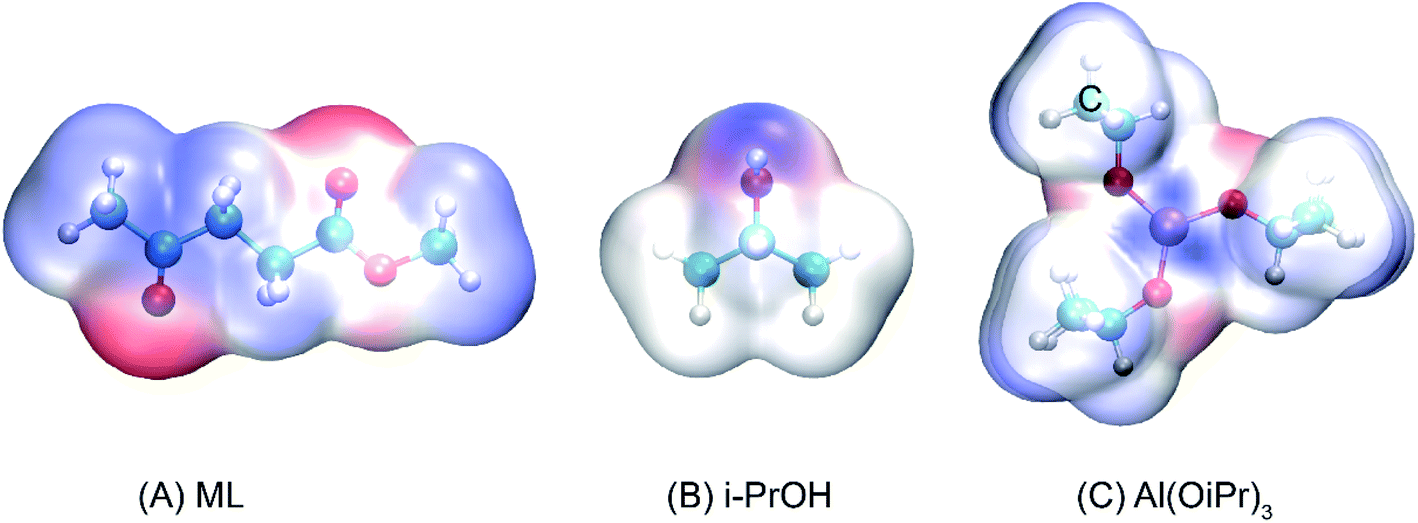DOI:
10.1039/D1RA08429A
(Paper)
RSC Adv., 2022,
12, 2788-2797
Probing the mechanism of the conversion of methyl levulinate into γ-valerolactone catalyzed by Al(OiPr)3 in an alcohol solvent: a DFT study†
Received
17th November 2021
, Accepted 6th January 2022
First published on 19th January 2022
Abstract
Biomass-derived γ-valerolactone (GVL) is a versatile chemical that can be used in various fields. As an efficient, cheap, and sustainable catalyst, Al(OiPr)3 has been successfully used in the conversion of methyl levulinate (ML) to GVL in the solvent isopropanol (IPA). However, the molecular mechanism of this conversion catalyzed by Al(OiPr)3 remains ambiguous. To investigate the mechanism of the conversion of ML to GVL catalyzed by Al(OiPr)3, the reaction pathways, including the transesterification, Meerwein–Ponndorf–Verley (MPV) hydrogenation, and ring-closure steps, were probed using density functional theory (DFT) calculations at the M062X-D3/def2-TZVP level. Among the elementary steps, it is found that ring-closure is the rate-determining step and that Al3+ can coordinate with the oxygen of 2-hydroxy-isopropyl levulinate (2HIPL) to catalyze the last ring-closure step. A four-centered transition state can be formed, and Al(OiPr)3 shows a strong catalytic effect in the two steps of the ester exchange reaction. The center of Al(OiPr)3 mainly coordinates with the carbonyl oxygen atom of the ester to catalyze the reaction. The present study provides some help in understanding the conversion mechanism of ML to GVL and designing more effective catalysts for use in biomass conversion chemistry.
1. Introduction
In light of environmental pollution and fossil resource depletion, lignocellulosic biomass has become a promising resource for the production of biofuels as a replacement for fossil fuels.1,2 γ-Valerolactone (GVL) is accessible from lignocellulosic biomass,3,4 and it has been identified as one of the most promising platforms for the sustainable production of fuels because of its outstanding physicochemical properties.5 As an ideal precursor for the production of high-value chemicals to alleviate dependence on fossil fuels, GVL can be used to produce valuable chemicals such as dimethyl adipate, valeric esters, and long-chain alkanes, among others.6–8 Furthermore, it could also be applied as an additive in food and drink because of its fruit flavour and low toxicity.9,10
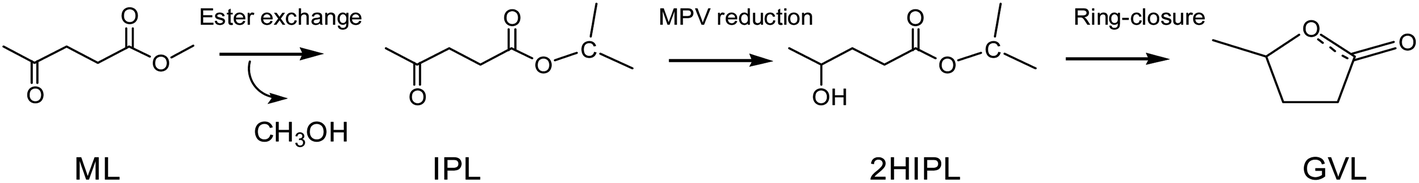
Generally, GVL is yielded from the catalytic hydrogenation–cyclization of levulinic acid (LA) or its esters in an alcohol solvent. The traditionally used hydrogen donor is H2, which requires high pressure to store and makes this process less safe.11 Alcohol solvents are common and cheap reagents that exist in liquid state at room temperature. As an energy-saving and cost-effective method, catalytic transfer hydrogenation (CTH) in alcohol solvents is regarded as an attractive approach compared to traditional hydrogenation using high-pressure and flammable H2.12–15 As shown in Scheme 1, the production of GVL in alcohols usually involves transesterification, Meerwein–Ponndorf–Verley (MPV) reduction, and a ring-closure reaction to finish the overall process.16,17 In particular, the use of secondary alcohols, such as isopropanol (i-PrOH) and 2-butanol, as a hydrogen donor has shown a better effect on MPV reduction.18,19 According to the previous reports, several heterogeneous catalysts containing noble metals (Ru, Ir, Rh, Pd, Re, etc.) have been developed and have shown excellent abilities to yield GVL.20–22 However, their high cost, difficult regeneration, and easy deactivation are highly challenging in practical applications.23,24 Zhang et al.25 reported that a 99.0% yield of GVL could be obtained from ethyl levulinate (EL) by using a ternary Cu/ZnO/Al2O3 catalyst in the CTH process with i-PrOH as the hydrogen donor. Moreover, He et al.26 utilized a series of Al–Zr mixed oxides to obtain an 83.2% yield at 220 °C in 4 h using i-PrOH as the hydrogen donor, and reported that the acid–base sites played a synergic role in the production of GVL from EL. Recently, the noble metal-free catalyst Al(OiPr)3 was shown to provide a high yield of GVL (up to 97.6%) under mild conditions (150 °C, 30 min); this catalyst has many advantages in the conversion of ML to GVL using i-PrOH as the solvent, such as its low price and easy availability.18 The C–O bond of carboxylic acid derivatives could be activated by chiral Al3+-based complexes, which also had a good effect on the MPV reduction.27 Efficient catalytic processes are usually required for the hydrogenation of keto compounds in MPV reduction. The conversion of GVL on Al-based catalysts has been studied experimentally to achieve high yields, but the reaction mechanism still remains ambiguous. Therefore, it is also essential to understand the molecular reaction mechanism to provide some basic guidelines for the design of more efficient catalysts in the future. In the conversion of ML to GVL in i-PrOH, the ML undergoes transesterification, MPV hydrogenation, and ring-closure steps. In this work, we combined all these basic elements to probe the detailed reaction mechanism from ML to GVL catalyzed by Al(OiPr)3. This will provide some help in understanding the mechanism of the conversion of ML to GVL from the viewpoint of theoretical calculations.
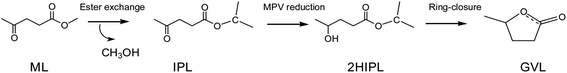 |
| | Scheme 1 A schematic representation of transesterification, hydrogenation, and ring-closure in the transformation of ML to GVL. | |
In this work, the detailed reaction mechanism of the conversion of ML into GVL catalyzed by Al(OiPr)3 in i-PrOH has been carried out using DFT calculations at the M062X-D3/def2-TZVP level. In the conversion of ML into GVL, there are three main steps, namely, transesterification, the MPV reduction reaction, and ring-closure to finish the process. To explore the detailed reaction mechanism of the transformation of ML to GVL and the roles of Al(OiPr)3 in the system, we have probed the conversion pathways of each step and studied the mechanism in the presence of Al(OiPr)3. Finally, a detailed reaction mechanism has been proposed based on the DFT calculations. These results should provide a better understanding of the mechanism of the transformation of ML to GVL catalyzed by Al(OiPr)3 in i-PrOH and give some basic guidelines to design more efficient catalysts.
