
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced hydrosilylation through hydrogen abstraction: an NMR and computational study of the structural effect of silane†
Thi-Hai-Yen Quacha,
Xavier Allonas *a,
Céline Croutxé-Barghorna,
Didier Le Nouenb and
Marco Sangermano*c
*a,
Céline Croutxé-Barghorna,
Didier Le Nouenb and
Marco Sangermano*c
aLaboratoire de Photochimie et d'Ingénierie Macromoléculaires, Institut Jean Baptiste Donnet, 3b rue Alfred Werner, 68093 Mulhouse Cedex, France. E-mail: Xavier.allonas@uha.fr
bLaboratoire d’Innovation Moléculaire et Applications, Institut Jean Baptiste Donnet, 3b rue Alfred Werner, 68093 Mulhouse Cedex, France
cPolitecnico di Torino, Dipartimento di Scienza Applicata e Tecnologia, C.so Duca degli Abruzzi 24, 10129 Torino, Italy. E-mail: marco.sangermano@polito.it
First published on 17th March 2022
Abstract
The hydrosilylation reaction, describing the addition of Si–H bonds to unsaturated bonds, is performed in the presence of catalysts, usually highly active platinum catalysts. This work focuses on the study of a photoinduced hydrosilylation by the use of benzophenone which promotes the addition reaction of olefin on different hydrosilanes. The reactivity of silanes towards addition onto the double bond during hydrosilylation appears to depend on their structure. It was observed that the consumption of Si–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C functional groups increases with the irradiation time, and reaches a maximum of approx. 51% in the case of diphenylsilane. The hydrosilylation products are determined with 1H NMR, HSQC, DEPT, COSY and 13C NMR. The main product corresponds to the single adduct of the silyl radical onto the double bond. Substitution of the Si–H bond by two or three phenyls groups (triphenylsilane, diphenysilane) enhances the yield of the reaction, although diphenylsilane was found to be more efficient than triphenylsilane because of its lower steric hindrance. The ketyl radical formed after hydrogen abstraction by the triplet state of benzophenone likely forms benzopinacol, a reaction which reduces the overall yield of the hydrosilylation reaction. All these experiments are in line with DFT calculations of the Gibbs free energy of the reactions involved. This sheds new light on the photoinduced hydrosilylation process and opens the way to more active combinations of photoinitiator/silane/vinylsilane systems.
C functional groups increases with the irradiation time, and reaches a maximum of approx. 51% in the case of diphenylsilane. The hydrosilylation products are determined with 1H NMR, HSQC, DEPT, COSY and 13C NMR. The main product corresponds to the single adduct of the silyl radical onto the double bond. Substitution of the Si–H bond by two or three phenyls groups (triphenylsilane, diphenysilane) enhances the yield of the reaction, although diphenylsilane was found to be more efficient than triphenylsilane because of its lower steric hindrance. The ketyl radical formed after hydrogen abstraction by the triplet state of benzophenone likely forms benzopinacol, a reaction which reduces the overall yield of the hydrosilylation reaction. All these experiments are in line with DFT calculations of the Gibbs free energy of the reactions involved. This sheds new light on the photoinduced hydrosilylation process and opens the way to more active combinations of photoinitiator/silane/vinylsilane systems.
1. Introduction
Hydrosilylation, also known as hydrosilation, is one of the most useful catalytic reactions leading to the formation of organosilanes and organosilicones. This reaction has a variety of applications in industry and in organic chemistry as intermediates. Hydrosilylation involves the terminal addition of silicon hydrides to unsaturated compounds such as alkenes.1,2|
R3SiH + R′–CHCH2 → R′–CH2–CH2–SiR3
| (1) |
The first hydrosilylation reaction was reported in 1947 by Sommer and used free radical chemistry; however, the reaction selectivity was quite low.3,4 Subsequently, a variety of transition metal catalysts for hydrosilylation have been developed.4–9 Among them, Speier's and Karstedt's catalysts are certainly the most popular and have been widely used in industry.4,10–12
Although the utility of platinum catalysts has been widely recognized for decades, the development of more efficient, more selective, and cheaper catalysts are still desired for a more economical production of organosilicon materials having superior properties.
With many advantages brought by the spatio-temporal control of the reaction, photochemical catalysts are potentially valuable candidates for such hydrosilylation reactions.13 Using benzophenone as a photoreducible aromatic ketone, Rowlands et al.1 suggested a chain reaction for photoinduced hydrosilylation (Scheme 1). The initiation reaction involves the absorption of light by benzophenone which turns into a first excited singlet state and, through fast intersystem crossing, forms a long-lived triplet state (1). The benzophenone triplet state then abstracts a hydrogen from a silane, leading to the formation of a ketyl radical and a silyl radical (2). During the propagation reaction, the silyl radical reacts with a double bond, leading to a carbon-centered radical (3). The latter can react with a second double bond (4), leading to a further carbon-centered radical. The silyl radical can also abstract a hydrogen from another silane (5), and this chain transfer reaction acts as an inhibition reaction. Interestingly, the ketyl radical formed in (2) may abstract a hydrogen from a silane, leading to the formation of benzhydrol (Bz) and a silyl radical (6). A termination reaction can take place through recombination between two radicals, particularly two ketyl radicals, forming benzopinacol (BPol) (7). The yield of the reaction has been reported to be 30%, due to important termination reactions.1
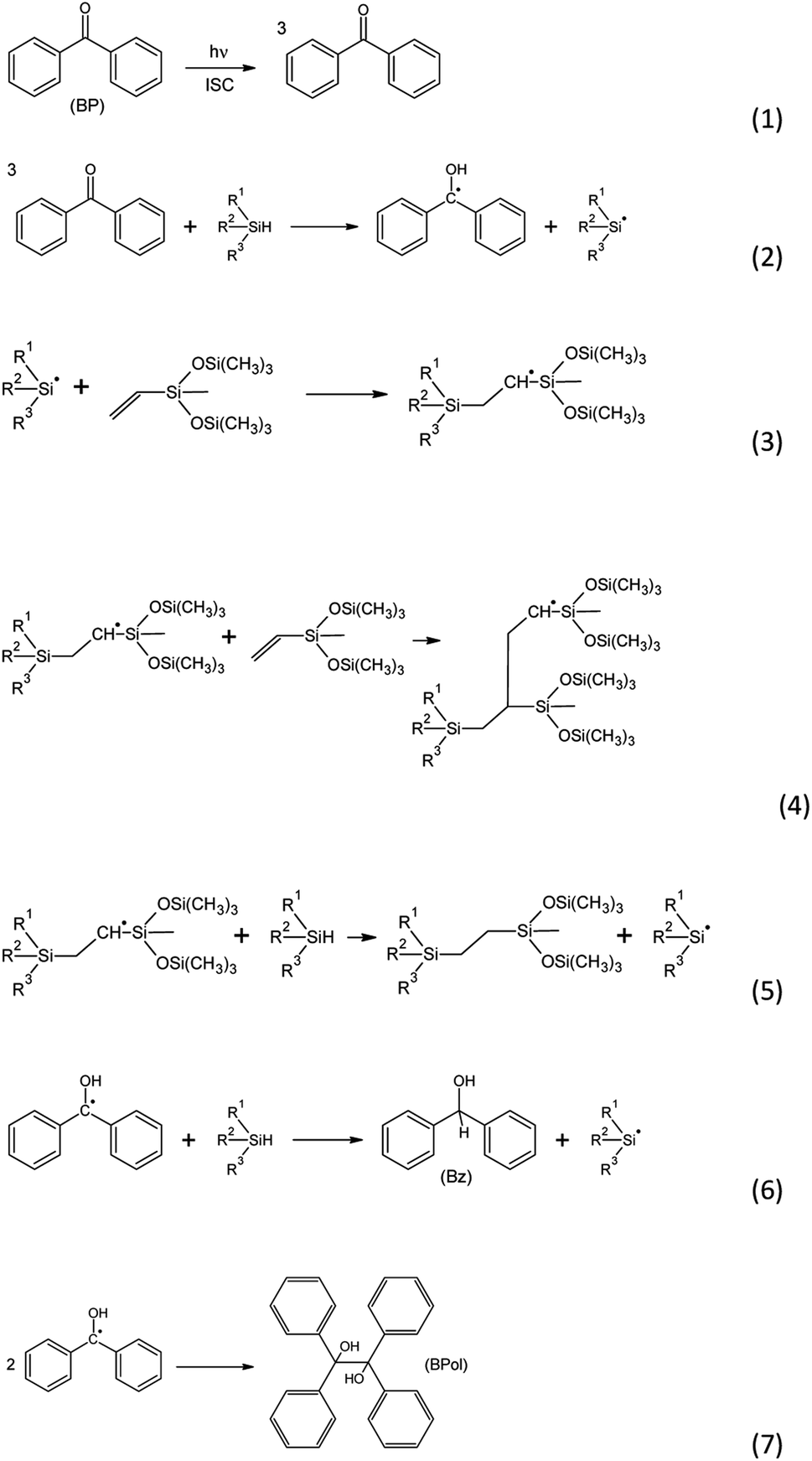 | ||
| Scheme 1 Mechanism of hydrosilylation with benzophenone (BP) as the photoinitiator. | ||
The aim of this paper is to provide a thorough investigation of the hydrosilylation reaction mediated by benzophenone, focusing particularly on the effect of silane. Different substituted hydrosilanes are used with methyl bis-(trimethylsilyloxy)-vinylsilane as the olefin. The photoreaction is observed at different reaction times by NMR spectroscopy.
