
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDBU mediated one-pot synthesis of triazolo triazines via Dimroth type rearrangement†
Ab Majeed Ganaia,
Tabasum Khan Pathana,
Nisar Sayyada,
Babita Kushwahaa,
Narva Deshwar Kushwahaa,
Andreas G. Tzakosb and
Rajshekhar Karpoormath *a
*a
aDepartment of Pharmaceutical Chemistry, Discipline of Pharmaceutical Sciences, College of Health Sciences, University of KwaZulu-Natal (Westville), Durban 4000, South Africa. E-mail: karpoormath@ukzn.ac.za
bSection of Organic Chemistry and Biochemistry, Department of Chemistry, University of Ioannina, Ioannina, 45110, Greece
First published on 12th January 2022
Abstract
Herein we report an efficient one-pot synthesis of [1,2,4]triazolo[1,5 a][1,3,5]triazines from commercially available substituted aryl/heteroaryl aldehydes and substituted 2-hydrazinyl-1,3,5-triazines via N-bromosuccinimide (NBS) mediated oxidative C–N bond formation. Isomerisation of [1,2,4]triazolo[4,3-a][1,3,5]triazines to [1,2,4]triazolo[1,5-a][1,3,5]triazines is driven by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) affording both isomers with good to excellent yields (70–96%).
Purines are nitrogen-containing heterocycles and are structural motifs in the nucleobases adenine and guanine of DNA as well as RNA. Purine nucleotides (ATP, GTP, cAMP, cGMP, NAD, FAD) also act as co-factors, substrates, or mediators in the functioning of numerous proteins.1 Therefore, bioisosteres of purines are widely explored and exploited by pharmaceutical chemists in developing new drug entities. Heterocycles containig the 1,3,5-triazine ring act as bioisosteres of purine, which exist as two isomers, namely [1,2,4]triazolo[4,3-a][1,3,5]triazine and [1,2,4]triazolo[1,5-a][1,3,5]triazine (Fig. 1), which have been extensively studied as adenosine receptor antagonists,2,3 as well as for other pharmacological activities1,4,5 (Fig. 2). Literature reports suggest that [1,2,4]triazolo[1,5-a][1,3,5]triazine has been exploited extensively in drug discovery as compared to its corresponding isomer. Further, from the literature it is evident that symmetrical disubstituted triazines,6–9 especially morpholine10–12 substituted, have displayed broad pharmacological activities.
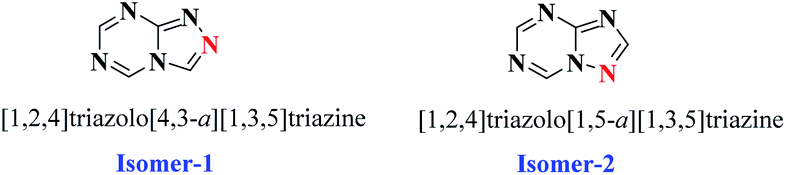 | ||
| Fig. 1 Structure of isomers of triazolo–triazine. | ||
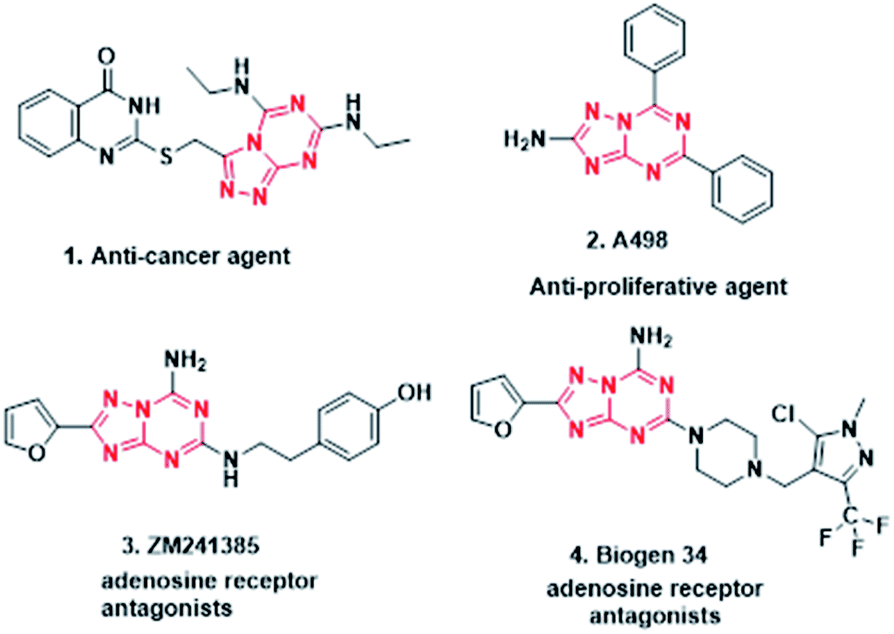 | ||
| Fig. 2 Biologically active molecules of [1,2,4]triazolo[4,3-a][1,3,5]triazine (1) and [1,2,4]triazolo[1,5-a][1,3,5]triazine (2, 3, 4). | ||
Various synthetic methods have been reported for the C–N bond formation by employing different starting materials13–15 via oxidative cyclisation,16 high temperature condition17–19 and/or metal-catalysed reactions.20 However, the current reported protocols were environmentally unfriendly as they suffered from drawbacks such as, multistep, and tedious procedures, use of carcinogenic solvents, high-temperature, expensive and toxic metal-catalysts, and other hazardous reagents.