2. Computational methods
All calculations were performed using the Gaussian 16 package,28 and the geometric structures were generated using CYL view.29 The geometry optimizations in this study were carried out using the M062X-D3 method30,31 with the basis set def2-TZVP.32 In the M062X-D3 method, the D3 term represents a dispersion correction.33,34 Zhao et al. tested a series of the M06 suite of density functionals and recommended the M06-2X functional for applications involving thermochemistry and noncovalent interactions.35 Def2-tzvp is the triple zeta valence basis set and has good quality for C, H, O, and Al, as has been reported in previous studies.36,37 The harmonic vibrational frequencies of the reaction complexes were calculated at the same level of theory and used to first determine whether the optimized structures were true minima or transition states (TSs). To verify the transition states connecting the reactants with the appropriate products, the intrinsic reaction coordinates (IRCs) were also calculated using the algorithm developed by Gonzalez and Schlegel.38 To account for the effects of the solution environment around the catalytic active site, the calculations were performed in an i-PrOH dielectric using the SMD solvation model39 at the M06-2X-D3/def2-TZVP level for the geometry evaluation. The activation energy (Ga), which is the energy barrier, and the reaction energy (Gr) of the systems are defined as follows:| | |
Ga = GTS − GR, Gr = Gp − GR
| (1) |
where GTS, GR, and GP represent the Gibbs free energies of the transition state, reactant, and product at 298.15 K and 100 kPa. To measure the reactive sites of the molecular surface, the electrostatic potential (ESP) of the reactants was analyzed and the standard Mulliken population analysis was used to obtain the charge density of each atom.40,41
3. Results and discussion
3.1 Transesterification reaction
The transesterification reaction is the main process to obtain some important energy sources, such as monoalkyl esters and glycerol, in alkyl alcohol.42,43 In the conversion of ML to GVL in i-PrOH, the content of isopropyl levulinate (IPL) is relatively high in the early stage of the reaction, and then follows a decreasing trend at different temperatures of 110, 120, 130, and 140 °C within 60 min.18 This indicates that the transesterification reaction of ML to IPL does indeed occur at the beginning of the transformation, and that the IPL would be further converted as the reaction proceeds. To investigate the reaction mechanism of the transesterification of ML to IPL in i-PrOH, the reaction pathways were probed using DFT calculations. As a useful tool to predict the reactive sites of a molecular surface, quantitative molecular surface analysis of ML, i-PrOH, and Al(OiPr)3 was carried out, the results are shown in Fig. 1, in which blue-colored surfaces represent positive electrostatic potential and red-colored surfaces represent negative electrostatic potential. The negatively charged area is susceptible to attack by electrophilic reagents. In the structures of ML and i-PrOH, the negative electrostatic potential is mainly distributed around the oxygen atoms, with a less-positive charge around the methyl group. The Al3+ center of Al(OiPr)3 can form strong H-bonds with the reactants. The differences in electrostatic potential play an important role in the formation of H-bonds to catalyze the reaction.
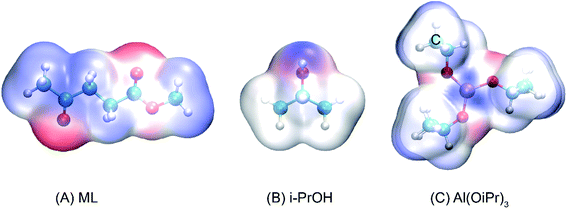 |
| | Fig. 1 Electrostatic potential surfaces of (A) ML, (B) i-PrOH, and (C) Al(OiPr)3 (isovalue = 0.001 a.u.). | |
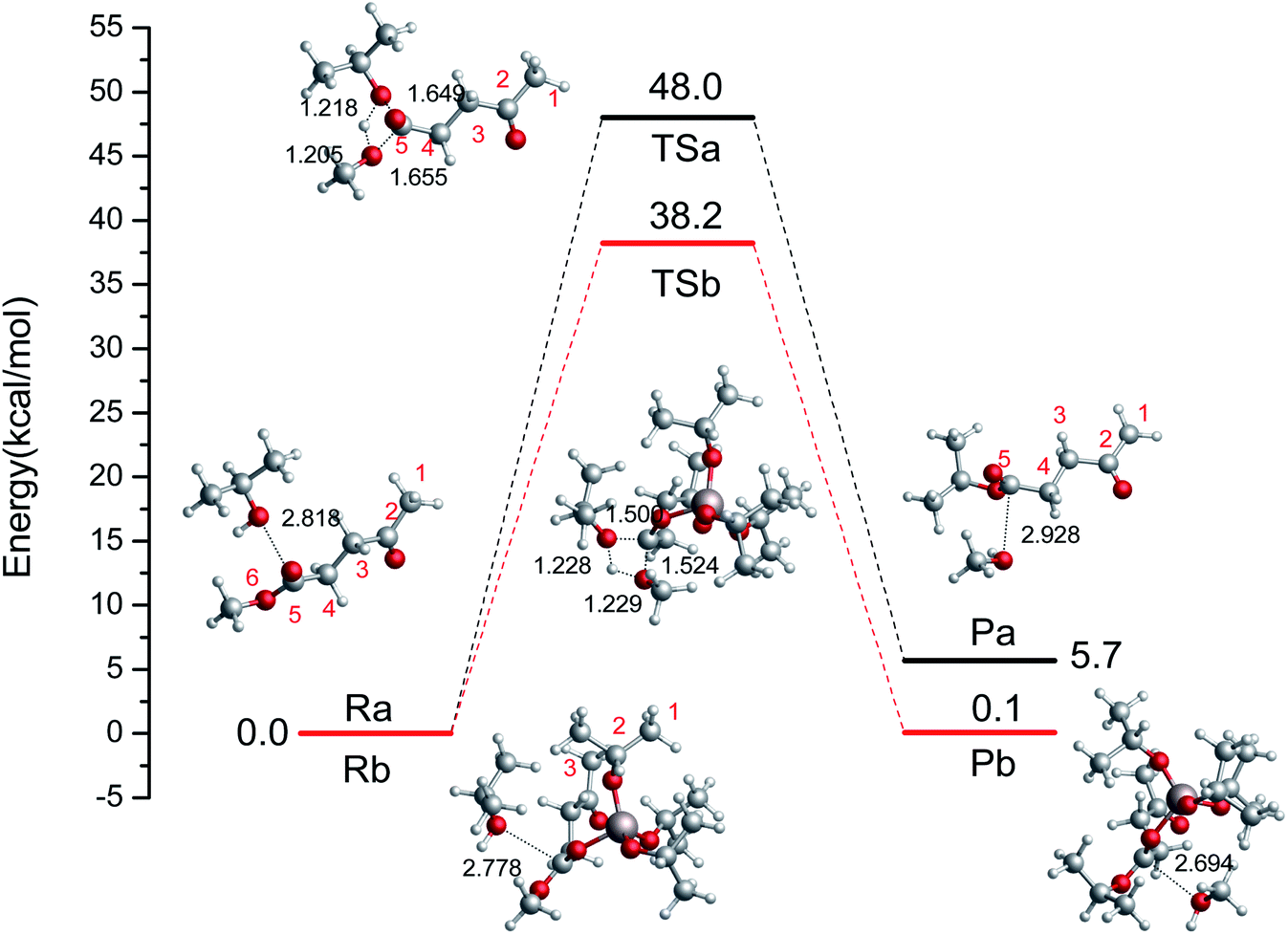
First, we probed the transesterification reaction of ML with i-PrOH as a model system without a catalyst (Ra → Pa), as shown in Fig. 2. Initially, H-bonds can be formed between the oxygen (i-PrOH) and the carbon of the ester group (ML) with a distance of 2.818 Å. Then, a four-center cyclic TS is formed between i-PrOH and ML with distances of 1.649, 1.218, 1.205, 1.655 Å (C–O, O–H, H–O, and O–C), respectively. The geometry of TSa is illustrated in Fig. 2. Finally, i-PrOH will add to the ML to release methanol. The reaction barrier to complete the transesterification in i-PrOH was 48.0 kcal mol−1, and this reaction is endothermic by 5.7 kcal mol−1. The oxygen atom of i-PrOH acts as a nucleophile and attacks the carbonyl carbon atom of the ester molecule.
