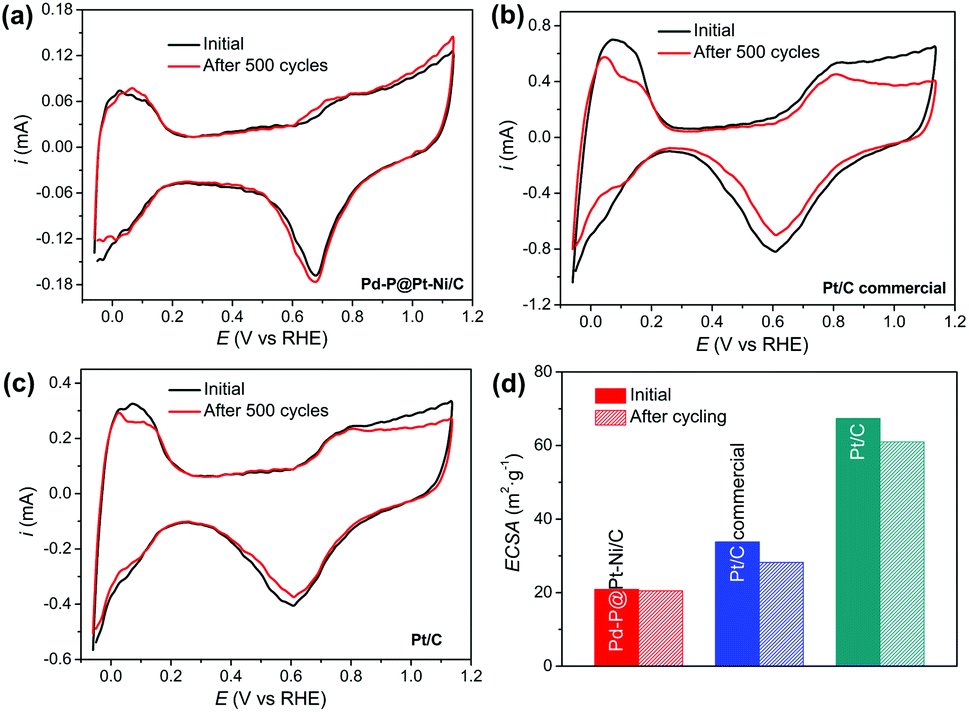 | ||
| Fig. 5 CV curves of (a) Pd–P@Pt–Ni/C NPs, (b) commercial Pt/C and (c) the as-prepared Pt/C before and after 500 potential cycles in N2-saturated 0.1 M HClO4 solution at a scan rate of 50 mV s−1. (d) Summary of the ECSA loss for different electrocatalysts. | ||
We further investigated the durability and antipoisoning capability by chronoamperometry measurement, which was recorded in 0.5 M H2SO4 + 1 M CH3OH at 0.85 V over a period of 3600 s (Fig. 6). The current density of Pd–P@Pt–Ni/C is higher than those of commercial Pt/C and the as-prepared Pt/C during the whole period, suggesting that the core–shell NPs are more active and stable than the reference electrocatalysts.
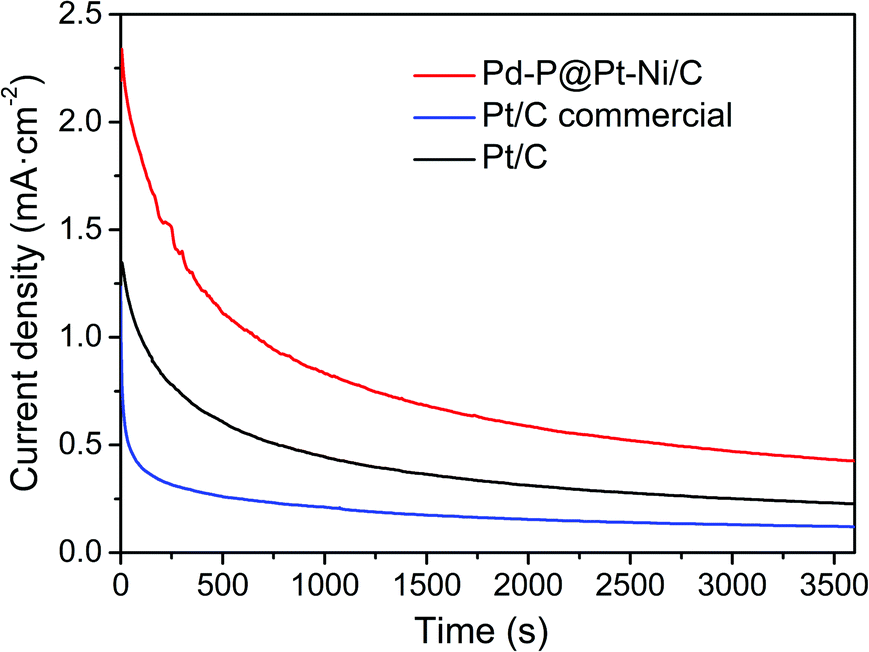 | ||
| Fig. 6 Chronoamperometry curves of Pd–P@Pt–Ni/C NPs, commercial Pt/C and the as-prepared Pt/C in 0.5 M H2SO4 + 1 M CH3OH solution at 0.85 V. | ||
In order to evaluate the applicability of Pd–P@Pt–Ni core–shell NPs, the electrocatalytic performances for formic acid oxidation reaction are also checked by cyclic voltammetry and chronoamperometry measurements. On one hand, Fig. S7(a)† indicates that the specific activity of Pd–P@Pt–Ni/C (1.46 mA cm−2) is 2.3 and 1.7 times higher than those of commercial Pt/C (0.63 mA cm−2) and the as-prepared Pt/C (0.86 mA cm−2), respectively. On the other hand, the core–shell NPs demonstrate better durability in formic acid oxidation reaction (Fig. S7(b), ESI†).
The observed excellent catalytic activity and durability of Pd–P@Pt–Ni/C NPs would be ascribed to the synergistic effects from both Pd–P core and Ni species in the shell. (i) To investigate the role of core part, the CV curves in MOR were also performed with our previously synthesized Pd–P@Pt/C electrocatalysts (Fig. S8, ESI†).48 Although it displays the similar specific activity (1.18 mA cm−2) with those of commercial Pt/C and the as-prepared Pt/C, the If/Ib ratio (0.88) is much higher than the values in reference catalysts. It is assumed that Pd–P alloy as the core part can change the electronic structure of Pt on the surface, exhibiting the favourable effect on the performance of Pt. The