
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved ORR/OER bifunctional catalytic performance of amorphous manganese oxides prepared by photochemical metal–organic deposition†
Fan Bai‡
 a,
Yuxiu He‡b,
Lincheng Xuac,
Yue Wanga,
Yan Wanga,
Zhanzhong Hao*c and
Fan Li*a
a,
Yuxiu He‡b,
Lincheng Xuac,
Yue Wanga,
Yan Wanga,
Zhanzhong Hao*c and
Fan Li*a
aFaculty of Environment and Life Sciences, Beijing University of Technology, Beijing, 100124, P. R. China. E-mail: vanadiumli@bjut.edu.cn
bBeijing Office of Metrohm China Ltd, Beijing 100085, P. R. China
cCollege of Chemistry, Baotou Teachers College, Bao Tou 014030, P. R. China. E-mail: 18438602389@163.com
First published on 18th January 2022
Abstract
Transition metal oxide nanomaterials or nanocomposites containing transition metal oxides have the potential to replace traditional catalysts for electrochemical applications, photocatalysis, and energy storage. Amorphous manganese oxide catalysts were prepared via photochemical metal–organic deposition (PMOD). Through XRD, SEM-EDS, Raman spectroscopy, FTIR spectroscopy, HRTEM-EDS, and XPS, we confirmed that amorphous manganese oxide catalysts were successfully prepared. Amorphous catalysts prepared with different photolysis times were compared in terms of their performance for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), and catalyst MnOx-PMOD48 showed the best performance because of its high Mn3+ proportion and electrochemically active surface area. MnOx-PMOD48 showed better ORR/OER performance than the crystalline MnOx and MnOx/Ti4O7 catalysts from our previous work. Following our previous work on crystalline manganese oxide catalysts, we added Ti4O7 during the PMOD process with 48 h of treatment and obtained the amorphous catalyst MnOx/Ti4O7-PMOD. MnOx/Ti4O7-PMOD was supported by Ti4O7 particles, which led to improved stability. The ORR/OER catalytic activity of MnOx/Ti4O7-PMOD was better than that of crystalline catalyst MnOx/Ti4O7-300, which was the best crystalline catalyst in our previous work. We also compared lithium-oxygen batteries assembled with MnOx/Ti4O7-PMOD and MnOx/Ti4O7-300. The battery performance tests confirmed that the amorphous manganese catalyst had better ORR/OER bifunctional catalytic performance than the crystalline manganese catalyst because of its high defect state with more abundant edge active sites and more surface-exposed catalytic active sites.
1. Introduction
New material technologies play a key role in clean energy and low-carbon technologies. Thermoelectric materials, including organic, inorganic, and hybrid organic–inorganic materials, are widely considered suitable materials for thermoelectric devices.1 Nanostructured metal oxides and their hybrids have attracted significant attention from researchers for photocatalytic, anticancer, and antibacterial applications.2,3 Nanocomposite membranes are at the heart of the process for pervaporation separation techniques.4,5 Carbon neutrality is the new mission for future generations and the planet as a whole. It is increasingly clear that reducing the use of fossil energy, turning to green energy, and emphasising low-carbon lifestyles and production techniques must be the new direction for mankind. Metal–air batteries are clean, low-carbon, safe, and efficient new energy systems with the advantages of high energy density and large output power.6 However, owing to factors like poor cyclic stability, large differences in charge–discharge voltage, and slow reaction kinetics, further application of metal–air batteries is greatly restricted. Many studies have shown that the slow reaction kinetics of the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) at the air electrode limit the performance improvement of metal–air batteries.7–9 Therefore, it is urgent to develop efficient and stable ORR/OER bifunctional catalysts to promote the development of metal–air batteries.Noble metals, such as platinum and iridium, are highly efficient and widely used ORR/OER catalysts.10–13 But the scarcity and high cost of noble metals are the main reasons that restrict their further widespread use, especially for replacing traditional energy sources with clean energy sources in every aspect of our lives.14 Mixed metal oxide nanocomposites (rare-earth-based) are a target for antibacterial, electrochemical, and photocatalytic research.15 Transition metals with large storage, low cost, and relatively high catalytic performance are very suitable substitutes for noble metals.16 Co/Ni metal nanostructures have been reported as efficient photocatalysts.17 Transition metal sulfides used in supercapacitors have also attracted broad attention.18 Among these transition metals, manganese is of particular interest because of its varied valencies and its ability to form a variety of oxides.19–21 Most of them, such as MnO2, Mn2O3, and Mn3O4, have been confirmed to be efficient ORR/OER catalysts.22 In particular, Mn3+ has been found to be the key to ORR/OER bifunctional catalysis.23–25 Manganese oxides have also been used in the Fischer–Tropsch (FT) process to improve the selectivity of long-chain carbon products.26 Manganese oxide remains stable over a wide pH range at a certain potential. Therefore, manganese oxide is a potentially efficient and stable catalyst.