2. Experimental
2.1. Materials
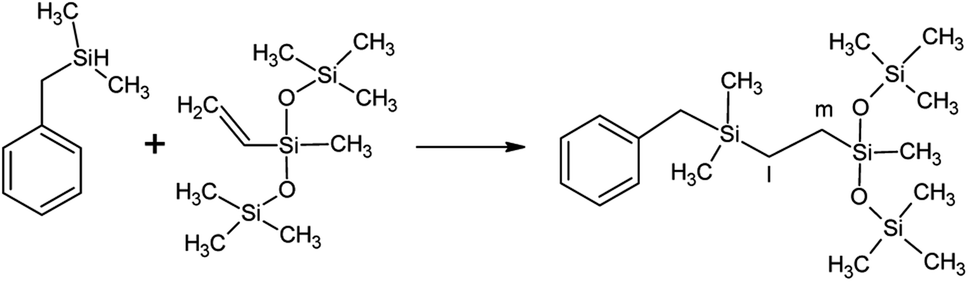
The reagents, shown in Scheme 2, were methyl-bis(trimethylsilyloxy)vinylsilane (A, SigmaAldrich), trimethylsilyl-terminated polymethylhydrosiloxane, 2-5 cSt (B, ABCR), benzyl(dimethyl)silane (C, ABCR), diphenylsilane (D, SigmaAldrich), triphenylsilane (E, Fluka) and benzophenone (BP, SigmaAldrich).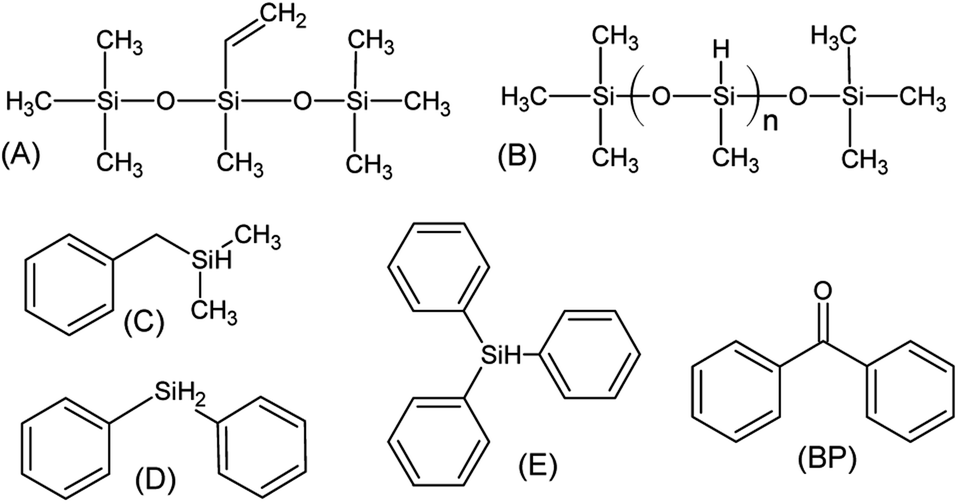 | ||
| Scheme 2 Compounds used in this study. | ||
2.2. Sample preparation
Samples containing a stoichiometric ratio of hydrosilane and olefin in the presence of benzophenone (10 mol% with respect to the Si–H content) were stirred at ambient temperature for 8 hours. These formulations are denoted as (A + B), (A + C), (A + D) and (A + E). In the case of triphenylsilane (E), tetrahydrofuran (THF) was added to ensure complete solubilization of the silane (approx. 50 mol% with respect to Si–H content). 0.05 ml of the formulation was introduced in an NMR glass tube which was then submitted to irradiation. The irradiation was stopped at given times in order to proceed to the NMR analysis. The reaction was initiated under UV radiation at 365 nm with an intensity of approx. 0.28 ± 0.01 W cm−2.2.3. Experimental techniques
The hydrosilylation reaction was monitored by 1H-NMR in the liquid state during the course of the irradiation. 1H-NMR spectra were recorded on an Advance 400 MHz Spectrophotometer (Bruker) using CDCl3 as the solvent. The chemical shifts are expressed in ppm and were calibrated using chloroform.Recording 1H-NMR spectra at various reaction times (0, 2, 5, 15, 37, 60 min) was aimed at determining the relative amounts of the different reactive groups in the samples. Thus, the integration of the corresponding peaks was calculated and referenced to that of the peak due to the proton in the Si–CH3 group at 0 ± 0.3 ppm. Since Si–CH3 groups are not involved in the hydrosilylation reaction, the intensity of this peak remains unchanged in the spectra.9 The proton integration of the Si–CH3 is normalized for (A + B), (A + C), (A + D) and (A + E) formulations.
3. Results and discussion
3.1. 1H-NMR spectra at the initial time
1H-NMR spectra at the initial time (t = 0; prior to UV irradiation) of different formulations are presented in Fig. 1. Chemical shifts δ (in ppm) of these formulations are located at (i) 7.4–7.9 (10H, BP); 5.6–6.3 (3H, –C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) CH2); 4.3–5.1 (1H, Si); 0–0.3 (33H, SiCH3) for the (A + B) formulation; (ii) 7.4–7.9 (10H, BP); 6.8–7.4 (5H, phenyl group of C); 5.6–6.3 (3H, –CCH2); 3.7–4.3 (1H, Si); 2.0–2.3 (2H, –CH2–Si of C); 0–0.3 (27H, SiCH3) for the (A + C) formulation; (iii) 7.2–7.9 (10H, BP + 5H, phenyl group of D); 5.6–6.3 (3H, –CCH2); 4.7–5.3 (1H, Si); 0–0.3 (21H, SiCH3) for (the A + D) formulation; (iv) 7.2–7.9 (10H, BP + 15H, phenyl group of E); 5.6–6.3 (3H, –CCH2); 5.5–5.6 (1H, Si
CH2); 4.3–5.1 (1H, Si); 0–0.3 (33H, SiCH3) for the (A + B) formulation; (ii) 7.4–7.9 (10H, BP); 6.8–7.4 (5H, phenyl group of C); 5.6–6.3 (3H, –CCH2); 3.7–4.3 (1H, Si); 2.0–2.3 (2H, –CH2–Si of C); 0–0.3 (27H, SiCH3) for the (A + C) formulation; (iii) 7.2–7.9 (10H, BP + 5H, phenyl group of D); 5.6–6.3 (3H, –CCH2); 4.7–5.3 (1H, Si); 0–0.3 (21H, SiCH3) for (the A + D) formulation; (iv) 7.2–7.9 (10H, BP + 15H, phenyl group of E); 5.6–6.3 (3H, –CCH2); 5.5–5.6 (1H, Si![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ); 0–0.3 (21H, SiCH3) for the (A + E) formulation. Integration of 1H-NMR signal for B shows that the number of repeating unit is n = 4 (Fig. SI1†).
