
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpontaneous in situ generation of photoemissive aurophilic oligomers in water solution based on the 2-thiocytosine ligand†
Daniel Blasco ,
María Rodríguez-Castillo,
M. Elena Olmos,
Miguel Monge and
José M. López-de-Luzuriaga*
,
María Rodríguez-Castillo,
M. Elena Olmos,
Miguel Monge and
José M. López-de-Luzuriaga*
Departamento de Química, Centro de Investigación en Síntesis Química (CISQ), Universidad de La Rioja, Madre de Dios 53, 26006 Logroño, Spain. E-mail: josemaria.lopez@unirioja.es
First published on 17th March 2022
Abstract
Complexes [Au(S-2-thiocytosinate)(PMe3)] (2, 2-thiocytosine = 4-amino-2-mercaptopyrimidine) and [Au(S-2-thiocytosine)(PMe3)](CF3CO2) (3) have been prepared by the reaction of [Au(acac)(PMe3)] (1, acac = acetylacetonate) or [Au(OCOCF3)(PMe3)] with 2-thiocytosine, respectively. The equimolecular mixture of complexes 1 and 3 also produces [{Au(PMe3)}2(μ-S,N1-2-thiocytosinate)](CF3CO2) (4), which features two distinct [Au(PMe3)]+ groups coordinated to the S and N1 atoms of the heterocycle. Complex 4 experiences a ligand redistribution process in water solution that liberates [Au(PMe3)2](CF3CO2) and a brightly coloured and luminescent species of [Aun(μ-S,N1-2-thiocytosinate)n] stoichiometry, presumably as a tetraauracycle (n = 4).
Introduction
A feature that attracts the attention of inorganic chemists towards the production of water-soluble gold(I) complexes is in some cases their dynamic behaviour in solution. This is because of aurophilicity as the origin of the self-aggregation of gold(I) centres at sub-van der Waals distances.1,2 This close approximation of coordinatively unsaturated gold(I) atoms is by no means innocent, as it induces (a) a deviation from the hardly-photoemissive ideal dicoordinate linearity of gold(I), and (b) the mixing of suitable “atomic orbitals” (fully gold(I)-centered molecular orbitals would be a more realistic image) into pairs of new AuI⋯AuI axis-centered molecular orbitals. Those new orbitals are now accessible by optical excitation of the sample, giving rise to one of the most appealing properties of gold(I) complexes, photoluminescence.3As devoted “aurophilic” chemists ourselves, we have considered in the recent years natural nucleobases as convenient ligands for the stabilization of molecular gold(I) compounds. In those, the co-existence of strong aurophilic interactions and numerous hydrogen bonds in an “orthogonal” fashion allows their effective self-aggregation in water and, hence, the design of low-molecular-weight gelators and/or luminophores.4 On the other side, thiol-functionalized nucleobases are not unfamiliar ligands for the stabilization of gold(I) complexes. Indeed, they are interesting polydentate S,N-donor heterocycles and, for this reason, they had been considered for achieving the main goals of nowadays' gold(I) chemistry (see Scheme 1). That resulted, for instance, in the discovery of diauracyclic 2-thiouracyl and 6-methyl-2-thiouracyl salts featuring tribochromic luminescence,5 or in the synthesis of a wide plethora of antiproliferative and/or antimicrobial complexes sharing the [Au(PR3)(SR)] scaffold of antiarthritic [Au(PEt3){S-(2,3,4,6-tetra-O-acetyl-1-thio-β-pyranosate)}] (trade name Auranofin®).6 The presence of amino and/or hydroxy groups in the nucleobases' moieties also confers potential water solubility to the (thiolate)gold(I) complexes, converting them in non-toxic “liquid gold” sources for the deposition of thin metal layers over glass and ceramics, showing an industrial application of such.7 Furthermore, surprising structural arrangements have to be expected for their X-ray structures, e.g., the astonishingly simple complex bis(S-2-pyridinethione)gold(I) perchlorate [Au(C5H5NS)2](ClO4), where five of the six cations that are present in the unit cell form a discrete pentamer by AuI⋯AuI interactions of ca. 3.3 Å, whereas the remaining sixth is segregated.8
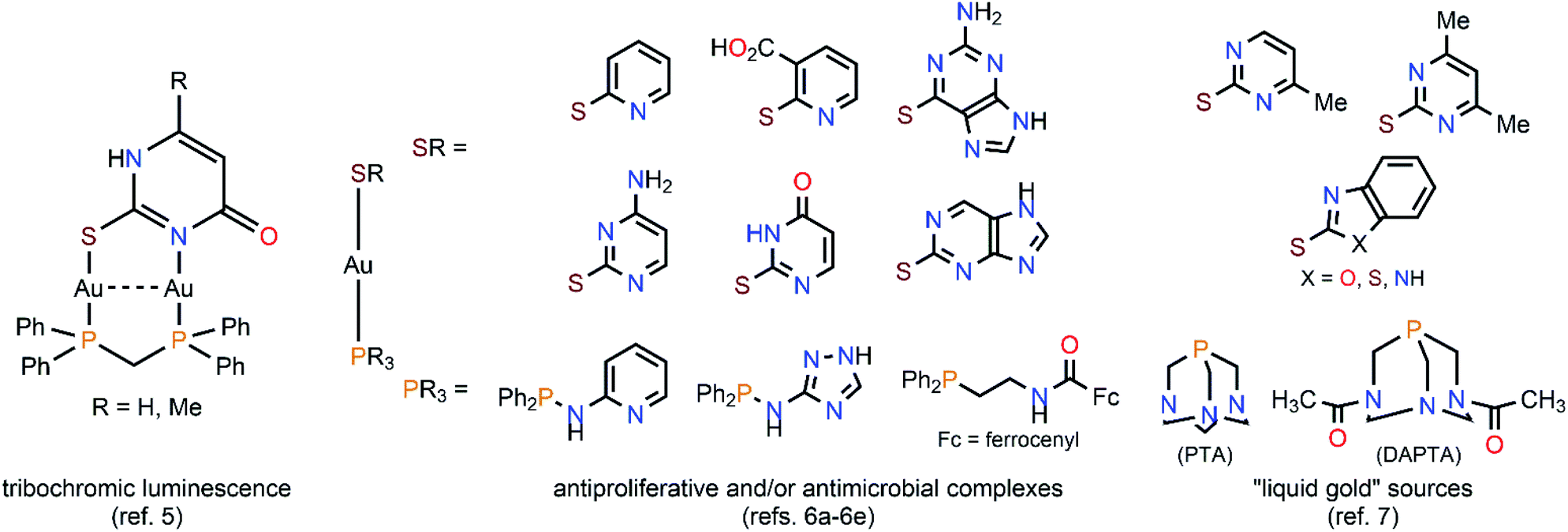 | ||
| Scheme 1 Selected examples of versatile (phosphine)(S,N-heterocycle)gold(I) complexes extracted from the literature. | ||
Hence, recalling both our reported results for extremely simple complexes featuring the natural nucleobase adenine (6-aminopurine), and those displayed by thiol-functionalized nucleobase-containing ones, we decided to investigate the properties of the products of the mono- and diauration of 2-thiocytosine (4-amino-2-mercaptopyrimidine). The choice of 2-thiocytosine responds to the existence of a previous report by H. Schmidbaur et al. that demonstrates its feasibility as a substrate for an analogous chemistry to that developed by us for adenine.9 Moreover, their study was completed with the crystal structure determination of [Au(S-2-thiocytosinate)(PR3)] (PR3 = PEt3, PPh3, PPh2py) and [{Au(S-2-thiocytosinate)}2(μ-dppm)] derivatives. As it will be illustrated, despite following conceptually identical synthetic strategies, the arrived properties of (2-thiocytosine)gold(I) complexes in water solution are dramatically different to those of (adenine)gold(I) ones.