d-band center position (which is relative to the Fermi level) of metal associates with its catalytic activity and durability in the electrochemical reaction by changing the binding energy of adsorbates.64 In this work, the surface strain originating from the deposition of Pt on a second metal with the smaller lattice constant has the influence on the d-band center position.65 The d-band becomes broader with shifting down the center by means of lateral compression, further resulting in a weaker binding of CO poisoning intermediate as well as good electrocatalytic properties.66 (ii) The positive effects due to nickel species in the shell could be explicated by the bi-functional mechanism and the electronic effect.67,68 In the electrochemical reaction process, platinum provides the active sites to adsorb and decompose the methanol molecules via C–H and O–H bond cleavages, yielding the poisoning intermediate CO to block the Pt surface.69 For the bi-functional mechanism, nickel oxide species generated by dissociative adsorption of H2O on the catalyst surface can facilitate the removal of CO residues and recover the Pt active sites for methanol oxidation.68 For the electronic effect, the electron transfer from Ni to Pt, as illustrated by the XPS analysis (Fig. 2), can modify the electronic structure of Pt in the as-synthesized core–shell NPs. The electron donation from Ni to Pt leads to the down-shift of the d-band center in Pt, resulting the same effect on the electrocatalytic properties of Pt as the Pd–P alloy core does. On the other hand, according to the CV curves of different electrocatalysts (Fig. 3), the oxygenated-species reduction peak of Pd–P@Pt–Ni/C NPs in the backward scan locates at a more positive position than those of commercial Pt/C and the as-prepared Pt/C, indicating the relatively easier desorption of OHad from the surface.70 The results further confirm the faster oxidation of adsorbed CO by the synergistic effect of Ni. Besides, the absence of any oxidized Pt species is also one of the reasons for the catalyst to exhibit high catalytic activity because the metallic Pt plays a critical role in electro-oxidation of methanol.
