
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSteady/transient state spectral researches on the solvent-triggered and photo-induced novel properties of metal-coordinated phthalocyanines†
Hongyu Cao *ac,
Meina Gonga,
Mengyan Wangb,
Qian Tangac,
Lihao Wangb and
Xuefang Zheng*bc
*ac,
Meina Gonga,
Mengyan Wangb,
Qian Tangac,
Lihao Wangb and
Xuefang Zheng*bc
aCollege of Life Science and Biotechnology, Dalian University, Dalian 116622, China. E-mail: caohongyu@foxmail.com
bCollege of Environmental and Chemical Engineering, Dalian University, Dalian 116622, China
cLiaoning Key Laboratory of Bio-Organic Chemistry, Dalian University, Dalian 116622, China. E-mail: dlxfzheng@126.com
First published on 17th February 2022
Abstract
The large conjugated system and the d orbitals of metallic phthalocyanines (MPcs) are easily triggered by solvent molecules and light to cause variations in photo-physical and photo-chemical properties. To clarify the novel properties of photo-excited MPcs in solvents, the steady/transient state spectral data were collected to investigate four MPc templates, including FePc, ZnPc, CoPc and AlClPc. The Q bands of FePc and CoPc were prone to redshift, while the peaks of ZnPc and AlClPc tended to blueshift in various solvents compared with that in DMSO. With xenon lamp irradiation, the characteristic absorption peak intensity of FePc and ZnPc in DMSO decreased gradually and then increased after being subjected to dark condition. The transient absorption spectra and kinetic data illustrated that photo-excited FePc and CoPc produced relatively short lifetime transient intermediates. The positive absorption at 650 nm in the Q bands of FePc and CoPc could be assigned to the d–π* electron transition in transient intermediates with unpaired d-orbital electron in the open shell configuration of Fe2+ and Co2+. The above studies were of great significance for the application of solvent-triggered and photo-induced novel properties of MPcs.
Introduction
The excellent thermal stability, chemical stability and unique optical properties, coupled with their extensive redox chemistry, make metal-coordinated phthalocyanines (MPcs) excellent candidates for electrocatalytic and photocatalytic processes in different fields, such as solar cells,1 photodynamic therapy (PDT),2 and chemical sensors.3 Influenced by the conjugate structure, MPcs usually aggregated into two types: face-to-face H-aggregates and head-to-tail J-aggregates. The degrees of aggregation depend on various factors, such as solvent, concentration, sidechain substituent, and the coordinated metal.4 The exploitation of the MPc aggregation and redox mechanism can be beneficial for its applications in the fields of photophysics,5 photochemistry,6 materials7 and catalysis.8 Transition metal phthalocyanine dual-metal-site catalysts9 and composite anchoring iron phthalocyanine (FePc)/TiO2 heterostructure10 demonstrated good photocatalytic activities for CO reduction or organic contaminant degradation.11 The redox and aggregation process mechanisms of CoPc or ZnPc were still unclear because the metal-based and ring-based one-electron redox processes were influenced by the axial coordinated ligand properties.12 Negrimovsky et al.13 proved that the increasing number of cationic groups led to the increased solubility of monomeric MPcs in dipolar aprotic organic solvents. Aluminium coordinated phthalocyanine (AlPc) and aluminium coordinated porphyrin have high binding affinity with ions. Studies on AlPc electrochemistry14 in aqueous and nonaqueous media were reported, but the photoinduced reaction was not included. The high electrocatalytic and photocatalytic activities of MPcs can be attributed to the unsaturated low-coordination environment of the central metal ions coordinated to four isoindole subunits.15 Current researches focus on the physical and chemical properties of metal phthalocyanines with side chains under non-photoexcited conditions. Although it has been shown that the central metal in MPcs played an important role in various photocatalytic and electrocatalytic reactions, the precise control by light or solvent effects is required to develop a deeper understanding. As a matter of fact, the photoirradiated MPcs displayed more complicated and diverse physical/chemical properties in solvent. Thus, with the application of steady-state/transient absorption spectroscopy, the photoexcited FePc, ZnPc, CoPc and AlClPc were investigated in solvents. Some new photophysical and photochemical properties of the photoexcited MPcs were detected, including the axial coordination variation and the aggregation of MPcs. The electron transition advantages of ferriporphyrin in heme proteins16 and the excellent optical adsorption properties of metal-coordinated porphyrins or phthalocyanines in photoelectric conversion applications17 were further understood based on the transient absorption bands and relaxation process of excited state intermediates. The photophysical and photochemical mechanisms were analysed, and the influence rules of external conditions were discussed to provide fundamental guidance for the practical application of MPcs in the field of photochemistry.Experimental