); 0–0.3 (21H, SiCH3) for the (A + E) formulation. Integration of 1H-NMR signal for B shows that the number of repeating unit is n = 4 (Fig. SI1†).
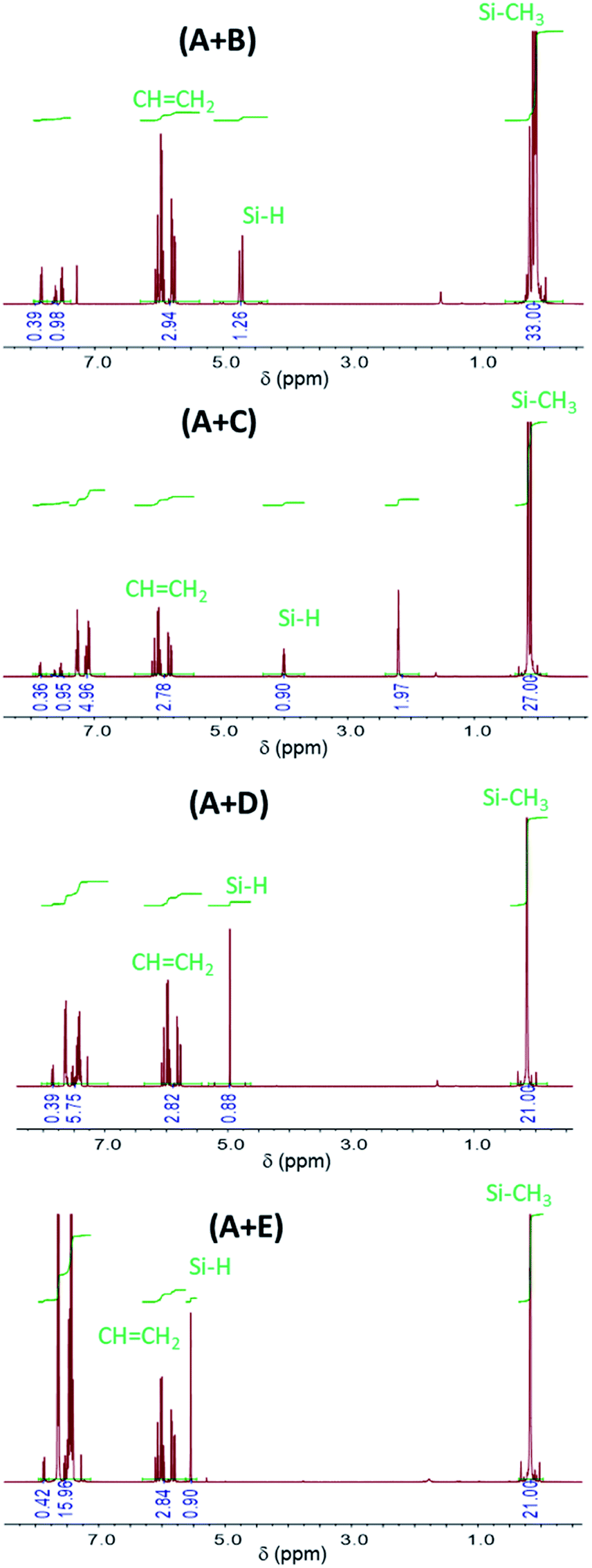 | ||
| Fig. 1 1H-NMR spectra of (A + B), (A + C), (A + D) and (A + E) formulations prior to UV irradiation (t = 0). | ||
3.2. Photoreaction of benzophenone
The absorption of light at 365 nm results in the excitation of benzophenone in its S0–S1 absorption band, leading to the n–π* triplet state. In the presence of a labile hydrogen, a photoreduction can occur, yielding ketyl and silyl radicals1,14–18 as shown in eqn (2). The rate constant of hydrogen abstraction was measured to be 3.1 107 M−1 s−1 for T in acetonitrile with a quantum yield of 0.75.18 BPol merely arises from the recombination of two ketyl radicals. This underlines the photoreaction of the benzophenone triplet state with the silanes which are the best hydrogen donors in the medium.Fig. 2 shows 1H NMR spectra for the formulation (A + B) at different irradiation times. At t = 0 min (prior to UV radiation), the characteristic peaks of the hydrogen atoms of the aromatic rings of BP can be observed in the range from 7.4 to 7.9 ppm. The a, b, c protons (see Fig. 2) are located at approx. 7.8 ppm, 7.5 ppm and 7.6 ppm, respectively. Under UV irradiation, these peaks (a, b, c) decrease and gradually disappear. Simultaneously, the appearance of news peaks can also be observed from 7.1 to 7.4 ppm. The peaks at approx. 7.2 ppm and 7.3 ppm can be attributed to the protons of BPol at positions d and e, respectively.19
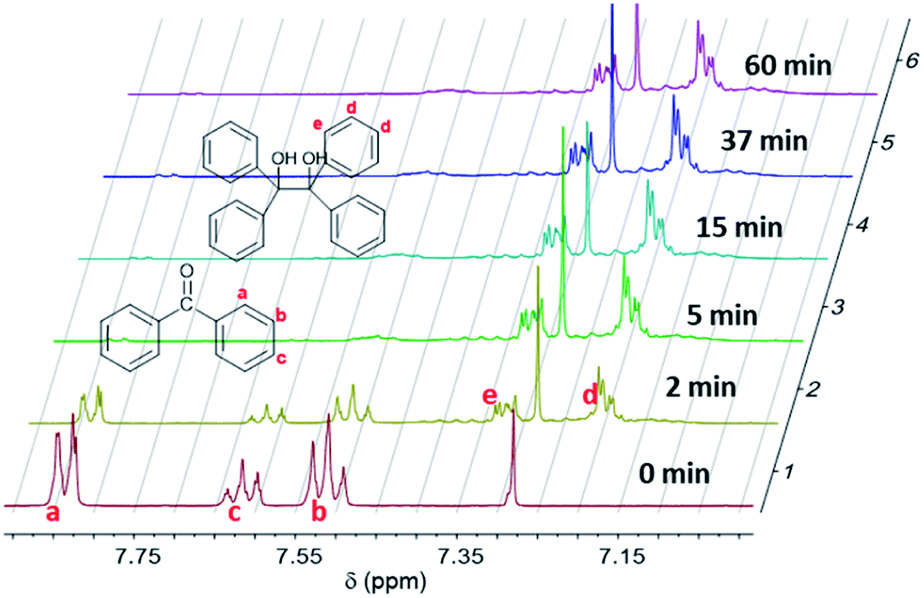 | ||
| Fig. 2 1H NMR spectra of formulation (A + B) at 0, 2, 5, 15, 37 and 60 min of UV irradiation. | ||
By using the Si–CH3 proton peak at approx. 0–0.3 ppm as a reference (Fig. 1), the residual amount of BP and BPol can be calculated as a function of the irradiation time, using protons a for BP and protons d for BPol (Fig. 2).