Results and discussion
Syntheses and characterization
The aforementioned report of H. Schmidbaur et al. provided a sensible procedure for the synthesis of [Au(S-2-thiocytosinate)(PMe3)] (2), involving the one-pot deprotonation of 2-thiocytosine and displacement of the chloride ligand from [AuCl(PMe3)] as precipitating NaCl.9 However, the overall yield of the process is poor (51%), probably due to the low solubility of complex 2 in the solvent (dichloromethane) selected for its purification. Furthermore, scarce chemical and spectroscopic, and no structural information of 2, is provided therein.Thus, we have implemented a two-step procedure for circumventing the experimental issues associated with the formation of NaCl as a by-product when synthesizing hydrophilic gold(I) complexes (as the ones discussed), analogous to that exploited by us and others, e.g., for the coordination of [Au(PTA)]+ (PTA = 1,3,5-triaza-7-phosphaadamantane) moieties to acidic substrates.4a,10 For doing so, [AuCl(PMe3)] was reacted with an slight excess of Tl(acac), affording pure [Au(acac)(PMe3)] (1) by complete ligand metathesis promoted by the progressive precipitation of insoluble TlCl in dichloromethane solution. Complex 1 is characterized by the strong IR stretching absorption of the acetylacetonate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (1629 cm−1). In the 1H NMR spectrum, the substitution of chloride by acetylacetone is evinced by the appearance of new singlet (CH3 protons resonance) and doublet (phosphorus-coupled CH proton resonance) signals. Also, the 31P{1H} singlet signal (−1.4 ppm) is downfield-shifted with respect to that of [AuCl(PMe3)] (−10.3 ppm). Apart of stabilizing the gold(I) complex, the acetylacetonate ligand is an efficient base; thus, [Au(acac)(PMe3)] is able to deprotonate 2-thiocytosine and to coordinate the remaining [Au(PMe3)]+ fragment to the thiolate position, simultaneously, rendering pure solid [Au(S-2-thiocytosinate)(PMe3)] (2), and easily-removable acetylacetone as the only by-product. If the ionic analog [Au(S-2-thiocytosine)(PMe3)](CF3CO2) (3) is desired, a tetrahydrofuran solution of the thermally unstable [Au(OCOCF3)(PMe3)] has to be prepared in situ, by reacting [AuCl(PMe3)] with stoichiometric Ag(OCOCF3) at ice bath temperature and under inert gas atmosphere.11 The addition of 2-thiocytosine to the filtered solution of [Au(OCOCF3)(PMe3)] forces the displacement of the trifluoroacetate ligand from the coordination sphere of gold(I) yielding complex 3, where [Au(PMe3)]+ binds to the sulphur atom of the incorporated ligand as revealed by X-ray diffraction (see X-ray crystal structure determination). Since complex 3 still retains an acidic proton, it is amenable of further functionalization through a deprotonation reaction. Thus, the mixture of [Au(S-2-thiocytosine)(PMe3)](CF3CO2) (3) and [Au(acac)(PMe3)] (1) in tetrahydrofuran solution leads to the steady precipitation of [{Au(PMe3)}2(μ-S,N1-2-thiocytosinate)](CF3CO2) (4). With this two-step synthetic procedure, the coordination of two consecutive [Au(PMe3)]+ units into a same molecular substrate is performed in a perfectly controlled manner avoiding undesired side-reactions.
O bonds (1629 cm−1). In the 1H NMR spectrum, the substitution of chloride by acetylacetone is evinced by the appearance of new singlet (CH3 protons resonance) and doublet (phosphorus-coupled CH proton resonance) signals. Also, the 31P{1H} singlet signal (−1.4 ppm) is downfield-shifted with respect to that of [AuCl(PMe3)] (−10.3 ppm). Apart of stabilizing the gold(I) complex, the acetylacetonate ligand is an efficient base; thus, [Au(acac)(PMe3)] is able to deprotonate 2-thiocytosine and to coordinate the remaining [Au(PMe3)]+ fragment to the thiolate position, simultaneously, rendering pure solid [Au(S-2-thiocytosinate)(PMe3)] (2), and easily-removable acetylacetone as the only by-product. If the ionic analog [Au(S-2-thiocytosine)(PMe3)](CF3CO2) (3) is desired, a tetrahydrofuran solution of the thermally unstable [Au(OCOCF3)(PMe3)] has to be prepared in situ, by reacting [AuCl(PMe3)] with stoichiometric Ag(OCOCF3) at ice bath temperature and under inert gas atmosphere.11 The addition of 2-thiocytosine to the filtered solution of [Au(OCOCF3)(PMe3)] forces the displacement of the trifluoroacetate ligand from the coordination sphere of gold(I) yielding complex 3, where [Au(PMe3)]+ binds to the sulphur atom of the incorporated ligand as revealed by X-ray diffraction (see X-ray crystal structure determination). Since complex 3 still retains an acidic proton, it is amenable of further functionalization through a deprotonation reaction. Thus, the mixture of [Au(S-2-thiocytosine)(PMe3)](CF3CO2) (3) and [Au(acac)(PMe3)] (1) in tetrahydrofuran solution leads to the steady precipitation of [{Au(PMe3)}2(μ-S,N1-2-thiocytosinate)](CF3CO2) (4). With this two-step synthetic procedure, the coordination of two consecutive [Au(PMe3)]+ units into a same molecular substrate is performed in a perfectly controlled manner avoiding undesired side-reactions.
The presence of 2-thiocytosine in complexes 2 and 3 is assessed by the observation of two well-resolved doublets in the aromatic region of the 1H NMR spectra, corresponding to the resonance of C6H and C5H protons (check Scheme 2 for the canonical numbering of 2-thiocytosine). The proton signals of PMe3 also appear as a doublet, displaying minimal differences in their chemical shifts that may relate to the neutral (2) and ionic (3) character of such similar gold(I) complexes. The 31P{1H} NMR spectra of complexes 2 and 3 show one singlet in each case, at −1.2 ppm (2), and −1.3 ppm (3). The number and multiplicity of the 1H NMR signals of complex 4 are identical to those described for 2 or 3, except for the fact that it appears to be equilibrated with [Au(PMe3)2]+ in, at least, methanol solution (check the residual 31P{1H} signal at 9.1 ppm in Fig. S16†). Moreover, only one [Au(PMe3)]+-related signal is recorded in both 1H (although with a higher integration than in 2 or 3) and 31P{1H} NMR spectra, evidencing an extremely fast exchange of the [Au(PMe3)]+ groups between the diverse N- and S-donor position of 2-thiocytosine, a process that takes place even at 200 K (Fig. S17†).