MPcs, DMSO, DMF, CH3OH and other organic reagents were purchased from Aladdin or Sigma, and all of the reagents were not further purified. The experimental water used was ultrapure water. MPcs were prepared in various solvents of the same concentration of 1 × 10−5 mol L−1.After three hours in dark condition, MPcs were irradiated for a certain time under a full wavelength xenon lamp with the energy of 150 W. The UV-visible spectra of MPcs were recorded on a UV-vis spectrophotometer (Jasco, Japan) using 1 cm quartz cuvettes. The slit width was 2 nm, the scanning wavelength range was 300–700 nm, the scanning rate was 400 nm min−1, and the response time was medium. The fluorescence spectra of the samples were measured with an FP-6500 after being irradiated using a full-wavelength xenon lamp. The fluorescence spectra and synchronous fluorescence spectra of the samples were measured using FP-6500 (Jasco, Japan) after being irradiated by a full-wavelength xenon lamp. The parameters of the fluorescence spectra were set with the excitation wavelength at 355 nm, and the parameters of the synchronous fluorescence spectra were also set with the delta wavelength of 20 nm, slit width of 5 × 5 nm, scanning rate of 500 nm min−1, and wavelength range of 300–700 nm. The transient absorption spectra and decay curves of the sample were measured using an LP-980 Laser flash photolysis (Edinburgh Instruments, Britain). The sample was placed in a quartz cuvette and excited by a fixed wavelength (355 nm) laser. The change in optical density (ΔOD) was as follows:
| ΔODt, λ = εT(λ)cT(t) − εG(λ)[cT(t) + cs(t)]d−ϕ(λ)cs(t) | (1.1) |
The fitting curve of the excited-state decay kinetics is as follows:
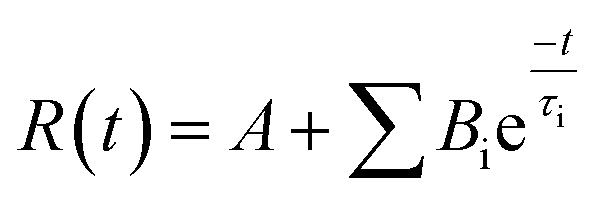 | (1.2) |
The laser flash photolysis used the absorption map in kinetic absorption mode for measurement (laser mode: 1 Hz; time range: 4000 ns; time shift: 15%; detection voltage range: 400 mV; repeated times of each measurement: 2 laser pulses; detector slit width: 2 nm; and detection wavelength range: 300–750 nm).
Results and discussions
Two strong absorption regions in the UV-vis spectra of MPcs were mainly caused by the π-electron transition on the conjugated system of phthalocyanine. The first absorption band located at 300 nm–500 nm is called the B band, which is assigned to the a2u → eg transition with an energy value of 3.8 eV. The second stronger absorption region located at 600 nm–700 nm is labelled as the Q band, where the electron transitioned from a1u to eg at a lower energy of about 1.8 eV.18 The central metals exerted significant effects on the position and intensity of characteristic absorption peaks in the UV-vis spectra of MPcs. MPcs have a simple planar molecular structure without any polar groups and side chains, but the delocalization of the π-bond and d orbitals of the metal centre in the molecule was easily affected by the solvent molecules, thus changing the shape and position of the B band and Q band.19The maximum absorption wavelengths of the MPcs were weakly correlative to the polarities of the solvents (Table 1 and Fig. S1†), but they were susceptible to the solvent molecules. The highest characteristic Q band of FePc was located at 653 nm in DMSO, while the peak was located at 659 nm in CH2OH and CH3CH2OH. Two broad characteristic absorption peaks of FePc could be observed in CHCl3 and C6H6. The Q band at a wavelength of 669 nm of FePc in C6H6 redshifted evidently (Δλ = 16 nm) than that in DMSO. The maximum absorption wavelength of ZnPc in DMSO was located at 672 nm, while the strongest peak of ZnPc in ethanol and CHCl3 was located at 665 nm and 684 nm, respectively. The Q band of CoPc located at 665 nm in CHCl3 redshifted significantly (Δλ = 9 nm) than that in DMSO. The maximum absorption peak of AlClPc red-shifted by 5 nm from 675 nm in DMSO to 680 nm in CHCl3.
| FePc | CoPc | ZnPc | AlClPc | |||||
|---|---|---|---|---|---|---|---|---|
| WL | Δλ | WL | Δλ | WL | Δλ | WL | Δλ | |
| a Take the spectral peaks of MPcs in DMSO solvent as the standard (0), “−” and “+” indicate the blueshifts and redshifts in other solvents, respectively. “WL” represents wavelength (nm). | ||||||||
| DMSO | 653 | 0 | 656 | 0 | 672 | 0 | 675 | 0 |
| DMF | 657 | +4 | 658 | +2 | 668 | −4 | 670 | −5 |
| CH3OH | 659 | +6 | 656 | 0 | 665 | −7 | 670 | −5 |
| CH3CH2OH | 659 | +6 | 656 | 0 | 667 | −5 | 670 | −5 |
| 1,4-Dioxane | — | — | 657 | +1 | 668 | −4 | 672 | −3 |
| EtOAc | — | — | 657 | +1 | 665 | −7 | 670 | −5 |
| CHCl3 | 655 | +2 | 665 | +9 | 684 | +12 | 680 | +5 |
| C6H6 | 669 | +16 | 660 | +4 | 670 | −2 | 673 | −2 |
The Q bands of FePc and CoPc were prone to redshift, while the peaks of ZnPc and AlClPc tend to blueshift in various solvents than that in DMSO (Table 1). The noticeable Q-band peak position shifts of FePc and CoPc with 3d electrons in various solvents indicated that the d–π* transitions were more susceptible to the solvent, and the solvent molecules were prone to be the axial coordinated ligand with the central metal, resulting in the differences in molecular photophysical properties. The full 3d10 electron configuration of Zn2+ in ZnPc, as well as the third periodic metal aluminium in AlClPc, formed the coordination bond with the inner N of phthalocyanine, which showed the opposite phenomena in various solvents (Table 1). The stable metal ions of ZnPc and AlClPc without the unpaired d-orbital electron or other single electron tended to keep the stable geometrical structures and the coordination numbers, which presented the relatively stable properties and avoided the interference of solvent molecules. In DMSO, the characteristic absorption peak intensities of FePc, CoPc and ZnPc (Fig. 1) gradually increased in the freshly prepared solutions. The Q-band characteristic peak of FePc was first located at 662 nm in the freshly prepared solutions. Then, the peak location blueshifted to 653 nm with an increase in the intensity in three hours (Table S1†). FePc in the freshly prepared solutions might be in an aggregation state, which led to an electron-rich effect in phthalocyanine ligand by the intermolecular π–π* stacking and formed similar configuration of FePc− (665 nm).20 The Q-band intensities of CoPc and ZnPc (656 nm and 672 nm, respectively) increased significantly without any wavelength shifts following the change in time under no light condition. The Q band of AlClPc was stably located at 675 nm with a small change in the intensity. The spectral curves revealed that CoPc and ZnPc in the freshly prepared solutions might be in a face-to-face H aggregation state, whereas AlClPc might be in a scattered state.