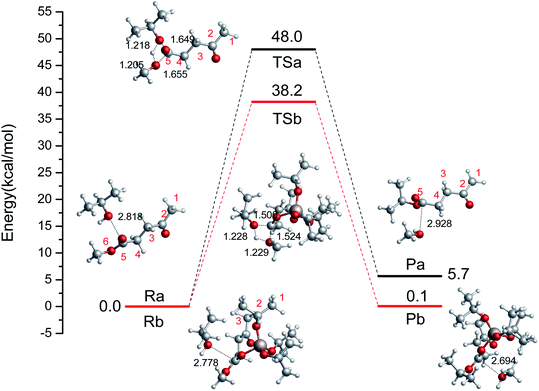 |
| | Fig. 2 The relative free energy diagram and optimized geometries of the reactants, TSs, and products without a catalyst (Ra–Pa) and with the catalyst Al(OiPr)3 (Rb–Pb) (bond distances in angstroms). | |
To investigate the reaction pathways catalyzed by Al(OiPr)3, the reaction energies and optimized geometries of the reactant, TS, and product complexes were calculated, and are shown in Fig. 2 (Rb → Pb). The Al3+ of Al(OiPr)3 mainly interacts with the oxygen of the ester group. A H-bond can be formed between Al(OiPr)3 and ML with a distance of 1.873 Å. The oxygen of i-PrOH attacks the carbon of ester group. A four-center cyclic TSb structure is formed between i-PrOH and ML. This process is completed with a 38.8 kcal mol−1 energy barrier. This means that Al(OiPr)3 exerts a catalytic effect to some extent by forming H-bonds with the oxygen of the ester group (ML). The R(Al–O) distance, Mulliken charges, and negative frequencies of the TS structures are listed in Table 1. The Mulliken charge of the carbonyl carbon (qcarbonyl C) in ML under Al(OiPr)3 catalysis is 0.0077 a.u.; the decrease in the charge of the carbonyl carbon atom will be beneficial for the transesterification reaction.
Table 1 Distance between the Al3+ center and the carboxyl oxygen atom of ML (R(Al–O)), the Mulliken charge densities of the carbonyl carbon (qcarbonyl C), and the negative frequencies of the TSs for the ester exchange step
| |
R(Al–O) [Å] |
qcarbonyl C [a.u.] |
Negative frequency of TS [cm−1] |
Barrier [kcal mol−1] |
| Uncatalyzed |
— |
0.3800 |
−1442.25 |
48.0 |
| Al(OiPr)3 |
1.874 |
0.0077 |
−1595.52 |
38.2 |
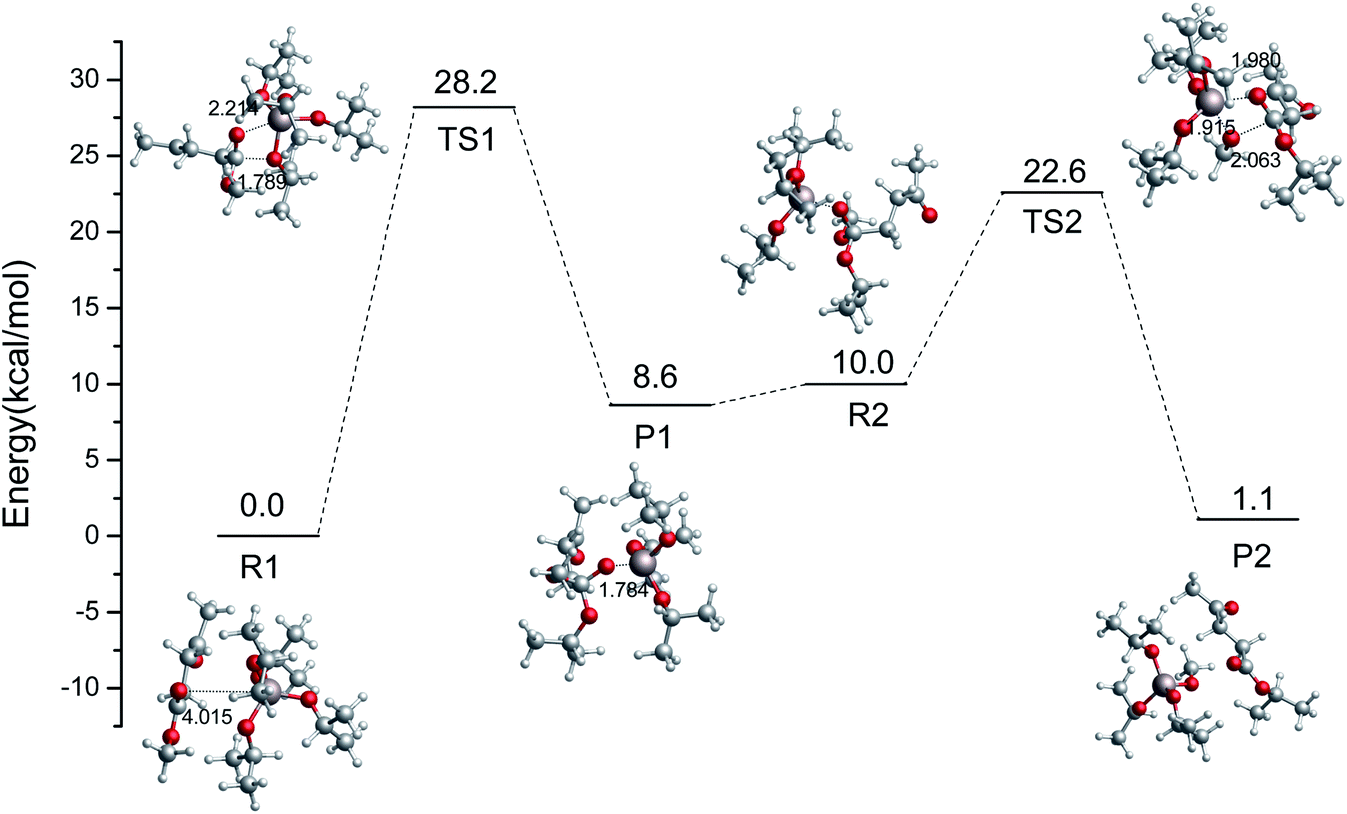
In the imidazolium-IL-catalyzed isomerization of glucose to fructose, the imidazolium cations play the dual roles of H-bond donor and proton shuttle.44 The role of Al3+-based catalysts was shown to be shortening the proton transfer distance.45 Similarly, another pathway catalyzed by Al(OiPr)3 is also probed in this section. The relative energy scheme and optimized geometries of the reactant, TS, and product complexes are plotted in Fig. 3. Firstly, the Al3+ forms a strong H-bond with the oxygen of the ester group in ML, and then the i-PrO˙ is added to the ester group carbon of ML. An intermediate (P1) is formed with a 28.2 kcal mol−1 energy barrier, and its formation is endothermic, with a reaction energy of 8.6 kcal mol−1. Then, the CH3O˙ will be coordinated with the Al3+ to finish the transesterification in the second step (just opposite to the former process). The energy barrier of the second step to finish the transesterification is 12.6 kcal mol−1, and this process is exothermic with a reaction energy of 8.9 kcal mol−1. Comparing the energy barriers in these two kinds of transesterification catalyzed by Al(OiPr)3 (Rb → Pb and R1 → P2), Al(OiPr)3 plays a critical role in the second pathway (R1 → P2) to catalyze the transesterification. Additionally, the activation potential energy of the reaction catalyzed by Al(OiPr)3 is much lower than that of the non-catalytic reaction, and Al(OiPr)3 can indeed affect the transesterification reaction to decrease the energy barrier.
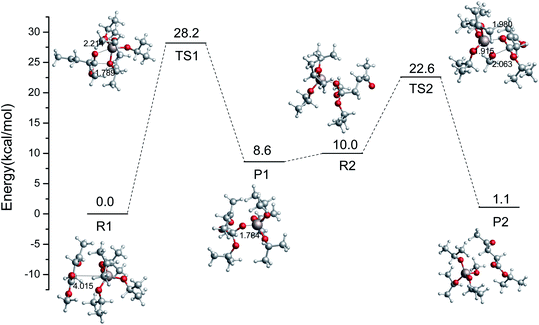 |
| | Fig. 3 Relative free energy diagram and optimized geometries for the ester exchange catalyzed by Al(OiPr)3 via another pathway (bond distances in angstroms). | |
3.2 Meerwein–Ponndorf–Verley (MPV) reduction
The catalytic hydrogenation of LA or its esters plays an important role in producing GVL with low energy consumption.46 The Meerwein–Ponndorf–Verley (MPV) reaction is very selective toward the keto functional group, which offers the possibility of catalytic hydrogenation of biomass derivatives in the liquid phase.17,47,48 In this section, we investigate the reaction pathways, including the energetics and structures, of the MPV hydrogenation of IPL to 2-hydroxy-isopropyl levulinate (2HIPL). To investigate the reaction barriers for the direct hydration of IPL using various hydrogen donors, we carried out computations without a catalyst. There are two pathways, i.e., an inter- and an intra-molecular pathway, to achieve the hydrogenation. The free energies of the hydrogen transfer reactions and the negative frequencies of the TSs are summarized in Table 2. The hydrogen of i-PrOH is directly transferred through a six-membered ring TS structure (TSc). The IRCs were calculated to verify the TSs connecting the reactants with the appropriate products in this work. The IRCs (maxpoint = 100, stepsize = 10) corresponding to TSc in Fig. 4A are listed in Fig. S1.† Initially, H-bonds can form between the hydroxyl of i-PrOH and IPL, and then the protons transfer to IPL to produce 2HIPL and isopropanone. As shown in Fig. 4(A), the protons of hydroxyl and CH in i-PrOH transfer to the carbonyl of ML to complete the inter-molecular hydrogenation, and the energy barrier of inter-molecular hydrogenation is 51.0 kcal mol−1. At the same time, IPL can undergo intra-molecular hydrogen transfer (Fig. 4(B)) to form GVL and isopropanone. However, a barrier of 51.0 kcal mol−1 must be overcome in this process, and it is exothermic by 7.1 kcal mol−1. The hydrogen donors can come from many sources, such as secondary alcohols or IPL. The energy barrier of inter-molecular hydrogenation is lower than that of intra-molecular hydrogenation. This further confirmed that the secondary alcohols are better than primary alcohols. The larger reaction barrier of intra-molecular hydrogenation implies that inter-molecular hydrogenation is preferred in the presence of hydrogen donors (solvents such as i-PrOH).
