
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective and efficient catalytic and photocatalytic oxidation of diphenyl sulphide to sulfoxide and sulfone: the role of hydrogen peroxide and TiO2 polymorph†
Paweł Mikrut ,
Aneta Święs,
Marcin Kobielusz*,
Lucjan Chmielarz and
Wojciech Macyk*
,
Aneta Święs,
Marcin Kobielusz*,
Lucjan Chmielarz and
Wojciech Macyk*
Faculty of Chemistry, Jagiellonian University, ul. Gronostajowa 2, 30-387 Kraków, Poland. E-mail: macyk@chemia.uj.edu.pl; kobielusz@chemia.uj.edu.pl
First published on 12th January 2022
Abstract
In this paper, we describe the role of anatase and rutile crystal phases on diphenyl sulphide (Ph2S) catalytic and photocatalytic oxidation. The highly selective and efficient synthesis of diphenyl sulfoxide (Ph2SO) and diphenyl sulfone (Ph2SO2) at titanium dioxide was demonstrated. Ph2S oxidation in the presence of hydrogen peroxide at anatase-TiO2 can take place both as a catalytic and photocatalytic reaction, while at rutile-TiO2 only photocatalytic oxidation is possible. The reaction at anatase leads mainly to Ph2SO2, whereas, in the presence of rutile a complete conversion to Ph2SO is achieved after only 15 min (nearly 100% selectivity). Studies on the mechanistic details revealed a dual role of H2O2. It acts as a substrate in the reaction catalysed only by anatase, but it also plays a key role in alternative photocatalytic oxidation pathways. The presented study shows the applicability of photocatalysis in efficient and selective sulfoxide and sulfone production.
Introduction
Conventional routes of synthesis of many important organic chemicals often require harsh operating conditions, such as high temperature and pressure. Moreover, sometimes the presence of toxic oxidation or reduction agents is required, far from the principles of green chemistry. The development of synthesis, which offers efficient organic compound production under mild reaction conditions, is highly desirable. Consequently, a photocatalytic organic synthesis, which utilizes economically sustainable and clean solar energy, has achieved tremendous attention in recent years.1 However, photocatalysis has been mostly considered as a non-selective process, involving activation of small molecules to initiate a chemical reaction (especially in water). These types of photocatalytic reactions include mainly processes governed by reactive oxygen species such as oxidation of alcohols,2 oxidation of aromatics3,4 or oxidation of side chains.5,6 Nevertheless, a carefully selected photocatalyst and synthesis conditions in a non-aqueous solvent can lead to highly selective photocatalytic fine product formation.Oxidation of organic sulphides (diphenyl sulphide, dimethyl sulphide) is one of the most important processes in organic chemistry. The organic sulfoxides and sulfones, which are the products of this reaction, were found to be very important substances for pharmacy, medicine, or substrates in drug synthesis. They can be used for the production of antibacterial, antifungal,7 antihypertensive agents,8 and vasodilators.9 Many catalytic systems, containing, e.g., V, Re, Ti, Mo, Te, W, Se, Fe elements, have been studied in a sulphide to sulfoxide oxidation.10–14 Titanium dioxide is used as a catalyst due to its desired physicochemical properties, high chemical stability, low cost of production, and lack of toxicity. Additionally, TiO2 doped with vanadium was found to be an active catalyst of Ph2S oxidation giving nearly 100% conversion of Ph2S with high selectivity to Ph2SO2.15 SiO2–TiO2 mesoporous xerogel catalyses oxidation to the final product, diphenyl sulfone, with the selectivity of 85%.16 Furthermore, it was confirmed that titanosilicate zeolites (inter alia, Ti-beta,17 Ti-MWW,18 Ti-FER, MTS-1 (ref. 19) and Ti-ITQ-6 (ref. 20)) present a good catalytic activity in this process. NaIO4, trichloroisocyanuric acid, sodium perborate, KMnO4, can be successfully applied as oxidizing agents.21 However, hydrogen peroxide, H2O2, seems to be the most suited oxidizer due to its low environmental impact (H2O is the only side product), nontoxicity, and high content of active oxygen.22–24 Aromatic sulphides can also be transformed using photocatalysts such as wide bandgap oxide semiconductors (TiO2, ZnO, etc.). Although, they undergo deep oxidation in the aqueous media, sulfoxides, and sulfones are the main reaction products in the presence of organic solvents.25 To the best of our knowledge, the air is the most common source of oxygen in the photocatalytic transformation of sulphides described in the literature. In this system, even when selectivity is high, the concentration of the final product is rather low. However, we showed recently that hydrogen peroxide is a crucial oxidation agent in the photocatalytic oxidation of Ph2S.26 No Ph2S conversion after 3 hours of the reaction in both catalytic and photocatalytic tests conducted in the absence of H2O2 over bare and doped TiO2 materials (concentrations of Ph2S oxidation products below the detection limit) was observed. Nevertheless, in the presence of hydrogen peroxide, the reaction can occur at P25 (anatase and rutile mixture) both on the catalytic and photocatalytic way, with significantly higher conversion of Ph2S under irradiation compared to the dark process. In our previous paper, we studied the influence of TiO2 modification (doping with vanadium, zinc, or tin) on the Ph2S oxidation. We suggested that the crystal phase composition has a higher influence on both catalytic and photocatalytic activity than the surface metal modifications.26 The goal of this work is to understand the difference between anatase and rutile in catalytic and photocatalytic Ph2S oxidation, describe the role of particular reactive oxygen species in these reactions, and learn how to control the reaction selectivity to different products.
Experimental
Materials
Titanium dioxide materials containing anatase phase [Hombikat N100 (N100), Sachtleben Chemie; Tronox AK-1 (TRX_A)], and rutile [Tronox TR (TRX_R); (CR-EL), Ishihara Sangyo] were studied.Crystal structure, morphology and phase composition
The phase compositions of materials were studied by the powder X-ray diffraction (XRD) using a Rigaku MiniFlex 600 X-ray diffractometer (Cu Kα radiation, 0.3 mm Ni filter) in 2θ range from 10 to 90°, at speed 3° min−1 and 0.05° step. The crystal size was calculated based on the Scherrer equation. To calculate the crystal size the most intense peaks were used, i.e., (101) and (110) for anatase and rutile, respectively.Materials morphology was examined by scanning electron microscopy (SEM) Tescan VEGA 3 with an LaB6 emitter. The measurements were performed on a carbon sheet.
The specific surface area (SSA) of studied materials was determined from nitrogen adsorption–desorption isotherms at 77 K using a Quantachrome Autosorb iQ-MP-AG-AG instrument. The Brunauer–Emmett–Teller model was applied.