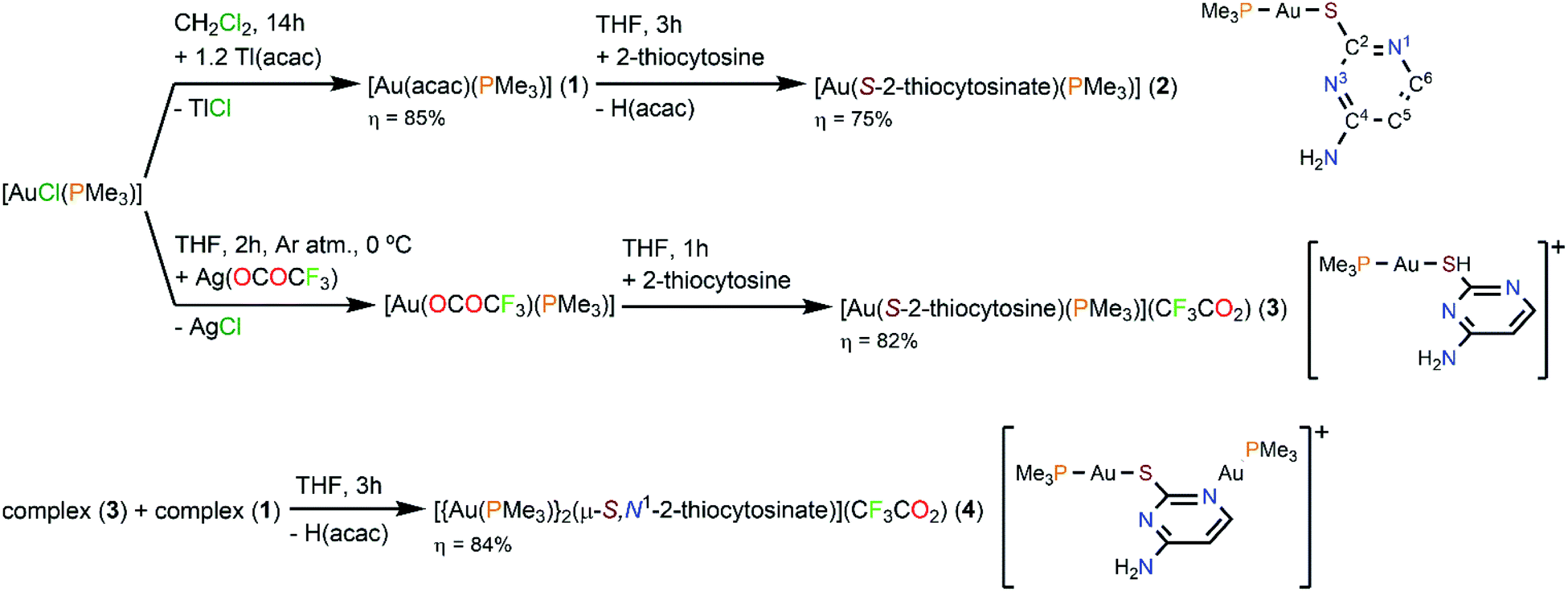 | ||
| Scheme 2 Syntheses of complexes 1–4. | ||
The ESI-MS(+) spectra of complexes 2–4 feature identical major peaks, corresponding to [Au(PMe3)2]+ (349.0 Da), [Au(S-2-thiocytosine)(PMe3)]+ (400.0 Da) and [{Au(PMe3)}2(μ-S,N1-2-thiocytosinate)]+ (672.0 Da), which represent extraordinary stable complex ions that, if not naturally present in the sample, are formed under the harsh ESI-MS measuring conditions. A peak corresponding to homoleptic [Au(S-2-thiocytosinate)2]− (449.0 Da) is also detected in the ESI-MS(−) spectra of 2 and 3.
Finally, molar conductivity measurements of complexes 3 (80.9 cm2 Ω−1 mol−1) and 4 (83.2 cm2 Ω−1 mol−1) in methanol solution pertain to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 electrolyte range, according with the proposed ion-pair stoichiometry.
:1 electrolyte range, according with the proposed ion-pair stoichiometry.
X-ray crystal structure determination
Suitable single crystals for the elucidation of the structure of complexes 2–4 by X-ray diffraction were obtained by slow diffusion of diethyl ether into solutions of 95% ethanol (for 2) or acetone (for 4), or n-hexane into acetone (for 3), all of them at room temperature. The interested reader is referred to Tables S1–S6† in the ESI for further data collection details and selected bond lengths and angles.Complex 2 crystallizes in the orthorhombic Pbca space group (Z = 8). Its asymmetric unit, which corresponds to one single [Au(S-2-thiocytosinate)(PMe3)] molecule (Fig. 1), reveals the binding of the phosphinegold(I) fragment (Au–P distance, 2.2564(7) Å) to the deprotonated sulphur atom (Au–S distance, 2.3222(7) Å) of 2-thiocytosine, in an almost linear fashion (S–Au–P angle, 177.14(3)°). The relative syn position of the C4–NH2 group with respect to the gold(I) center is also noteworthy, although the H⋯AuI distance is considerably long (4.518 Å) to suggest any kind of stabilizing interaction between them. The aforementioned work of H. Schmidbaur et al.9 and another one of A. Laguna, M. C. Gimeno et al.6a provided valuable structural information of several [Au(S-2-thiocytosinate)(PR3)] examples. In all cases, the coordination sphere of gold(I) is identical to that of 2, but displaying notably shorter Au–S distances despite bearing bulkier phosphine ligands: PR3 = PEt3, 2.291(3)–2.307(2) Å; PPh3, 2.303(1)–2.308(1) Å; PPh2py, 2.3068(8)–2.3083(8) Å; PPh2NHpy, 2.3130(8) Å; μ-dppm, 2.3052(9)–2.309(1) Å. However, the most significant fact is that the supramolecular network of these five examples is exclusively built-up by hydrogen bonds only (a long intramolecular AuI⋯AuI interaction of 3.3317(2) Å is present in dinuclear [{Au(S-2-thiocytosinate)}2(μ-dppm)]), giving rise to intricate 3-D structures. The authors explain the remarkable absence of aurophilic interactions as a result of competing weak forces that include hydrogen bonding rather than by the steric hindrance of the phosphines. Complex [Au(S-2-thiocytosinate)(PMe3)] (2) is not an exception, because no AuI⋯AuI distances within the interaction range are found. Instead, the crystalline structure grows along the b axis by a combination of C4N–H⋯N1, C4N–H⋯S and C5–H⋯S hydrogen bonds (Fig. S24†), and through the other two axes by weaker C6–H⋯N3 and PC–H⋯N bonds.
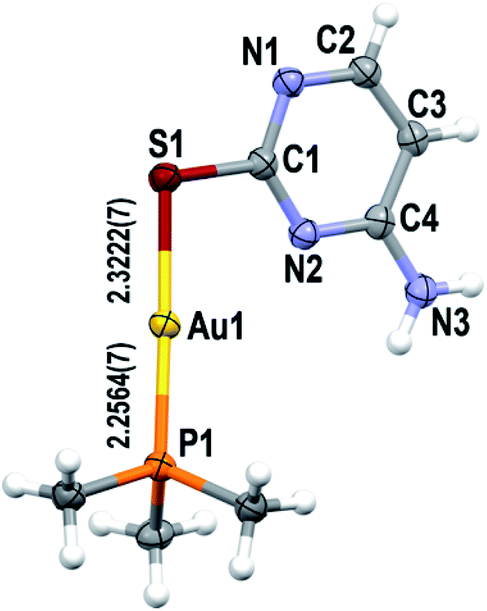 | ||
| Fig. 1 Asymmetric unit of complex 2, with the labelling scheme for the atom positions (50% probability ellipsoids) and selected bond distances. Colour code: C, grey; H, white; Au, yellow; N, blue; P, orange; S, brown. | ||
The ionic analogue [Au(S-2-thiocytosine)(PMe3)](CF3CO2) (3) also crystallizes in the Pbca space group of the orthorhombic system (Z = 8). The molecular structure of the gold(I) cation is isostructural to that of complex 2 (Fig. 2), so no reiterative description will be provided in that sense. However, the poor quality of the collected data precludes the report of precise bond distances and angles (the corresponding cif file is available as ESI†).