Table 2 Computed relative energies (kcal mol−1) for the different hydrogen transfer reaction pathways in different ways and the negative frequencies of the TSs
| |
Activation energy [kcal mol−1] |
Reaction energy [kcal mol−1] |
Negative frequency of TS [cm−1] |
| (A) Inter- |
34.6 |
−0.2 |
−1453.74 |
| (B) Intra- |
51.0 |
−7.1 |
−812.46 |
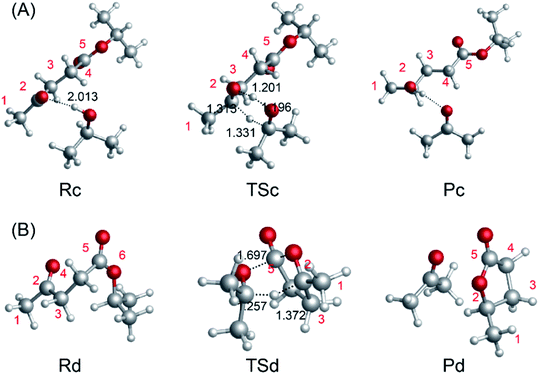 |
| | Fig. 4 (A) Intermolecular hydrogen transfer between IPL and i-PrOH to form 2HIPL and isopropanone. (B) Intramolecular hydrogen transfer involving IPL to form GVL and isopropanone. | |
To investigate the pathways of the MPV reaction catalyzed by Al(OiPr)3, the relative energies and the corresponding structures are shown in Fig. 5. Assary et al. reported the reaction pathways of MPV reduction catalyzed by Sn, Zr, and Al model catalysts.49 The free energy of MPV hydrogenation from IPL to 2HIPL catalyzed by Al(OiPr)3 at the MP2/6-311+G(3df, 3pd)//B3LYP/6-31+G(d)49 and M06-2X-D3/def2-tzvp levels are compared in Fig. S2.† As shown in Fig. S2,† the activation barriers at the M06-2X-D3/def2-tzvp and MP2/6-311+G(3df,3pd)//B3LYP/6-31+G(d) level for the pathways reported by Assary et al. are less than 1 kcal mol−1.49 DFT-D3 provides a good description of π-stacking interactions for multiatomic systems.50,51 The small difference in the energies is mainly caused by the structures optimized at the B3LYP and M06-2X-D3 levels, but both of them are reasonable to calculate the energies. Furthermore, Cohen et al. studied the mechanistic details of the MPV reduction of ketones via experiments and DFT calculations, and found that a six-membered ring TS is the most favorable pathway.52
 |
| | Fig. 5 The computed free energy profile with schematic diagrams of geometries for the hydrogenation of IPL to 2HIPL by Al(OiPr)3 in i-PrOH medium (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH3CHCH3). CH3CHCH3). | |
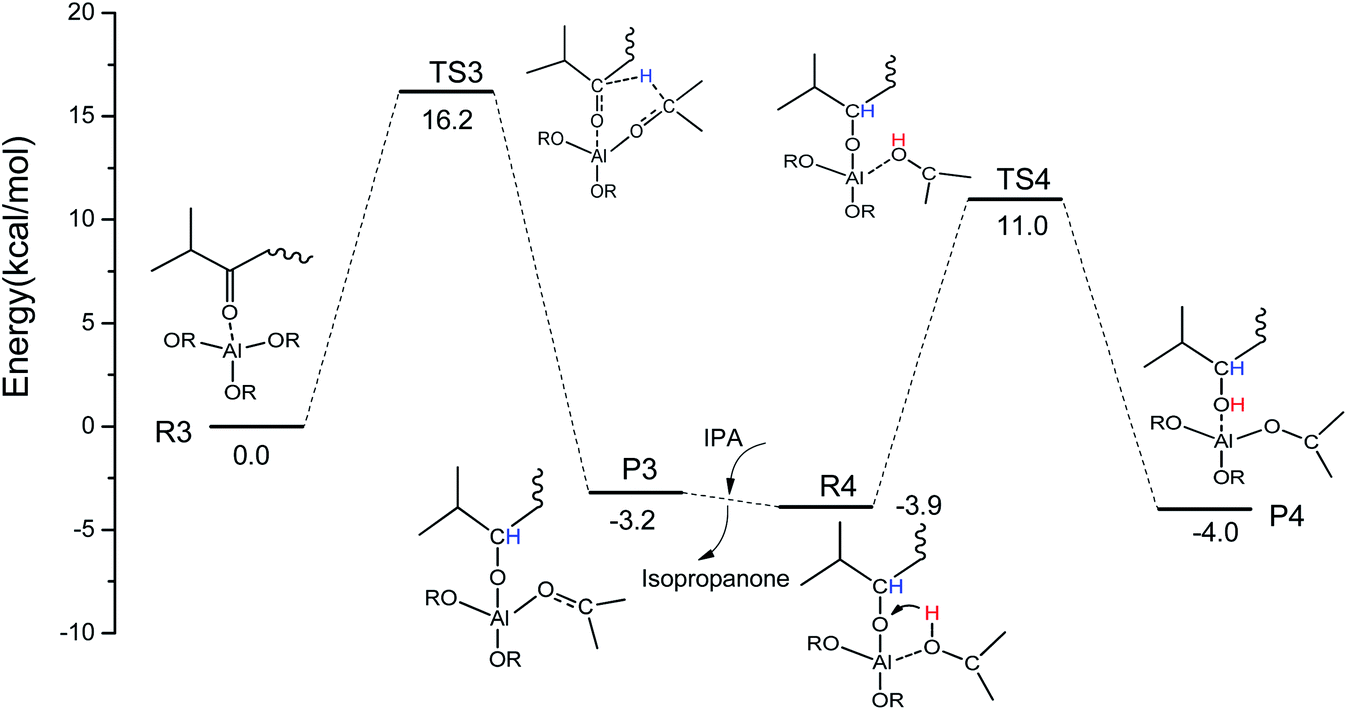
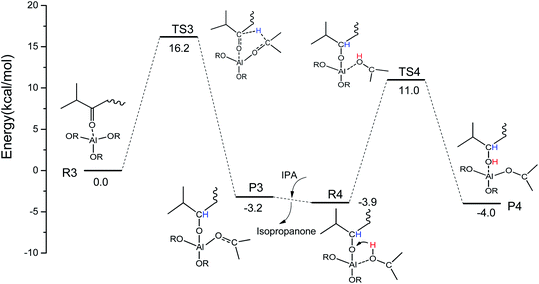
Some similar reaction mechanisms for the MPV reaction of ketones catalyzed by Al(OiPr)3 in i-PrOH solvent with a two-step intermolecular hydrogen transfer mechanism were explored using DFT calculations. In the first step (R3 → P3), H-bonds are formed between Al(OiPr)3 and the oxygen of the carbonyl group (IPL). The oxygen of the carbonyl group (IPL) can be activated by Al(OiPr)3. Then, the hydrogen proton of C2 of Al(OiPr)3 is transferred to the carbon of the carbonyl group (IPL) via a six-membered ring TS structure (TS3). This process requires an energy barrier of 16.2 kcal mol−1 to be overcome, and is exothermic by about 3.2 kcal mol−1. The intermediate product P3 is formed to complete the first hydrogenation. In the solvent i-PrOH, isopropanone will be removed, and the second step (R4 → P4) will be continuous. The interaction position between Al(OiPr)3 and IPL changes to form R4 due to the molecular flexibility. For the second hydrogenation of IPL (R4 → P4), the H of the hydroxyl of i-PrOH coordinated with Al3+ transfers to the O of the carbonyl group (IPL) via a six-membered ring TS structure (TS4). This process presents a 14.9 kcal mol−1 energy barrier to complete the MPV hydrogenation. The energy barrier of the first hydrogenation is a bit higher than that of the second. Additionally, a six-membered ring TS is formed between Al(OiPr)3 and IPL in the MPV hydrogenation. The oxygen of IPL can be activated by coordination with Al(OiPr)3.