Fig. 3 shows the corresponding data plotted as a function of irradiation duration for formulations (A + B), (A + C), (A + D), (A + E). Under UV irradiation, the BP signal decreases significantly within 2 min through a pseudo-first order rate law. Simultaneously, a remarkable increase of the BPol signal is observed (50–75% of formation within 2 min) until a plateau is reached after 5 min of irradiation for the (A + B), (A + C) and (A + D) formulations, and after 15 for the (A + E) formulation. Table 1 shows that 81 to 93% of the disappearance of BP results in the formation of BPol, which agrees with the termination reaction (7). Therefore, this termination reaction is almost quantitative, leading to the disappearance of the photoinitiator BP. It should be noted that further UV dissociation of BPol is prevented by the absorption of the UV-C light by vinylsilane and is, therefore, not taken into account.
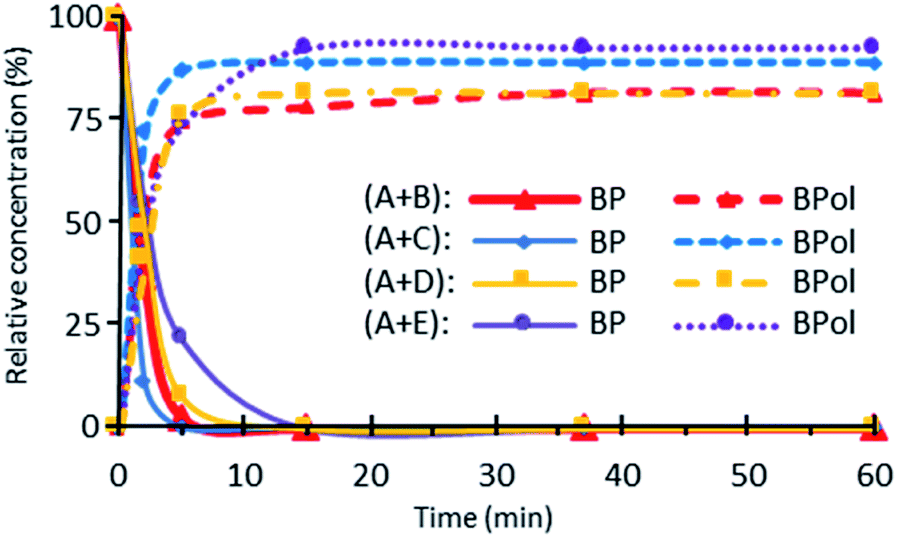 | ||
| Fig. 3 Changes in BP and BPol signals with irradiation time for (A + B), (A + C), (A + D) and (A + E) formulations. | ||
| Irradiation time (min) | B | C | D | E | ||||
|---|---|---|---|---|---|---|---|---|
| % BP | % BPol | % BP | % BPol | % BP | % BPol | % BP | % BPol | |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 63 | 55 | 89 | 72 | 51 | 41 | 45 | 48 |
| 5 | 97 | 74 | 100 | 87 | 92 | 76 | 79 | 74 |
| 15 | 100 | 78 | 100 | 89 | 100 | 81 | 100 | 93 |
| 37 | 100 | 82 | 100 | 89 | 100 | 81 | 100 | 93 |
| 60 | 100 | 82 | 100 | 89 | 100 | 81 | 100 | 93 |
It is, therefore, interesting to investigate the effect of the different functionalities and molecular structures of the hydrosilanes. In addition, the silyl radical formed is expected to react with a double bond, leading to the formation of an adduct. For these reasons, detailed NMR studies of the reactions were conducted and the results will be discussed in the following sections.
3.3. Reaction profiles of SiH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C groups
C groups
Proton NMR spectra of the starting (t = 0) and final (t = 60 min) reaction mixtures of (A + B), (A + C), (A + D) and (A + E) formulations show a decrease in the peak area of the signals corresponding to the Si–H and CC groups (Fig. 4 and SI1†). This evidences the reaction between the hydrosilane and the vinyl compound in the presence of BP and UV irradiation. However, in none of the spectra do the Si–H or CC peaks disappear completely, indicating that the hydrosilylation process