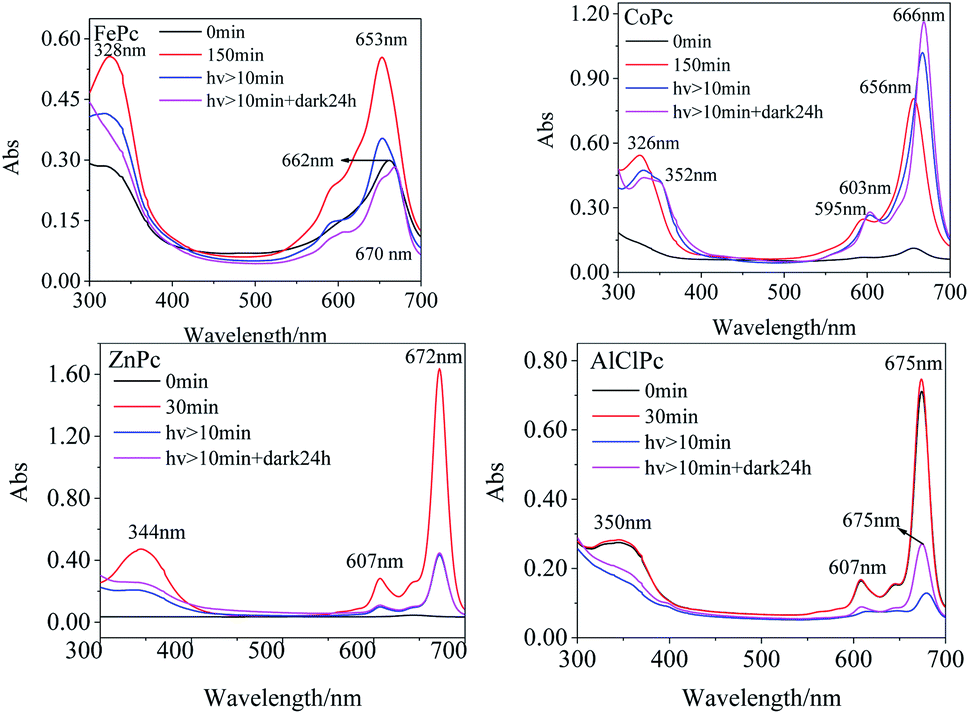 | ||
| Fig. 1 UV-vis spectra of photo-excited FePc, CoPc, ZnPc and AlClPc in DMSO. | ||
The 653 nm absorption intensity of FePc decreased after irradiating with a xenon lamp and redshifted to 670 nm, which could be assigned to the characteristic peak of Fe3+Pc−.21 The variations in spectra illustrated the metal–ligand charge transfer (MLCT) process in photo-excited FePc. The susceptible spectra of FePc under light and dark conditions reflected the active electron transition between the iron ion and ligand, which generated the excellent and special properties of the photo-excited FePc.
After CoPc was irradiated using a xenon lamp for at least 10 min (Fig. 1), the B-band peak redshifted from 326 nm to 332 nm with a decrease in the absorption intensity, while a new peak appeared at 352 nm with a gradual increase in intensity. The Q band redshifted from 596 nm/656 nm to 603 nm/666 nm (Δλmax = 10 nm), and the intensity at 666 nm significantly enhanced. The phenomena of nascent peak and redshifted Q-band in the spectra might attribute to the fact that the long-time xenon lamp excitation changed the axial coordination of Co in CoPc with small molecules, such as O2. The Q-band peak intensity increased gradually with an irradiation using the xenon lamp, indicating that CoPc did not aggregate in the photo-excited process.
In DMF solution, the characteristic absorption peak intensities of the four MPcs decreased gradually with irradiation time. The absorption peaks of FePc, CoPc and AlClPc redshifted, while the peak location of ZnPc remained unchanged and reached a stable state after a period (Fig. 2). The unique characteristics might be that the saturated electron configuration of Zn2+ was too stable to coordinate with small molecules as the axial ligand. After the dark treatment, the absorption intensity of the four metal-coordinated phthalocyanines recovered at varying degrees.