3.3 Ring-closure step
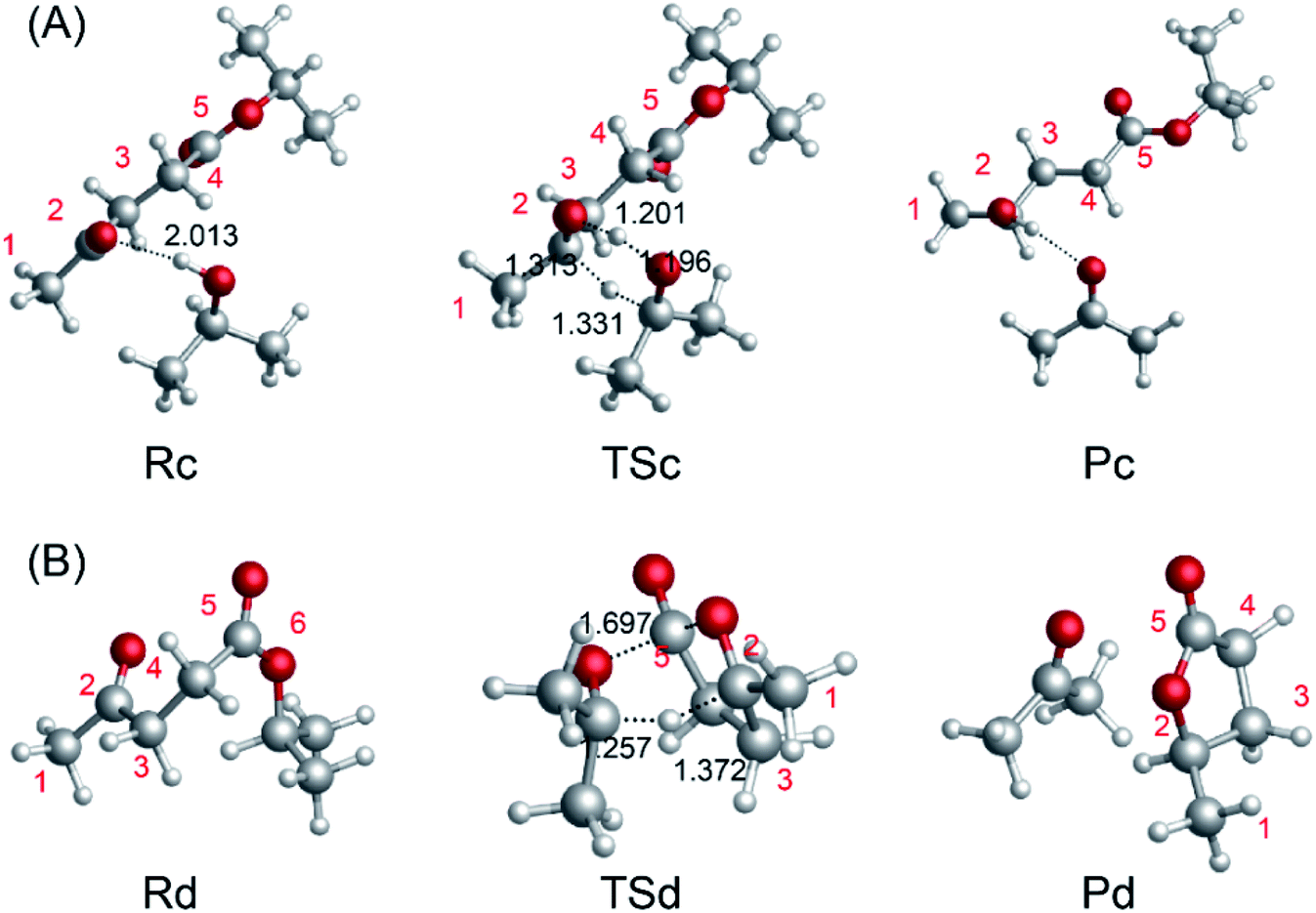
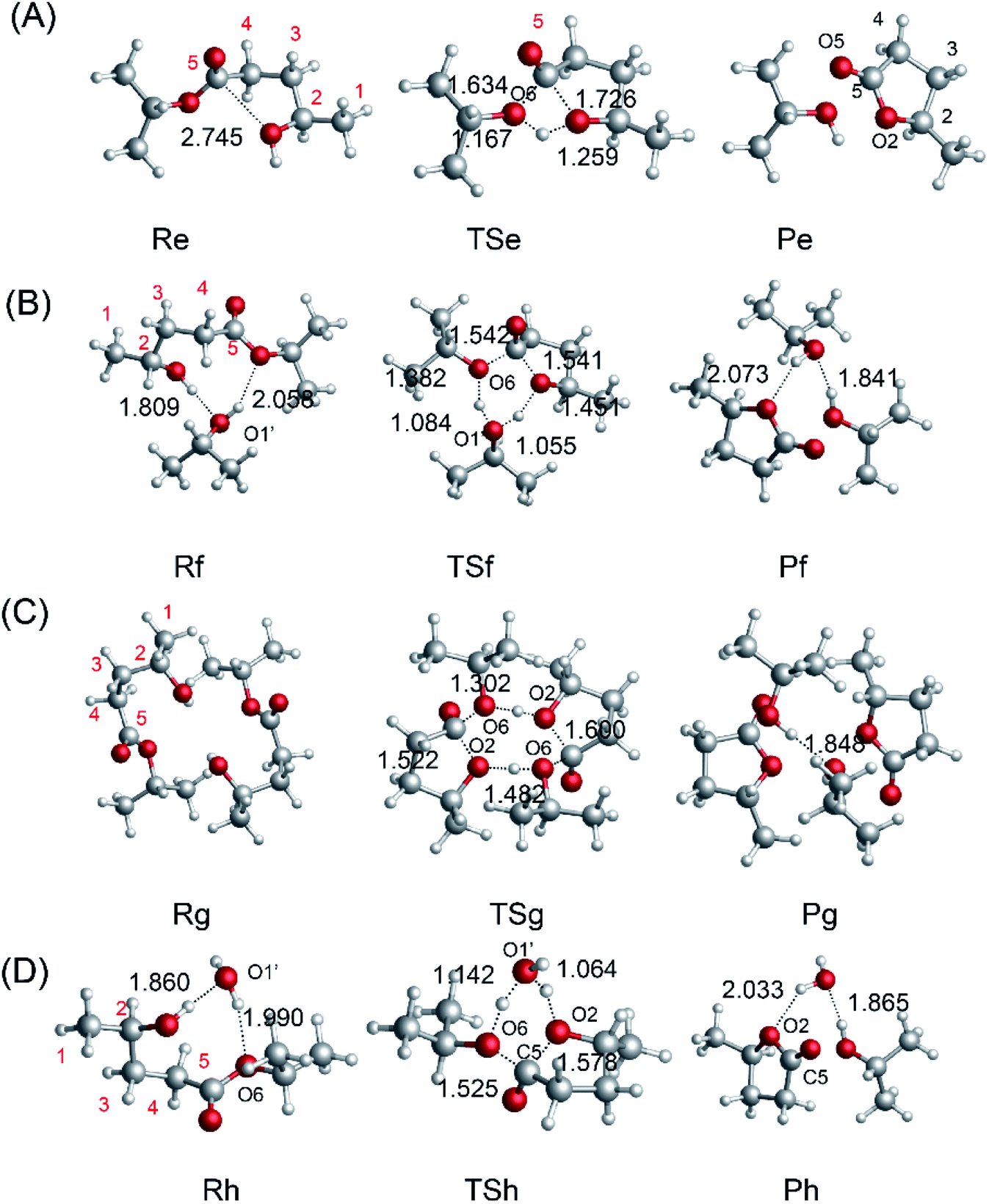
To investigate the mechanism of the ring-closure step to give GVL, we probed the reaction pathway without a catalyst and those catalyzed by i-PrOH, 2HIPL, and H2O. To compare the reactions catalyzed by the different catalysts quantitatively, the reaction of GVL and i-PrOH without any catalyst was first investigated and is shown in Fig. 6(A). To provide GVL, the O2 of 2HIPL approaches C5, and the H of the hydroxyl transfers to O6. The transition state involved in this process is TSe, in which the ester group C5–O6 bond (1.634 Å) is almost broken due to the attack of the oxygen of the hydroxyl with a C5–O2 distance of 1.726 Å. About 46.9 kcal mol−1 is required to cross the transition-state energy barrier. The reaction without a catalyst is an endothermic process of about 1.8 kcal mol−1. In this process, catalysts such as i-PrOH, 2HIPL, and H2O can participate in the reaction as a proton transfer mediator. To investigate the roles of the other catalysts, the corresponding reaction pathways were also studied, as shown in Fig. 6(B–D). Taking the reaction pathway catalyzed by i-PrOH as an example, the H transfers to the O of i-PrOH, and the H of the hydroxyl of i-PrOH transfers to O6 of 2HIPL. This process must overcome a 37.4 kcal mol−1 energy barrier, and is endothermic by about 2.4 kcal mol−1. The oxygen of the catalysts plays the role of a proton shuttle to carry out the ring-closure reaction. Similarly to Rf-Pf (Fig. 6B), the ring-closure steps catalyzed by 2HIPL and H2O were also calculated, and are shown in Fig. 6(C and D). As shown in Table 3, the barriers of the four kinds of ring-closure steps were calculated to be 46.9, 37.4, 62.8, and 39.4 kcal mol−1, respectively. Comparing the activation energy of the different catalysts, their proton transfer ability follows the order i-PrOH > H2O > 2HIPL. This means that i-PrOH plays a substantial role as a proton shuttle to promote the reaction.