Conclusion
In summary, we have reported Pd–P@Pt–Ni core–shell NPs through a seed-mediated growth method as the electrocatalysts in methanol oxidation reaction. The XRD and XPS analyses indicate that the insertion of Ni atoms into the shell part and the change of electronic properties of Pt, respectively. In the electrochemical measurements, the current density of Pd–P@Pt–Ni core–shell NPs is much higher than those of commercial Pt/C and the as-prepared Pt/C. In addition, the larger If/Ib ratio reveals the stronger tolerance of the catalyst to carbonaceous species accumulation. Considering the durability of electrocatalyst, the less ECSA loss and the lower decay of current density in chronoamperometry compared to the reference catalysts demonstrate that Pd–P@Pt–Ni core–shell NPs are highly stable and tolerant. The enhanced electrocatalytic performances can be attributed to the synergistic effects from core part and nickel species, such as bi-functional mechanism and electronic effect. It is expected that the present work will provide a platform for the development of a wide array of useful anode electrocatalysts for methanol oxidation reaction.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Natural Science Foundation of Fujian Province (Grant No. 2020J05155), the National Natural Science Foundation of China (Grant No. 21676222, U1705252), the Quanzhou City Science & Technology Program of China (Grant No. 2019C106), and the Scientific Research Foundation of Quanzhou Normal University (Grant No. H18001, H17015).Notes and references
- C. T. Asset, R. Chattot, M. Fontana, B. Mercier-Guyon, N. Job, L. Dubau and F. Maillard, ChemPhysChem, 2018, 19, 1552–1567 CrossRef PubMed.
- Y. Wang, S. Zou and W.-B. Cai, Catalysts, 2015, 5, 1507–1534 CrossRef CAS.
- M. T. Anwar, X. Yan, M. R. Asghar, N. Husnain, S. Shen, L. Luo and J. Zhang, Int. J. Energy Res., 2019, 43, 2694–2721 CrossRef CAS.
- S. Huang, A. Shan and R. Wang, Catalysts, 2018, 8, 538 CrossRef.
- Y. Liu, Z. Li, S. Xu, Y. Xie, Y. Ye, X. Zou and S. Lin, J. Colloid Interface Sci., 2019, 554, 640–649 CrossRef CAS PubMed.
- X. L. Tian, L. Wang, P. Deng, Y. Chen and B. Y. Xia, J. Energy Chem., 2017, 26, 1067–1076 CrossRef.
- C. Shang and E. Wang, Phys. Chem. Chem. Phys., 2019, 21, 21185–21199 RSC.
- W. Huang, H. Wang, J. Zhou, J. Wang, P. N. Duchesne, D. Muir, P. Zhang, N. Han, F. Zhao, M. Zeng, J. Zhong, C. Jin, Y. Li, S.-T. Lee and H. Dai, Nat. Commun., 2015, 6, 10035 CrossRef CAS PubMed.
- J. Wang, B. Li, T. Yersak, D. Yang, Q. Xiao, J. Zhang and C. Zhang, J. Mater. Chem. A, 2016, 4, 11559–11581 RSC.
- Y.-J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- Y. Yang, C. Tan, Y. Yang, L. Zhang, B.-W. Zhang, K.-H. Wu and S. Zhao, ChemCatChem, 2021, 13, 1587–1594 CrossRef CAS.
- G. Oskueyan and M. Mansour Lakouraj, J. Appl. Electrochem., 2019, 49, 755–765 CrossRef CAS.
- M. F. R. Hanifah, J. Jaafar, M. H. D. Othman, A. F. Ismail, M. A. Rahman, N. Yusof, F. Aziz and N. A. A. Rahman, J. Alloys Compd., 2019, 793, 232–246 CrossRef CAS.
- Y. Wang, X. Jiang, G. Fu, Y. Li, Y. Tang, J.-M. Lee and Y. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 34869–34877 CrossRef CAS PubMed.
- N. K. Chaudhari, Y. Hong, B. Kim, S.-I. Choi and K. Lee, J. Mater. Chem. A, 2019, 7, 17183–17203 RSC.
- L. S. Parreira, R. M. Antoniassi, I. C. Freitas, D. C. de Oliveira, E. V. Spinacé, P. H. C. Camargo and M. C. dos Santos, Renewable Energy, 2019, 143, 1397–1405 CrossRef CAS.
- H. Liu, D. Yang, Y. Bao, X. Yu and L. Feng, J. Power Sources, 2019, 434, 226754 CrossRef CAS.
- Y. Qin, H. Zhuo, X. Liang, K. Yu, Y. Wang, D. Gao and X. Zhang, Dalton Trans., 2019, 48, 10313–10319 RSC.
- H. Wang, S. Liu, H. Zhang, S. Yin, Y. Xu, X. Li, Z. Wang and L. Wang, New J. Chem., 2020, 44, 15492–15497 RSC.