efficiency reaches a maximum. The changes in the Si–H and CC content is followed by a calculation of the corresponding consumption from the peaks area. Fig. 5 shows the change in consumption for SiH and CC groups at various reaction times for the (A + B), (A + C), (A + D) and (A + E) formulations. From Fig. 5, a significant increase in the consumption of both Si–H and CC functional groups can be observed during the first minute of irradiation. Interestingly, this rate of consumption corresponds quite well with the fate of BP. After BP disappears entirely, the SiH and CC consumptions reach quasi-plateau values.
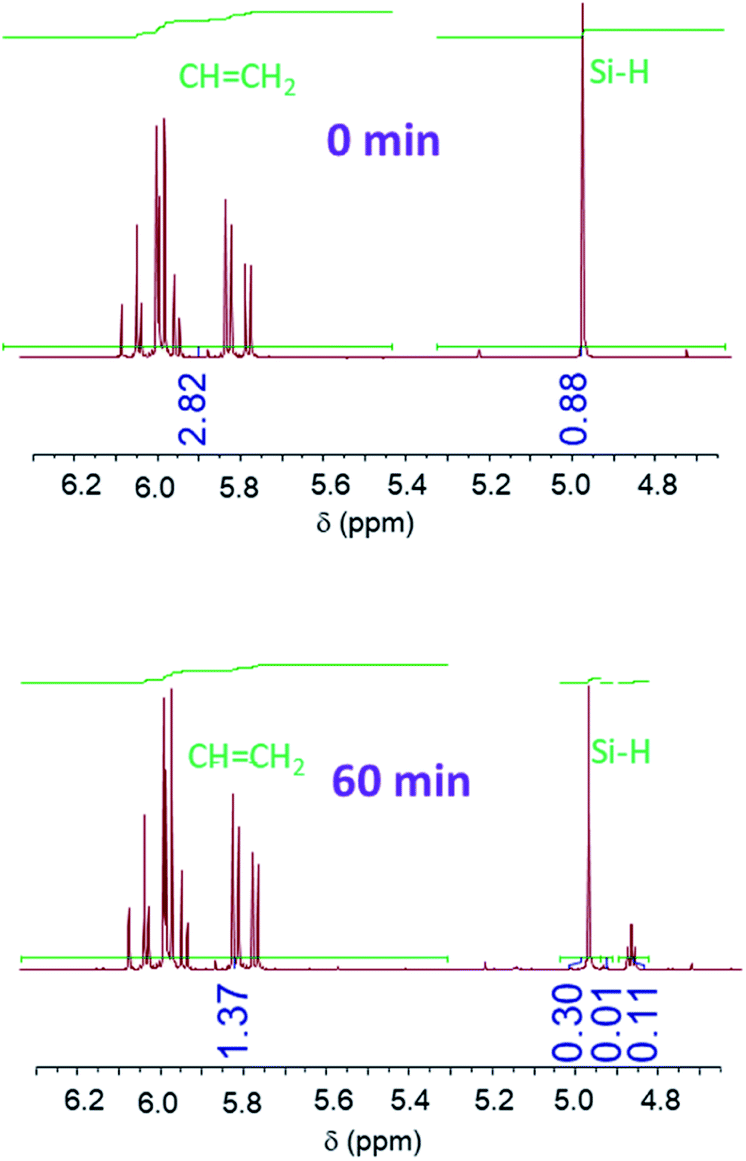 | ||
| Fig. 4 1H NMR spectra of formulation (A + D) at reaction times t = 0 and t = 60 min. | ||
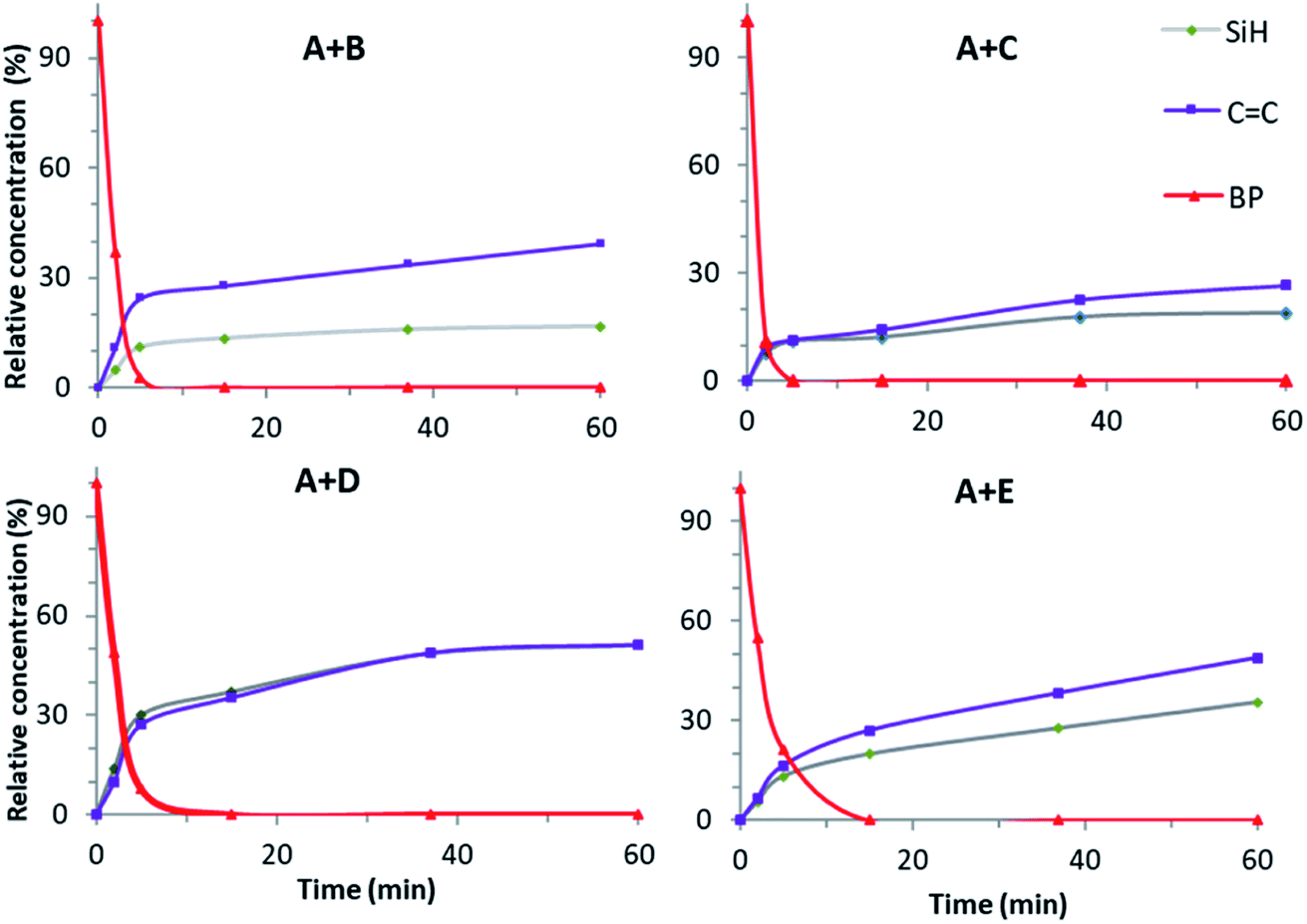 | ||
| Fig. 5 Change in the ratio (in%) of the BP, SiH and CC groups at various reaction times for the (A + B), (A + C), (A + D) and (A + E) formulations. | ||
For the (A + D) and (A + E) systems, the consumption of Si–H reaches approx. 50% after 60 min of photoreaction, in contrast to the (A + C) and (A + B) systems which hardly reach 20%. Interestingly, the consumption of the CC groups is higher than that of Si–H for the (A + B) and (A + E) systems. The difference in reactivity between these formulations can be ascribed to the differences in the molecular structures of the hydrosilanes (see below). The relatively low value of the final consumption is merely ascribed to the disappearance of BP, as discussed above.
3.4. Hydrosilylation products
Apart from the decrease of the SiH and CC NMR peaks, new signals are observed that can be attributed to the hydrosilylation products (Fig. 6 and SI2†). The rates of formation of these products match quite well with the decrease of the SiH signal. The main products are observed in the range 0.4–0.8 ppm, 0.3–0.5 ppm, 0.5–1.4 ppm and 0.3–1.4 ppm for the (A + B), (A + C), (A + D) and (A + E) formulations, respectively.
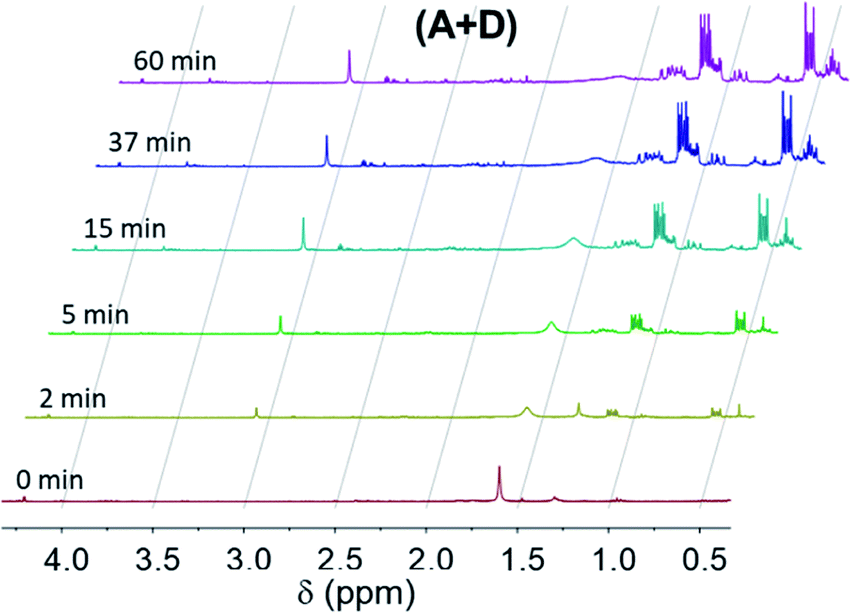 | ||
| Fig. 6 Proton NMR spectra obtained for the (A + D) formulation during irradiation. | ||
The most reactive formulations, namely (A + E) and (A + D), were investigated by advanced NMR methods, i.e., heteronuclear single quantum coherence spectroscopy (HSQC), distortionless enhancement by polarization transfer (DEPT), two-dimensional nuclear magnetic resonance spectroscopy (COSY) and 13C NMR, to identify the main hydrosilylation product (Fig. 7 and 8) in these two formulations.