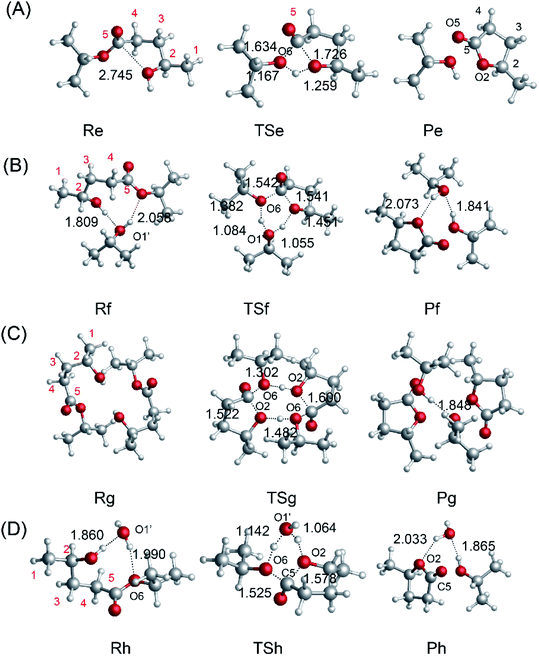 |
| | Fig. 6 Optimized geometries for the reactants, transition states, and products with (A) no catalyst, (B) i-PrOH, (C) 2HIPL, and (D) H2O. The unit of chemical bond lengths is Å, 1 Å = 0.1 nm. | |
Table 3 Computed relative energies (kcal mol−1) for the reaction of the ring-closure step catalyzed by different catalysts and the negative frequencies of the TSs
| Catalyst |
Activation energy [kcal mol−1] |
Reaction energy [kcal mol−1] |
Negative frequency of TS [cm−1] |
| — |
46.9 |
1.8 |
−1282.42 |
| i-PrOH |
37.4 |
2.4 |
−394.48 |
| 2HIPL |
62.8 |
−2.9 |
−462.71 |
| H2O |
39.4 |
−0.1 |
−782.58 |
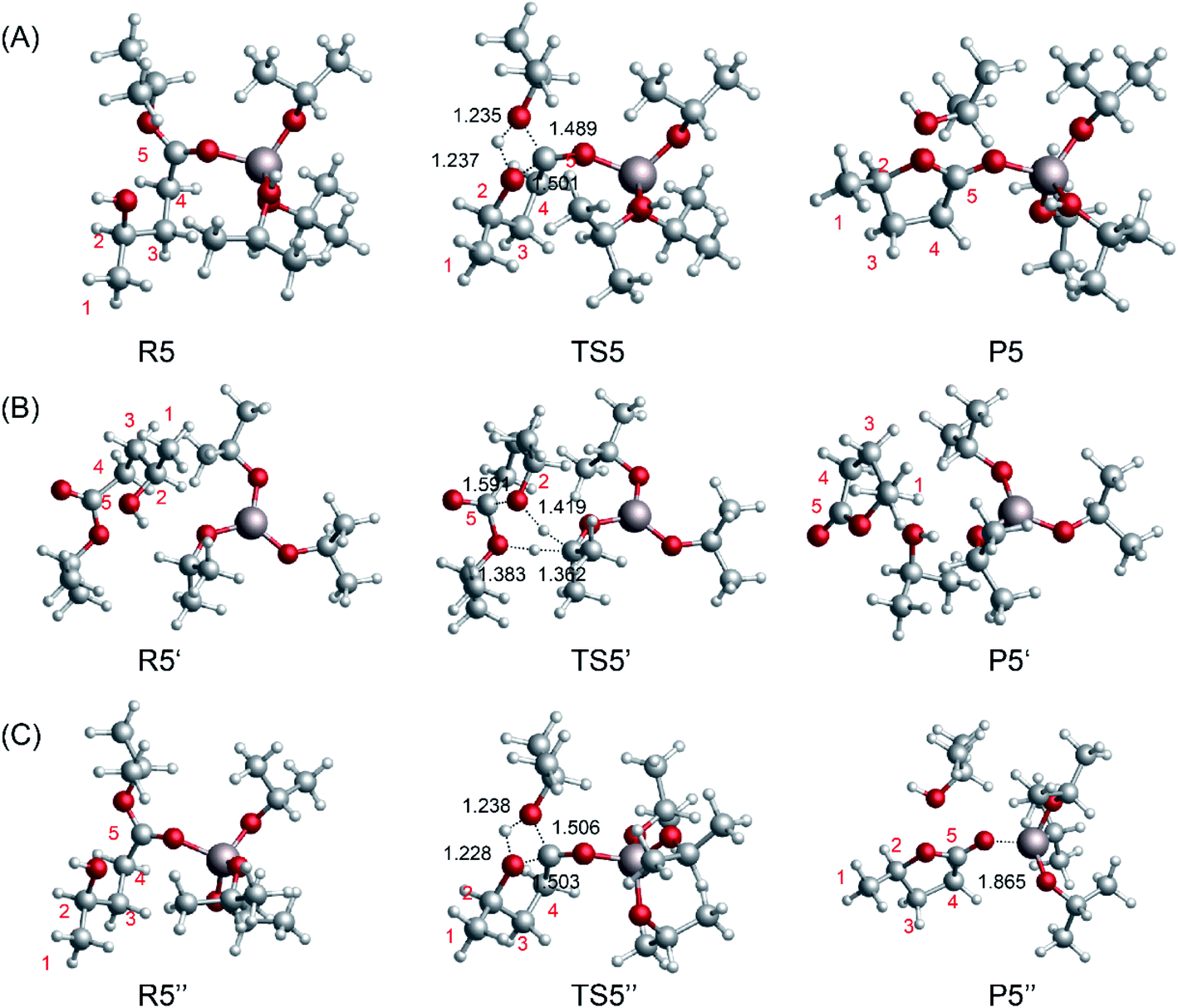
To compare the catalytic effects of the Al-based catalysts, the reaction pathways were probed using DFT calculations. The structures of the different catalytic pathways are shown in Fig. 7. Some studies have reported that the bifunctional Lewis acid and Bronsted base sites of Al-based catalysts play an important role in sugar isomerization.45,53 For R5 → P5 (Fig. 7A), the O5 of 2HIPL can coordinate with the Al3+ of [Al(OiPr)2(i-PrOH)]+1, and the O2–C5 bond is closed to produce GVL with a 37.1 kcal mol−1 energy barrier. This reaction is endothermic by about 1.1 kcal mol−1. For R5′ → P5′ (Fig. 7B), the proton of O2H of 2HIPL transfers to the C2 of i-PrO˙ coordinated with Al3+, and the H2 of i-PrO˙ migrates to the O6 of 2HIPL. The O2–C5 bond is closed to produce GVL with an 81.4 kcal mol−1 energy barrier. The reaction then proceeds via the formation of the product P5, which involves GVL, i-PrOH, and Al(OiPr)3. This reaction is exothermic by about 1.6 kcal mol−1. Al3+ located at a distance of about 0.1852 nm forms a hydrogen bond with the ketone oxygen in 2HIPL. These results show that Al(OiPr)3 has poor proton transfer ability, and may not catalyze the ring-closure reaction in this way. In the other pathway of the ring-closure step catalyzed by Al(OiPr)3 (R5′′ → P5′′) (Fig. 7C), the carbonyl O5 of 2HIPL is activated by coordination with Al(OiPr)3. The H2 of the hydroxyl (2HIPL) migrates to O6, and a C5–O2 bond is formed via a four-membered ring TS. The energy barrier of this process is 40.1 kcal mol−1. Comparing this value with the energy barriers of the processes catalyzed by the other catalysts discussed above, the Al-based catalysts are found to play an important role in the ring-closure reaction.
