
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-Kolbe electrolysis of N-protected-α-amino acids: a standardized method for the synthesis of N-protected (1-methoxyalkyl)amines†
A. Walęcka-Kurczykab,
J. Adamek‡
 ab,
K. Walczaka,
M. Michalakc and
A. Październiok-Holewa*ab
ab,
K. Walczaka,
M. Michalakc and
A. Październiok-Holewa*ab
aDepartment of Organic Chemistry, Bioorganic Chemistry and Biotechnology, Silesian University of Technology, B. Krzywoustego 4, 44-100 Gliwice, Poland. E-mail: agnieszka.pazdzierniok-holewa@polsl.pl
bBiotechnology Center of Silesian University of Technology, B. Krzywoustego 8, 44-100 Gliwice, Poland
cInstitute of Organic Chemistry, Polish Academy of Sciences, 01-224 Warsaw, Poland
First published on 12th January 2022
Abstract
Here, we report a standardized method for the synthesis of N-protected (1-methoxyalkyl)amines by the electrochemical decarboxylative α-methoxylation of α-amino acid derivatives using the commercially available, easy-to-use, compact ElectraSyn 2.0 setup. The use of equipment with a standardized power source, electrodes, and other accessories allows this experimental procedure to be easily transferred to any laboratory in the world. A simple workup and chromatography-free purification produced the products in excellent yields above 90%.
Introduction
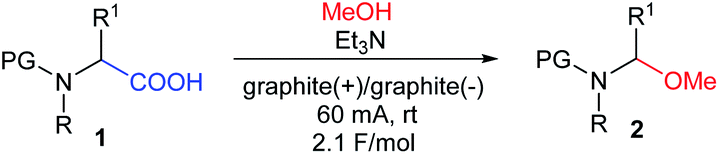
Synthetic organic electrochemistry is one of the most important alternatives to classical chemical approaches. It can be used to obtain structurally diverse compounds under mild and green conditions in high yields and, more importantly, good regio- and chemoselectivity.1–6 Thus, it is no wonder that the number of articles describing the use of electrochemical synthetic methods has continually increased over the last decade.Among the many electrochemical oxidation reactions, the reaction of compounds containing a carboxyl group deserves special attention.7,8 Depending on the structure of the substrate and the conditions, reactions can occur via two pathways. The one-electron oxidation of carboxylate anions followed by decarboxylation and the formation of radicals that can easily dimerize is called Kolbe electrolysis, which is considered to be the oldest known electro-organic reaction.9 A high current density increases the concentration of radicals on the anode surface, which promotes the formation of dimers; however, if the oxidation potential of the radical intermediates is low enough (due to their structure), a second electron may be detached, which generates stabilized carbocations. This process is known as non-Kolbe electrolysis.10 This route occurs for compounds with an electron-donating group in the α position to the carboxyl group, e.g., N-protected α-amino acids. Electrolysis can be carried out in protic (nucleophilic) solvents, e.g., MeOH, H2O, or AcOH, in which newly-formed carbocations are quickly captured by solvent molecules. The first step in the decarboxylative α-methoxylation of N-protected α-amino acids 1 is the formation of highly reactive N-acyliminium-type cations, which easily subsequently react with nucleophilic MeOH to give N-protected (1-methoxyalkyl)amines 2 (Fig. 1).
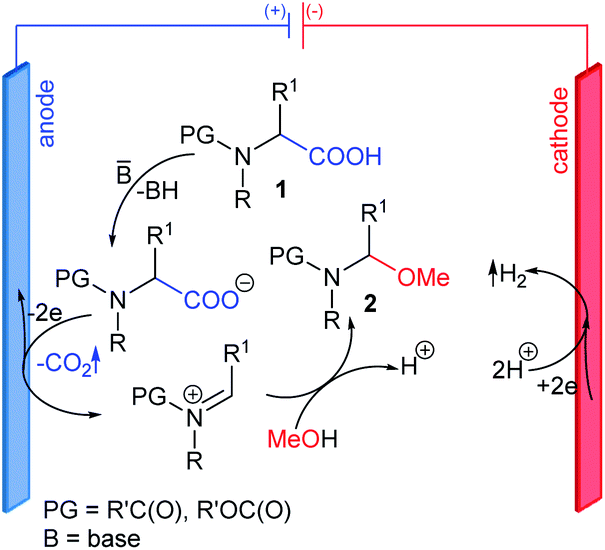 | ||
| Fig. 1 Electrochemical decarboxylative α-methoxylation of N-protected α-amino acids. | ||