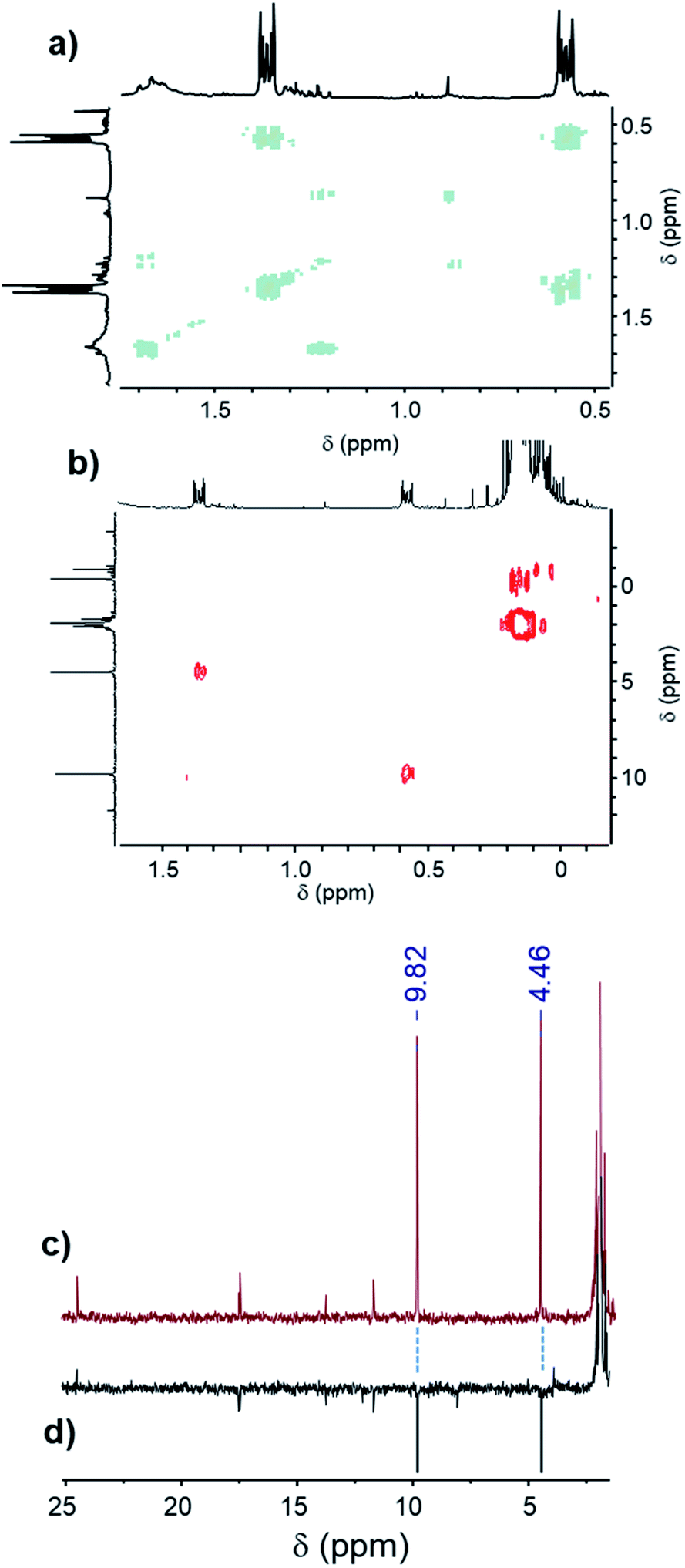 | ||
| Fig. 7 (a) COSY spectrum, (b) HSQC spectrum, (c) 13C NMR spectrum and (d) DEPT spectrum of the (A + E) formulation after 60 min of UV irradiation. | ||
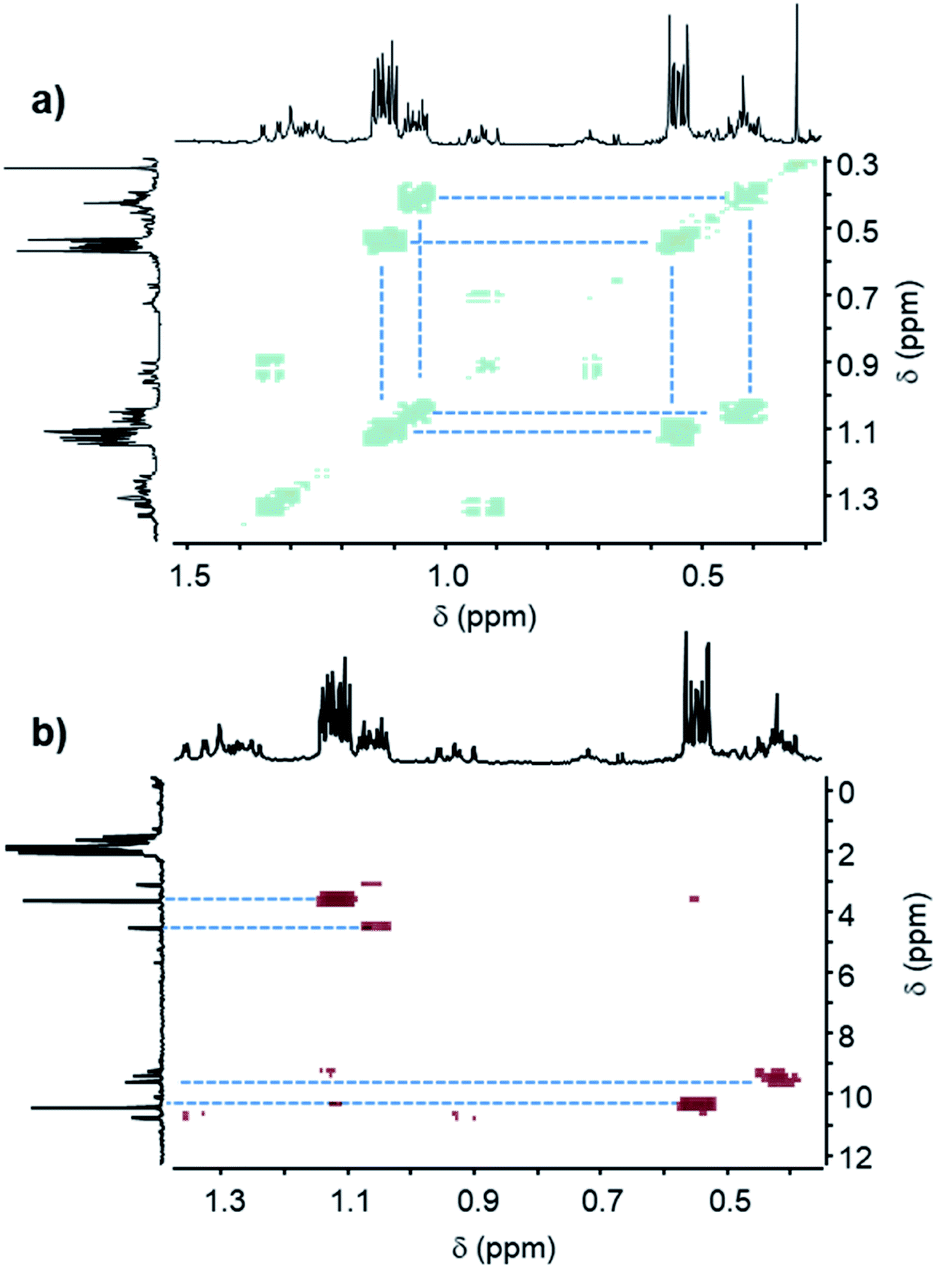 | ||
| Fig. 8 (a) COSY spectrum and (b) HSQC spectrum of the (A + D) formulation after 60 min of UV irradiation. | ||
In the case of the triphenylsilane (A + E) formulation, the COSY spectrum (Fig. 7a) shows strong coupling between the two main signals at 0.57 ppm and 1.36 ppm in the 1H NMR spectrum. These two signals are coupled with two signals at 9.82 ppm and 4.46 ppm of the 13C NMR spectrum (Fig. 7b and c). They are characteristic of the –CH2–CH2– bond (as confirmed by the DEPT spectrum, Fig. 7d) arising from the coupling of the triphenylsilyl radical with the double bond.20,21 The signals at 0.57 ppm and 1.36 ppm of the 1H NMR spectrum can be assigned to the protons located at the f and g positions of the product, as shown in Scheme 3.