Studies on reaction conversion and product selectivity
Catalytic and photocatalytic oxidation of diphenyl sulphide (Ph2S, 98%, Sigma Aldrich) to diphenyl sulfoxide (Ph2SO, 96%, Sigma Aldrich) and sulfone (Ph2SO2, 97%, Sigma Aldrich), in the presence of hydrogen peroxide (30%, Sigma Aldrich) was investigated. The reaction mixture consisted of 0.4 mmol dm−3 of diphenyl sulphide, 10 cm3 of acetonitrile (99.9%, Sigma Aldrich) and 0.1 mmol dm−3 of bromobenzene (≥99.5%, Sigma Aldrich) used as the internal standard. In a typical experiment, 5 mg of the tested material powder was sonicated for 2 min in 10 cm3 of the reaction mixture, then 2 mmol dm−3 of 30% hydrogen peroxide was added (suspension concentration of 0.5 g dm−3). The prepared suspension was placed in a round quartz cuvette (5 cm dia., 1 cm optical path) and irradiated for 3 hours with a xenon lamp (XBO-150, Instytut Fotonowy) equipped with 10 cm 0.1 mol dm−3 CuSO4 aqueous solution filter and a 320 nm cut-off filter. To determine the apparent quantum efficiency the photon flux in the range of 300 to 400 nm was measured with the StellarNet spectroradiometer. In the case of the catalytic studies, the reaction tests were performed in the dark in order to avoid a light-assisted conversion of Ph2S. The progress of the reactions was monitored by HPLC analysis of the reaction mixture using a mixture of acetonitrile/water in the volume ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 as the eluent. The samples of the reaction mixtures were filtered using a 0.22 μm nylon membrane filter and analysed by the PerkinElmer Flexar chromatograph equipped with the COL-Analytical C18 column (150 mm × 4.6 mm i.d., 5 μm pore size). The column was maintained at 25 °C during the analysis and the UV detector was set at 254 nm.
:30 as the eluent. The samples of the reaction mixtures were filtered using a 0.22 μm nylon membrane filter and analysed by the PerkinElmer Flexar chromatograph equipped with the COL-Analytical C18 column (150 mm × 4.6 mm i.d., 5 μm pore size). The column was maintained at 25 °C during the analysis and the UV detector was set at 254 nm.
Electrochemical measurements
The electrochemical measurements were carried out in a three-electrode system with Ag/Ag+ electrode [AgNO3 (10 mmol dm−3) in 0.1 mol dm−3 Bu4NClO4 in acetonitrile], platinum wire, and carbon electrode as the reference, counter, and working electrodes, respectively. As the electrolyte 0.1 mol dm−3 LiClO4 solution in acetonitrile was used. The potential of the reference electrode relative to the standard hydrogen electrode (SHE) was set at +0.62 V based on the cyclic voltammetry measurement for ferrocene (scan speed 10 mV s−1) and the literature value for Fc+/Fc couple. The electrode potential was controlled by an electrochemical analyser (Bio-Logic, SP-150). The cyclic voltammetry (CV) was collected at room temperature, with a scan rate of 60 mV s−1, from −0.1 V to 2.0 V vs. Ag/Ag+.Hydroxyl radicals generation
The hydroxyl radicals generation by studied materials was examined in the reaction of terephthalic acid (TA) hydroxylation. TiO2 (0.5 g dm−3) suspended in 16 cm3 of the TA solution (Aldrich, 98%; 3 × 10−3 mol dm−3 dissolved in 0.01 mol dm−3 NaOH, pH = 7.6) was irradiated with a xenon lamp (XBO-150, Instytut Fotonowy). To avoid excitation of TA a 320 nm cut-off filter was used as well as a near infra-red and IR filter (10 cm optical path, 0.1 mol dm−3 solution of CuSO4). Samples of 1.5 cm3 were collected during irradiation, then centrifuged to separate the photocatalyst powder. In the reaction of non-fluorescent TA with hydroxyl radicals, hydroxyterephthalic acid (TAOH) is formed. TAOH was monitored by emission spectroscopy. TAOH shows a broad emission band at λmax = 425 nm (when excited at λexc = 315 nm). Moreover, TAOH generation in the presence of hydrogen peroxide (2 mmol dm−3) was investigated.Results and discussion
Morphology and phase composition
Materials in the form of white powders were used for the catalytic tests. Scanning electron microscopy revealed that anatase and rutile materials form aggregates in size ca. 1 μm and below 0.5 μm, respectively (Fig. 1). The crystal structure of the materials was confirmed using X-ray diffraction (XRD) (Fig. S1†). The crystal size of anatase and rutile materials is ca. 20 and 50 nm, respectively. In XRD of all samples only one polymorph is present, only CR-EL material contains a small admixture of anatase, up to 1%.27 Porosimetry measurements showed that the anatase samples have a much higher specific surface area than the rutile materials (Table 1). | ||
| Fig. 1 SEM images of studied materials. | ||
| Material | Phase | Specific surface area/m2 g−1 | Crystal size/nm ± 0.1 |
|---|---|---|---|
| a (Photo)catalytic conversion of diphenyl sulphide in the presence of anatase and rutile. | |||
| N100 | Anatase | 97 ± 2 | 18.1 |
| TRX_A | Anatase | 90 ± 2 | 17.3 |
| TRX_R | Rutile | 5.5 ± 0.1 | 48.2 |
| CR-EL | Rutile | 8 ± 0.1 | 46.9 |
Ph2SO2 was the main product of the photocatalytic Ph2S oxidation at anatase materials. In the case of N100 material 100% conversion of Ph2S was achieved after 45 minutes of irradiation (Fig. 2a). In the case of the reaction conducted at TRX_A material, total Ph2S oxidation was reached after 1 hour of irradiation (Fig. 2b). A slightly better selectivity to Ph2SO2 was observed in the case of N100 (100% after 90 minutes of irradiation) than for TRX_A material. Diphenyl sulphide oxidation at anatase materials in the dark was tested as well. The reaction progress was observed for both anatase samples. Additionally, the reaction products were not observed in the absence of TiO2 in the reaction mixture, even when the mixture was heated to 40 °C. Nevertheless, after 3 hours of irradiation in the same reaction mixture (without TiO2, with H2O2) a slight conversion (ca. 7%) of Ph2S to Ph2SO as the final product took place (Fig. S2a†). We suggest that the residual oxidation of diphenyl sulphide is possible due to H2O2 photolysis, although 320 nm cut-off filter was used. The absorbance of the initial reaction mixture is negligible above 320 nm (Fig. S2b†), however, direct photolysis of H2O2 initiated at any wavelength lower than 380 nm was reported by Cataldo.28
 | ||
| Fig. 2 Concentration of reactants during Ph2S oxidation: at anatase materials (a) N100, (b) TRX_A upon irradiation λ > 320 nm (solid lines) and in the dark (dotted lines); at rutile material (c) TRX_R, (d) CR-EL upon irradiation. | ||
These results clearly show that the oxidation of Ph2S in the presence of anatase materials is possible both as catalytic and photocatalytic processes. However, a significantly higher conversion of Ph2S for photocatalytic conditions compared to the dark processes was observed (Fig. S3a and b†). A similar positive effect of light was observed when Ph2SO as a substrate was used (Fig. S10†). Both photocatalytic and catalytic oxidation of Ph2S at anatase materials leads mainly to Ph2SO2 production with the selectivity of almost 100% after 3 h of tests (Fig. 2a, b and S3c, d†). Nevertheless, for both anatase materials, during the first hour more sulfoxide is formed upon irradiation than under purely catalytic conditions.