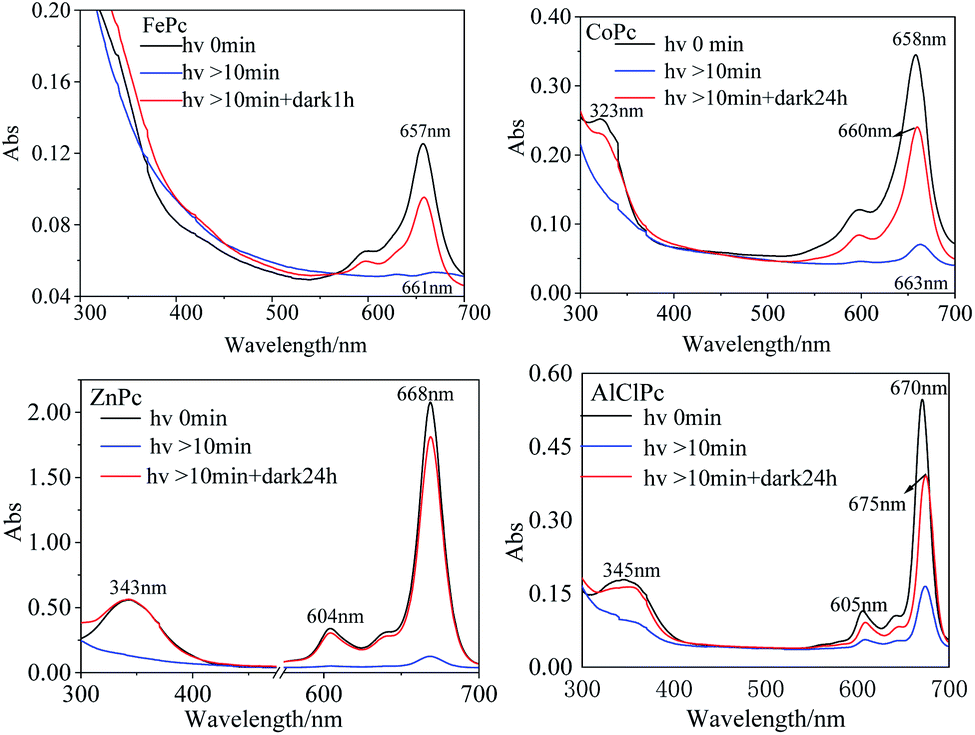 | ||
| Fig. 2 UV-vis spectra of photo-excited and dark-treated FePc, CoPc, ZnPc and AlClPc in DMF. | ||
In general, the aggregation process followed the first-order reaction:
A(t) = A0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−ket) exp(−ket)
| (1.3) |
| Solvent | ke (min−1) | |||
|---|---|---|---|---|
| FePc | CoPc | ZnPc | AlClPc | |
| DMSO | 3.10 × 10−3 | — | 1.47 × 10−2 | 1.19 × 10−1 |
| DMF | — | 2.80 × 10−2 | 1.40 × 10−1 | 1.50 × 10−2 |
Synchronous fluorescence (Δλ = 20 nm) showed that the fluorescence peaks of FePc were located at 326 nm, 346 nm, 410 nm and 563 nm (Fig. 3) in DMSO. After xenon lamp irradiation, the fluorescence peak at 326 nm and 346 nm gradually vanished, and finally a new fluorescence peak with a higher intensity appeared at 340 nm. The 410 nm fluorescence peak assigned to the large conjugate system of the MPc varied gradually with the photo-irradiation time. Four emission peaks at 326 nm, 346 nm, 557 nm and 620 nm were observed in the synchronous fluorescence (Δλ = 20 nm) spectrum of CoPc (Fig. 3). The fluorescence peaks at 326 nm and 346 nm faded away, and a new fluorescence peak generated at 340 nm along with a gradual increase in intensity under the light irradiation, which was similar to that of FePc. The same peak merging phenomena about the 340 nm peaks could be observed in the synchronous fluorescence spectra of ZnPc and AlClPc. However, the 410 nm fluorescence peak intensity of ZnPc and AlClPc remained unchanged under light. The fluorescence peak merging phenomena might be due to an increase in the molecular symmetry and the degeneracy of π-orbital energy levels of the photo-excited MPcs.22 The 410 nm fluorescence variations were assigned to Fe and Co ions that coordinated with O2 or other small molecules in solvents, resulting in the reduction of unpaired d-orbital electrons. Interestingly, strong synchronous fluorescence (Fig. 3) and fluorescence peaks (Fig. S2†) near 700 nm could be found in the spectra of ZnPc and AlClPc, but they could not be observed in FePc and CoPc. The notable features indicated that the unpaired d electrons of Fe and Co led to the electron delocalization and internal energy conversion in the molecules.