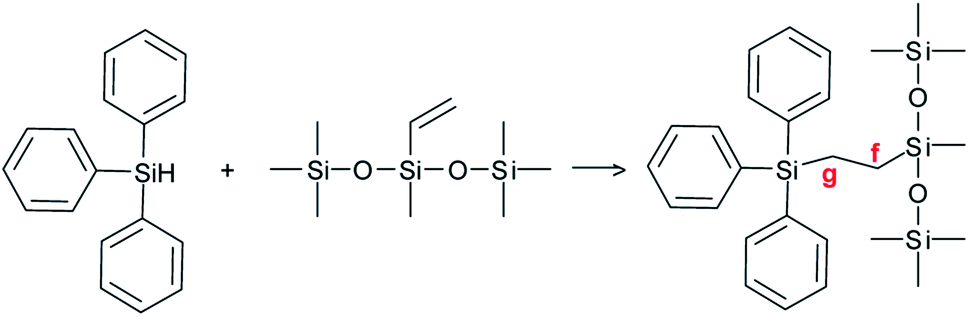 | ||
| Scheme 3 Reaction product in the case of triphenylsilane (E). | ||
In the case of diphenylsilane (D), beside the two main signals at 0.54 ppm and 1.12 ppm, two smaller signals can be detected at 0.42 ppm and 1.05 ppm in the 1H NMR spectrum, which are strongly coupled together as shown by the COSY spectrum (Fig. 8a). These proton peaks at 0.54 ppm, 1.12 ppm, 0.42 ppm and 1.05 ppm correspond to the four carbon peaks at 10.44 ppm, 3.65 ppm, 9.59 ppm and 4.54 ppm of the 13C NMR spectra, respectively (Fig. 8b, see Fig. SI3†). The DEPT spectrum (see Fig. SI3†) indicates that this molecular group corresponds to a CH2 bond.
Furthermore, the COSY spectrum (see Fig. SI3†) shows strong coupling between the proton at 4.86 ppm, which is assigned to the SiH group, and the proton at 1.12 ppm, which arises from the CH2 bond. Therefore, the two main proton signals at 0.54 ppm and 1.12 ppm can be attributed to positions h and i (Scheme 4), and the two smaller proton signals at 0.42 ppm and 1.05 ppm can be assigned to the proton located at positions j and k (Scheme 4).
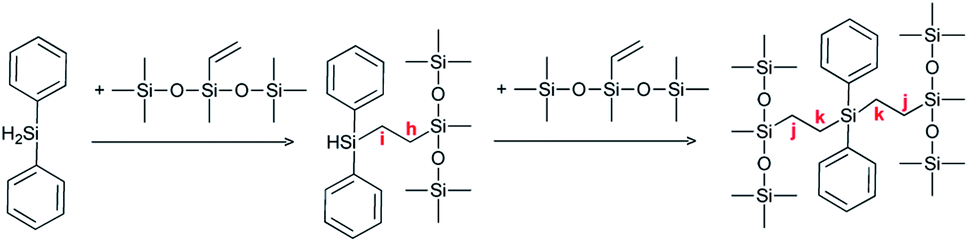 | ||
| Scheme 4 Reaction product in the case of diphenylsilane (D). | ||
The low consumption renders a more difficult identification of the reaction products for systems (A + B) and (A + C). COSY experiments were performed for these two formulations. As can be seen in Fig. 9 for the (A + C) formulation, there is proton coupling between the signals at 0.45 ppm and 0.3 ppm. These two signals can be attributed to the proton at positions l and m, respectively (Scheme 5).
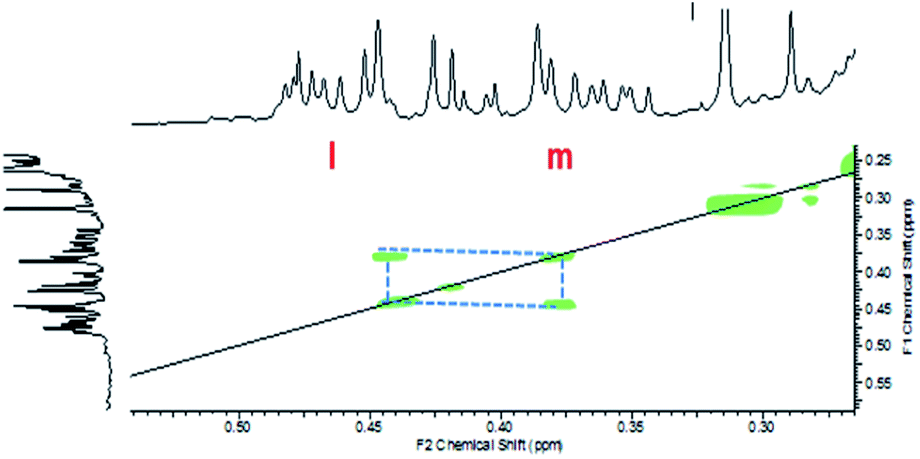 | ||
| Fig. 9 COSY spectrum of formulation (A + C) after 60 min of UV irradiation. | ||
 | ||
| Scheme 5 Main reaction product between A and C. | ||
Proton coupling for the (A + B) formulation was also observed, although the weak signal, due to the low efficiency of the reaction, does not allow for the integration of the peaks.
3.5. Computational study of mechanistic behavior
Gibbs free energies ΔG for the different reactions in Scheme 1 were computed at the B3LYP/6-31G*//B3LYP/6-311++G** level of theory and corrected for zero-point energy and temperature (298 K) at the B3LYP/6-31G* level. These computational data are shown in Fig. 10 and confirm the experiments. Indeed, hydrogen abstraction (reaction (2) in Scheme 1) is energetically favorable, with ΔG being between −16.6 and −51.3 kJ mol−1, depending on the structure of the silane. The lowest values of ΔG are obtained for D and E as a result of the stronger stabilization of the unpaired electron by the phenyl groups as can be seen from the spin density distribution (see Fig. 10). The actual rate constant of hydrogen abstraction from E to 3BP was measured as 3.1 107 M−1 s−1.17 One can expect a lower rate constant for B and C, a fact which could account for the lowest efficiency of the reaction in these cases, knowing that the triplet state of benzophenone has a limited lifetime of approx. 5–10 μs.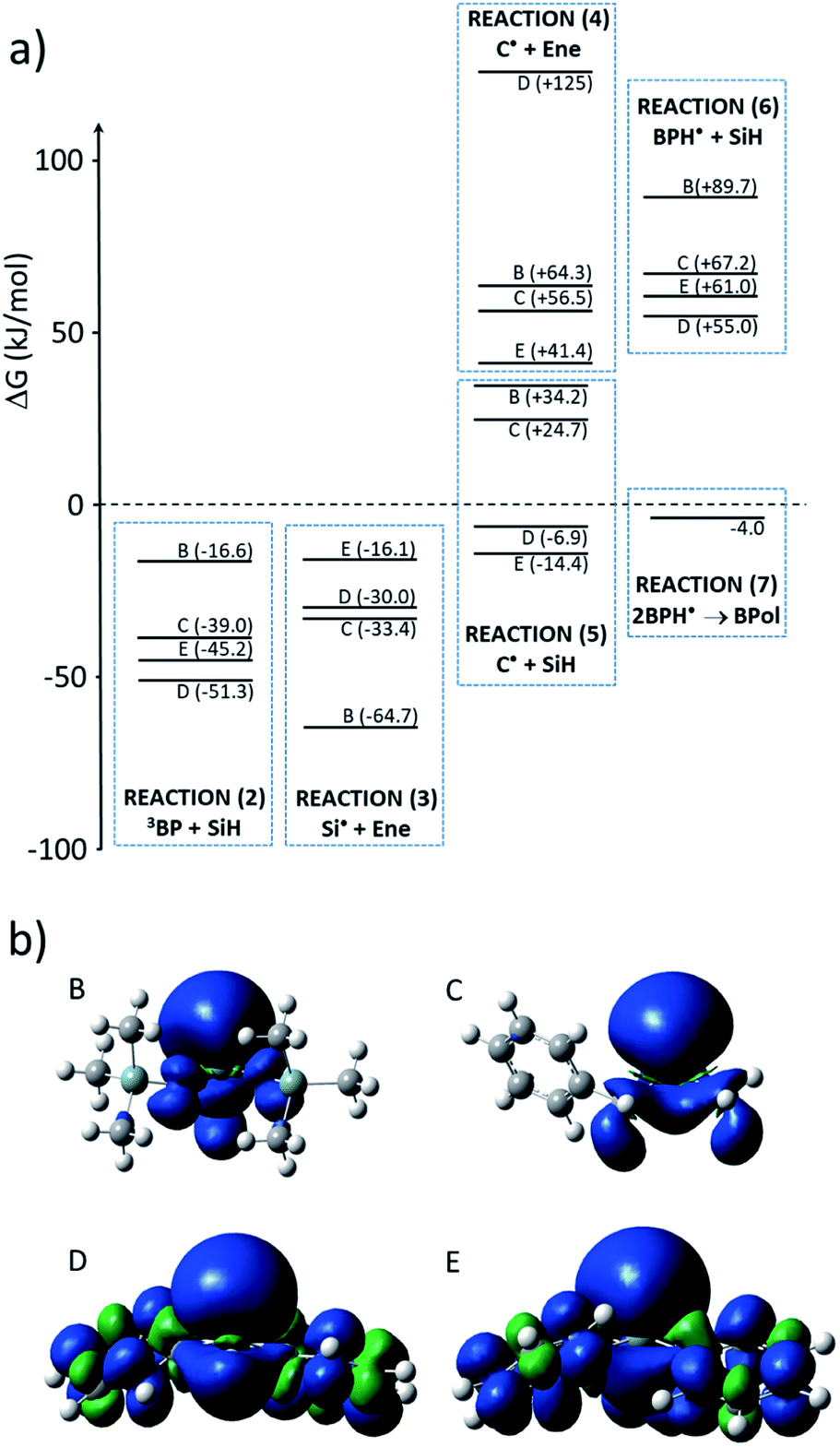 | ||
| Fig. 10 (a) Values of the Gibbs free energy ΔG (kJ mol−1) accompanying the different reactions (with respect to Scheme 1) and (b) spin density of the different silyl radicals. | ||
After the formation of silyl radicals through reaction (2), the addition of these radicals onto the double bond through reaction (3) is energetically favorable, with ΔG values between −16.1 and −64.7 kJ mol−1. Interestingly, the radicals which are the most stabilized in reaction (2), i.e., D and E, lead to less exergonic reactions in the addition reaction (3). It is, therefore, not surprising that the best efficiency was found for these two silanes.