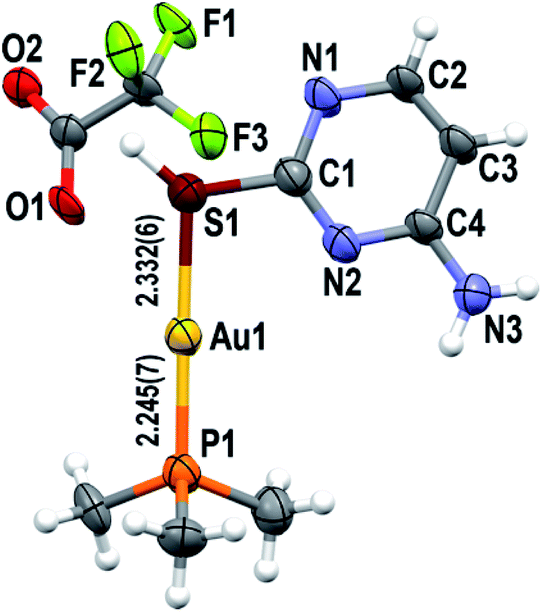 | ||
| Fig. 2 Asymmetric unit of complex 3, with the labelling scheme for the atom positions (50% probability ellipsoids) and selected bond distances. Colour code: C, grey; H, white; Au, yellow; F, green; N, blue; O, red; P, orange; S, brown. | ||
Lastly, the crystalline structure of dinuclear [{Au(PMe3)}2(μ-S,N1-2-thiocytosinate)](CF3CO2)·0.25H2O (4·0.25H2O) has been determined. This complex crystallizes in the monoclinic P21/c space group (Z = 8), with two formula units integrating the asymmetric unit. The cationic part of the formula (Fig. 3, left) features one [Au(PMe3)]+ fragment coordinated to the S-donor position of 2-thiocytosine (as in complexes 2 and 3), in a relative syn fashion with respect to the C4–NH2 group, and with Au–S bond distances of 2.331(3) and 2.324(3) Å. The second [Au(PMe3)]+ unit, by its side, coordinates to the endocyclic N1 position, with identical Au–N bond distances of 2.091(10) Å for both cations in the asymmetric unit. The Au–P bond distances are clearly affected by the nature of the second donor atom bound to gold(I), shortening from 2.251(3), 2.254(3) Å (phosphorus trans to sulphur) to 2.235(4), 2.238(3) Å (phosphorus trans to endocyclic nitrogen). It should be highlighted that, in contrast to the structures of complexes 2 and 3 and the others reported, intermolecular aurophilic interactions are found in here, being also master forces for the polymer-like growth of the supramolecular assembly along the c axis (Fig. 3, right). This way, each S-bound gold(I) atom interacts with a N-bound one of the closest symmetry-related cation, with repetitive AuI⋯AuI distances of 3.0371(7) or 3.1317(7) Å, one for each of the analogous 1-D aurophilic chains that grow starting from the two different cations present in the asymmetric unit. It is also surprising that despite [Au(PMe3)]+ fragments could be disposed in close proximity since they coordinate to vicinal positions of 2-thiocytosine, they avoid intramolecular AuI⋯AuI contacts. Trifluoroacetate anions play a more important structural role in here rather than balancing the total electric charge of the 1-D aurophilic chains. Each one acts as a double acceptor of two C4N–H⋯O hydrogen bonds, with the particularity that only two cation–anion pairs are involved per repetitive motif. This gives rise to a square-like adduct which joins pairs of 1-D aurophilic chains all along their length (Fig. S25†), expanding the crystalline structure into a complex 2-D folded sheet-like pattern (Fig. S26†).
 | ||
| Fig. 3 Molecular structure of the cation of 4·0.25H2O (left, 50% probability ellipsoids), and its polymer expansion through aurophilic interactions along the c axis (right, trifluoroacetate anions and water molecules have been omitted for clarity), with the labelling scheme for the atom positions and selected bond distances. Colour code: C, grey; H, white; Au, yellow; N, blue; P, orange; S, brown. | ||
Solid-state optical properties
The DRUV-Vis absorption spectra of complexes 2–4 (Fig. S4†) are featureless and, in general, resemble that of free 2-thiocytosine, suggesting an intraligand origin for all of them. From those, only the band-edge of complex 4, also the only one where polymeric intermolecular AuI⋯AuI interactions are observed within its crystalline structure, is notably red-shifted with respect to that of the ligand.From the three complexes 2–4, only [Au(S-2-thiocytosinate)(PMe3)] (2) and [{Au(PMe3)}2(μ-S,N1-2-thiocytosinate)](CF3CO2) (4) display visible photoluminescence when irradiated with 365 nm UV light at room temperature (see Fig. 4). In both cases, the yellowish emission corresponds to a non-structured band with respective maxima at 515 nm (for 2, λex = 370 nm, ϕ = 1.7%) and 531 nm (for 4, λex = 406 nm, ϕ = 6.6%), and associated long lifetimes of 512 ns (2) and 550 ns (4) that suggest a phosphorescent deactivation process of the optically accessible excited states. When the temperature is lowered to 77 K, a poorly resolved vibronic profile with negligible energy shifting arises for 2 (spacings of 1425, 1334 and 569 cm−1 between consecutive peaks), suggesting the ligand participation in the emission as the spacings' energies match those of CC or CN stretching modes. Upon cooling, complex 4 shifts its greenish emission up to 494 nm (λex = 362 nm). Also, complex 3 turns photoluminescent, displaying an asymmetric broad emission with a maximum at 458 nm (λex = 357 nm, τ = 722 ns) and a low-energy tail extending up to 800 nm (Table 1).