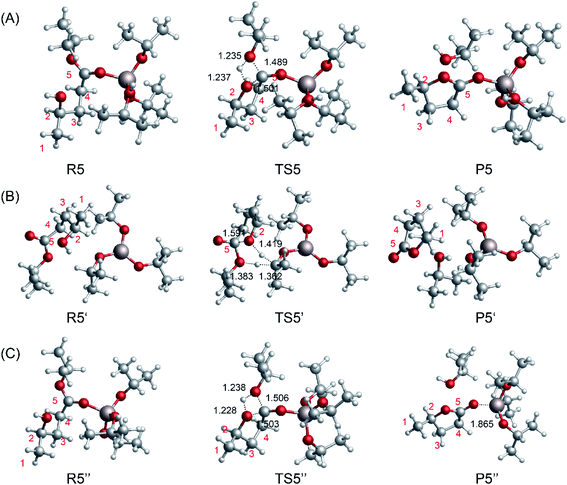 |
| | Fig. 7 Optimized geometries for the reactants, transition states, and products of the ring-closure step from 2HIPL to GVL catalyzed by the Al-based catalysts (A) [Al(OiPr)2(i-PrOH)]+1 and (B and C) Al(OiPr)3. The unit of chemical bond lengths is Å, 1 Å = 0.1 nm. | |
3.4 Overall reaction process
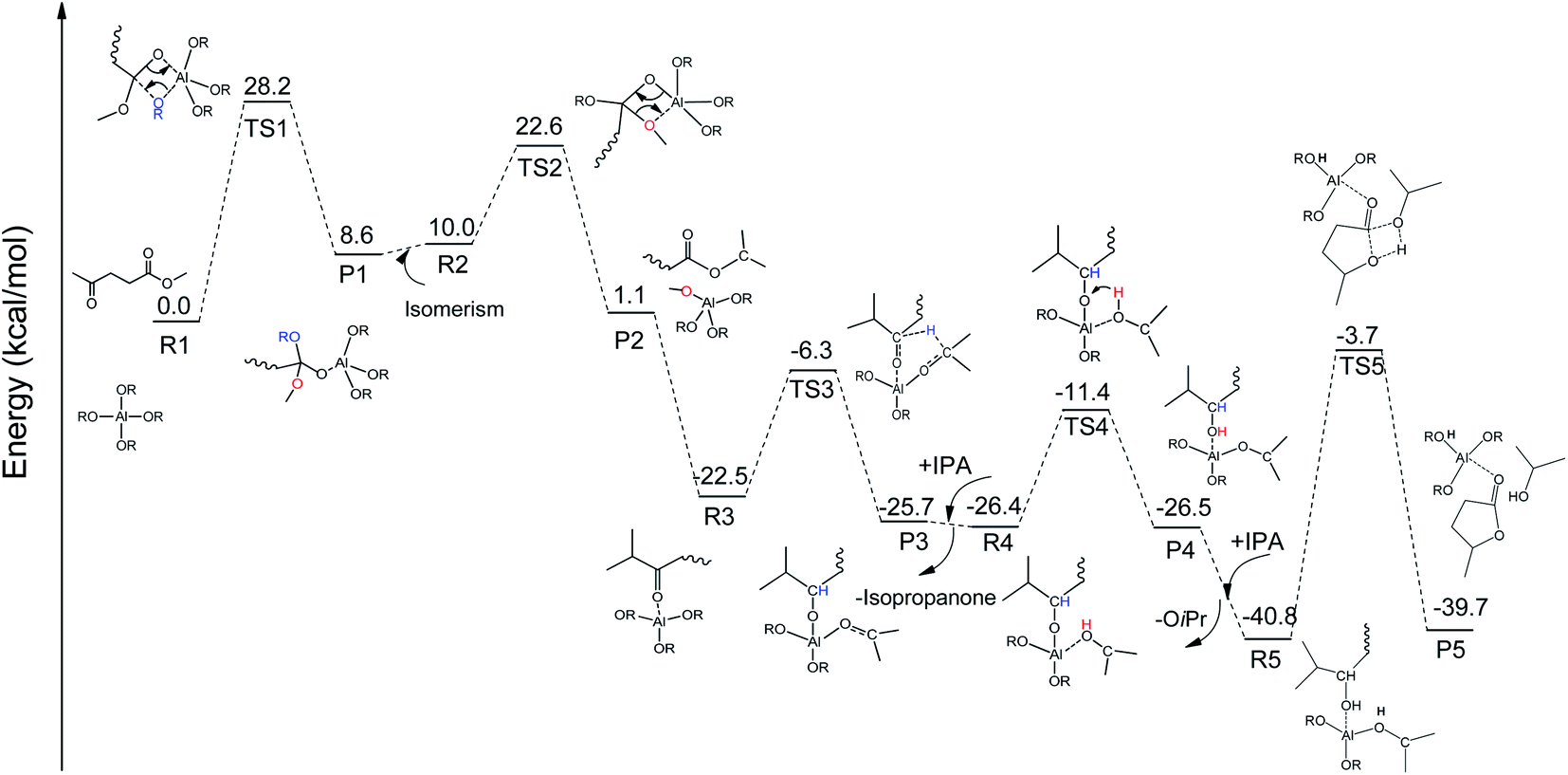
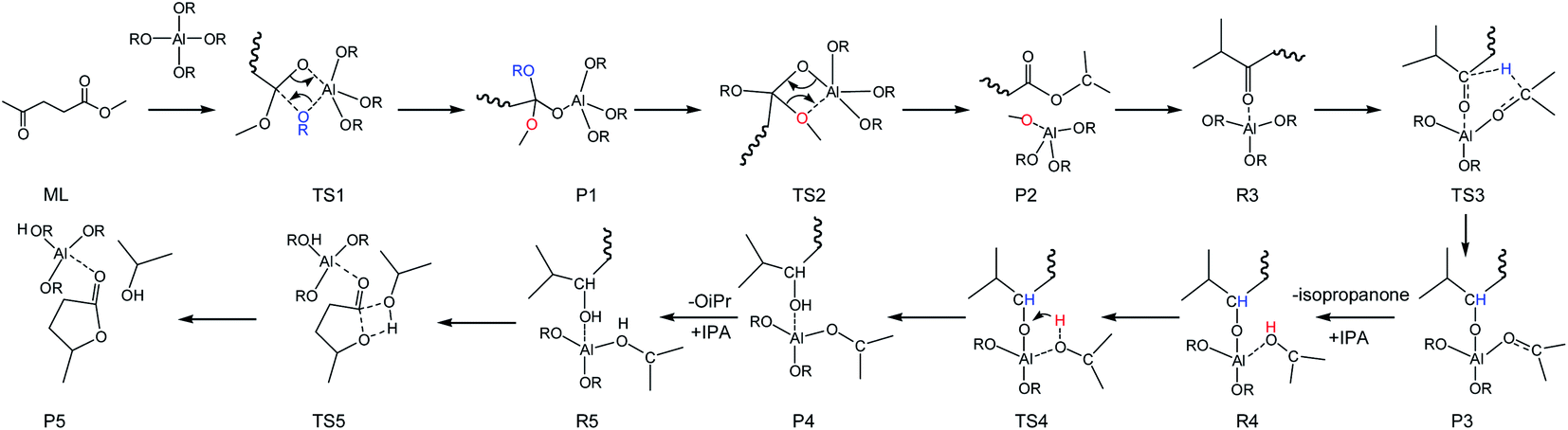
To integrate all the basic steps, the lowest reaction pathways were combined. As shown in Fig. 8, the mechanism consists of three processes, namely, ester exchange, Meerwein–Ponndorf–Verley (MPV) hydrogenation, and the ring-closure step. The proposed reaction mechanism for the transformation of ML to GVL catalyzed by Al(OiPr)3 in i-PrOH solvent is presented in Scheme 2. The ester exchange process involves two elementary steps. In the first step, Al(OiPr)3 mainly interacts with the oxygen of ML to form the intermediate P1 via transition state TS1 with a barrier of 28.2 kcal mol−1. The second step along the reaction coordinate is the isomerization of P1 to R2, with R2 being higher than P1 by about 1.4 kcal mol−1. The methoxy group is then eliminated from R2, and transition state TS2 with a four-membered ring is formed. This process is just the reverse of the first step. Approximately 12.6 kcal mol−1 of energy is needed to cross the energy barrier. This reaction is exothermic by about 8.9 kcal mol−1. The subsequent MPV process involves hydride transfer from the keto carbon of the Al(OiPr)3 to the keto carbon of the intermediate isopropyl levulinate (IPL) (R3 → TS3) through transition state TS3 with an energy barrier of about 16.2 kcal mol−1. In solution, the isopropanone will be replaced by an i-PrOH molecule, the i-PrOH in intermediate P3, to form the complex R4. This process (P3 → R4) is exothermic by only 0.7 kcal mol−1. The release of the 2-hydroxy-isopropyl levulinate (2HIPL) (R4 → P4) requires a proton from the i-PrOH molecule. The protonation occurs via transition state TS4, which leads to the formation of 2HIPL and an isopropanone molecule (P4) with a 15 kcal mol−1 energy barrier. In the last process, the ring-closure step (R5 → P5), the O5 of 2HIPL can coordinate with Al3+ to catalyze the reaction. The rate-determining step is the ring-closure with a 37.1 kcal mol−1 energy barrier. The roles of Al(OiPr)3 involve mainly coordination by the oxygen atoms of the reactants. A six-membered ring can be formed between Al(OiPr)3 and IPL in the MPV reaction.