Results of photocatalytic oxidation of Ph2S at rutile materials are depicted in Fig. 2c and d. Photocatalytic conversion of Ph2S at rutile materials was significantly faster compared to both photocatalytic and catalytic conditions applied to anatase materials. The total conversion of Ph2S was observed after 15 and 30 min of irradiation for TRX_R and CR-EL, respectively (Fig. 2c and d). Based on these data and the measured photon flux the apparent quantum efficiency of Ph2S to Ph2SO conversion within the first 15 min of irradiation was ≥2%. Oxidation of Ph2S at rutile materials led to Ph2SO as the main product. High selectivity to Ph2SO at the beginning of the photocatalytic reaction was noticed (ca. 100%). During the reaction course the selectivity to Ph2SO was decreasing, while the production of Ph2SO2 became the dominant process. However, upon further irradiation, the production of Ph2SO increased again, hence diphenyl sulfoxide was the main product of the long term photocatalytic oxidation at rutile (Fig. 2c and d). Contrary to anatase, rutile induced no conversion in the absence of light, neither for Ph2S nor Ph2SO used as a substrate (Fig. S4 and S11a†).
Interestingly, the total concentration of analyzed reactants (Ph2S, Ph2SO, Ph2SO2) decreased by ca. 5% during the experiments with rutile material under illumination. Moreover, in the case of photocatalytic reaction over CR-EL material, at chromatograms the appearance of low amounts of a new compound, not detected for any other material described above was observed. To elucidate the role of H2O2 in this reaction a new portion of H2O2 was added after 90 min of irradiation (Fig. S5†). No increase of the selectivity to Ph2SO2 was observed. Furthermore, in our previous work we showed that after almost complete conversion of Ph2S into both Ph2SO and Ph2SO2, hydrogen peroxide was not totally consumed,20 pointing at a sufficient amount of H2O2 used in the reaction. As the Ph2SO2 concentration decreases after prolonged irradiation (Fig. 2c), either its reduction to Ph2SO or transformation to other products could be considered. A photocatalytic reduction of Ph2SO2 directly by electrons from the conduction band does not seem plausible, since 3 hours of irradiation of Ph2SO2 in the presence of H2O2 and CR-EL did not result in any Ph2SO formation (data not shown). Moreover, the initial concentration of Ph2SO2 remained unchanged, what excludes a possibility of Ph2SO2 transformation to any other product. Taking these observations into account it was postulated that a decrease in Ph2SO2 and increase in Ph2SO concentrations, observed after 45 min of the test, are possibly the result of Ph2SO2 disproportionation resulting a new unidentified compound. A detailed analysis of this product is still needed, however, we suggest that it could be one of many products identified by Vosooghian et al. in their meticulous analysis.29
It is worth comparing the presented results with our earlier work where we tested the activity of doped titanium dioxide. Here we conclude, that rutile is not acting as a catalyst (i.e., it is not active in the dark), however, we previously reported that materials with higher amounts of this polymorph show a higher activity in the oxidation of Ph2S. Therefore, it can be concluded that the presence of rutile has an indirect effect on the catalytic process – the material itself is not active, but the coexistence of rutile and anatase enhances the catalytic activity of the materials. Such anatase/rutile composites appeared also very active in photocatalytic transformations, both of Ph2S and Ph2SO, due to a synergistic effect of these phases reported in numerous publications on P25 and similar materials.30–34 N100 and TRX_A appear more active catalytically, therefore, in a similar time window the second oxidation product, Ph2SO2, becomes the main product both in the dark and upon irradiation. The pure rutile phase, despite of its catalytic inactivity, appears the most active photomaterial. In this case a mixture of Ph2SO and Ph2SO2 is obtained.
Reaction mechanism
Research studies on sulphides oxidation describe both catalytic and photocatalytic processes. The mechanism of catalytic oxidation involves H2O2 activation on the catalyst surface. Al-Maksound et al. studied catalytic oxidation of various sulphides using H2O2 and self-synthesized TiO2 (75–80% of anatase).22 The authors showed high selectivity to sulfones. Rodríguez-Padrón et al. studied microwave-assisted catalytic oxidation of sulphides using H2O2 and commercial anatase, as well as self-synthesized protein-templated anatase TiO2 in ethanol solution.35 The authors showed that Ph2S oxidation over reference anatase leads to sulfone production, while at protein-templated TiO2 mainly sulfoxide was produced. Radko et al. studied catalytic oxidation of sulphides in the presence of titanosilicates and found that the dimethyl sulphide and diphenyl sulphide conversion increased with the increasing titanium content in the zeolite structure.20 In our current study catalytic Ph2S oxidation with H2O2 at anatase TiO2 to Ph2SO2 was observed. Contrary to that, no catalytic oxidation of Ph2S at rutile materials was observe. We suggest that the opposite results can be attributed to the various activation paths of hydrogen peroxide (and maybe Ph2S) on the anatase/rutile surfaces. However, the differences can also originate from a low specific surface area of rutile materials, and therefore a low number of active sites. In order to elucidate the effect of specific surface area, the catalytic test with self-synthesized rutile material (SSA of ca. 25 m2 g−1) was performed. Although the specific surface area was 3–5 times higher in this case, no catalytic oxidation of Ph2S was observed as well.Published articles describing photocatalytic oxidation of sulphides postulate that the photocatalytic oxidation is initiated by the formation of a surface bound radical cation generated as a result of oxidation with the photogenerated hole.29 Also, superoxide radical anion (O2˙−) plays a key role in the reaction.36–38 Zhang et al. suggested that O2˙− reacts with generated organosulfur radical.39 Lang et al. studied Ph2S visible light induced oxidation in the presence of a dye (alizarin red S) and TEMPO as the redox mediator to increase the dye stability.40–44 The authors reported Ph2S conversion to Ph2SO, pointing at O2˙− importance in the reaction mechanism. Li et al. confirmed significance of superoxide radical anions and sulphide cation radicals by experiments involving radical scavengers and 18O2.45 Vosooghian et al. showed that oxidation of diphenyl sulphide was effective only in the oxygen rich atmosphere (detectable amounts of products were neither observed in the presence of atmospheric oxygen nor under argon flux).29 In the presence of photosensitizers able to produce singlet oxygen, it is postulated that 1O2 may participate in the Ph2S oxidation.46–49 The mechanism described in the literature (illustrated in Fig. 3, blue color) cannot fully explain our results, which show that no oxidation products are formed in the absence of hydrogen peroxide. In other words, the mechanism involving Ph2S˙+ formation and its reaction with either O2˙− or 1O2 should lead to the products (Ph2SO, Ph2SO2), which have not been detected.