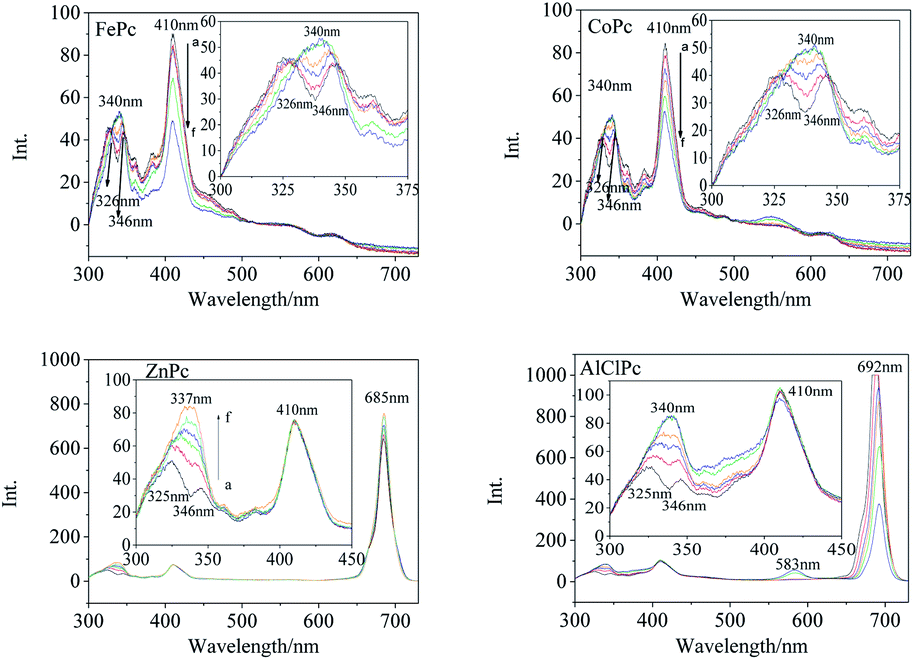 | ||
| Fig. 3 Synchronous fluorescence spectra (Δλ = 20 nm) of FePc, CoPc, ZnPc and AlClPc after xenon lamp irradiation in DMSO (a–f indicates the irradiation duration of the xenon lamp, i.e., 0, 5, 10, 15, 30 and 60 minutes). | ||
The transient spectra of the laser-excited MPcs displayed more significant differences. Two evident negative peaks at 384 nm and 404 nm (Fig. 4) indicated that one intermediate of FePc was generated by laser excitation. A larger εT value of the intermediate than the εG value of the ground state would result in a transient absorption peak with a positive ΔOD value (eqn (1.1)). A positive peak at 650 nm and a negative absorption peak at 670 nm in the Q band also illustrated the generation of transient intermediates of FePc. The transient spectrum of ZnPc was classified as a typical characteristic absorption peak of T1–Tn transition in MPc.24 The two negative peaks (607 and 672 nm) in the Q band were matched with the ground state absorption peaks in the steady absorption spectra. The ground state bleaching peak recovery process synchronized with an attenuation of the ground state positive spectral peak (400 nm–600 nm). A wide range of transient spectral bands and a relatively slow relaxation process of ZnPc demonstrated a long-life excited state intermediate. This might be one of the reasons why it could play an important role in the field of photoelectric conversion.25 Two positive peaks at 586 nm and 650 nm in the Q band could be observed (Fig. 4) in CoPc transient absorption. The weak absorption peak at 586 nm could be assigned to the excited state intermediate of CoPc. The transient negative peak with a ΔOD value of −0.29 a.u. at 682 nm corresponded to the AlClPc fluorescence peak, which was assigned to the fluorescence term Φ(λ) of AlClPc. In addition, three positive transient absorption peaks at 418 nm, 580 nm and 627 nm, which were attributed to the excited state (εt > εG), proved the generation of stable triple-excited intermediate of AlClPc after laser excitation.
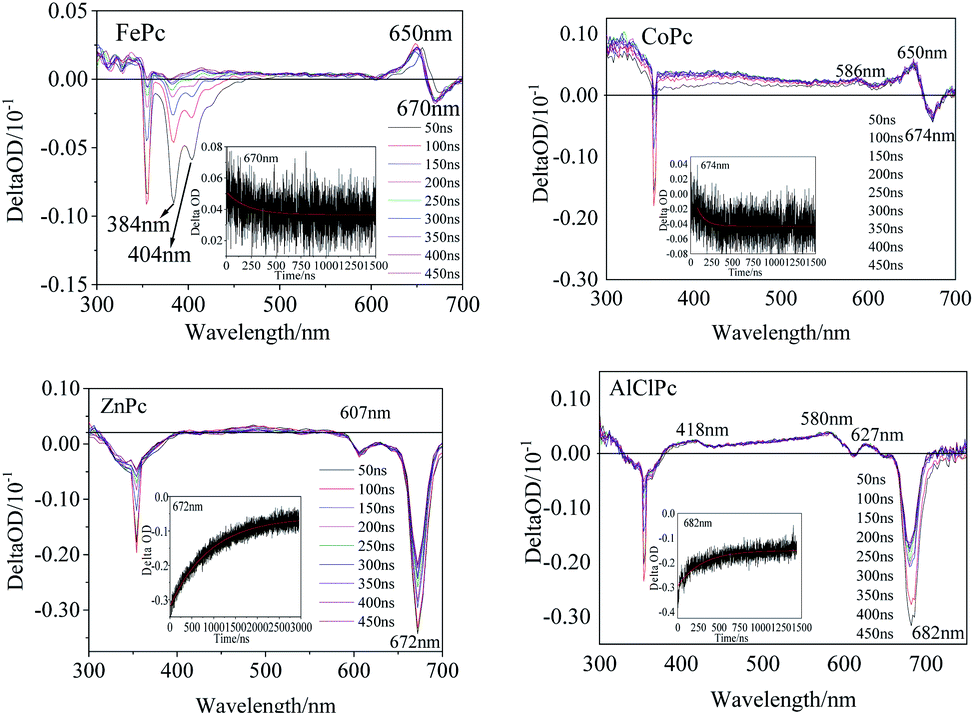 | ||
| Fig. 4 Transient absorption maps of FePc, CoPc, ZnPc and AlClPc in DMSO. | ||
The excitation decay kinetic curves of the characteristic peak in the transient spectra of the four MPcs (Fig. 4 and S3†) fitted using eqn (1.2), and the data are listed in Table 3. The decay time of FePc at 384 nm, 404 nm, 650 nm and 670 nm were 194 ns, 295 ns, 213 ns and 759 ns, respectively. The decay lifetime in the same order of magnitude confirmed the same intermediate of FePc. The short decay time of the transient peak at 674 nm in CoPc transient spectra was 194 ns. The positive absorption at 650 nm in the Q band of FePc and CoPc could be assigned to the d–π* in transient intermediates with unpaired d-orbital electron in the open shell configuration of Fe2+ and Co2+. Contrarily, the negative ΔOD values of ZnPc and AlClPc at Q band decayed with time in the kinetics curves. Moreover, interestingly, the long kinetic lifetimes of ZnPc at 607 nm and 672 nm were 709 ns and 918 ns, respectively. The kinetic times of ZnPc and AlClPc were clearly longer than that of FePc and CoPc, indicating that the electron transition and relaxation processes were distinctly different between them irrespective of the presence of an unpaired d-orbital electron in the molecules.