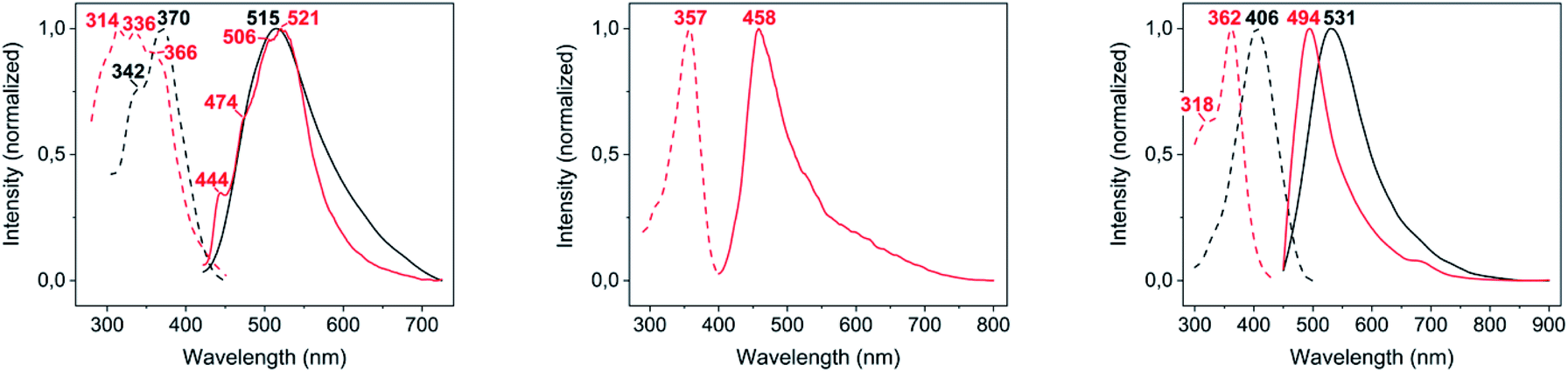 | ||
| Fig. 4 Excitation (dashed lines) and emission (solid lines) spectra in the solid state of complexes 2–4 (from left to right), at room temperature (black lines) and 77 K (red lines). | ||
Computational studies
The photoluminescent emission that gold(I) thiolates commonly display has been traditionally assigned to a spin-forbidden (phosphorescent) charge transfer from sulphur to gold(I) (3LMCT) or to the gold(I)···gold(I) axis (3LMMCT), in the basis of the observation of large Stokes shifts in their luminescence spectra and long emission lifetimes.12A possible origin of the photoluminescent emission of complexes 2 and 4 in the solid state is proposed in here with the aid of DFT and TD-DFT calculations. For modelling complex 2, a single molecule of [Au(S-2-thiocytosinate)(PMe3)] was considered, whereas for complex 4, which possesses a more intricate supramolecular structure in terms of gold(I)-related interactions, two consecutive [{Au(PMe3)}2(μ-S,N1-2-thiocytosinate)]+ cations were picked from the X-ray structure. Both respective models 2a and 4a were freely optimized (DFT-D3/PBE level of theory) and then employed as inputs for further TD-DFT calculations including singlet-to-singlet and, as possible phosphors, singlet-to-triplet vertical excitations. A comparison between the calculated transitions and the experimental absorption and excitation profiles is shown in Fig. 5; for a full description of the labelled ones (transition, wavelength, oscillator strength, involved orbitals, their shapes and population analysis), the ESI† should be checked.
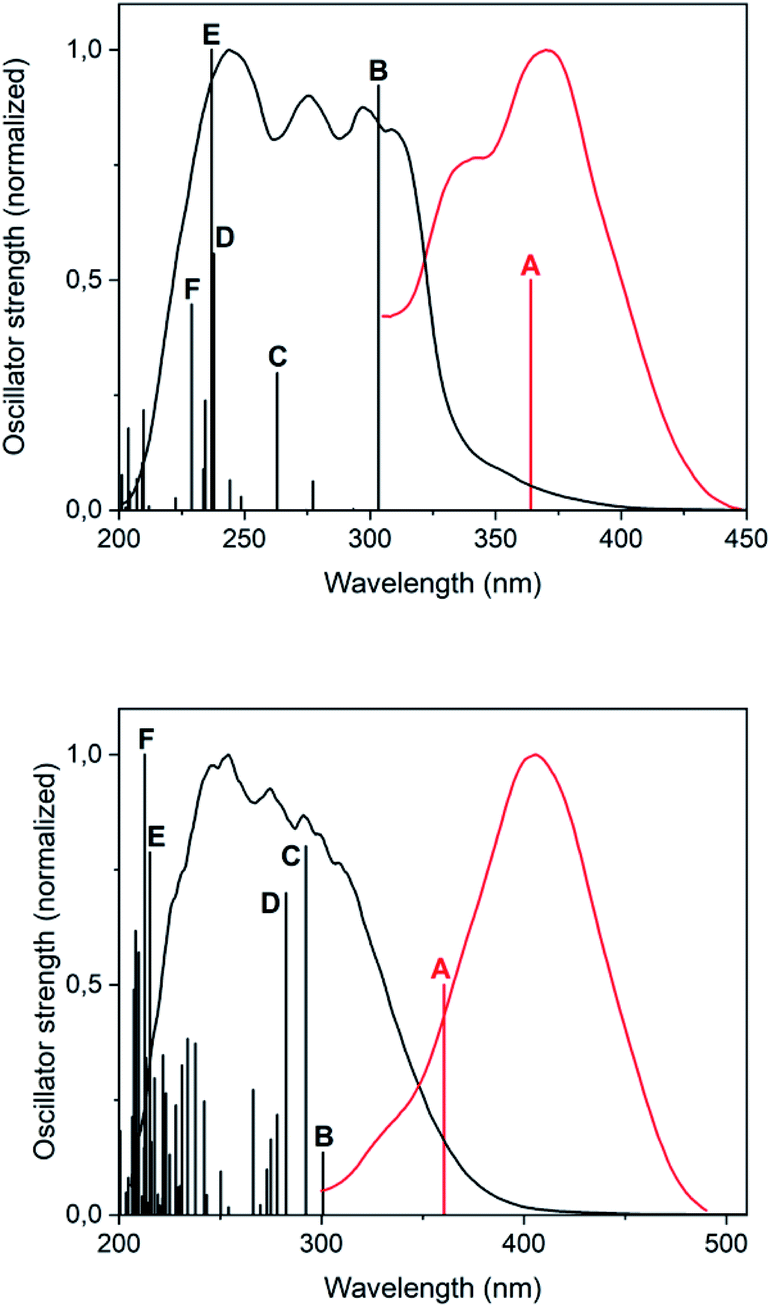 | ||
| Fig. 5 Superimposition of the TD-DFT singlet-to-singlet (black bars) and first singlet-to-triplet (red bar, arbitrary oscillator strength of 0.5) transitions with the absorption (black line, anhydrous KBr mull) and excitation (red line, pure solid sample) spectra, for models 2a (top) and 4a (bottom). | ||
Regarding the superimposition of model 2a (Fig. 5, top), the excitation and absorption spectra only overlap in the low energy region of the latter, what accords with the speculated spin forbiddance of the excitation transition. Also, the calculated singlet-to-triplet transition energy nicely matches with that of the overlap and, according to the population analysis, is composed of the mixture of 3LMCT and 3IL charge transfers from HOMO to LUMO, and a second 3IL contribution from HOMO to LUMO+4. Whereas this qualitative description provided for model 2a also serves for 4a, the orbitalic origin of the forbidden singlet-to-triplet transition of the latter is less evident, since any relevant MO surface is clearly located over definite areas of the model. It can be roughly concluded, however, that a metal-perturbed intraligand transition (3IL) is responsible for the vertical excitation of the model.