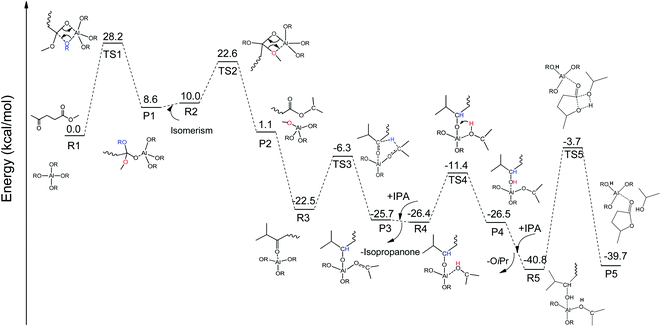 |
| | Fig. 8 The free energy profile of the formation of GVL from ML catalyzed by Al(OiPr)3 in i-PrOH solvent with the relevant TSs noted corresponding to Scheme 2 (RCH3CHCH3). | |
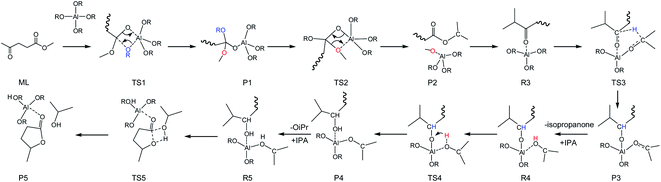 |
| | Scheme 2 Overall reaction pathway for the conversion of ML into GVL in i-PrOH solvent (RCH3CHCH3). | |
4. Conclusions
GVL is a promising renewable platform that can be produced from lignocellulose via LA/ML hydrogenation. In this paper, DFT calculations have been used to probe the details of the mechanism for the conversion of ML to GVL catalyzed by Al(OiPr)3 in i-PrOH solvent. Comprehensive quantum chemical computations of the reaction via different pathways were carried out, and there are three main processes (ester exchange, MPV hydrogenation, and the ring-closure step) involved. Al(OiPr)3 mainly interacts with the oxygen of the carbonyl group ML to activate the reaction. In the ester exchange step, a four-membered ring transition state is formed between i-PrOH and ML, and two steps are required to complete this process. Al(OiPr)3 can show a great catalytic effect via the formation of strong H-bonds, compared to when no catalyst is used. In the MPV hydrogenation reaction, IPL undergoes two proton additions with the catalyst Al(OiPr)3 via a TS with six-centered-ring geometry. O5 of 2HIPL can coordinate with Al-based catalysts to reduce the barrier of the ring-closure step, which is rate-determining. The present theoretical study provides a clear elementary-step mechanistic profile of the transition from ML to GVL catalyzed by Al(OiPr)3 and gives some basic guidance for the design of more efficient catalysts in the future.
Abbreviations
| IPL | Isopropyl levulinate |
| 2HIPL | 2-Hydroxy-isopropyl levulinate |
| MPV | Meerwein–Ponndorf–Verley |
| GVL | γ-Valerolactone |
| LA | Levulinic acid |
| i-PrOH, IPA | Isopropanol alcohol |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 32071626) and the Research Fund for the Quzhou University (No. BSYJ202015 and BSYJ202113).
References
- H.-J. Lee, W.-S. Lim and J.-W. Lee, J. Ind. Eng. Chem., 2013, 19, 2010–2015 CrossRef CAS.
- D. Kim, K. Lee and K. Y. Park, J. Ind. Eng. Chem., 2016, 42, 95–100 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516 RSC.
- J. Q. Bond, A. A. Upadhye, H. Olcay, G. A. Tompsett, J. Jae, R. Xing, D. M. Alonso, D. Wang, T. Zhang, R. Kumar, A. Foster, S. M. Sen, C. T. Maravelias, R. Malina, S. R. H. Barrett, R. Lobo, C. E. Wyman, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2014, 7, 1500–1523 RSC.
- K. Yan, Y. Yang, J. Chai and Y. Lu, Appl. Catal., B, 2015, 179, 292–304 CrossRef CAS.
- C. E. Chan-Thaw, M. Marelli, R. Psaro, N. Ravasio and F. Zaccheria, RSC Adv., 2013, 3, 1302–1306 RSC.
- Y. Yang, X. Wei, F. Zeng and L. Deng, Green Chem., 2016, 18, 691–694 RSC.
- K. Yan, Y. Yang, J. Chai and Y. Lu, Appl. Catal., B, 2015, 179, 292–304 CrossRef CAS.
- V. Fábos, M. Y. Lui, Y. F. Mui, Y. Y. Wong, L. T. Mika, L. Qi, E. Cséfalvay, V. Kovács, T. Szűcs and I. T. Horváth, ACS Sustainable Chem. Eng., 2015, 3, 1899–1904 CrossRef.
- C. Xiao, T.-W. Goh, Z. Qi, S. Goes, K. Brashler, C. Perez and W. Huang, ACS Catal., 2016, 6, 593–599 CrossRef CAS.
- U. Omoruyi, S. Page, J. Hallett and P. W. Miller, ChemSusChem, 2016, 9, 1–12 CrossRef PubMed.
- X. Tang, H. Chen, L. Hu, W. Hao, Y. Sun, X. Zeng, L. Lin and S. Liu, Appl. Catal., B, 2014, 147, 827–834 CrossRef CAS.
- A. M. R. Galletti, C. Antonetti, V. De Luise and M. Martinelli, Green Chem., 2012, 14, 688 RSC.
- J. Song, L. Wu, B. Zhou, H. Zhou, H. Fan, Y. Yang, Q. Meng and B. Han, Green Chem., 2015, 17, 1626–1632 RSC.
- H. Zhou, J. Song, H. Fan, B. Zhang, Y. Yang, J. Hu, Q. Zhu and B. Han, Green Chem., 2014, 16, 3870–3875 RSC.
- X. Tang, H. Chen, L. Hu, W. Hao, Y. Sun, X. Zeng, L. Lin and S. Liu, Appl. Catal., B, 2014, 147, 827–834 CrossRef CAS.
- A. Corma, M. E. Domine and S. Valencia, J. Catal., 2003, 215, 294–304 CrossRef CAS.
- T. Zhao, Z. Ju, Y. Zhang, L. Han and W. Xiao, Chem. Eng. J., 2020, 390, 124505 CrossRef.
- M. Chia and J. A. Dumesic, Chem. Commun., 2011, 47, 12233–12235 RSC.
- S. Wang, H. Huang, V. Dorcet, T. Roisnel, C. Bruneau and C. Fischmeister, Organometallics, 2017, 36(16), 3152–3162 CrossRef CAS.
- S. Li, Y. Wang, Y. Yang, B. Chen, J. Tai, H. Liu and B. Han, Green Chem., 2019, 21, 770–774 RSC.
- J. M. Nadgeri, N. Hiyoshi, A. Yamaguchi, O. Sato and M. Shirai, Appl. Catal., A, 2014, 470, 215–220 CrossRef CAS.
- H. Zhou, J. Song, X. Kang, J. Hu, Y. Yang, H. Fan, Q. Meng and B. Han, RSC Adv., 2015, 5, 15267–15273 RSC.
- H. Li, Z. Fang and S. Yang, ACS Sustainable Chem. Eng., 2016, 4(1), 236–246 CrossRef CAS.
- C. Zhang, Z. Huo, D. Ren, Z. Song, Y. Liu, F. Jin and W. Zhou, J. Energy Chem., 2019, 32, 189–197 CrossRef.
- J. He, H. Li, Y.-M. Lu, Y.-X. Liu, Z.-B. Wu, D.-Y. Hu and S. Yang, Appl. Catal., A, 2016, 510, 11–19 CrossRef CAS.
- P. Nandi, A. Solovyov, A. Okrut and A. Katz, ACS Catal., 2014, 4, 2492–2495 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- H. Guernon and C. Y. Legault, Organometallics, 2013, 32, 1988–1994 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 119, 525 Search PubMed.
- N. Y. Acelas and E. Flórez, Adsorption, 2018, 24, 243–248 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853–5860 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- R. Fu, T. Lu and F.-W. Chen, Acta Phys.-Chim. Sin., 2014, 30, 628–639 CAS.
- J. S. Murray and P. Politzer, in Encyclopedia of Computational Chemistry, John Wiley & Sons, Ltd, 2002, DOI: DOI:10.1002/0470845015.cca014.
- A. Cesar Huppes da Silva, S. Claudino da Silva, E. L. Dall'Oglio, P. T. de Sousa and C. Alberto Kuhnen, Fuel, 2013, 104, 379–385 CrossRef.
- Y. Asakuma, K. Maeda, H. Kuramochi and K. Fukui, Fuel, 2009, 88, 786–791 CrossRef CAS.
- Y. Jing, J. Gao, C. Liu and D. Zhang, J. Phys. Chem. B, 2017, 121, 2171–2178 CrossRef PubMed.
- Z. Ju, Y. Zhang, T. Zhao, W. Xiao and X. Yao, ACS Sustainable Chem. Eng., 2019, 7, 14962–14972 CrossRef CAS.
- S. Song, S. Yao, J. Cao, L. Di, G. Wu, N. Guan and L. Li, Appl. Catal., B, 2017, 217, 115–124 CrossRef CAS , S0926337317305015..
- A. Corma, M. E. Domine, L. Nemeth and S. Valencia, J. Am. Chem. Soc., 2002, 124, 3194–3195 CrossRef CAS PubMed.
- S. H. Liu, S. Jaenicke and G. K. Chuah, J. Catal., 2002, 206, 321–330 CrossRef CAS.
- R. S. Assary, L. A. Curtiss and J. A. Dumesic, ACS Catal., 2013, 3, 2694–2704 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- D. Josa, J. Rodríguez-Otero, E. M. Cabaleiro-Lago and M. Rellán-Piñeiro, Chem. Phys. Lett., 2013, 557, 170–175 CrossRef CAS.
- R. Cohen, C. R. Graves, S. T. Nguyen, J. M. L. Martin and M. A. Ratner, J. Am. Chem. Soc., 2004, 126, 14796–14803 CrossRef CAS PubMed.
- S. Saravanamurugan, M. Paniagua, J. A. Melero and A. Riisager, J. Am. Chem. Soc., 2013, 135, 5246–5249 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: xyz coordinates of all structures. See DOI: 10.1039/d1ra08429a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Shaokeng Fenga,
Lanhui Ren
a,
Shaokeng Fenga,
Lanhui Ren