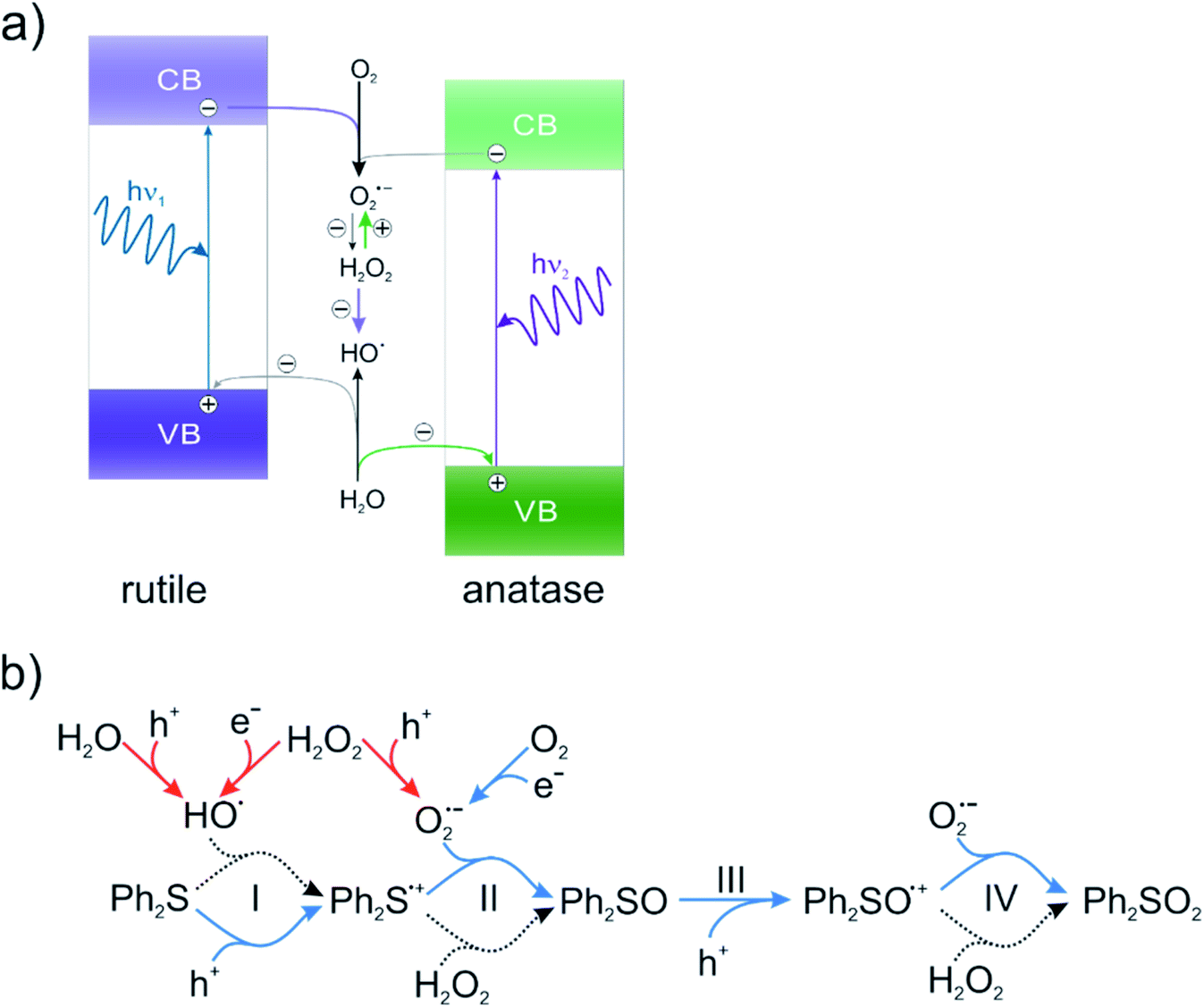 | ||
| Fig. 3 (a) Main reactive oxygen species generation pathway in the presence of rutile and anatase photocatalysts, based on ref. 31. Violet and green arrows depict main processes characteristic for rutile and anatase, respectively. (b) Plausible pathway of Ph2S oxidation at TiO2. Blue color shows the paths described in the literature so far. Red color shows pathways of H2O2 oxidation and reduction. Dotted black lines show discussed processes, which complete the mechanism of Ph2S oxidation at TiO2 in the presence of H2O2. | ||
According to our knowledge the role of H2O2 in the mechanism and its influence on the efficiency of photocatalytic Ph2S oxidation have not been reported in the literature. The presence of H2O2 dramatically influences the generation of HO˙ and O2˙−, both at rutile and anatase material (Fig. 3, red color). Photocatalytic production of hydroxyl radicals at rutile is inefficient, however, the generation of this reactive species is strongly enhanced in the presence of H2O2, as the result of its reduction. On the other hand, a similar effect of H2O2 on anatase photoactivity is not observed. Addition of H2O2 leads to increasing O2˙− production at anatase, as the result of H2O2 oxidation with holes.50 Generally, rutile has better reduction properties compared to anatase, meanwhile, anatase is a better oxidant.31 Using various TiO2 phase compositions in the combination with H2O2 can be an effective way to control the selectivity of the reaction in the system, in which a fine product formation is highly dependent on the generated ROS type. The HO˙ generation was monitored in the reaction of terephthalic acid oxidation. Hydroxyl radicals are generated as the result of a hole reaction with surface hydroxyl groups or adsorbed water molecules. In the reaction of TA with hydroxyl radicals, highly-fluorescent hydroxyterephthalic acid (TAOH) is formed. TAOH generation in the presence of hydrogen peroxide (2 mmol dm−3) was also investigated. The TAOH formation is depicted in Fig. S6.† In the absence of hydrogen peroxide anatase shows a more efficient HO˙ generation compared to the rutile (TAOH concentration amounted 57 μmol dm−3 and 29 μmol dm−3 respectively). However, after addition of H2O2 to the system containing anatase, TAOH generation decreased (10 μmol dm−3), while it significantly increased for rutile material (452 μmol dm−3). Undoubtedly, combination of hydrogen peroxide and rutile significantly enhances HO˙ generation.