Furthermore, there is very less reported research on these heterocycles, and that can be because of the unavailability of the efficient and cheaper methods. Thus, there is a need to develop new versatile synthetic method for the synthesis of disubstituted triazolotriazine heterocycles of pharmacological interest.
In 1970, for the first time Kobe21 et al. (scheme a) reported the synthesis of [1,2,4]triazolo[4,3-a][1,3,5]triazine utilizing lead tetraacetate in benzene under reflux conditions (Fig. 3). However, no further Isomerization was carried out and resulted low to moderate yield. Deshpande22 et al. reported (scheme b) the reaction of 2-hydrazinyl-1,3,5-triazine with various substituted benzoic acids. The product formed was treated with P2O5, refluxed in xylene for 10h to yield [1,2,4]triazolo[4,3-a][1,3,5]triazine. Further, Isomerization of the resulting product was carried out in 2% methanolic-NaOH solution resulting in poor yields. Recently, Stefano23 et al. (scheme c) reported, a multistep protocol by reacting the intermediate with bis(methyl-sulfanyl)methylenecyanamide at 180 °C under N2 for 3h resulting in low yield due to the formation of several side products. In addition no isomerization studies were carried out, and only the [1,2,4]triazolo[1,5-a][1,3,5]triazine analogs were reported.
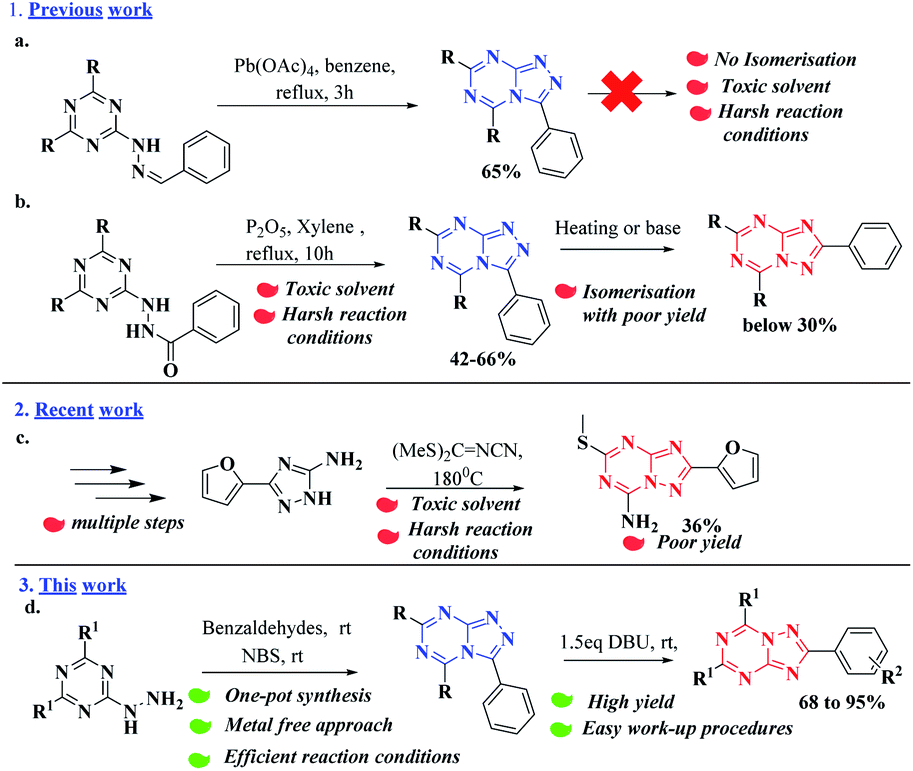 | ||
| Fig. 3 Different approaches for the synthesis of triazolo triazines. | ||
Herein, we report an economical one-pot synthesis of [1,2,4]triazolo[1,5-a][1,3,5]triazine analogs via Dimroth type rearrangement of [1,2,4]triazolo[4,3-a][1,3,5]triazine derivatives. This one pot novel methodology was carried out by reacting readily available, inexpensive starting materials such as substituted aryl/heteroaryl benzaldehydes and substituted 2-hydrazinyl-1,3,5-triazine in methanol (a mild solvent)24 at room temperature giving excellent yields of the desired product. For cyclization reaction, an eco-friendly reagent NBS25 was used and the resulting product was treated with DBU to yield its corresponding desired isomer. To the best of our knowledge this is the first report for the greener synthesis of symmetric disubstituted triazolotriazine heterocycles via Dimroth type rearrangement. We believe that this simple, yet novel methodology could be further exploited by the researchers in pharmaceutical industries and academics settings in drug discovery.