N-Protected (1-methoxyalkyl)amines 2 are one of the most relevant α-amidoalkylating agents used for α-amidoalkylation reactions.11 They can be easily transformed into reactive α-amido sulfones,12 N-[1-(benzotriazol-1-yl)alkyl]amides,13 or 1-(N-acylamino)alkyltriarylphosphonium salts,14 which have recently been introduced to organic synthesis. All of the above mentioned compounds (including N-protected (1-alkoxyalkyl)amines) play an important role in the synthesis or modification of a wide range of organic compounds, including natural and/or bioactive products with particular importance in pharmaceutical chemistry.11 The main reason for this is the ease of forming new C–C and C-heteroatom bonds (e.g., C–P, C–S, C–N) via both intermolecular α-amidoalkylation and intramolecular cyclization, leading to carbo- and heterocyclic compounds.11,15–19
Previous electrochemical methods
The most general and effective strategies for the synthesis of N-protected (1-alkoxyalkyl)amines are based on electrochemical reactions such as the decarboxylative α-alkoxylation of N-protected α-amino acids or the electrochemical oxidation of amides, lactams, or carbamates; however, among the above routes, the first one is of particular importance (Fig. 1).11In 1951, Linstead et al. reported the decarboxylative α-alkoxylation of glycine and alanine derivatives in an alcoholic solution with the addition of metallic sodium to provide the corresponding N-(1-alkoxyalkyl)amides in good yields.20 After this pioneering work, the electrochemical decarboxylative α-methoxylation of N-acylated α-amino acids was explored by several research groups. However, the described methods were initially limited to relatively simple N-acyl-α-amino acids, such as derivatives of glycine, alanine, valine, leucine, phenylalanine, or proline.20–22 Next, the reaction was expanded to more complex compounds, such as oxazolidine-2-one-4-carboxylic acids derived from threonine,23 2,5-piperazinediones derived from aminomalonic acid,24 and di-, tri-, or tetrapeptides.25 From 2000–2003, the Matsumura and Onomura groups applied this electrochemical process for the stereoselective functionalization of N,O-acetals derived from serine.26–28
Electrochemical alkoxylation is usually carried out in MeOH in the presence of a base. Sodium methanolate (MeONa),22,26–28 inorganic bases,21,24 such as KOH, NaOAc, LiH, CsCO3 or organic bases Et3N or (i-Pr)2EtN,25 are typically used to generate carboxylate ions from the corresponding α-amino acids. Additionally, in 2007, Tajima et al. showed that the decarboxylative α-methoxylation of alanine and proline derivatives could be accomplished using solid-supported bases (e.g., 3-(1-piperidino)propyl-functionalized silica gel (SiO2-Pip)).29
In 2012, we conducted extensive research in the electrochemical decarboxylative α-methoxylation of N-protected α-amino acids. We developed an efficient method for transforming structurally diverse N-protected α-amino acids 1 to N-protected (1-methoxyalkyl)amines 2 in the presence of MeONa or SiO2-Pip.14 The latter approach (in the presence of SiO2-Pip) was particularly useful since no aqueous workup was required to isolate the product. The electrolysis of N-protected α-amino acids 1 in MeOH was performed under stirring at a current density of 0.3 A dm−2 at 10 °C until 2.4–3.75 F mol−1 charge had passed (Fig. 2A). Then, the solid-supported base was filtered off, and pure N-protected (1-methoxyalkyl)amines 2 were obtained after evaporating volatiles.14
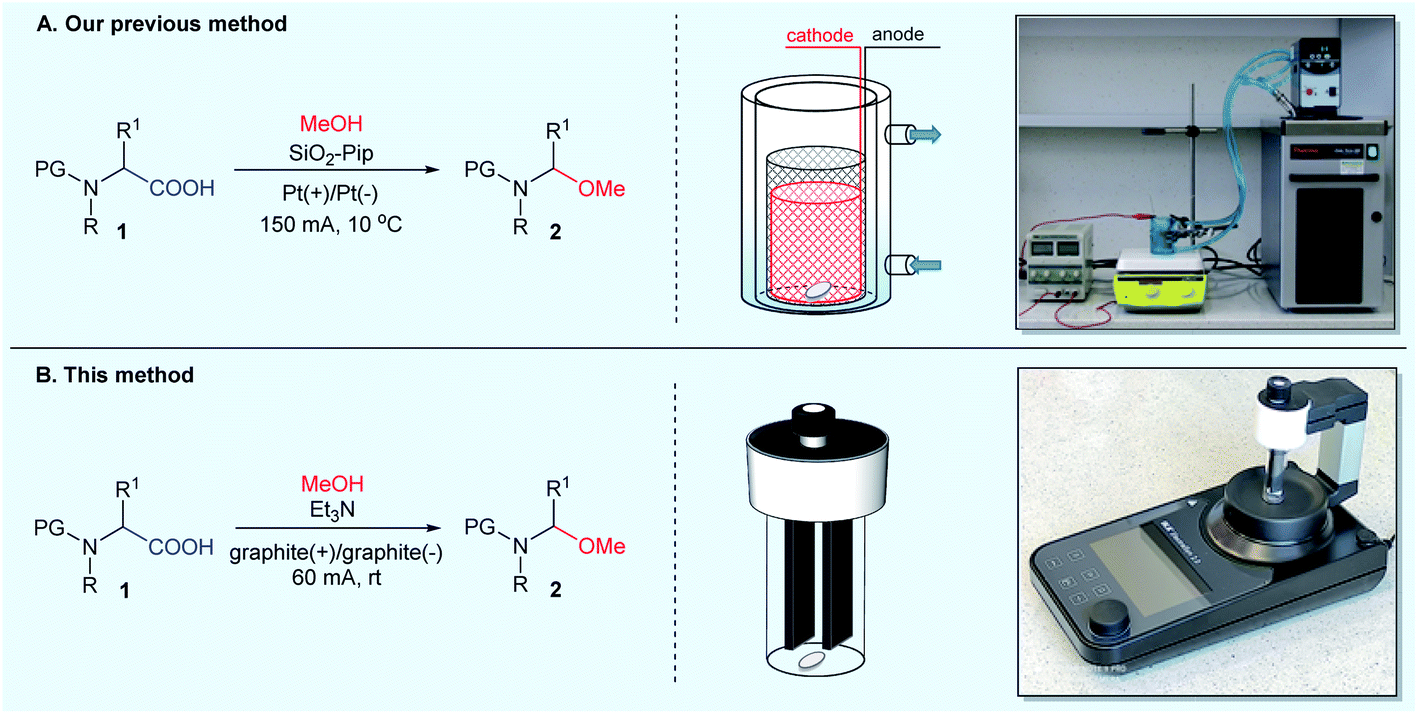 | ||