The presence of hydrogen peroxide can lead to the increased concentration of photogenerated O2˙− and/or HO˙ radicals, whereas only the influence of O2˙− on the oxidation of Ph2S is known. In order to determine the possible role of hydroxyl radicals in the reaction, the Fenton process was induced in the presence of Ph2S. In the Fenton reaction (production of HO˙ in the reaction of H2O2 with iron(II) cations) the conversion of Ph2S and efficient production of Ph2SO were observed (Fig. 4a). Slight oxidation of Ph2SO (Ph2SO2 formation) in the Fenton process was also noticed. However, an inefficient Ph2SO2 generation might originate from the transformation of other species that contaminate the original Ph2SO sample (Fig. 4b). This shows that the hydroxyl radical and/or hydrogen peroxide (the reactant in this reaction) is able to oxidize Ph2S to Ph2SO. Recently, we described that the addition of tert-butyl alcohol (hydroxyl radical scavenger) significantly diminishes the Ph2S conversion.26 Furthermore, isotopic oxygen (H218O) experiments, reported by Li et al. revealed that the oxygen atoms of sulfoxide (obtained photocatalytically from phenyl sulphide in water containing mixture, in the absence of H2O2) originated mainly from water.51 We concluded that hydroxyl radicals play a significant role in the first step of diphenyl sulphide oxidation, yielding Ph2SO. Similarly, in our actual system HO˙ is involved in the oxidation of Ph2S to Ph2SO, but the further oxidation to Ph2SO2 is not possible.
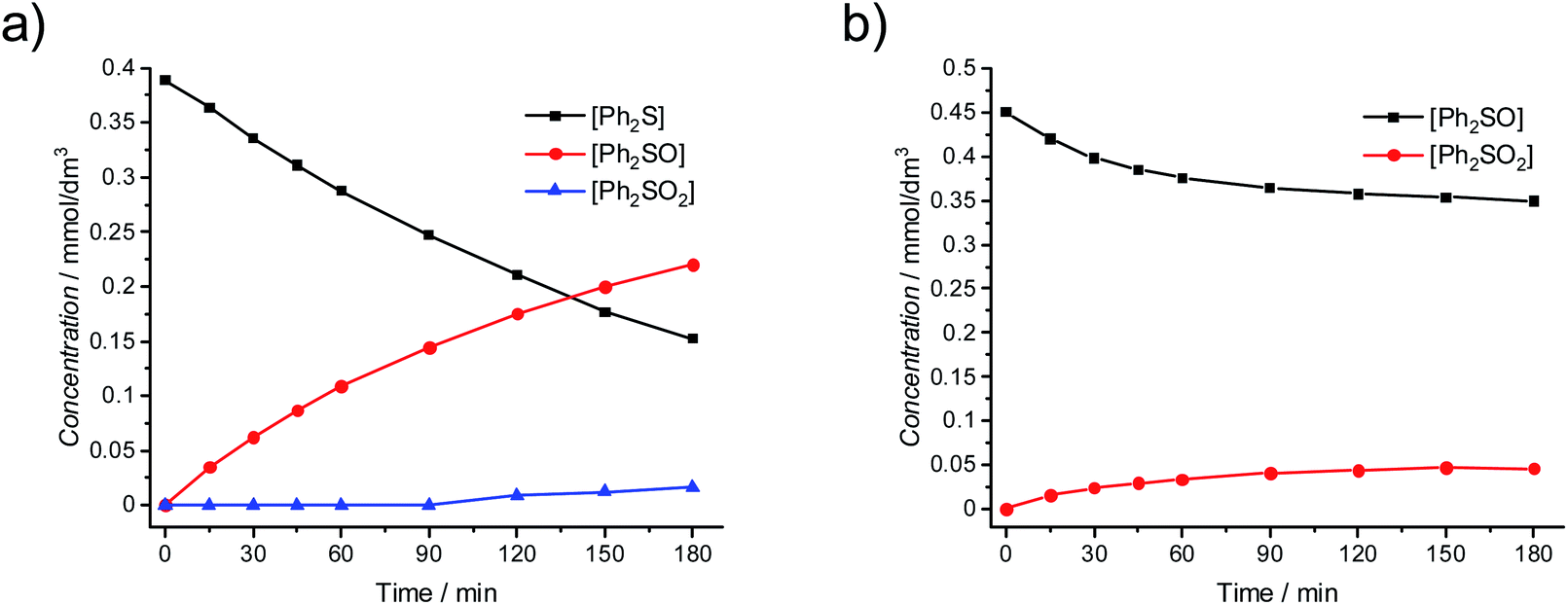 | ||
| Fig. 4 Oxidation of (a) Ph2S by the Fenton reagent (0.4 mmol dm−3 Ph2S, 2 mmol dm−3 H2O2, 0.2 mmol dm−3 Fe2+); (b) Ph2SO by the Fenton reagent (0.4 mmol dm−3 Ph2SO). | ||
In order to explain why the Fenton reaction leads only to Ph2SO generation, cyclic voltammetry measurements were performed (Fig. 5). In the case of Ph2S, two oxidation peaks 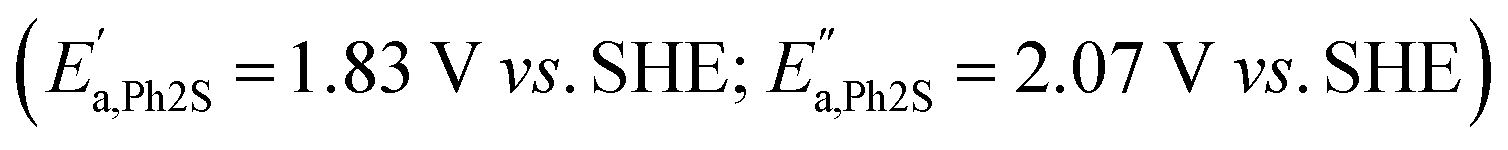 and one reduction peak
and one reduction peak  were observed in the cyclic voltammogram. In the case of Ph2SO only one oxidation peak was detected
were observed in the cyclic voltammogram. In the case of Ph2SO only one oxidation peak was detected  . Finally, the CV of Ph2SO2 has shown neither oxidation nor reduction peaks in the explored potential range. In the case of Ph2S cyclic voltammogram, the first oxidation at 1.83 V vs. SHE can presumably be attributed to Ph2S˙+ formation. Furthermore, based on the CV measurements the formation of Ph2SO as an irreversible process was found. The second oxidation process at 2.07 V vs. SHE is a partially reversible reaction
. Finally, the CV of Ph2SO2 has shown neither oxidation nor reduction peaks in the explored potential range. In the case of Ph2S cyclic voltammogram, the first oxidation at 1.83 V vs. SHE can presumably be attributed to Ph2S˙+ formation. Furthermore, based on the CV measurements the formation of Ph2SO as an irreversible process was found. The second oxidation process at 2.07 V vs. SHE is a partially reversible reaction 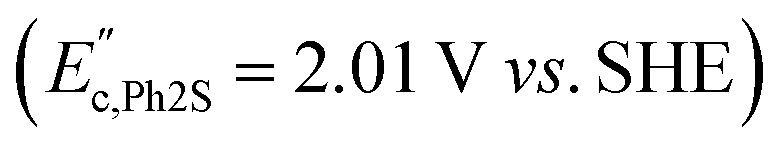 with oxidation and reduction halfwaves separated by 0.06 V, pointing at one electron reversible process. Based on these considerations we conclude that oxidation of Ph2S to Ph2SO can be achieved by HO˙ (HO˙/H2O = 2.27 V vs. SHE, pH = 7 (ref. 52)). CV of Ph2SO reveals that the potential of sulfoxide oxidation is too high to enable its oxidation by hydroxyl radicals. Nevertheless, Ph2SO can be still directly oxidized to Ph2SO˙+ by TiO2 VB holes (step III, Fig. 3).53
with oxidation and reduction halfwaves separated by 0.06 V, pointing at one electron reversible process. Based on these considerations we conclude that oxidation of Ph2S to Ph2SO can be achieved by HO˙ (HO˙/H2O = 2.27 V vs. SHE, pH = 7 (ref. 52)). CV of Ph2SO reveals that the potential of sulfoxide oxidation is too high to enable its oxidation by hydroxyl radicals. Nevertheless, Ph2SO can be still directly oxidized to Ph2SO˙+ by TiO2 VB holes (step III, Fig. 3).53