Formation of the Schiff base was initiated reacting 4,4′-(6-hydrazinyl-1,3,5-triazine-2,4-diyl)dimorpholine 1a and unsubstituted benzaldehyde as shown in Table 1. The reaction (entry 1) was carried out using N-bromosuccinimide (NBS) as an oxidant followed by the addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in ethanol at room temperature for 16h,resulting in desired product 2a with 68% yield. The product appeared as a clean blue spot on the TLC, when visualized under UV (254 nm). Interestingly, the formation of isomer-1 was very straightforward; hence, optimization was achieved for its conversion into isomer-2. Solvent optimization (entry 1–6) was performed using different solvents, starting with DMF or DCM (entry 2, 3) which did not yield expected results. Yield improvement was recorded by the use of MeOH as a solvent, although i-PrOH (entry 4, 5) resulted in a trace amount of the products. The same reaction was performed in water (entry 6), but it did not give valued results. Next, base optimization (entry 7–11) was carried out using MeOH as the solvent. The first reaction (entry 7) was performed without base, which showed no further isomerization. Inorganic bases such as 2%NaOH and K2CO3 (entry 8,9) were also used but displayed no significant results. Triethylamine and DABCO (1,4-diazabicyclo[2.2.2]octane) were unable to give the desirable product 2a.
|
||||||
|---|---|---|---|---|---|---|
| Entry no. | Solvent | Oxidant | Base | Base equiv. | Time (hour) | Yield% |
| a Conditions: 1a (1 mmol), benzaldehyde (1 mmol), solvent, rt, 20 min, then oxidant (1 mmol), 5 min, rt, then DBU, stir at rt till completion of the reaction. | ||||||
| 1 | EtOH | NBS | DBU | 1.0 | 16 | 68 |
| 2 | DCM | NBS | DBU | 1.0 | 16 | 0 |
| 3 | DMF | NBS | DBU | 1.0 | 16 | 0 |
| 4 | MeOH | NBS | DBU | 1.0 | 16 | 72 |
| 5 | i-PrOH | NBS | DBU | 1.0 | 16 | Trace |
| 6 | H2O | NBS | DBU | 1.0 | 16 | 0 |
| 7 | MeOH | NBS | — | — | 16 | 0 |
| 8 | MeOH | NBS | 2%NaOH | 1.0 | 16 | Trace |
| 9 | MeOH | NBS | K2CO3 | 1.0 | 16 | 0 |
| 10 | MeOH | NBS | TEA | 1.0 | 16 | 0 |
| 11 | MeOH | NBS | DABCO | 1.0 | 16 | 0 |
| 12 | MeOH | NBS | DBU | 1.5 | 2 | 85 |
| 13 | MeOH | NBS | DBU | 2.0 | 1.5 | 80 |
| 14 | MeOH | NCS | DBU | 1.5 | 2 | 56 |
| 15 | MeOH | NIS | DBU | 1.5 | 2 | 61 |
| 16 | MeOH | IBD | DBU | 1.5 | 2 | 63 |
| 17 | MeOH | KI/I2 | DBU | 1.5 | 2 | 53 |
| 18 | MeOH/H2O | NBS | DBU | 1.5 | 2 | 70 |
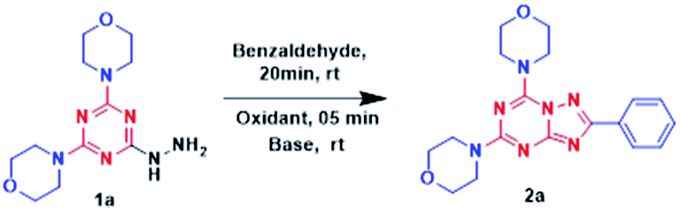
Meanwhile, equivalents of DBU were adjusted (entry 12, 13) to attain the highest yield %. Reaction with 1.5 eq. showed drastic improvement in yields in 2 h whereas 2.0 eq. resulted in good yields with less reaction time. Considering the yield factor (entry 12), the oxidant optimization was achieved (entry 14–17). Using N-chlorosuccinimide (NCS) and N-iodosuccinimide (NIS) (entry 14, 15) offered 56% and 61% yield respectively. When the reaction was performed using phenyliodine(III) diacetate (PIDA) and KI/I2 (entry 16, 17) it provided 2a in 63% and 53% yields, respectively. Finally, methanol–water and ethanol–water systems in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were used, which gave approximately 70% yield, concluding that entry 12 gives the best result, demanding alcoholic solvents especially methanol as a key factor for Dimroth type rearrangement. Additionally, it was supported by the observation that reaction goes well in methanol, a little worse in ethanol (Table 1), and practically does not go in isopropanol, as the bulky i-PrO− cannot attack the sterically hindered C5 atom in isomer-1 from the side of the phenyl ring. It was also observed that isomerisation did not proceed in presence of acid (HCl or AcOH) at room temperature in methanol solvent. Therefore, MeO− as well as DBU were responsible for the rearrangement.