| Fig. 2 Electrochemical methods for the synthesis of N-protected (1-methoxyalkylamines) 2: (A) alkoxylations were conducted in an undivided cylindrical glass electrolyzer with a thermostatic jacket, equipped with a magnetic stirrer, and using a DC power supply. To overcome the high resistance of the electrolyte, a cylindrical mesh anode (Pt) and cathode (Pt) were arranged concentrically 2.5 ± 0.5 mm from each other; (B) methoxylations were carried out using commercially available, compact setup – IKA ElectraSyn 2.0. | ||
All of the above electro-organic methods produced N-protected (1-methoxyalkyl)amines in good to excellent yields and chemoselectivity, but they also have some limitations. When using bases such as MeONa or KOH, aqueous workup was necessary, which resulted in a lower reaction efficiency, especially for products derived from polar, hydrophilic amino acids. The application of more expensive, albeit reusable, solid-supported bases, such as SiO2-Pip, simplified the workup procedure; however, as we observed, during electrolysis in the presence of SiO2-Pip, platinum electrodes were mechanically degraded, which generated additional costs (see Note A). For non-“electro-curious” chemists, the collection or purchase of equipment and the subsequent construction of homemade electrochemical setups is often a great technical inconvenience; especially that even the detailed copying of self-made installations does not guarantee the reproducibility necessary to obtain the expected results. The necessity to optimize the reaction conditions each time makes electrochemical methods problematic from a synthetic point of view.
The ideal solution for electrochemical reactions seems to be the commercially available, easy-to-use, compact setup, like ElectraSyn 2.0 (designed by IKA®). A synthetic method performed on equipment with a standardized power source, electrodes, and other accessories would allow the successful transfer of an experimental procedure from one lab to another every time.
Herein, we report our results of the application of the ElectraSyn 2.0 setup for improving the electrochemical decarboxylative α-methoxylation of N-protected α-amino acids.
Results and discussion
In preliminary experiments, we screened various reaction conditions for the decarboxylative α-methoxylation of N-Piv-α-alanine 1a as a model N-acyl-α-amino acid (Table 1). An initial set of experiments was performed to evaluate the influence of the electrode material and the type of base on the reaction. All experiments were carried out in an electrochemical reactor (ElectraSyn 2.0) equipped with a glass vial (5 mL), a magnetic stirrer, and a set of electrodes (Fig. 2B). Reactions were performed in constant current mode on a 0.4 mmol scale, using 0.1 M N-Piv-α-alanine 1a in MeOH in the presence of 0.075 equiv. of base. In contrast to our previous method, electrochemical decarboxylative α-methoxylation was carried out at room temperature, not at 10 °C.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Electrode | Base | I [mA] | Q [F mol−1] | Conv.b [%] | 2ac [%] |
| a Conditions: substrate 1a (0.4 mmol), base (0.03 mmol, 0.075 equiv.), MeOH (4 mL), room temperature. Entries 1–4 and 7 were performed in a 5 mL vial on IKA Electrasyn 2.0.b Determined by integration of signals in the 1H NMR spectrum.c Isolated yield.d Due to the low conversion of substrate 1a, the product was not isolated.e The reaction on a scale of 0.75 mmol of 1a in a 10 mL vial.f The reaction on a scale of 1.6 mmol of 1a in a 20 mL vial. | ||||||