- K. Guo, Y. Liu, M. Han, D. Xu and J. Bao, Chem. Commun., 2019, 55, 11131–11134 RSC.
- S. Yin, R. D. Kumar, H. Yu, C. Li, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, ACS Sustainable Chem. Eng., 2019, 7, 14867–14873 CrossRef CAS.
- L. S. Mpeta, S. S. Gwebu, O. A. Arotiba and N. W. Maxakato, Electrocatalysis, 2019, 10, 672–679 CrossRef CAS.
- R. Cao, T. Xia, R. Zhu, Z. Liu, J. Guo, G. Chang, Z. Zhang, X. Liu and Y. He, Appl. Surf. Sci., 2018, 433, 840–846 CrossRef CAS.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- X. Yan, S. Yu, Y. Tang, D. Sun, L. Xu and C. Xue, Nanoscale, 2018, 10, 2231–2235 RSC.
- Y. Xiong, Y. Ma, J. Li, J. Huang, Y. Yan, H. Zhang, J. Wu and D. Yang, Nanoscale, 2017, 9, 11077–11084 RSC.
- C. Tan, Y. Sun, J. Zheng, D. Wang, Z. Li, H. Zeng, J. Guo, L. Jing and L. Jiang, Sci. Rep., 2017, 7, 6347 CrossRef PubMed.
- X. Sun, D. Li, Y. Ding, W. Zhu, S. Guo, Z. L. Wang and S. Sun, J. Am. Chem. Soc., 2014, 136, 5745–5749 CrossRef CAS PubMed.
- X. Zhao, S. Chen, Z. Fang, J. Ding, W. Sang, Y. Wang, J. Zhao, Z. Peng and J. Zeng, J. Am. Chem. Soc., 2015, 137, 2804–2807 CrossRef CAS PubMed.
- L.-L. Shen, G.-R. Zhang, S. Miao, J. Liu and B.-Q. Xu, ACS Catal., 2016, 6, 1680–1690 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- P. Yang, X. Yuan, H. Hu, Y. Liu, H. Zheng, D. Yang, L. Chen, M. Cao, Y. Xu, Y. Min, Y. Li and Q. Zhang, Adv. Funct. Mater., 2018, 28, 1704774 CrossRef.
- D. Gao, S. Li, Y. Lv, H. Zhuo, S. Zhao, L. Song, S. Yang, Y. Qin, C. Li, Q. Wei and G. Chen, J. Catal., 2019, 376, 87–100 CrossRef CAS.
- W. Gong, Z. Jiang, R. Wu, Y. Liu, L. Huang, N. Hu, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2019, 246, 277–283 CrossRef CAS.
- Y. Wu, M. Zhao, J.-P. Cao, J. Xu, T. Jin and N. Asao, Chem. Commun., 2020, 56, 8984–8987 RSC.
- C. Baldizzone, S. Mezzavilla, H. W. P. Carvalho, J. C. Meier, A. K. Schuppert, M. Heggen, C. Galeano, J.-D. Grunwaldt, F. Schüth and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 14250–14254 CrossRef CAS PubMed.
- S. W. Kang, Y. W. Lee, M. Kim, J. W. Hong and S. W. Han, Chem.–Asian J., 2011, 6, 909–913 CrossRef CAS PubMed.
- L. Calvillo, V. Celorrio, R. Moliner, A. B. Garcia, I. Caméan and M. J. Lazaro, Electrochim. Acta, 2013, 102, 19–27 CrossRef CAS.
- S. C. Zignani, E. Antolini and E. R. Gonzalez, J. Power Sources, 2009, 191, 344–350 CrossRef CAS.
- T.-Y. Jeon, S. K. Kim, N. Pinna, A. Sharma, J. Park, S. Y. Lee, H. C. Lee, S.-W. Kang, H.-K. Lee and H. H. Lee, Chem. Mater., 2016, 28, 1879–1887 CrossRef CAS.
- K. Hyun, J. H. Lee, C. W. Yoon, Y.-H. Cho, L.-H. Kim and Y. Kwon, Synth. Met., 2014, 190, 48–55 CrossRef CAS.
- D. Wang, L. Zhuang and J. Lu, J. Phys. Chem. C, 2007, 111, 16416–16422 CrossRef CAS.
- G. M. Burke, D. E. Wurster, M. J. Berg, P. Veng-Pedersen and D. D. Schottelius, Pharm. Res., 1992, 9, 126–130 CrossRef CAS PubMed.