| WL (nm) | τ (ns) | χ2 | SD (ns) | |
|---|---|---|---|---|
| a “WL/nm” represents “Wavelength/nm” and “SD/ns” indicates “standard deviation/ns”. | ||||
| FePc | 384 | 194 | 1.03 | 5 |
| 404 | 295 | 0.97 | 8 | |
| 650 | 213 | 0.80 | 7 | |
| 670 | 759 | 0.90 | 15 | |
| CoPc | 586 | 322 | 0.87 | 14 |
| 650 | 296 | 0.82 | 29 | |
| 674 | 94 | 0.85 | 13 | |
| ZnPc | 607 | 709 | 0.937 | 27 |
| 672 | 918 | 0.957 | 6 | |
| AlClPc | 418 | 456 | 1.18 | 30 |
| 580 | 885 | 1.00 | 28 | |
| 682 | 292 | 0.99 | 6 | |
Four possible mechanisms for the photo-induced properties of metal-coordinated phthalocyanines in different solutions were proposed: (1) photolysis of the phthalocyanines; (2) photo-induced redox reaction of the central metal ions; (3) the variation of the metal coordination modes; and (4) photo-induced aggregation of MPcs. After being illuminated by a xenon lamp and then treated in the dark for 24 h, the peak intensity of ZnPc in DMF recovered to the initial level. The reversible photo-induced process denied the ZnPc (or other MPcs) photolysis speculation. The assumption that the variations in photo-physical and photo-chemical properties were caused by the photo-induced oxidation or reduction of MPcs was also excluded for quite stable metal ion valence states in ZnPc and AlClPc. In the UV-vis absorption spectrum and transient absorption spectrum, the Q-band characteristic peak varied significantly, which was the reflection of the metal coordination modes and aggregation states, including the axial coordinated ligands of metal with solvent or other small molecules.
FePc in an aggregation state possessed electron-rich effect in the phthalocyanine ligand by the intermolecular π–π stacking and formed FePc− (665 nm).20 The Q band of the light-irradiated FePc redshifted to 670 nm, which proved the production of Fe3+Pc−.21 The spectral variations illustrated the metal–ligand charge transfer (MLCT) process in the photo-excited FePc. The active electron transition between the iron ion and the ligand illustrated the excellent and special properties of the photo-excited FePc, which could explain the important roles of ferriporphyrin in heme proteins, such as cytochromes and myoglobin.16
Zn2+ and Fe2+ were both positive bivalent metal ions and coordinated with phthalocyanine to form single-core monolayer planar complexes, but the axial coordination ability of FePc and ZnPc were quite dissimilar. With the outermost electron configuration of 3d10, zinc ion in ZnPc was too stable to coordinate with the small molecule as the axial ligand. The saturation electron configuration of Zn2+ in ZnPc was beneficial to generate long-life intermediates, which further provided an important basis for the selection of metal-coordinated porphyrinoids in photoelectric conversion application. The spectra of AlClPc revealed that the coordination bond Al–Cl was destroyed by photo-excitation, which promoted the intermolecular π–π stacking and easily caused the aggregation of AlClPc. The Al–Cl coordinated bond breaking led to the redshift of the maximum absorption wavelength, while the aggregation of AlClPc reduced the absorption intensity (Fig. 5).