| 1 | Pt (+/−) | SiO2-Pip | 10 | 3.8 | 13 | —d |
| 2 | Pt (+/−) | Et3N | 60 | 3.8 | 50 | —d |
| 3 | Pt (+/−) | Et3N | 60 | 7.6 | >99 | 93 |
| 4 | Graphite (+/−) | Et3N | 60 | 2.1 | >99 | 95 |
| 5e | Graphite (+/−) | Et3N | 60 | 2.1 | >99 | 95 |
| 6f | Graphite (+/−) | Et3N | 60 | 2.1 | >99 | 97 |
| 7 | Graphite (+/−) | SiO2-Pip | 10 | 2.1 | >99 | 92 |
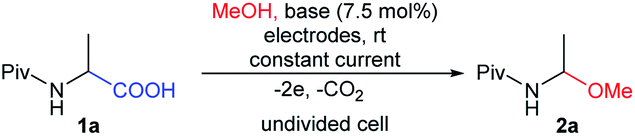
In the first experiment, we used Pt-plated electrodes (anode and cathode) and SiO2-Pip as a base, similar to our previous method. Under these conditions, a constant current of only 10 mA was achieved, which was 15 times lower than the value obtained using our self-made setup. Electrolysis was conducted until 3.8 F mol−1 charge had passed (Table 1, entry 1). After this time, the progress of the reaction was determined by 1H NMR. Unfortunately, the reaction mixture contained mainly unreacted substrate 1a and only a small amount of the expected product 2a (Table 1, entry 1). Next, with Pt-plated electrodes, changing the base from SiO2-Pip to Et3N increased the conductivity of the electrolyte, which allowed the process to be carried out at a current of 60 mA. Upon a charge consumption of 3.8 F mol−1, the substrate conversion was still unsatisfactory (Table 1, entry 2). Increasing the electric quantity from 3.8 F mol−1 to 7.6 F mol−1 produced the expected product 2a with a high yield of 93% (Table 1, entry 2 vs. entry 3). However, the best results were achieved by replacing the Pt-plated electrodes with graphite ones (anode and cathode). In conjunction with Et3N as the base and at a constant current of 60 mA, product 2a was obtained in an excellent yield of 95%. A total charge of just 2.1 F mol−1 was necessary to perform this reaction (Table 1, entry 4). Next, the scalability of this process was explored by conducting the reaction using 0.75 mmol and 1.6 mmol of substrate 1a (Table 1, entry 5 and 6). In both cases, no decrease in the yield of product 2a was observed. We also demonstrated that the decarboxylative α-methoxylation of N-Piv-alanine 1a using graphite electrodes could be carried out effectively also in the presence of the solid-supported base, SiO2-Pip (Table 1, entry 7). Similar to electrolysis with Pt-plated electrodes, the maximum current did not exceed 10 mA, which significantly affected the extension of the electrolysis time (Table 1, entry 1 vs. entry 7).
Next, with the optimal electrolysis conditions (with an optimal set of electrolysis parameters) in hand, a new procedure was tested on most of our previously reported N-protected α-amino acids (Fig. 2B, Table 2). Hydrophobic aliphatic and aromatic N-protected α-amino acids (alanine, valine, phenylalanine, norvaline, norleucine, cyclohexylglycine, phenylalanine, and phenylglycine), α-amino acids with functionalized side chains (tyrosine, serine, asparagine, glutamine, aspartic acid, and glutamic acid), and cyclic proline appeared to be suitable substrate. The expected N-protected (1-methoxyalkyl)amines 2a–u were isolated in excellent yields (94–99%), which were comparable or higher than the yields obtained using our previous method. To verify the repeatability of this non-Kolbe electrolysis, the methoxylation of N-Cbz-α-alanine 1b was carried out five times, obtaining the expected product 2b in 99%, 99%, 97%, 98%, 99% yields, respectively. For a comparison of the yields obtained using both experimental procedures, see the ESI.†