:1 were used, which gave approximately 70% yield, concluding that entry 12 gives the best result, demanding alcoholic solvents especially methanol as a key factor for Dimroth type rearrangement. Additionally, it was supported by the observation that reaction goes well in methanol, a little worse in ethanol (Table 1), and practically does not go in isopropanol, as the bulky i-PrO− cannot attack the sterically hindered C5 atom in isomer-1 from the side of the phenyl ring. It was also observed that isomerisation did not proceed in presence of acid (HCl or AcOH) at room temperature in methanol solvent. Therefore, MeO− as well as DBU were responsible for the rearrangement.
The detailed plausible reaction mechanism also supports the product formation and is described in Fig. 4. The reaction involves a Schiff base formation (II) by 1a and benzaldehydes, the addition of NBS results in oxidative cyclization reaction to produce isomer 1. The addition of DBU in isomer-1 initiates a famous Dimroth type rearrangement, protonation of III results in ring-opening with the formation of unstable intermediate V. It undergoes tautomerism by 1,3 proton shift, bond rotation and proton abstraction by methoxide ion facilitating the intramolecular cyclization to afford the isomer 2. This reaction mechanism is supported by the formation of the Schiff base, isomer-1, and its conversion to isomer-2, which were easily monitored by TLC, isolated, and characterized (ESI†). In addition to that, a single crystal of compound 2e was obtained, which further supports the Dimorth type of rearrangement (ESI†).
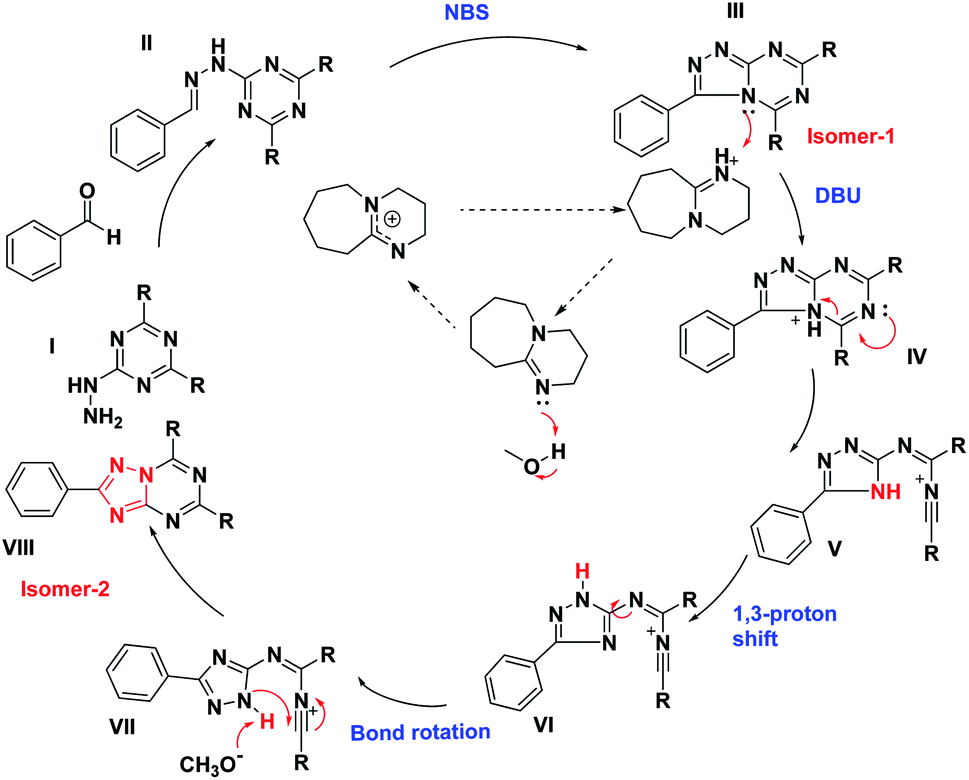 | ||
| Fig. 4 Plausible mechanism of the reaction pathway. | ||
Having in hand the optimized conditions, the substrate scope was further explored by using different aldehydes (Fig. 5). The reaction was carried out using 1a with benzaldehydes having electron-donating groups (2a-f) followed by NBS and DBU additions. The reaction was allowed to stir at room temperature till reaction completion, as monitored by TLC, which would approximately take 2 h. The reaction could smoothly give final products in excellent reaction yields (79 to 96%). Electron withdrawing groups such as chlorobenzaldehydes (2g-i) despite the position of substitution gave excellent yields (93–95%). With the 4-bromo and different fluoro-benzaldehydes, the reaction resulted in 2j (89%) and 2k-m (84–90%) with very good yields. Also, reaction afforded 70% and 77% yields with heterocyclic aldehydes such as 3-pyridinecarboxyaldehyde (2n) and thiophene-2-carbaldehyde (2o), respectively.