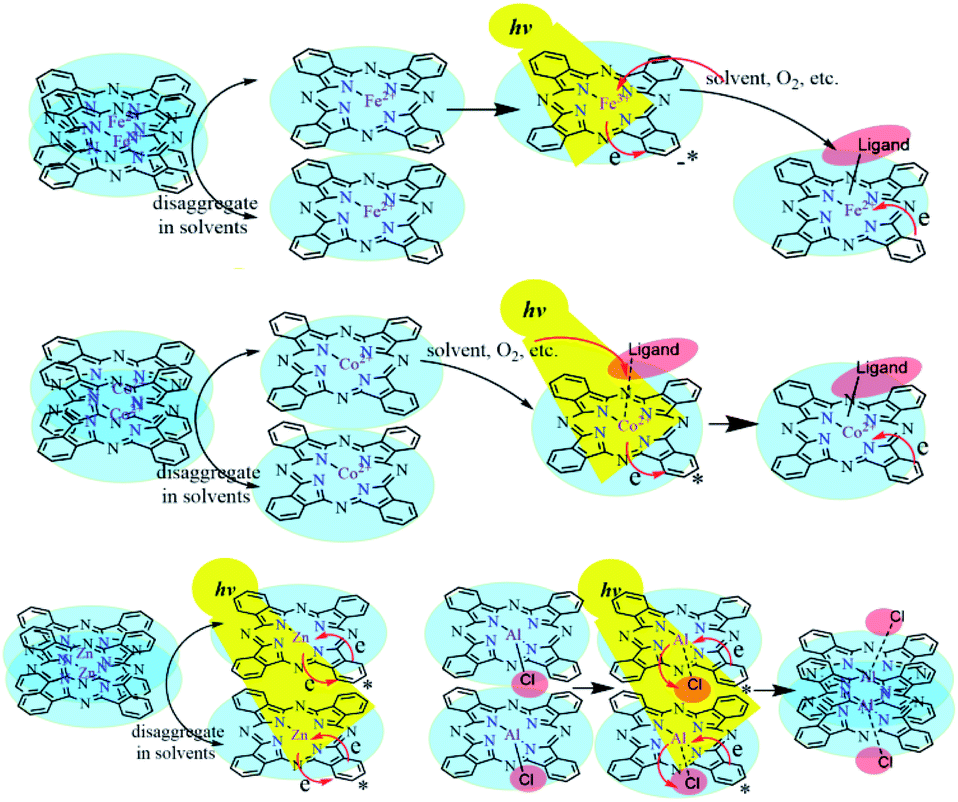 | ||
| Fig. 5 The solvent-triggered and photo-induced reaction or aggregation mechanisms of the four metal-coordinated phthalocyanines. | ||
Conclusions
The noticeable Q-band shifts of FePc and CoPc with 3d electrons in various solvents indicated that the d–π* transition was more susceptible to solvent, and the solvent molecules were prone to the axial coordinated ligand with the central metal. The aggregation rates of MPcs in DMSO followed the order AlClPc > ZnPc > FePc > CoPc, which revealed that the solvents significantly affected the photochemical properties of the MPcs. The decay times of ZnPc and AlClPc were clearly longer than that of FePc and CoPc, indicating that the electron transition and relaxation processes were different from those of FePc and CoPc irrespective of the presence of an unpaired d-orbital electron. The Q band of the light-irradiated FePc redshifted to 670 nm. It proved the production of Fe3+Pc− and the active metal–ligand charge transfer (MLCT) process in the photo-excited FePc, which could be applied to explain the important function of ferriporphyrin in heme proteins, such as cytochromes and myoglobin. A wide range of transient absorption bands and a relatively slow relaxation process of ZnPc demonstrated a long-life excited state intermediate, which could be used in the field of photoelectric conversion. The solvent-triggered and photo-induced MPcs would significantly affect the presence or absence of axial coordinated ligands and aggregation states, and furthermore, generate many unique properties for the photo-physical and photo-chemical applications of MPcs.Author contributions
Hongyu Cao contributed to supervision and review and editing. Meina Gong and Mengyan Wang performed the formal analysis and prepared the original draft. Qian Tang and Lihao Wang were involved in the formal analysis and validation. Xuefang Zheng contributed to the supervision and conceptualization.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (no. 21601025, 21571025, and 21601024), the Scientific Research Projects in Universities of Education Department of Liaoning Province (LJKQZ2021168), the Scientific Research Platform Project of Dalian University (202101ZD01) and the Excellent Youth Team for Scientific Research, Innovation and Entrepreneurship of Dalian University (XQN202004).Notes and references
- K. Takahashi, N. Kuraya, T. Yamaguchi, T. Komura and K. Murata, Three-layer organic solar cell with high-power conversion efficiency of 3.5%, Sol. Energy Mater. Sol. Cells, 2000, 61, 403–416 CrossRef CAS.
- S. Belali, A. R. Karimi and M. Hadizadeh, Novel nanostructured smart, photodynamic hydrogels based on poly(N-isopropylacrylamide) bearing porphyrin units in their crosslink chains: a potential sensitizer system in cancer therapy, Polymer, 2017, 109, 93–105 CrossRef CAS.
- L. Breloy, O. Yavuz, I. Yilmaz, Y. Yagci and D. L. Versace, Design, synthesis and use of phthalocyanines as a new class of visible-light photoinitiators for free-radical and cationic polymerizations, Polym. Chem., 2021, 12, 4291–4316 RSC.
- M. Choi, P. Li and D. Ng, A Direct Comparison of the Aggregation Behavior of Phthalocyanines and 2, 3-Naphthalocyanines, Tetrahedron, 2000, 56, 3881–3887 CrossRef CAS.
- A. Ng, X. Y. Li and D. Ng, Synthesis and Photophysical Properties of Nonaggregated Phthalocyanines Bearing Dendritic Substituents, Macromolecules, 1999, 32, 5292–5298 CrossRef CAS.
- H. Isago, C. C. Leznoff, M. F. Ryan, R. A. Metcalfe, R. Davids and A. B. P. Lever, Aggregation Effects on Electrochemical and Spectroelectrochemical Properties of [2, 3, 9, 10, 16, 17, 23, 24-Octa (3, 3-dimethyl-1-butynyl) phthalo-cyaninato] cobalt (II) Complex, Bull. Chem. Soc. Jpn., 1998, 71, 1039–1047 CrossRef CAS.