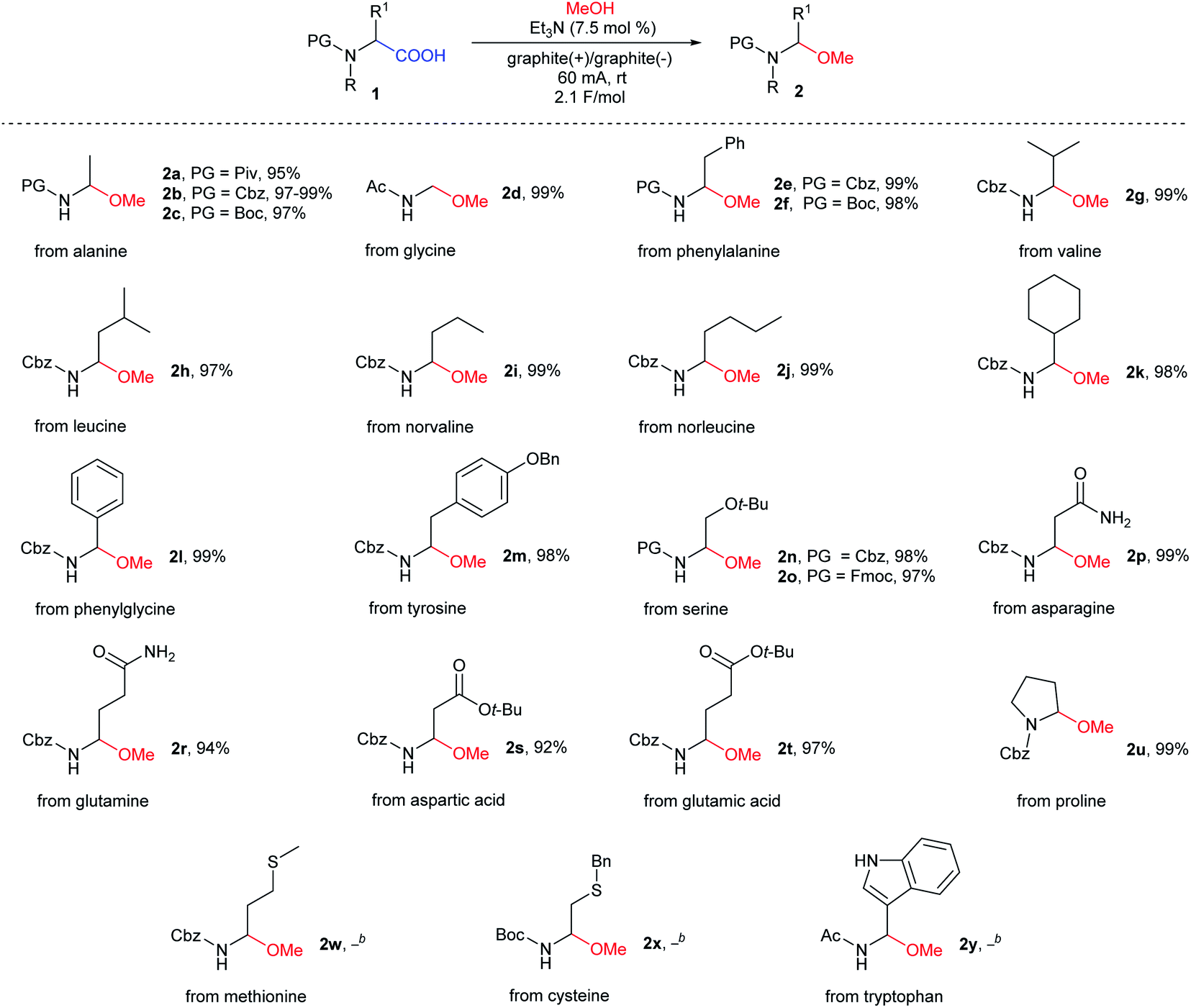
Similar to previous studies, this reaction proved to be tolerant to various amine protecting groups. This time, however, the reaction workup only involved the evaporation of volatiles and drying of residues. In the case of methionine 2w, cysteine 2x, and tryptophan 2y derivatives, the reaction proceeded with the formation of a complex mixture (Table 2). This was ascribed to the presence of easily oxidized fragments, e.g., sulfide-type groups 2w–x or sensitive nature of indole derivative 2y.
Procedure and characterization
General methods
Melting points were determined in capillaries, in a Stuart Scientific SMP3 melting point apparatus and were uncorrected. Infrared spectra (IR) were recorded on a Nicolet 6700 FT-IR spectrophotometer (ATR method). 1H NMR spectra were acquired on either a Varian 600 or Agilent NMR Magnet-400 instrument, at 600 or 400 MHz, respectively. Data are recorded as follows: chemical shift (δ, ppm) with tetramethylsilane (TMS) as the internal standard, multiplicity (s = singlet; d = doublet; t = triplet; q = quartet; qu = quintet; m = multiplet; br = broad), coupling constant (Hz), integration. 13C NMR spectra were measured on either a Varian 600 or 400 MHz at, as appropriate, 150 or 100 MHz. Chemical shifts were reported in ppm from the solvent resonance employed as the internal standard. Data for 13C NMR are reported in terms of chemical shift (δ, ppm). High resolution mass spectroscopy (HRMS) analyses were obtained using a commercial apparatus Waters Corporation Xevo G2 QTOF instrument (ESI source).General procedure for the electrochemical synthesis of N-protected (1-methoxyalkyl)amines 2
(1) A 5 mL IKA ElectraSyn vial, equipped with a magnetic stir bar is charged sequentially with N-protected α-amino acid 1 (0.4 mmol, 1 equiv.), MeOH (4 mL), and Et3N (4.2 μL, 0.03 mmol, 0.075 equiv.).
(2) The vial is then closed with a cap with two Graphite SK-50 electrodes attached, and the mixture is stirred at 500 rpm for 4 minutes (Note B).
(3) Next, the vial is put into the ElectraSyn set up, and electrolysis is carried out at room temperature. The mixture is stirred at 600 rpm in constant-current mode (60 mA) until 2.1 F mol−1 of charge is passed (time: 22 min 29 s).
(4) After completion of the reaction, the reaction mixture is quantitatively transferred to a round-bottom flask, and the volatiles are removed under reduced pressure using rotary evaporator.
(5) Finally, the residue is dried under vacuum (10–15 mmHg) to yield desired product, N-protected (1-methoxyalkyl)amine 2.
The original procedure was repeated for randomly selected N-protected α-amino acids (2a, e, h, l, m, t) by J. Adamek (at 0.4 mmol scale of appropriate substrate 1).
Repeat: white solid (59.9 mg, 94% yield). 1H NMR (400 MHz, CDCl3) δ 5.75 (br s, 1H), 5.29 (dq, J1 = 12.0 Hz, J2 = 6.0 Hz, 1H), 3.31 (s, 3H), 1.33 (d, J = 5.6 Hz, 3H), 1.22 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 178.4, 77.7, 55.6, 38.8, 27.5, 21.6.
Repeat: white solid (113.9 mg, 99% yield). 1H NMR (400 MHz, CDCl3) δ 7.36–7.21 (m, 10H), 5.18–5.13 (m, 1H), 5.08 (s, 2H), 5.00 (br d, J = 9.6 Hz, 1H), 3.34 (s, 3H), 2.93 (d, J = 6.0 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 155.9, 136.2, 135.8, 129.7, 128.5, 128.4, 128.2, 128.0, 126.8, 83.5, 66.8, 55.7, 41.6.