In our previous work, we prepared crystalline manganese oxide to test its catalytic performance.27 Catalysts used in practical applications all contain defects. Srinivas showed that a smaller crystal size leads to the occurrence of oxygen vacancies at the surface and grain boundaries, which enhances catalytic performance.17 For transition metal oxides, the increase in surface defects can provide more active sites to improve catalytic activity and ameliorate the problem of poor conductivity.28,29 It is worth studying how to control the increase in defects during the preparation process to improve the catalytic activity. The synthesis of amorphous catalytic materials, or materials with high defects, can help to provide more active sites in another way. Several physical and chemical methods have been reported for preparing 2-dimensional (2D) film nanomaterials.30 Photochemical metal–organic deposition (PMOD) has been used to successfully prepare manganese oxide amorphous materials.31,32 The PMOD strategy has also been used in the synthesis of amorphous oxides of lead, iron, cobalt, and nickel, and related studies have confirmed that the catalytic performance of amorphous catalysts is superior to that of the corresponding crystalline materials.33–37 Although the conductivity of metal oxide nanomaterials or amorphous materials can be improved by changing their bandgap energy, it is still not as good as that of metallic or carbon materials. To improve the electrical conductivity, metal oxide nanomaterials or amorphous materials can be combined with other conductive carriers. In energy storage supercapacitors and solar cells, polyaniline combines with graphene and improves the conductivity by working as an organic conductive polymer.38 Apart from improving the conductivity, SiO2 carriers have been used to improve the surface area of the nanomaterial.39
In this study, we synthesised amorphous manganese oxide catalysts using the PMOD method and introduced carbon particles to improve the electrical conductivity. Different catalysts were synthesized by controlling the irradiation time, and their phases and catalytic activities were compared to explore the essential relationship between the synthesis strategy and catalytic activity. The optimum amorphous catalyst was compared with the crystalline catalysts prepared in our previous work. The disappearance of defects from amorphous catalytic materials is an important cause of catalyst deactivation. To address this issue, we referred to our experience in the preparation of crystalline catalysts and introduced Ti4O7 as a support material. We then compare the resulting catalyst with the optimal crystalline catalyst supported by Ti4O7 from our previous work. Finally, we concluded that amorphous MnOx/Ti4O7-PMOD is the best catalyst for our system.
2. Experimental
2.1 Sample synthesis
Amorphous manganese oxides were synthesised via PMOD. Manganese(II) 2-ethylhexanoate (7.1114 g, 6% manganese, Alfa Aesar), carbon powder XC-72 (0.3000 g, >99%, Alfa Aesar), and (A) none or (B) 0.3790 g Ti4O7 (>95%, Titanium Energy Technology) were dispersed in 5 mL n-hexane (>97%, TongGuang Fine Chemicals) in a watch glass and treated with an ultrasonic cleaner for 10 min. Three watch glasses of A were placed in a dark chamber with 254 and 185 nm ultraviolet (UV) light sources and one glass each was irradiated for 24, 48, and 72 h. One watch glass of B was placed in the same chamber and irradiated for 48 h. After UV irradiation, all the watch glasses were dried in a drying oven at 60 °C for 2 h. Amorphous manganese oxide MnOx-PMOD samples prepared with different UV treatment durations were named MnOx-PMOD24, MnOx-PMOD48, and MnOx-PMOD72. The amorphous MnOx composited with Ti4O7 was named MnOx/Ti4O7-PMOD. More details are provided in the ESI.†2.2 Characterization methods
Phase analysis of the MnOx-PMOD samples was performed using X-ray diffraction (XRD, Bruker D8 advance) at a scan rate of 10° min−1 from 10° to 80°. The morphology and microstructure were characterised using scanning electron microscopy (SEM, JEOL JSM-7900) and high-resolution transmission electron microscopy (HRTEM, FEI Tecnica G2 F20). Valence state analysis of the surface was carried out by X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi) with an Al Kα radiation source. To confirm the consumption of the manganese source, Fourier transform infrared (FTIR) spectroscopy was performed using a Bruker Vertex 70 spectrometer. Raman spectroscopy was performed using a Renishaw inVia™ Qontor instrument to confirm the formation of manganese oxides.2.3 Electrochemical measurements