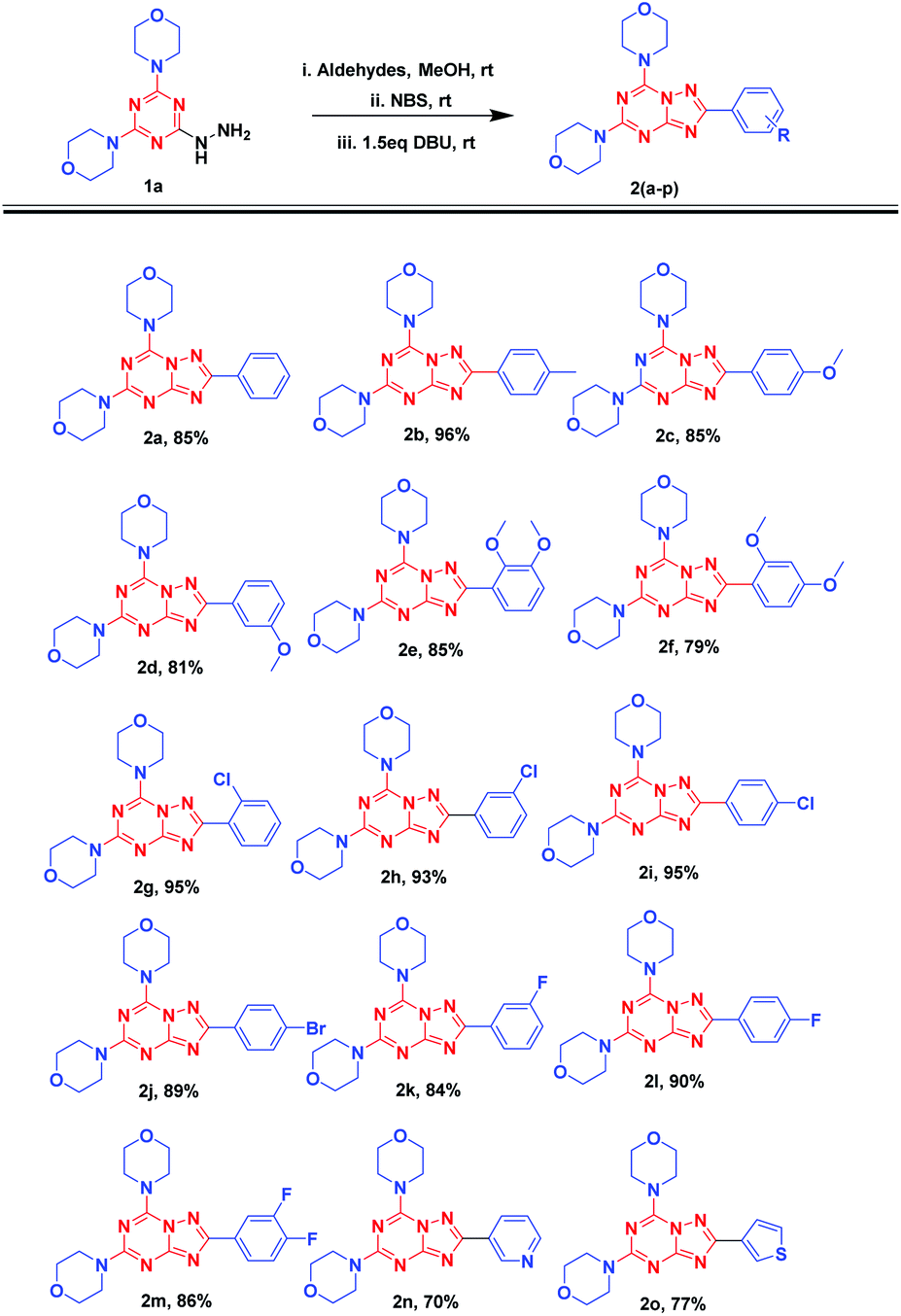 | ||
| Fig. 5 Substrate scope for different aldehydes. | ||
The gram scale reaction was performed to further validate this synthetic procedure. The reaction was carried out using 1a (1 g) with benzaldehyde (0.38 g) and stirred for 30 min. The NBS (1.26 g, 1 eq.) was added slowly and stirred till new spot appeared on TLC followed by the slow addition of DBU (1.5 eq.) resulted in isomerisation to give 2a in good yields (1.1 g, 84%) (ESI†).