Repeat: colorless oil (100.0 mg, 99% yield). 1H NMR (600 MHz, CDCl3) δ 7.36–7.31 (m, 5H), 5.12 (s, 2H), 4.99–4.92 (m, 2H), 3.35 (s, 3H), 1.75–1.69 (m, 1H), 1.58–1.53 (m, 1H), 1.40–1.36 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 156.0, 136.3, 128.5, 128.2, 128.1, 82.3, 66.8, 55.4, 44.6, 24.5, 22.6, 22.4.
Repeat: white crystals (106.8 mg, 98% yield). 1H NMR (400 MHz, CDCl3) δ 7.42–7.32 (m, 10H), 5.89 (d, J = 10.0 Hz, 1H), 5.35 (br d, J = 11.2 Hz, 1H), 5.17 (s, 2H), 3.47 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 155.9, 139.1, 136.1, 128.6, 128.56, 128.55, 128.2, 128.1, 125.8, 84.0, 67.1, 55.7.
Repeat: white crystals (155.0 mg, 98% yield) 1H NMR (400 MHz, CDCl3) δ 7.43–7.6.89 (m, 14H), 5.11–5.08 (m, 1H), 5.09 (s, 2H), 5.03 (s, 2H), 4.98 (br d, J = 10 Hz, 1H), 3.33 (s, 3H), 2.86 (d, J = 5.6 Hz 2H), 13C NMR (150 MHz, CDCl3) δ 157.7, 155.9, 137.0, 136.2, 130.7, 128.54, 128.51, 128.2, 128.0, 127.9, 127.4, 114.8, 83.5, 70.0, 66.8, 55.7, 40.7.
Repeat: colorless oil (126.0 mg, 97% yield). 1H NMR (400 MHz, CDCl3) δ 7.37–7.30 (m, 5H), 5.20 (br d, J = 9.6 Hz, 1H), 5.12 (s, 2H), 4.93–4.87 (m, 1H), 3.34 (s, 3H), 2.37–2.26 (m, 1H), 1.95–1.85 (m, 2H), 1.43 (s, 9H).13C NMR (100 MHz, CDCl3) δ 172.3, 156.0, 136.2, 128.5, 128.2, 128.1, 82.9, 80.6, 66.8, 55.6, 30.9, 30.4, 28.0.
Reagents and solvents
• N-Cbz-L-alanine (1b) (Sigma-Aldrich, CAS Registry No. 1142-20-7, 98% purity).• N-Boc-L-alanine (1c) (Sigma-Aldrich, CAS Registry No. 15761-38-3, ≥99.0% purity).
• N-Ac-glycine (1d) (Sigma-Aldrich, CAS Registry No. 543-24-8, 99% purity)
• N-Cbz-L-phenylalanine (1e) (Sigma-Aldrich, CAS Registry No. 1161-13-3, 99% purity)
• N-Boc-L-phenylalanine (1f) (Sigma-Aldrich, CAS Registry No. 13734-34-4, ≥99.0% purity).
• N-Cbz-L-valine (1g) (Fluorochem, CAS Registry No. 1149-26-4, 98.6% purity).
• N-Cbz-D,L-leucine (1h) (Fluka Analytical, CAS Registry No. 3588-60-1, ≥98% purity).
• N-Cbz-D,L-norvaline (1i) (ChemCruz, CAS Registry No. 21691-43-0, ≥98% purity)
• N-Cbz-D,L-norleucine (1j) (ChemCruz, CAS Registry No. 15027-13-1, ≥98% purity).
• N-Cbz-cyclohexyl-L-glycine (1k) (Carl ROTH, CAS Registry No. 69901-75-3, ≥98% purity).
• N-Cbz-phenylglycine (1l) (Trimen Chemicals, CAS Registry No. 53990-33-3, ≥98% purity).
• N-Cbz-O-benzyl-L-tyrosine (1m) (Sigma-Aldrich, CAS Registry No. 16677-29-5, ≥98.0% purity).
• Cbz-O-tert-butyl-L-serine (1n) (Sigma-Aldrich, CAS Registry No. 1676-75-1, ≥98% purity).
• Fmoc-O-tert-butyl-L-serine (1o) (Sigma-Aldrich, CAS Registry No. 71989-33-8, ≥98% purity).
• Cbz-L-asparagine (1p) (Acros Organics, CAS Registry No. 2304-96-3, 98% purity).
• N-Cbz-L-glutamine (1r) (Fluka, CAS Registry No. 2650-64-8, ≥99.0% purity).
• N-Cbz-L-aspartic acid 4-tert-butyl ester (1s) (Fluka Analytical, CAS Registry No. 5545-52-8, ≥98% purity).
• N-Cbz-L-glutamic acid 5-tert-butyl ester (1t) (Acros Organics, CAS Registry No. 3886-08-6, ≥98% purity).
• N-Cbz-L-proline (1u) (Acros Organics, CAS Registry No. 1148-11-4, 99% purity).
• N-Cbz-D,L-methionine (1w) (Fluka, CAS Registry No. 4434-61-1, ≥98% purity).