By contrast, the propagation reaction (4), corresponding to the addition of the carbon-centered radical onto the double bond, is undoubtedly endergonic. This explains why the corresponding species were not detected in our experiments and were reported in literature to be minor products.1
The termination reaction (5) by hydrogen abstraction from a silane is a mandatory step to achieve cyclic behavior, allowing for a step-growth mechanism of the photoinduced hydrosilylation. Interestingly, as can be seen from Fig. 10, this reaction is exergonic only for E and B, and is endergonic for B and BM. This merely explains the efficiency of the reaction with D and E, for which 30 to 50% of the Si–H bonds are consumed, in contrast to the reaction with B or C.
Hydrogen abstraction from the silane by the ketyl radical, i.e., reaction (6), is found to be endergonic. Therefore, secondary hydrogen abstraction and the formation of benzhydrol (Bz, Scheme 1) is not expected to take place, a fact that suppress a pathway for the formation of silyl radicals. The termination reaction through the formation of benzopinacol (BPol) would preferably occur, as the formation of BPol from two ketyl radicals is computed to be ΔG = −4.0 kJ mol−1, a process which is energetically more favorable, despite the steric hindrance.
Additional coupling of two carbon-centered radicals was not computed for sake of computation time. However, the recombination of carbon radicals is known to be exergonic. This may present an additional pathway for the termination of the process, considering that this recombination would be sterically hindered for D and E, and favored for B and C. This is also in line with experimental yields of the reaction.
4. Conclusion
Based on NMR spectroscopy and a computational approach, the reaction between different hydrosilanes and methylbis(trimethylsilyloxy) vinylsilane using benzophenone as the photoinitiator was studied. The benzophenone triplet state abstracts hydrogen from the silane leading to a silyl radical which further adds onto a double bond. The influence of the molecular structure and functionality of hydrosilane on the hydrosilylation is highlighted. The main hydrosilylation products (especially with the A + E and A + D formulations) were determinated using HSQC, DEPT, COSY and 13C NMR. The photoreaction efficiency of triphenylsilane and diphenylsilane is higher than that of polymethylhydrosiloxane trimethylsilyl terminated 2-5 cSt and benzyl(dimethyl)silane. This mainly results from the more efficient hydrogen abstraction from the silane by the propagating carbon-centered radical, due to the lower bond dissociation energy of the phenyl-substituted silanes with respect to the aliphatic ones. However, it is difficult for a step-growth mechanism to take place due to termination reactions which are favored with respect to the hydrogen abstraction from a silane by the radical adduct. A possible improvement would be to suppress the formation of BPol. Therefore, these results shed some light on the main drawbacks in the hydrosilylation reaction under light, and can contribute to the development of new photoinitiators for improving the process.Conflicts of interest
There are no conflicts to declare.References
- C. C. Rowlands, J. C. Evans, P. Hupfield and S. E. Cray, J. Chem. Soc., Faraday Trans., 1990, 86, 3221–3227 RSC.
- J. L. Speier, Adv. Organomet. Chem., 1979, 17, 407 CrossRef CAS.
- L. H. Sommer, E. W. Pietrusza and F. C. Whitmore, J. Am. Chem. Soc., 1947, 69, 188 CrossRef CAS.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603 RSC.
- M. Okamoto, H. Kiya, H. Yamashita and E. Suzuki, Chem. Commun., 2002, 15, 1634–1635 RSC.
- J. Li, C. Yang, L. Zhang and T. Ma, J. Organomet. Chem., 2011, 696, 1845–1849 CrossRef CAS.
- Y. Bai, J. Peng, H. Yang, J. Li, G. LaI and X. Li, Chin. J. Chem. Eng., 2012, 20, 246–253 CrossRef CAS.
- W. Hu, H. Xie, H. Yue, P. Prinsen and R. Luque, Catal. Comm., 2017, 97, 51–55 CrossRef CAS.
- A. Nyczyk, C. Paluszkiewicz, M. Hasik, M. Cypryk and P. Pospiech, Vib. Spectrosc., 2012, 59, 1–8 CrossRef CAS.
- J. L. Speier, J. A. Webster and G. H. Barnes, J. Am. Chem. Soc., 1957, 79, 974 CrossRef CAS.
- B. D. Karstedt, US Pat., US3775452 (A), General Electric Company, 1973 Search PubMed.
- B. Marciniec, J. Gulinski, W. Urbaniak and Z. W. Kornetka, Comprehensive Handbook on Hydrosilylation, Pergamon Press, Oxford, NewYork, Seoul, Tokyo, 1992 Search PubMed.
- Z. Huang, Z. Chen, Y. Jiang, N. Li, S. Yang, G. Wang and X. Pan, J. Am. Chem. Soc., 2021, 143, 19167–19177 CrossRef CAS PubMed.
- H. Leaver and G. C. Ramsey, Tetrahedron, 1969, 25, 5669 CrossRef.
- C. R. Morgan, F. Magnotta and A. D. Ketley, J. Polym. Sci., 1977, 15, 627 CAS.
- C. Chatgilialoglu, J. Scaiano and K. Ingold, Organometallics, 1982, 1, 466–469 CrossRef CAS.
- J. Lalevée, X. Allonas and J. P. Fouassier, J. Org. Chem., 2007, 72, 6434–6439 CrossRef PubMed.
- J. Lalevée, A. Dirani, M. El-Roz, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 2003–2010 CrossRef.
- G. Verma, Chem. Sci. J., 2017, 8, 1000165 Search PubMed.
- H. Shin and B. Moon, J. Polym. Sci. A: Polym. Chem., 2018, 56, 527–536 CrossRef CAS.
- D. M. Haddleton, N. Risangud, Z. Li, A. Anastasaki, P. Wilson and K. Kempe, RSC Adv., 2015, 5, 5879–5885 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08099g |
| This journal is © The Royal Society of Chemistry 2022 |