Encouraged from the potency of the reaction to generate in good to excellent yields [1,2,4]triazolo[1,5-a][1,3,5]triazine analogues, we shifted our investigation to isolate [1,2,4]triazolo[4,3-a][1,3,5]triazine derivatives (isomer-1) as described in Fig. 6. Different aldehydes were reacted with hydrazinyl-1,3,5-triazine analogs (1a, 3a, and 4a) and followed by the addition of NBS, stirred at room temperature till the completion of reaction as monitored by TLC. Compound 1a on reaction with 4-bromobenzaldehyde and NBS resulted in 1b with 90% yield. Similarly, 3a with 4-bromobenzaldehydes and thiophene-2-carbaldehyde afforded 3b and 3c with 84% and 83% yields, respectively. The same reaction was carried out with 4a and 4-bromobenzaldehyde bearing an electron-withdrawing group gave 3d with 87% yield. Further, 4a with aldehydes bearing electron-donating groups resulted 4c and 4d, with 88% and 84% yields, respectively (Fig. 7).
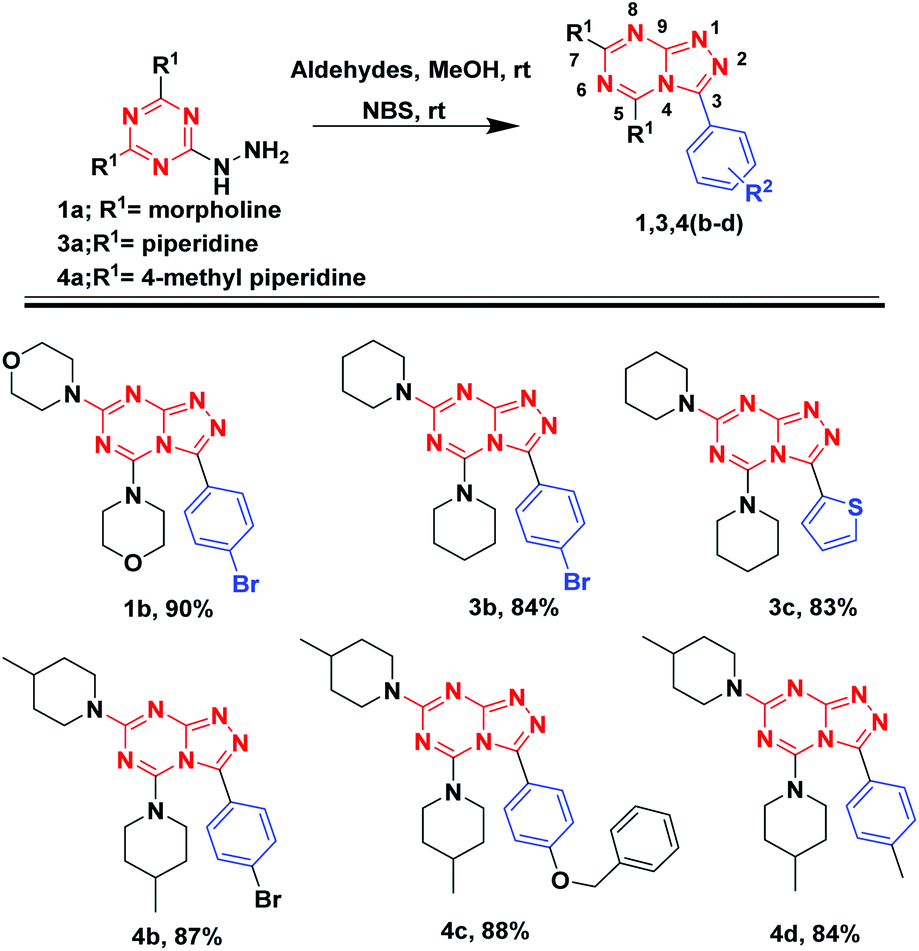 | ||
| Fig. 6 Substrate scope for [1,2,4]triazolo[4,3-a][1,3,5]triazine analogues. | ||
 | ||
| Fig. 7 Substrate scope for different disubstituted triazinyl-hydrazine and aldehydes. | ||
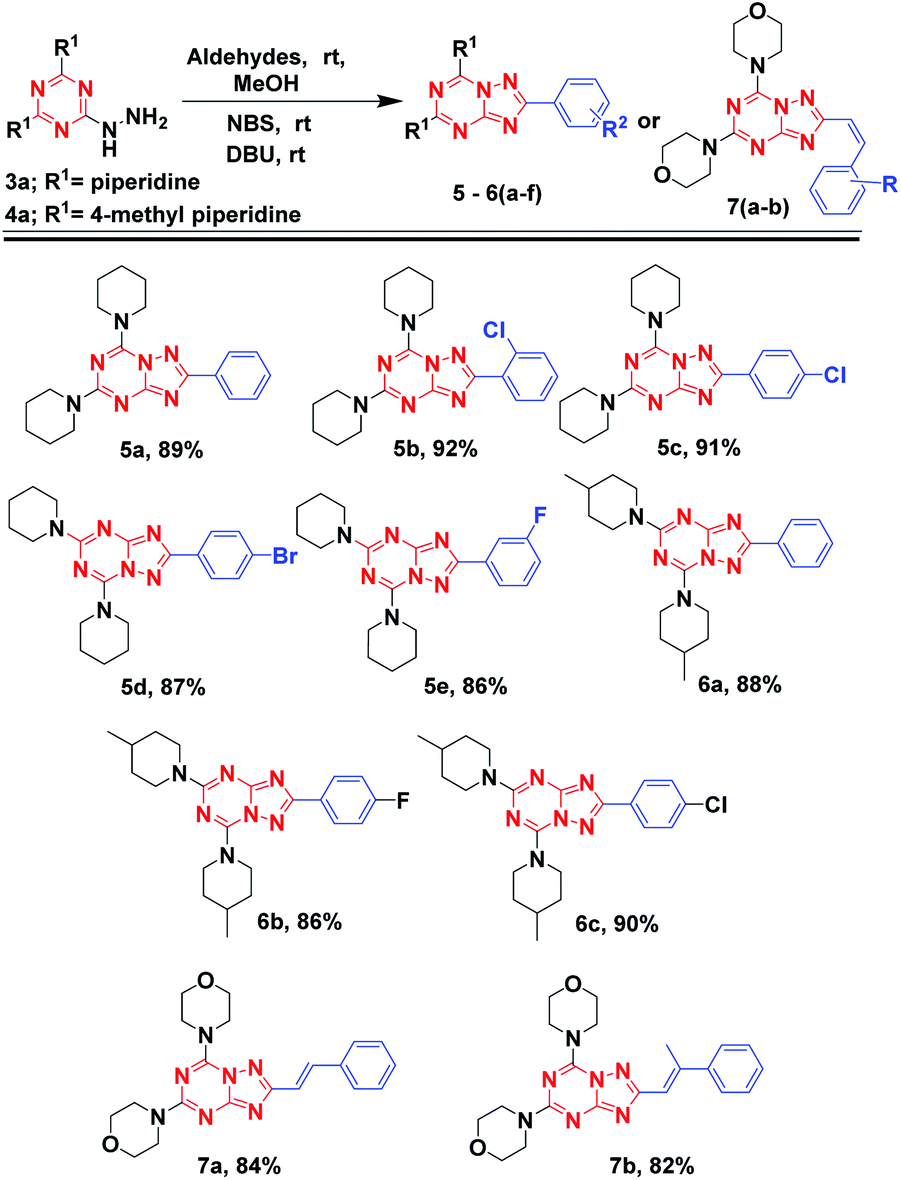
With these promising results, we explored the scope of aldehydes and 2-hydrazinyl-1,3,5-triazine analogs for the synthesis of [1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives. Compound 3a, when reacted with different aldehydes and employing the optimized protocols gave 5a-e with excellent yields (86–92%) in 2–4 h. Similarly compounds 6a, 6b and 6c, synthesized from 4a also afforded excellent yields 88%, 86% and 90% respectively. Further, substituted cinnamaldehydes were also explored and reacted with 1a followed by the addition of NBS and DBU resulted in 7a and 7b in appreciable yields of 84% and 82%, respectively.