Absorption and emission properties in aqueous solution
Among the series of complexes 2–4, only the trifluoroacetate salts, namely [Au(S-2-thiocytosine)(PMe3)](CF3CO2) (3) and [{Au(PMe3)}2(μ-S,N1-2-thiocytosinate)](CF3CO2) (4), are freely water-soluble. This probably may be because the numerous and varied hydrogen bonds that are found in the crystalline structure of neutral [Au(S-2-thiocytosinate)(PMe3)] (2) preclude its solvation by usual organic solvents or water. Strikingly, those complexes 3 and 4 give rise to brightly coloured solutions of a vivid yellow hue, even at concentrations as low as 1 mM, with its colour intensity gradually augmenting upon aging (see Fig. S32†).The poor thermodynamic stability of complex 3 in water solution precluded the completion of a full photophysical study (grey decomposition is observed after few hours); however, aqueous solutions of 4 are perfectly stable even for days, allowing the collection of absorption data for extremely long periods of time. For this reason, variable absorption spectra of a diluted aqueous solution of 4 (0.1 mM) were collected at constant intervals of 900 s (15 min) for 12 h (720 min), which are superimposed in Fig. 6. In the initial measurement, only three high-energy absorptions are registered at 205, 241 and 280 nm, matching the energies of the corresponding absorption bands of free 2-thiocytosine (Fig. S33†). Thus, those absorptions may be assigned to spin-allowed intraligand transitions (1IL) with negligible contributions of the phosphinegold(I) fragments. Immediately afterwards, a new band arises at 405 nm with no appreciable energy-shifting within the considered time interval of 12 h. The appearing of this absorption in the violet-to-blue region of the visible spectrum would be responsible for the vivid yellow colour of the solution, and its increase with respect to time for the attractive color saturation. The original absorption at 241 nm is not static either, as its absorbance decreases at a more pronounced rate; thus, an isosbestic point near 280 nm appears, suggesting chemical equilibria between coexisting entities in solution. Besides, yellow solutions of 4 are photoluminescent, emitting orange light when irradiated with a 365 nm UV hand lamp (see inset in Fig. 7). Neither special solvent handling (argon bubbling, etc.) is needed for achieving such luminescence, which is even observed in aerated water solutions. The emission profile that is recorded from a 5 mM aged solution is structureless, centered at 603 nm, and associated to a lifetime of 129 ns which, in fact, is notably shorter than that of complex 4 in the solid state. The nature of the radiant process cannot be securely elucidated from the lifetime value itself due to the high spin–orbit coupling effect of gold, although it is likely that is associated to a fluorescent one. The excitation profile shows two different bands associated with a dynamic equilibrium within the experimental timescale, although the emission energy is apparently independent from the excitation wavelength (Fig. S34†).
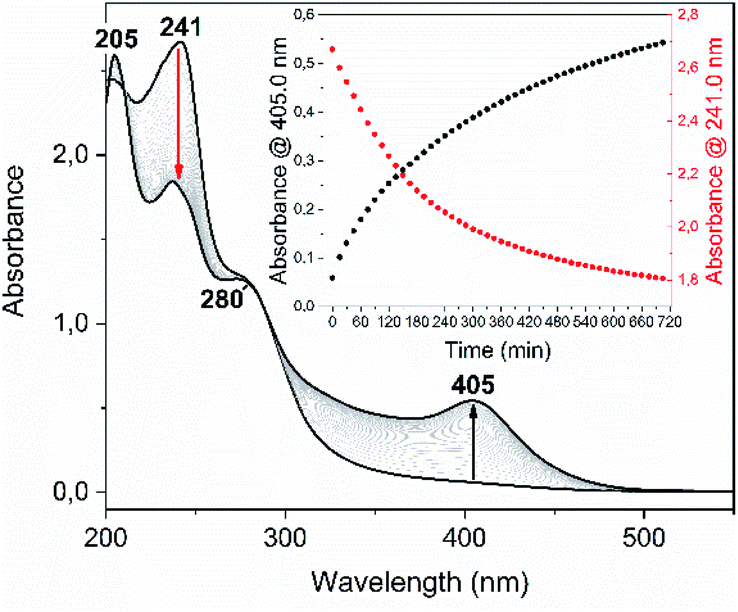 | ||
| Fig. 6 UV-Vis spectra collection of complex 4 (0.1 mM in distilled water), recorded at constant intervals of 900 s (15 min) for 12 h (720 min) and at room temperature. Inset: absorbance at 405.0 nm (black dots) and 241.0 nm (red dots) versus time. | ||
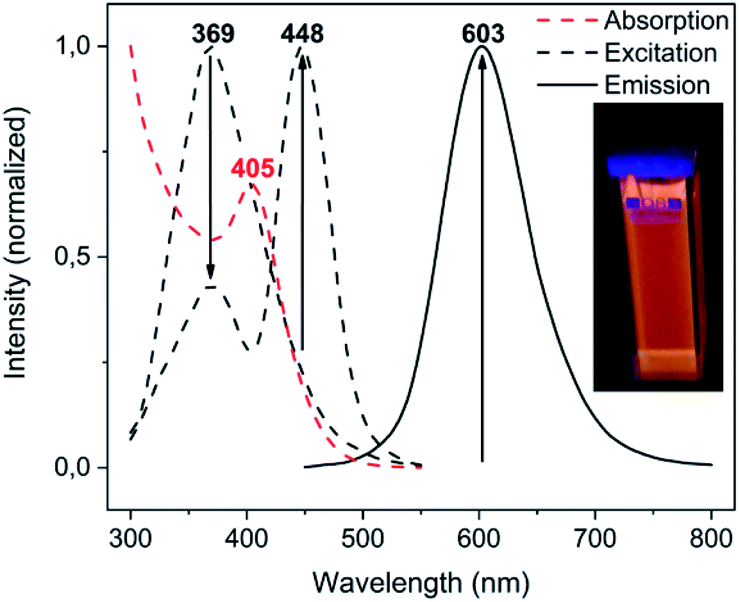 | ||
| Fig. 7 Luminescence spectra of an aqueous solution of complex 4 (0.5 mM), as freshly prepared and after 3 h. The arrows denote the direction of the excitation and emission bands' intensity variation. Inset: sample under 365 nm UV light. | ||
These results prompted us to deepen in its study through time-resolved 1H and 31P{1H} NMR. For this purpose, a solution of complex 4 in deuterium oxide was monitored between constant dwell times for 3 h (180 min). The most clarifying fact that revealed such experiment, which is depicted in the ESI,† is the increasing appearance of the homoleptic cation [Au(PMe3)2]+, which is easily identified by its characteristic 1H triplet-like resonance that exhibits in deuterium oxide,13 and by a singlet in the 31P{1H} NMR spectrum.
The steady formation of [Au(PMe3)2](CF3CO2) leaves a second neutral species of empirical formula [Au(μ-S,N1-2-thiocytosinate)] (Scheme 3). This product is insoluble in all common organic solvents and precludes single crystal seeding methods. Therefore, its exact structure and nuclearity remain non-elucidated. The photophysical properties of complex 4, however, give a clue of the effective establishing of additional AuI⋯AuI interactions. In a similar example, N. W. Mitzel et al. demonstrated that certain rigid bis((phosphine)gold(I))-substituted 1,8-diethynylanthracenes rearrange towards diauracyclization for releasing torsional strain.14 That does not seem to be the underlying reason in here (much less bulkier phosphines are coordinating to both gold(I) atoms) but, according to the numerous examples of eight-membered diauracycles that are findable in the literature,15 such a cyclization process may also happen in here. A wide range of solid-state structures has been described in the literature for neutral homoleptic digold(I) complexes, spanning from discrete dimers to higher nuclearities' forms, including tetramers, triangular or arrow tip-shaped hexamers, and infinite arrays of 1-D polymers. Even, some of them present an additional degree of self-assembly through intermolecular AuI⋯AuI interactions.15c This evinces the potential polymorphic character of [Aun(L∧L)n] complexes (L∧L = anionic ditopic ligand) and, in particular, those where n = 2. Thus, to propose a likely molecular geometry for [Aun(μ-S,N1-2-thiocytosinate)n], the ground state energies and the first singlet-to-singlet vertical transitions of two additional tetragold(I) models, namely [Au2(μ-S,N1-2-thiocytosinate)2]2 (4b) and [Au4(μ-S,N1-2-thiocytosinate)4] (4c, Chart 1), were computed and compared. The energetic difference of 34.61 kJ mol−1 favouring model 4c indicates it to be a more stable species, which may be attributed to the additional aurophilic interactions that are established within the square tetragold(I) cluster. Also, the most intense among the lowest-lying singlet-to-singlet transitions of models 4b and 4c, which are summarized in the ESI,† are suggested to have a different origin. For model 4b, the HOMO → LUMO transition has a major 1MLCT character and a minor 1IL contribution, whereas for model 4c, 1LMCT and 1IL contributions are identified in the symmetry-related HOMO-2 → LUMO and HOMO-1 → LUMO transitions.