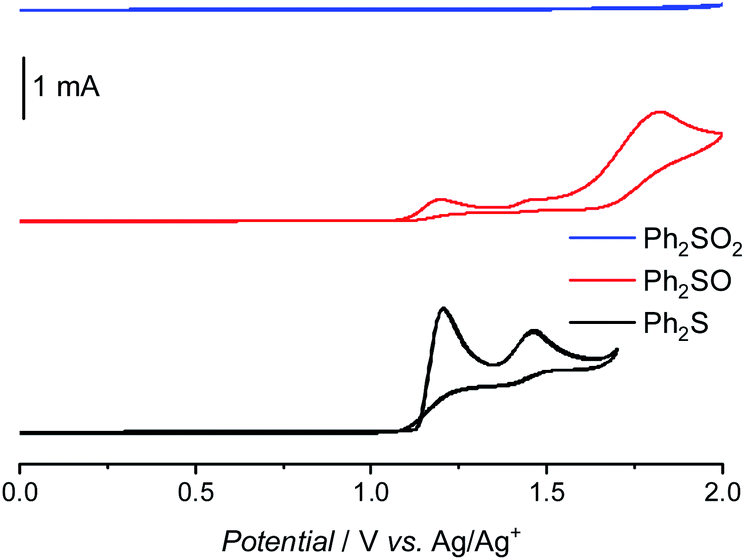 | ||
| Fig. 5 Cyclic voltammograms of diphenyl sulphide, sulfoxide, and sulfone in acetonitrile. | ||
To summarize, the electrochemical and Fenton experiments showed that Ph2S can be easily oxidized by HO˙ to Ph2S˙+. However, holes are required for further oxidation of Ph2SO to the Ph2SO˙+ radical, which explains why the Fenton reaction led only to Ph2SO. The Fenton process does not generate O2˙−, however, Ph2SO as a product of the Fenton reaction was effortlessly detected. It is noteworthy that in the Fenton reaction the concentration of hydrogen peroxide was the same that in the photocatalytic system. The results suggest that H2O2 is involved in the Ph2S˙+ to Ph2SO conversion (Fig. 3). Our hypothesized mechanism explains the results of photocatalytic oxidation of Ph2S at anatase and rutile materials. In the case of rutile, a very fast conversion of Ph2S was observed, due to a highly efficient H2O2 reduction, and thus effective HO˙ formation (Fig. S6†). These radicals oxidize Ph2S within first few minutes of the reaction. Our study on HO˙ generation shows that in the presence of rutile and H2O2 ca. 400 μmol dm−3 of TAOH is generated after 15 min of irradiation (in aqueous solution). This concentration of hydroxyl radicals trapped as TAOH is equal to the initial concentration of Ph2S in the tested reaction. These results clearly show that a fast conversion of Ph2S at rutile CR-EL is a purely photocatalytic process, being a result of an efficient H2O2 reduction. At anatase, which preferably oxidizes H2O2, HO˙ generation is much less effective, therefore, the oxidation rate is lower.
The presented results indicate that hydrogen peroxide and superoxide radical anion play a similar role, oxidizing the radical cations, Ph2S˙+ and Ph2SO˙+, to sulfoxide and sulfone, respectively. However, to unequivocally confirm these conclusions, the influence of oxygen on the reaction was tested. The studied reaction mixtures were purged with argon or oxygen before (15 min) and during the experiments. For the dark reaction at TRX_A, the observed conversion and selectivity were independent on the presence of oxygen (Fig. S7†). This result suggests that catalytic oxidation of Ph2S involves only the activation of hydrogen peroxide activation, without any contribution of O2 (alternatively, the contribution of oxygen is low, with a negligible influence on the products formation). However, when TRX_A was used as a photocatalyst (under irradiation), the influence of O2 on the reaction progress was significant. A decrease of the Ph2S conversion from ca. 60% under ambient conditions to ca. 35% under argon atmosphere and increased conversion to ca. 75% under the oxygen rich atmosphere was observed (Fig. S8a†). Under the oxygen free conditions, the Ph2S conversion decreased, but photocatalytic oxidation of Ph2S to Ph2SO was still observed. Moreover, changes in the selectivity to Ph2SO2 at TRX_A were observed. The selectivity to Ph2SO2 increased in the following order: under argon, ambient, and oxygen-rich conditions (Fig. S8b†). In the case of the photocatalytic reaction at CR-EL, no significant changes in the Ph2S conversion under various conditions were observed (Fig. S9a†). Nevertheless, remarkable changes in the selectivity to Ph2SO were observed. Under oxygen-rich conditions a decrease in the production of Ph2SO, and an increase in the Ph2SO2 production after first 15 min of the reaction, were observed (Fig. S9b†). Moreover, the point of the second increase of the Ph2SO production appeared earlier (after 30 min of irradiation) for oxygen-rich conditions.