• N-Boc-S-benzyl-L-cysteine (1x) (Acros Organics, CAS Registry No. 5068-28-0, ≥98% purity).
• N-Ac-D,L-tryptophan (1y) (Sigma-Aldrich, CAS Registry No. 87-31-1, 98% purity).
• Triethylamine (Alfa-Aesar, CAS Registry No. 121-44-8, 99% purity).
• Methanol, p.a. ACS, ISO, Ph. Eur (S.WITKO CHS, Chemsolve, CAS Registry No. 67-56-1, 99.8% purity)
Reagent setup
• N-Piv-D,L-alanine 1a was synthesized by described protocol33 (see ESI†).• The reagents 1b–y, Et3N and MeOH, purchased from commercial suppliers were used without further purification.
Equipment
• ElectraSyn 2.0 with 5 mL vial and two graphite SK-50 electrodes (IKA®).

Notes
Note A: a photo of the degraded platinum electrodes.

Note B: good mixing of the reagents ensures rapidly achieving stable electrolysis parameters. Only in the case of Cbz-L-asparagine (1p), the electrolysis was carried out for the suspension, which disappeared during the reaction progress.
Conclusions
Herein, we have described an improved method for the non-Kolbe electrolysis of N-protected-α-amino acids using an ElectraSyn 2.0 setup. As we have demonstrated, this easy-to-use and compact apparatus allows us to conduct electrochemical decarboxylative methoxylation under mild conditions in excellent yields. The necessity of building a homemade electrochemical setup is no longer a limitation of this transformation. Moreover, the use of standardized equipment ensures the repeatability of the reactions performed in laboratories all over the world. Besides, a simple, low-waste workup procedure and the possibility of using clean electricity are in line with the trends of developing sustainable and “green” organic synthesis procedures.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by Silesian University of Technology (Poland) Grant BK No. 04/020/BK_22/1035.References
- H. Lund and O. Hammerich, Organic Electrochemistry, CRC Press, New York, 4th edn, 2001 Search PubMed
.
- M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, Organic Electrochemistry: Molecular Syntheses with Potential, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Organic electrosynthesis: a promising green methodology in organic chemistry, Green Chem., 2010, 12, 2099–2119 RSC
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, A Survival Guide for the “Electro-curious”, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed
- D. Pollok and S. R. Waldvogel, Electro-organic synthesis – a 21st century technique, Chem. Sci., 2020, 11, 12386–12400 RSC
- S. Torii and H. Tanaka, in Organic Electrochemistry, ed. H. Lund and O. Hammerich, CRC Press, New York, 2001, ch. 14, pp. 499–543 Search PubMed
- M. C. Leech and K. Lam, Electrosynthesis Using Carboxylic Acid Derivatives: New Tricks for Old Reactions, Acc. Chem. Res., 2020, 53, 121–134 CrossRef CAS PubMed
- H. Kolbe, Untersuchungen über die Elektrolyse organischer Verbindungen, Justus Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef
- E. Klocke, A. Matzeit, M. Gockeln and H. J. Schäfer, Influences on the Selectivity of the Kolbe versus the Non-Kolbe Electrolysis in the Anodic Decarboxylation of Carboxylic Acids, Chem. Ber., 1993, 126, 1623–1630 CrossRef CAS
- R. Mazurkiewicz, A. Październiok-Holewa, J. Adamek and K. Zielińska, in Adv. Heterocycl. Chem., ed. A. R. Katritzky, Academic Press, 2014, vol. 111, ch. 2, pp. 43–94 Search PubMed
- J. Adamek, R. Mazurkiewicz, A. Październiok-Holewa, M. Grymel, A. Kuźnik and K. Zielińska, 1-(N-Acylamino)alkyl Sulfones from N-Acyl-α-amino Acids or N-Alkylamides, J. Org. Chem., 2014, 79, 2765–2770 CrossRef CAS PubMed
- J. Adamek, R. Mazurkiewicz, A. Październiok-Holewa, A. Kuźnik, M. Grymel, K. Zielińska and W. Simka, N-[1-(Benzotriazol-1-yl)alkyl]amides from N-acyl-α-amino acids or N-alkylamides, Tetrahedron, 2014, 70, 5725–5729 CrossRef CAS
- R. Mazurkiewicz, J. Adamek, A. Październiok-Holewa, K. Zielińska, W. Simka, A. Gajos and K. Szymura, α-Amidoalkylating Agents from N-Acyl-α-amino Acids: 1-(N-Acylamino)alkyltriphenylphosphonium Salts, J. Org. Chem., 2012, 77, 1952–1960 CrossRef CAS PubMed
- A. R. Katritzky, X. Lan, J. Z. Yang and O. V. Denisko, Properties and Synthetic Utility of N-Substituted Benzotriazoles, Chem. Rev., 1998, 98, 409–548 CrossRef CAS PubMed