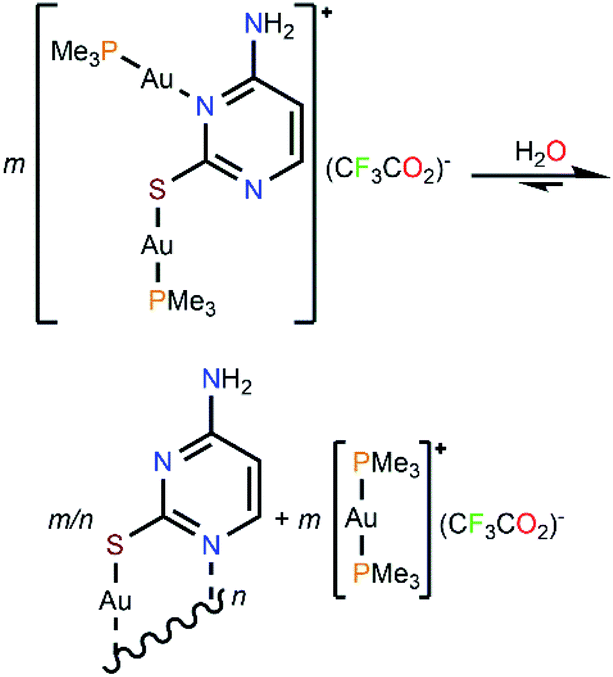 | ||
| Scheme 3 Proposed rearrangement process for complex 4 in aqueous solution. | ||
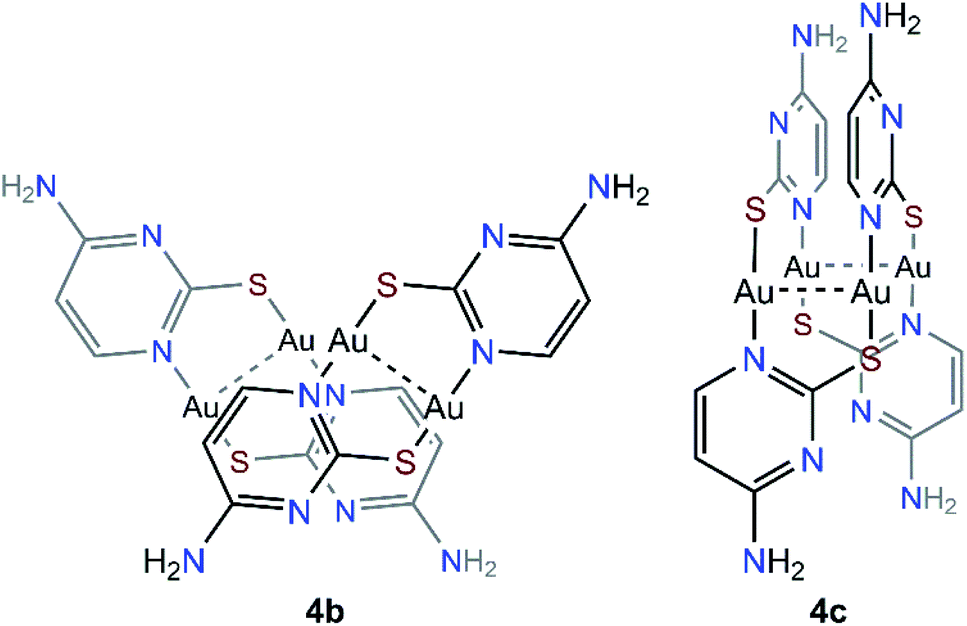 | ||
| Chart 1 Theoretical models [Au2(μ-S,N1-2-thiocytosinate)2]2 (4b) and [Au4(μ-S,N1-2-thiocytosinate)4] (4c). | ||
Conclusions
The products of the mono- and diauration of 2-thiocytosine show different photoluminescence behaviour depending on the ionic charge (complexes 2 versus 3) or the number of gold atoms coordinated to the thiol-functionalized nucleobase (complexes 2 and 3 versus 4). Likewise, the nature of the emission-leading excitation is fully different (3LMCT + 3IL for 2, metal-perturbed 3IL for 4). When left free in fluid solution, gold(I) complexes rearrange themselves by, for instance, ligand exchange, to reach a more stable form. In the present case, this implies the water-promoted breakage of the structure of [{Au(PMe3)}2(μ-S,N1-2-thiocytosinate)](CF3CO2) (4) towards ionic [Au(PMe3)2](CF3CO2) and neutral [Aun(μ-S,N1-2-thiocytosinate)n] species. The excellent properties of water molecules for solvating ionic pairs or hydrogen-bond donors/acceptors may explain this spontaneous process, rather than structural strain loss.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Grant PID2019-104379RB-C22 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”.Notes and references
- (a) H. Schmidbaur and A. Schier, A briefing on aurophilicity, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC; (b) H. Schmidbaur and A. Schier, Aurophilic interactions as a subject of current research: an up-date, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- (a) J. C. Lima and L. Rodríguez, Supramolecular gold metallogelators: the key role of metallophilic interactions, Inorganics, 2015, 3, 1–18 CrossRef CAS; (b) A. Pinto, N. Svahn, J. C. Lima and L. Rodríguez, Aggregation induced emission of gold(I) complexes in water or water mixtures, Dalton Trans., 2017, 46, 11125–11139 RSC; (c) N. Mirzadeh, S. H. Privér, A. J. Blake, H. Schmidbaur and S. K. Bhargava, Innovative molecular design strategies in materials science following the aurophilicity concept, Chem. Rev., 2020, 120, 7551–7591 CrossRef CAS PubMed; (d) G. Romo-Islas and R. Gavara, Recent progress on supramolecular luminescent assemblies based on aurophilic interactions in solution, Inorganics, 2021, 9, 1–36 CrossRef.
- (a) J. M. Forward, J. P. Fackler Jr and Z. Assefa, in Optoelectronic properties of inorganic compounds, ed. J. P. Fackler Jr, Plenum Press, 1999, ch. 6, pp. 195–229 Search PubMed; (b) J. M. López-de-Luzuriaga, in Modern supramolecular gold chemistry: gold-metal interactions and applications, ed. A. Laguna, Wiley-VCG, 2008, ch. 6, pp. 347–401 Search PubMed.