- D. K. Singh, V. Ganesan, D. K. Yadav and M. Yadav, Metal (Mn, Fe, Co, Ni, Cu, and Zn) Phthalocyanine Immobilized Mesoporous Carbon Nitride Materials as Durable Electrode Modifiers for the Oxygen Reduction Reaction, Langmuir, 2020, 36, 12202–12212 CrossRef CAS PubMed.
- S. Baranton, C. Coutanceau, E. Garnier and J. M. Léger, How does α-FePc catalysts dispersed onto high specific surface carbon support work towards oxygen reduction reaction (orr), J. Electroanal. Chem., 2006, 590, 100–110 CrossRef CAS.
- X. Wan, Z. Zhang, H. Niu, Y. Yin, C. Kuai, J. Wang, C. Shao and Y. Guo, Machine-Learning-Accelerated Catalytic Activity Predictions of Transition Metal Phthalocyanine Dual-Metal-Site Catalysts for CO Reduction, J. Phys. Chem. Lett., 2021, 12, 6111–6118 CrossRef CAS PubMed.
- B. Popanda, M. Roda, R. Sota and M. Zakrzyk, Metallophthalocyanines as optical active dopants in borate glass, Dyes Pigm., 2021, 193, 109496 CrossRef CAS.
- J. Fei, Z. Han, Y. Deng, T. Wang, J. Zhao, C. Wang and X. Zhao, Enhanced photocatalytic performance of iron phthalocyanine/TiO2 heterostructure at joint fibrous interfaces, Colloids Surf., A, 2021, 625, 126901 CrossRef CAS.
- İ. Koç, M. Çamur, M. Bulut and A. R. Özkaya, Electrochemical and in situ spectroelectrochemical investigations into the redox and aggregation behaviours of phthalocyanines bearing octyl 4-phenyloxyacetate moieties, Can. J. Chem., 2010, 88, 375–382 CrossRef.
- V. Negrimovsky and K. Volkov, Synthesis of water-soluble cobalt and zinc phthalocyanines with cationic substituents, J. Porphyrins Phthalocyanines, 2013, 17, 750–755 CrossRef CAS.
- Z. Ou, J. Shen and K. M. Kadish, Electrochemistry of Aluminum Phthalocyanine: Solvent and Anion Effects on UVVisible Spectra and Reduction Mechanisms, Inorg. Chem., 2006, 45, 9569–9579 CrossRef CAS PubMed.
- S. Yang, Y. Yu, X. Gao, Z. Zhang and F. Wang, Recent advances in electrocatalysis with phthalocyanines, Chem. Soc. Rev., 2021, 50, 12985 RSC.
- H. Y. Cao, Y. Q. Ma, L. X. Gao, Q. Tang and X. F. Zheng, Photo induced reaction of myoglobins with energy transferred from excited free tryptophan, RSC Adv., 2020, 10, 43853–43858 RSC.
- V. Cuesta, R. Singhal, de la C. Pilar, G. D. Sharma and F. Langa, Near-IR Absorbing D-A-D Zn-Porphyrin-based Small Molecule Donors for Organic Solar Cells with Low Voltage Loss, ACS Appl. Mater. Interfaces, 2019, 11, 7216–7225 CrossRef CAS PubMed.
- X. Xu, M. Hu, Y. Wang and G. Yang, Synthesis and spectroscopic properties of metal phthalocyanines, Chin. J. Chem. Educ., 2019, 40, 38–41 Search PubMed.
- A. Ogunsipe, D. Maree and T. Nyokong, Solvent effects on the photochemical and fluorescence properties of zinc phthalocyanine derivatives, J. Mol. Struct., 2003, 650, 131–140 CrossRef CAS.
- D. W. Clack and J. R. Yandle, Electronic spectra of the negative ions of some metal phthalocyanines, Inorg. Chem., 1972, 11, 1738–1742 CrossRef CAS.
- P. C. Minor, M. Gouterman and A. Lever, Electronic spectra of phthalocyanine radical anions and cations, Chem. Informationsdienst, 1985, 16, 1900–1984 Search PubMed.
- T. Gośliński, T. Osmalek, K. Konopka, M. Wierzchowski, P. Fita and J. Mielcarek, Photophysical properties and photocytotoxicity of novel phthalocyanines potentially useful for their application on photodynamic therapy, J. Polyhedron, 2011, 30, 1538–1546 CrossRef.
- X. W. Zou, J. Ke, J. Y. Hao, X. Y. Yan and Y. M. Tian, A new method for synthesis of ZnO flower-like nanostructures and their photocatalytic performance, Phys. B: Condens. Matter., 2022, 624, 413395 CrossRef CAS.
- X. F. Zhang and W. Guo, Imidazole functionalized magnesium phthalocyanine photosensitizer: modified photophysics, singlet oxygen generation and photooxidation mechanism, J. Phys. Chem. A, 2012, 116, 7651–7657 CrossRef CAS PubMed.
- Z. H. Ma, M. Y. Wang, H. Y. Cao, Q. Tang, L. H. Wang and X. F. Zheng, Transient Absorption and Decay Kinetic Properties of Photo-excited Metal Coordinated Tetraphenylporphyrin, Chem. J. Chinese U., 2021, 42, 767–775 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08345g |
| This journal is © The Royal Society of Chemistry 2022 |