- A. R. Katritzky, K. Manju, S. K. Singh and N. K. Meher, Benzotriazole mediated amino-, amido-, alkoxy- and alkylthio-alkylation, Tetrahedron, 2005, 61, 2555–2581 CrossRef CAS
- M. Petrini, α-Amido Sulfones as Stable Precursors of Reactive N-Acylimino Derivatives, Chem. Rev., 2005, 105, 3949–3977 CrossRef CAS PubMed
- E. Marcantoni, A. Palmieri and M. Petrini, Recent synthetic applications of α-amido sulfones as precursors of N-acylimino derivatives, Org. Chem. Front., 2019, 6, 2142–2182 RSC
- J. Adamek, A. Węgrzyk, J. Kończewicz, K. Walczak and K. Erfurt, 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications, Molecules, 2018, 23, 2453 CrossRef PubMed
- R. P. Linstead, B. R. Shephard and B. C. L. Weedon, Anodic syntheses. Part V. Electrolysis of N-acylamino-acids. A novel alkoxylation reaction, J. Chem. Soc., 1951, 2854–2858, 10.1039/JR9510002854
- Y. Matsumura, T. Tanaka, G. N. a. a. Wanyoike, T. Maki and O. Onomura, Memory of chirality in the non-Kolbe reaction of N-arylcarbonylated L-prolines, J. Electroanal. Chem., 2001, 507, 71–74 CrossRef CAS
- H. Yamazaki, H. Horikawa, T. Nishitani and T. Iwasaki, A Facile Synthesis of Optically Pure Amines by Reduction of N-Acyl-α-methoyalkylamines Derived from α-Amino Acids Using Triethylsilane, Chem. Pharm. Bull., 1990, 38, 2024–2026 CrossRef CAS
- A. Zietlow and E. Steckhan, Diastereoselective Amidoalkylation Reactions of Electrochemically Methoxylated Chiral 2-Oxazolidinones with Organocopper Reagents, J. Org. Chem., 1994, 59, 5658–5661 CrossRef CAS
- G. Kardassis, P. Brungs and E. Steckhan, Electrogenerated chiral cationic glycine equivalents — Part 1: The 6-methoxy derivative of cyclo(L-Pro-Gly), Tetrahedron, 1998, 54, 3471–3478 CrossRef CAS
- D. Seebach, R. Charczuk, C. Gerber, P. Renaud, H. Berner and H. Schneider, Elektrochemische Decarboxylierung von L-Threonin- und Oligopeptid-Derivaten unter Bildung von N-Acyl-N,O-acetalen: Herstellung von Oligopeptiden mit Carboxamid-oder Phosphonat-C-Terminus, Helv. Chim. Acta, 1989, 72, 401–425 CrossRef CAS
- Y. Matsumura, Y. Shirakawa, Y. Satoh, M. Umino, T. Tanaka, T. Maki and O. Onomura, First Example of Memory of Chirality in Carbenium Ion Chemistry, Org. Lett., 2000, 2, 1689–1691 CrossRef CAS PubMed
- Y. Matsumura, G. N. a. a. Wanyoike, O. Onomura and T. Maki, Enantioselective substitution of N-acylated α-amino acids by electrochemical oxidation, Electrochim. Acta, 2003, 48, 2957–2966 CrossRef CAS
- G. N. a. a. Wanyoike, O. Onomura, T. Maki and Y. Matsumura, Highly Enhanced Enantioselectivity in the Memory of Chirality via Acyliminium Ions, Org. Lett., 2002, 4, 1875–1877 CrossRef CAS PubMed
- T. Tajima, H. Kurihara and T. Fuchigami, Development of an Electrolytic System for Non-Kolbe Electrolysis Based on the Acid−Base Reaction between Carboxylic Acids as a Substrate and Solid-Supported Bases, J. Am. Chem. Soc., 2007, 129, 6680–6681 CrossRef CAS PubMed
- M. Terada, K. Machioka and K. Sorimachi, Activation of Hemiaminal Ethers by Chiral Brønsted Acids for Facile Access to Enantioselective Two-Carbon Homologation Using Enecarbamates, Angew. Chem., Int. Ed., 2009, 48, 2553–2556 CrossRef CAS PubMed
- M. Chorev, S. A. MacDonald and M. Goodman, Retro-inverso isomerization of peptides: side reactions in the synthesis of N,N'-diacyl-1,1-diamino-2-phenylethane derivatives, J. Org. Chem., 1984, 49, 821–827 CrossRef CAS
- A. Walęcka-Kurczyk, K. Walczak, A. Kuźnik, S. Stecko and A. Październiok-Holewa, The Synthesis of α-Aminophosphonates via Enantioselective Organocatalytic Reaction of 1-(N-Acylamino)alkylphosphonium Salts with Dimethyl Phosphite, Molecules, 2020, 25, 405 CrossRef PubMed
- A. K. Das, P. P. Bose, M. G. B. Drew and A. Banerjee, The role of protecting groups in the formation of organogels through a nano-fibrillar network formed by self-assembling terminally protected tripeptides, Tetrahedron, 2007, 63, 7432–7442 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental procedure for the preparation of N-pivaloylalanine 1a and characterization data. Table with comparison of yields for both (previous and current) procedures. 1H and 13C NMR spectra of products 2a–u. 1H and 13C NMR spectra of products 2 obtained in repeated syntheses. See DOI: 10.1039/d1ra08124a |
| ‡ Author who checked the methodologies. |
| This journal is © The Royal Society of Chemistry 2022 |