- (a) D. Blasco, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, D. Pascual and M. Rodríguez-Castillo, Cooperative Au(I)···Au(I) interactions and hydrogen bonding as origin of a luminescent adeninate hydrogel formed by ultrathin molecular nanowires, Inorg. Chem., 2018, 57, 3805–3817 CrossRef CAS PubMed; (b) D. Blasco, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos and M. Rodríguez-Castillo, Balancing ionic and H-bonding interactions for the formation of Au(I) hydrometallogels, Dalton Trans., 2019, 48, 7519–7526 RSC; (c) D. Blasco, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, D. Pascual and M. Rodríguez-Castillo, Time-dependent molecular rearrangement of [Au(N9-adeninate)(PTA)] in aqueous solution and aggregation-induced emission in a hydrogel matrix, Inorg. Chem., 2021, 60, 3667–3676 CrossRef CAS PubMed; (d) D. Blasco, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, M. Rodríguez-Castillo, H. Amaveda, M. Mora, V. García Sakai and J. A. Martínez-González, Inorg. Chem. Front., 2021, 8, 3707–3715 RSC.
- Y.-A. Lee and R. Eisenberg, Luminescence tribochromism and bright emission in gold(I) thiouracilate complexes, J. Am. Chem. Soc., 2003, 125, 7778–7779 CrossRef CAS PubMed.
- (a) M. F. Fillat, M. C. Gimeno, A. Laguna, E. Latorre, L. Ortego and M. D. Villacampa, Synthesis, structure and bactericide activity of (aminophosphane)gold(I) thiolate complexes, Eur. J. Inorg. Chem., 2011, 9, 1487–1495 CrossRef; (b) L. A. Mullice, H. J. Mottram, A. J. Hallett and S. J. A. Pope, Gold(I) complexes incorporating emissive mercapto-pteridine ligands: syntheses, X-ray structure, luminescence and preliminary cytotoxic evaluation, Eur. J. Inorg. Chem., 2012, 18, 3054–3060 CrossRef; (c) H. Goitia, Y. Nieto, M. D. Villacampa, C. Kasper, A. Laguna and M. C. Gimeno, Antitumoral gold and silver complexes with ferrocenyl-amide phosphines, Organometallics, 2013, 32, 6069–6078 CrossRef CAS; (d) L. Ortego, F. Cardoso, S. Martins, M. F. Fillat, A. Laguna, M. Meireles, M. D. Villacampa and M. C. Gimeno, Strong inhibition of thioredoxin reductase by highly cytotoxic gold(I) complexes. DNA binding studies, J. Inorg. Biochem., 2014, 130, 32–37 CrossRef CAS PubMed; (e) E. Abas, R. Pena-Martinez, D. Aguirre-Ramírez, A. Rodriguez-Dieguez, M. Laguna and L. Grasa, New selective thiolate gold(I) complexes inhibit the proliferation of different human cancer cells and induce apoptosis in primary cultures of mouse colon tumors, Dalton Trans., 2020, 49, 1915–1927 RSC.
- E. Vergara, S. Miranda, F. Mohr, E. Cerrada, E. R. T. Tiekink, P. Romero, A. Mendía and M. Laguna, Gold(I) and palladium(II) thiolato complexes containing water-soluble phosphane ligands, Eur. J. Inorg. Chem., 2007, 18, 2926–2933 CrossRef.
- R. Usón, A. Laguna, M. Laguna, J. Jiménez, M. P. Gómez, A. Sáinz and P. G. Jones, Gold complexes with heterocyclic thiones as ligands. X-ray structure determination of [Au(C5H5NS)2]ClO4, J. Chem. Soc., Dalton Trans., 1990, 11, 3457–3463 RSC.
- J. D. E. T. Wilton-Ely, A. Schier, N. W. Mitzel, S. Nogai and H. Schmidbaur, Hydrogen-bonded networks: (phosphine)gold(I) 4-amino-2-pyrimidine-thiolates, J. Organomet. Chem., 2002, 643–644, 313–323 CrossRef CAS.
- E. Aguiló, R. Gavara, C. Baucells, M. Guitart, J. C. Lima, J. Llorca and L. Rodríguez, Tuning supramolecular aurophilic structures: the effect of counterion, positive charge and solvent, Dalton Trans., 2016, 45, 7328–7339 RSC.
- M. Preisenberger, A. Schier and H. Schmidbaur, (Phosphine)gold(I) trifluoromethanesulfonates, trifluoroacetates and trichlorothioacetates, J. Chem. Soc., Dalton Trans., 1999, 10, 1645–1650 RSC.
- (a) J. M. Forward, D. Bohmann, J. P. Fackler Jr and R. J. Staples, Luminescence studies of gold(I) thiolates, Inorg. Chem., 1995, 34, 6330–6336 CrossRef CAS; (b) W. B. Jones, J. Yuan, R. Narayanaswamy, M. A. Young, R. C. Elder, A. E. Bruce and M. R. M. Bruce, Solid state EXAFS and luminescence studies of neutral, dinuclear gold(I) complexes. Gold(I)–gold(I) interactions in the solid state, Inorg. Chem., 1995, 34, 1996–2001 CrossRef CAS; (c) S. D. Hanna and J. I. Zink, Analysis of the vibronic structure in the emission and absorption spectra of (μ-1,1-dicyanoethylene-2,2-dithiolato-S,S′)bis(triphenylphosphine)digold(I) and assignment of the emissive state, Inorg. Chem., 1996, 35, 297–302 CrossRef CAS PubMed.
- H. Schmidbaur and R. Franke, Organogold-chemie. X. Methylgold(I) und gold(I)-halogenide als komplexbildende zentren für trimethylphosphin und trimethylphosphit, Chem. Ber., 1972, 105, 2985–2997 CrossRef CAS.
- P. Niermeier, L. Wickemeyer, B. Neumann, H.-G. Stammler, L. Goett-Zink, T. Kottke and N. W. Mitzel, Aurophilicity in action: stepwise formation of dinuclear Au(I) macrocycles with rigid 1,8-dialkynylanthracenes, Dalton Trans., 2019, 48, 4109–4113 RSC.
- (a) L. Hao, R. J. Lachicotte, H. J. Gysling and R. Eisenberg, Novel luminescent Au(I) pyrimidinethiolate dimer having an unusual π-stacking structure, Inorg. Chem., 1999, 38, 4616–4617 CrossRef CAS PubMed; (b) W. J. Hunks, M. C. Jennings and R. J. Puddephatt, Supramolecular gold(I) thiobarbiturate chemistry: combining aurophilicity and hydrogen bonding to make polymers, sheets, and networks, Inorg. Chem., 2002, 41, 4590–4598 CrossRef CAS PubMed; (c) M.-R. Azani, O. Castillo, M. L. Gallego, T. Parella, G. Aullón, O. Crespo, A. Laguna, S. Álvarez, R. Más-Ballesté and F. Zamora, The structural diversity triggered by intermolecular interactions between AuIS2 groups: aurophilia and beyond, Chem.–Eur. J., 2012, 18, 9965–9976 CrossRef CAS PubMed (and references therein).; (d) J. Grote, B. Neumann, H.-G. Stammler and N. W. Mitzel, Diversity of aggregation motifs in gold(I) dithiocarboxylate complexes, Dalton Trans., 2018, 47, 4701–4706 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentation, syntheses, spectroscopic characterization, structural characterization, optical properties, computational studies, NMR kinetic studies, atomic coordinates of computational optimizations (xyz format). CCDC 2123504 and 2123505. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ra00368f |
| This journal is © The Royal Society of Chemistry 2022 |