The first stable oxidation product, Ph2SO, can be further oxidized by holes from the valence band of TiO2 to Ph2SO˙+. This intermediate is further transformed to Ph2SO2 in the reaction with superoxide radical (O2˙−) formed as the result of either oxygen reduction or H2O2 oxidation. To confirm this path a possibility of Ph2SO oxidation, both in the dark and upon irradiation in the presence of hydrogen peroxide and TiO2, was studied. In the case of anatase material, an efficient conversion of Ph2SO to Ph2SO2 under both catalytic and photocatalytic conditions was observed, with a much better efficiency upon irradiation (Fig. S10†). In the case of rutile, no conversion was observed in the dark, however, irradiation induced this reaction (Fig. S11†). Clearly, the higher rate of the photocatalytic reaction in the case of anatase material is the result of a more efficient O2˙− production at anatase than on the rutile in the presence of H2O2. Furthermore, the possibility of Ph2SO oxidation upon irradiation in the presence of TiO2, but in the absence of H2O2, was tested (Fig. S12†). Low conversion of Ph2SO under such conditions was observed – ca. 13% and 9% after 3 hours of irradiation in the presence of anatase and rutile, respectively. In a similar test, performed under anaerobic conditions (Ar-saturated solution and addition of Fe3+ as an electron acceptor), the conversion of Ph2SO to Ph2SO2 was not observed. Abovementioned results clearly show that O2˙− radicals can easily react with Ph2SO˙+, yielding Ph2SO2. We suggest that the formation of Ph2SO2 is possible in the presence of O2˙− and Ph2SO+ (hole oxidized Ph2SO), however, such reaction has a low effectivity. In order to enhance the production of Ph2SO2, the presence of hydrogen peroxide is necessary. The full mechanism of the photocatalytic Ph2S oxidation in the presence of hydrogen peroxide is shown in Fig. 3.
Conclusions
In summary, we demonstrated that Ph2S oxidation in the presence of hydrogen peroxide at anatase-TiO2 can take place both as a catalytic and photocatalytic reaction. In contrast, at rutile materials only photocatalytic oxidation is possible, and no reaction progress was observed in the dark. The reaction at anatase leads mainly to Ph2SO2 as the product, whereas, the same reaction in the presence of rutile leads mainly to Ph2SO formation. In the case of rutile the total conversion of Ph2S was observed already after 15 min (compared to at least 1 h required for a catalytic conversion) with nearly 100% selectivity towards Ph2SO. It is the first reported photocatalytic conversion of Ph2S using H2O2 in the presence of pure rutile. Moreover, the efficiency of this reaction under the tested conditions is also much higher compared to the systems described in literature so far (apparent quantum yield of ca. 2%, concentration of products in the absence of H2O2 below the detection limit). The conversion level and the reaction selectivity can be controlled by the choice between rutile and anatase polymorphs, as well as by irradiation time (in the case of rutile).Catalytic oxidation of Ph2S involves only the activation of hydrogen peroxide at the catalyst surface, without any contribution of dioxygen. Whereas, the presence of O2 in the photocatalytic reaction increases the Ph2S conversion due to generation of O2˙−. Studies on the mechanistic details revealed the dual role of H2O2 (Fig. 3). At the same time, it plays the role of a substrate in the reaction catalysed only by anatase, but it also plays a key role in alternative oxidation pathways available through photocatalysis. Hydrogen peroxide can be either oxidized or reduced photocatalytically, resulting in the generation of O2˙− and HO˙ radicals, respectively. Hydroxyl and superoxide radicals play an important role in various reaction steps. Hydroxyl radicals can oxidize Ph2S to Ph2S˙+, however, Ph2SO oxidation to Ph2SO˙+ requires a stronger oxidant, i.e. holes. The cation radical intermediates, Ph2S˙+ and Ph2SO˙+, react with superoxide anions or directly with H2O2 yielding stable sulfoxide (Ph2SO) and sulfone (Ph2SO2).
The presented studies prove the applicability of photocatalysis in an efficient and selective synthesis of sulfoxide and sulfone through oxidation of organic sulphides.
Author contributions
Conceptualization P. M., M. K.; methodology P. M., M. K.; formal analysis P. M., M. K., A. Ś.; investigation P. M., M. K., A. Ś.; resources, L. C., W. M.; data curation P. M., M. K.; writing P. M., M. K., A. Ś., L. C. and W. M.; visualization P. M., M. K.; supervision M. K., W. M.; project administration W. M.; funding acquisition W. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work on catalytic and photocatalytic properties of various TiO2 polymorphs was supported by the Foundation for Polish Science (FNP) within the TEAM project (POIR.04.04.00-00-3D74/16). Selectivity and mechanistic studies were supported by the National Science Centre (NCN, Poland) within the Solar-Driven Chemistry project (2019/01/Y/ST5/00027).References
- M. Kobielusz, P. Mikrut and W. Macyk, in Advances in Inorganic Chemistry, Elsevier, 2018, vol. 72, pp. 93–144 Search PubMed.
- T. Jedsukontorn, V. Meeyoo, N. Saito and M. Hunsom, Chem. Eng. J., 2015, 281, 252–264 CrossRef CAS.
- T. D. Bui, A. Kimura, S. Ikeda and M. Matsumura, J. Am. Chem. Soc., 2010, 132, 8453–8458 CrossRef CAS PubMed.
- F. F. Karam, M. I. Kadhim and A. F. Alkaim, Int. J. Chem. Sci., 2015, 13, 650–660 CAS.
- V. Augugliaro, G. Camera-Roda, V. Loddo, G. Palmisano, L. Palmisano, F. Parrino and M. A. Puma, Appl. Catal. B Environ., 2012, 111, 555–561 CrossRef.
- K. Schoumacker, C. Geantet, M. Lacroix, E. Puzenat, C. Guillard and J.-M. Herrmann, J. Photochem. Photobiol. A, 2002, 152, 147–153 CrossRef CAS.
- M. Sovova and P. Sova, Ceska Slov. Farm., 2003, 52, 82–87 CAS.
- B. Kotelanski, R. J. Grozmann and J. N. Cohn, Clin. Pharmacol. Ther., 1973, 14, 427–433 CrossRef CAS PubMed.
- S. Padmanabhan, R. C. Lavin and G. J. Durant, Tetrahedron: Asymmetry, 2000, 11, 3455–3457 CrossRef CAS.
- K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 35, 8315–8327 CrossRef.
- A. Mahdian, M. H. Ardakani, E. Heydari-Bafrooei and S. Saeednia, Appl. Organomet. Chem., 2021, 35, e6170 CrossRef CAS.
- S. Mirfakhraei, M. Hekmati, F. H. Eshbala and H. Veisi, New J. Chem., 2018, 42, 1757–1761 RSC.