Finally, we investigated both the isomers (3b and 5d) spectroscopically, to elucidate the change in proton chemical shifts during rearrangement. Interestingly, it was observed that the proton chemical shift for 5d at the 7th position (Fig. 6) was not affected. However, 5th position of 5d suffers a downfield shift due to change in its electronic environment as compared to its corresponding isomer 3b. In general, the proton chemical shifts for the substitution at 5th position and for the aromatic region was found to be more for isomer-2 as compared to isomer-1.
Conclusions
We have developed a simple yet novel, one-pot, facile, rapid, efficient, and greener approach for the synthesis of pharmacologically important triazolo triazines. Economical and commercially available benzaldehydes were reacted with symmetrical disubstituted triazinyl-hydrazines to obtain a Schiff base followed by in situ reaction initiated by NBS to form isomer-1 [1,2,4]triazolo[4,3-a][1,3,5]triazine analogues, further addition of DBU yielded isomer-2 [1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives via Dimroth type of rearrangement. Operational simplicity, short reaction time and high product yield make this method especially striking for library generation in drug discovery process. Thus, we believe that these heterocycles could lead to hit in medicinal chemistry, if explored and exploited on different biological targets. Further applications of this methodology in developing drug like candidates is underway.Author contributions
All authors contributed equally in the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the Discipline of Pharmaceutical Sciences, College of Health Sciences, University of KwaZulu-Natal (UKZN), Durban, South Africa for providing all the necessary facilities and financial support. R. Karpoormath is also thankful to National Research Foundation-South Africa (NRF-SA) for funding this project (Grant No. 103728, 112079 and 129247). The authors would also like to acknowledge fellow UKZN colleagues Dr Vuyisa Mzozoyana (NMR spectroscopy) and S. J. Zamisa (X-ray crystallography).References
- F. Phei, L. Lim and A. V. Dolzhenko, Eur. J. Med. Chem., 2014, 85, 371–390, DOI:10.1016/j.ejmech.2014.07.112.