- M. Brun, A. Berthet and J. C. Bertolini, J. Electron Spectrosc. Relat. Phenom., 1999, 104, 55–60 CrossRef CAS.
- C. Liang, L. Ding, C. Li, M. Pang, D. Su, W. Li and Y. Wang, Energy Environ. Sci., 2010, 3, 1121–1127 RSC.
- A. S. Aricò, A. K. Shukla, H. Kim, S. Park, M. Min and V. Antonucci, Appl. Surf. Sci., 2001, 172, 33–40 CrossRef.
- W. Eberhardt, P. Fayet, D. M. Cox, Z. Fu, A. Kaldor, R. Sherwood and D. Sondericker, Phys. Rev. Lett., 1990, 64, 780–783 CrossRef CAS PubMed.
- J. Xu, M. Zhao, S.-i. Yamaura, T. Jin and N. Asao, J. Appl. Electrochem., 2016, 46, 1109–1118 CrossRef CAS.
- A. Dutta and J. Ouyang, ACS Catal., 2015, 5, 1371–1380 CrossRef CAS.
- B. Narayanamoorthy, K. K. R. Datta, M. Eswaramoorthy and S. Balaji, ACS Catal., 2014, 4, 3621–3629 CrossRef CAS.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- S. Guo, S. Dong and E. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS PubMed.
- B. Y. Xia, H. B. Wu, X. Wang and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 13934–13937 CrossRef CAS PubMed.
- J. Chang, L. Feng, C. Liu, W. Xing and X. Hu, Energy Environ. Sci., 2014, 7, 1628–1632 RSC.
- L. Wang, Y. Nemoto and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 9674–9677 CrossRef CAS PubMed.
- F. Xie, L. Ma, M. Gan, H. He, L. Hu, M. Jiang and H. Zhang, Appl. Surf. Sci., 2019, 481, 1425–1434 CrossRef CAS.
- T. C. Deivaraj, W. Chen and J. Y. Lee, J. Mater. Chem., 2003, 13, 2555–2560 RSC.
- Z. Cui, H. Chen, M. Zhao, D. Marshall, Y. Yu, H. Abruña and F. J. DiSalvo, J. Am. Chem. Soc., 2014, 136, 10206–10209 CrossRef CAS PubMed.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS.
- J. Sanetuntikul, K. Ketpang and S. Shanmugam, ACS Catal., 2015, 5, 7321–7327 CrossRef CAS.
- E. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255–2259 CrossRef CAS PubMed.
- J. B. Raoof, R. Ojani and S. R. Hosseini, J. Solid State Electrochem., 2012, 16, 2699–2708 CrossRef CAS.
- S. Chen, Z. Wei, X. Qi, L. Dong, Y.-G. Guo, L. Wan, Z. Shao and L. Li, J. Am. Chem. Soc., 2012, 134, 13252–13255 CrossRef CAS PubMed.
- K. Jiang, H.-X. Zhang, S. Zou and W.-B. Cai, Phys. Chem. Chem. Phys., 2014, 16, 20360–20376 RSC.
- L. A. Kibler, A. M. El-Aziz, R. Hoyer and D. M. Kolb, Angew. Chem., Int. Ed., 2005, 44, 2080–2084 CrossRef CAS PubMed.
- U. B. Demirci, J. Power Sources, 2007, 173, 11–18 CrossRef CAS.
- R. Mu, Q. Fu, H. Xu, H. Zhang, Y. Huang, Z. Jiang, S. Zhang, D. Tan and X. Bao, J. Am. Chem. Soc., 2011, 133, 1978–1986 CrossRef CAS PubMed.
- K.-W. Park, J.-H. Choi, B.-K. Kwon, S.-A. Lee, Y.-E. Sung, H.-Y. Ha, S.-A. Hong, H. Kim and A. Wieckowski, J. Phys. Chem. B, 2002, 106, 1869–1877 CrossRef CAS.
- J. M. Léger, J. Appl. Electrochem., 2001, 31, 767–771 CrossRef.
- H.-H. Li, C.-H. Cui, S. Zhao, H.-B. Yao, M.-R. Gao, F.-J. Fan and S.-H. Yu, Adv. Energy Mater., 2012, 2, 1182–1187 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization of synthetic condition and characterization results. See DOI: 10.1039/d1ra07998k |
| This journal is © The Royal Society of Chemistry 2022 |