- W.-Y. Zhou, M. Chen, P.-Z. Zhang, A.-Q. Jia and Q.-F. Zhang, Russ. J. Org. Chem., 2021, 57, 816–824 CrossRef CAS.
- J. Přech, R. E. Morris and J. Čejka, Catal. Sci. Technol., 2016, 6, 2775–2786 RSC.
- M. Radko, A. Kowalczyk, E. Bidzińska, S. Witkowski, S. Górecka, D. Wierzbicki, K. Pamin and L. Chmielarz, J. Therm. Anal. Calorim., 2018, 132, 1471–1480 CrossRef CAS.
- A. M. Cojocariu, P. H. Mutin, E. Dumitriu, A. Aboulaich, A. Vioux, F. Fajula and V. Hulea, Catal. Today, 2010, 157, 270–274 CrossRef CAS.
- I. Martausová, D. Spustová, D. Cvejn, A. Martaus, Z. Lacný and J. Přech, Catal. Today, 2019, 324, 144–153 CrossRef.
- Y. Kon, T. Yokoi, M. Yoshioka, Y. Uesaka, H. Kujira, K. Sato and T. Tatsumi, Tetrahedron Lett., 2013, 54, 4918–4921 CrossRef CAS.
- Z. Kang, G. Fang, Q. Ke, J. Hu and T. Tang, ChemCatChem, 2013, 5, 2191–2194 CrossRef CAS.
- M. Radko, M. Rutkowska, A. Kowalczyk, P. Mikrut, A. Święs, U. Díaz, A. E. Palomares, W. Macyk and L. Chmielarz, Microporous Mesoporous Mater., 2020, 110219 CrossRef CAS.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edn, Wiley, 2001 Search PubMed.
- W. Al-Maksoud, S. Daniele and A. B. Sorokin, Green Chem., 2008, 10, 447–451 RSC.
- K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469–2476 CrossRef CAS.
- V. Hulea, F. Fajula and J. Bousquet, J. Catal., 2001, 198, 179–186 CrossRef CAS.
- A. V. Vorontsov and P. G. Smirniotis, in Environmentally Benign Photocatalysts: Applications of Titanium Oxide-based Materials, ed. M. Anpo and P. V. Kamat, Springer New York, New York, NY, 2010, pp. 579–621 Search PubMed.
- M. Radko, A. Kowalczyk, P. Mikrut, S. Witkowski, W. Mozgawa, W. Macyk and L. Chmielarz, RSC Adv., 2020, 10, 4023–4031 RSC.
- M. Kobielusz, A. Nitta, W. Macyk and B. Ohtani, J. Phys. Chem. Lett., 2021, 12, 3019–3025 CrossRef CAS PubMed.
- F. Cataldo, Front. Chem., 2014, 23, 99 Search PubMed.
- H. Vosooghian and M. H. Habibi, Int. J. Photoenergy, 2007, 2007, 7 CrossRef.
- K. Yaemsunthorn, M. Kobielusz and W. Macyk, ACS Appl. Nano Mater., 2021, 4, 633–643 CrossRef CAS.
- M. Buchalska, M. Kobielusz, A. Matuszek, M. Pacia, S. Wojtyła and W. Macyk, ACS Catal., 2015, 5, 7424–7431 CrossRef CAS.
- B. Ohtani, O. Prieto-Mahaney, D. Li and R. Abe, J. Photochem. Photobiol. Chem., 2010, 216, 179–182 CrossRef CAS.
- H. Cheng, J. Wang, Y. Zhao and X. Han, RSC Adv., 2014, 4, 47031–47038 RSC.
- K. Connelly, A. Wahab and H. Idriss, Mater. Renew. Sustain. Energy, 2012, 1, 3 CrossRef.
- D. Rodríguez-Padrón, A. R. Puente-Santiago, F. Luna-Lama, A. l. Caballero, M. J. Muñoz-Batista and R. Luque, ACS Sustain. Chem. Eng., 2019, 7, 5329–5337 CrossRef.
- W. Sheng, J.-L. Shi, H. Hao, X. Li and X. Lang, J. Colloid Interface Sci., 2020, 565, 614–622 CrossRef CAS PubMed.
- H. Hao, X. Li and X. Lang, Appl. Catal. B Environ., 2019, 259, 118038 CrossRef CAS.
- J.-L. Shi and X. Lang, Chem. Eng. J., 2020, 392, 123632 CrossRef CAS.
- P. Zhang, Y. Wang, H. Li and M. Antonietti, Green Chem., 2012, 14, 1904–1908 RSC.
- X. Lang, W. Hao, W. R. Leow, S. Li, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 5000–5005 RSC.
- X. Lang, J. Zhao and X. Chen, Angew. Chem., 2016, 128, 4775–4778 CrossRef.
- H. Hao, Z. Wang, J. L. Shi, X. Li and X. Lang, ChemCatChem, 2018, 10, 4545–4554 CrossRef CAS.
- X. Ma and X. Lang, Sustain. Energy Fuels, 2020, 4, 1754–1763 RSC.
- H. Hao, X. Li and X. Lang, Appl. Catal. B Environ., 2019, 259, 118038 CrossRef CAS.
- C. Li, N. Mizuno, K. Murata, K. Ishii, T. Suenobu, K. Yamaguchi and K. Suzuki, Green Chem., 2020, 22, 3896–3905 RSC.
- H. Wang, S. Jiang, S. Chen, D. Li, X. Zhang, W. Shao, X. Sun, J. Xie, Z. Zhao and Q. Zhang, Adv. Mater., 2016, 28, 6940–6945 CrossRef CAS PubMed.
- J. Li, Y. Chen, X. Yang, S. Gao and R. Cao, J. Catal., 2020, 381, 579–589 CrossRef CAS.
- Q. Li, X. Lan, G. An, L. Ricardez-Sandoval, Z. Wang and G. Bai, ACS Catal., 2020, 10, 6664–6675 CrossRef CAS.
- R. S. Davidson and J. E. Pratt, Tetrahedron lett, 1983, 24, 5903–5906 CrossRef CAS.
- T. Hirakawa, K. Yawata and Y. Nosaka, Appl. Catal. Gen., 2007, 325, 105–111 CrossRef CAS.
- B. Zhang, J. Li, B. Zhang, R. Chong, R. Li, B. Yuan, S.-M. Lu and C. Li, J. Catal., 2015, 332, 95–100 CrossRef CAS.
- M. Kaneko and I. Okura, Photocatalysis: science and technology, Springer, 2002 Search PubMed.
- C. Maheu, L. Cardenas, E. Puzenat, P. Afanasiev and C. Geantet, Phys. Chem. Chem. Phys., 2018, 20, 25629–25637 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08364c |
| This journal is © The Royal Society of Chemistry 2022 |