- X. Yang, T. J. M. Michiels and C. De Jong, et al., J. Med. Chem., 2018, 61(17), 7892–7901, DOI:10.1021/acs.jmedchem.8b00860.
- M. Jörg, L. T. May and F. S. Mak, et al., J. Med. Chem., 2015, 58(2), 718–738, DOI:10.1021/jm501254d.
- H. Bera, B. J. Tan, L. Sun, A. V. Dolzhenko, W. K. Chui and G. N. C. Chiu, Eur. J. Med. Chem., 2013, 67, 325–334, DOI:10.1016/j.ejmech.2013.06.051.
- S. Redenti, I. Marcovich, T. DeVita, C. Pérez, R. De Zorzi, N. Demitri, D. I. Perez, G. Bottegoni, P. Bisignano, M. Bissaro, S. Moro, A. Martinez, P. Storici, G. Spalluto, A. Cavalli and S. Federico, ChemMedChem, 2019, 14, 310 CrossRef CAS PubMed.
- H. Zstk, G. W. Rewcastle and S. A. Gamage, et al., J. Med. Chem., 2011, 54(20), 7105–7126, DOI:10.1021/jm200688y.
- A. M. Venkatesan, C. M. Dehnhardt and E. D. Delos Santos, et al., J. Med. Chem., 2010, 53(6), 2636–2645, DOI:10.1021/jm901830p.
- D. Rageot, T. Bohnacker and E. Keles, et al., J. Med. Chem., 2019, 62(13), 6241–6261, DOI:10.1021/acs.jmedchem.9b00525.
- D. Rageot, T. Bohnacker and A. Melone, et al., J. Med. Chem., 2018, 61(22), 10084–10105, DOI:10.1021/acs.jmedchem.8b01262.
- A. P. Kourounakis, D. Xanthopoulos and A. Tzara, Med. Res. Rev., 2020, 40, 709–752, DOI:10.1002/med.21634.
- F. Arshad, M. F. Khan, W. Akhtar, M. M. Alam, L. M. Nainwal, S. K. Kaushik, M. Akhter, S. Parvez, S. M. Hasan and M. Shaquiquzzaman, Eur. J. Med. Chem., 2019, 167, 324–356 CrossRef CAS PubMed.
- A. Kumari and R. K. Singh, Morpholine as ubiquitous pharmacophore in medicinal chemistry, Bioorg. Chem., 2020, 96, 103578 CrossRef CAS PubMed.
- A. V. Zavodskaya, V. V. Bakharev and V. E. Parfenov, et al., Tetrahedron Lett., 2015, 56(9), 1103–1106, DOI:10.1016/j.tetlet.2015.01.151.
- A. V. Dolzhenko, A. V. Dolzhenko and W. K. Chui, Heterocycles, 2006, 68(8), 1723–1759, DOI:10.3987/REV-06-607.
- D. V. Kalinin, S. A. Kalinina and A. V. Dolzhenko, Heterocycles, 2013, 87(1), 147–154, DOI:10.3987/COM-12-12601.
- Y. Wu, Y. Yang and Y. Qi, et al., Tetrahedron Lett., 2018, 59(48), 4216–4220, DOI:10.1016/j.tetlet.2018.10.019.
- L. Y. Zhang, C. Zhang, T. Wang, Y. L. Shi, M. T. Ban and D. M. Cui, J. Org. Chem., 2019, 84(2), 536–543, DOI:10.1021/acs.joc.8b02150.
- G. L. Hoang, K. Søholm Halskov and J. A. Ellman, J. Org. Chem., 2018, 83(16), 9522–9529, DOI:10.1021/acs.joc.8b01249.
- A. V. Dolzhenko, S. A. Kalinina and D. V. Kalinin, RSC Adv., 2013, 3(36), 15850, 10.1039/C3RA41932K.
- M. Wang, Y. Meng, W. Wei, J. Wu, W. Yu and J. Chang, Adv. Synth. Catal., 2018, 360, 86 CrossRef CAS.
- J. Kobe, B. Stanovnik and M. Tisler, Tetrahedron, 1970, 26, 3357–3368 CrossRef CAS.
- R. J. Deshpande and A. V. Rama Rao, Synthesis, 1974, 863–865 CrossRef CAS , published online .
- S. Crosignani, Patent Application, Publication Pub. No: US 2020/0102319A1(43), 2020 Search PubMed.
- D. Prat, A. Wells and J. Hayler, et al., Green Chem., 2015, 18(1), 288–296, 10.1039/c5gc01008j.
- M. H. Shinde and U. A. Kshirsagar, Green Chem., 2016, 18, 1455–1458 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2053179. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra07749j |
| This journal is © The Royal Society of Chemistry 2022 |