A three-electrode system was used in this study. A 5 mm-diameter glassy carbon (GC) controlled by a rotating ring and disk electrode (RRDE, IPS) was used as the working electrode, with a Hg/HgO reference electrode and carbon rod counter electrode. Cyclic voltammetry (CV), linear sweep voltammetry (LSV), and electrochemical impedance spectroscopy (EIS) were performed to evaluate the electrocatalytic performance. The electrode material was prepared by dispersing 2.0 mg MnOx-PMOD sample in 1 mL anhydrous ethanol and 0.5 mL 0.2% Nafion ethanol solution and then forming a slurry using an ultrasonic dispersing instrument. The electrode material slurry (20 μL) was added dropwise onto the GC working electrode using a microsyringe. The three-electrode system was installed in a customised electrolytic cell and submerged in 0.1 mol L−1 KOH solution saturated with argon for activation and then saturated with oxygen for electrochemical measurement. For chronoamperometry (CA) and polarisation curve tests, a GC piece (10 × 10 mm) was used as the working electrode. The above-mentioned formulation was used for the electrode material slurry; however, the slurry amount was changed to 200 μL, and all electrochemical tests were performed using a Metrohm Autolab PGSTAT302N electrochemical workstation without iR compensation.2.4 Li–O2 battery tests
The MnOx-PMOD sample, conductive carbon black (Super P, >99%, Alfa Aesar), and an agglomerant (polyvinylidene fluoride, PVDF) were mixed in a mortar at a mass ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:1. Subsequently, N-methyl-2-pyrrolidinone (>99%, Aladdin) was added to configure the slurry. Carbon paper was used as the current collector substrate to spray the slurry. The whole electrode material was dried at 120 °C for 12 h in an oven and then cut into pieces 14 mm in diameter for assembling the battery. Finally, a CR2032 coin lithium–oxygen battery was assembled in an argon-filled glove box (Braun, Lab Star) and tested in a customised pure oxygen-filled battery box for charge and discharge tests (Neware, CT-4008). More details regarding the carbon paper electrode are provided in the ESI.†
:6:1. Subsequently, N-methyl-2-pyrrolidinone (>99%, Aladdin) was added to configure the slurry. Carbon paper was used as the current collector substrate to spray the slurry. The whole electrode material was dried at 120 °C for 12 h in an oven and then cut into pieces 14 mm in diameter for assembling the battery. Finally, a CR2032 coin lithium–oxygen battery was assembled in an argon-filled glove box (Braun, Lab Star) and tested in a customised pure oxygen-filled battery box for charge and discharge tests (Neware, CT-4008). More details regarding the carbon paper electrode are provided in the ESI.†
3. Results and discussion
3.1 Phase identification and morphology analysis
The blue, red, and black curves in Fig. 1A show the XRD patterns of the amorphous manganese oxide MnOx-PMOD samples obtained with different UV light decomposition times. Protrusions were observed around 2θ = 28.9°, and no peaks were found in any of the three patterns. The samples prepared by PMOD did not show crystallisation and were still high-defect-state amorphous or primary crystallisation materials. From Fig. S1,† the absorption peaks at 2961 cm−1, 2876 cm−1, 2937 cm−1, and 2853 cm−1 can be assigned to manganese(II) 2-ethylhexanoate from the organic manganese source used herein.35,36 With increasing UV photolysis time, the intensities of the organic manganese absorption peaks decreased, indicating that the organic manganese source was consumed. We found a peak at approximately 650 cm−1 in all three Raman spectra, as shown in Fig. S2.† Compared with the standard Raman spectrogram, this peak corresponds to the Mn–O bond, and the peak intensity increases with increasing UV photolysis time. The FTIR and Raman results together show that the organic manganese source was converted to amorphous manganese oxides.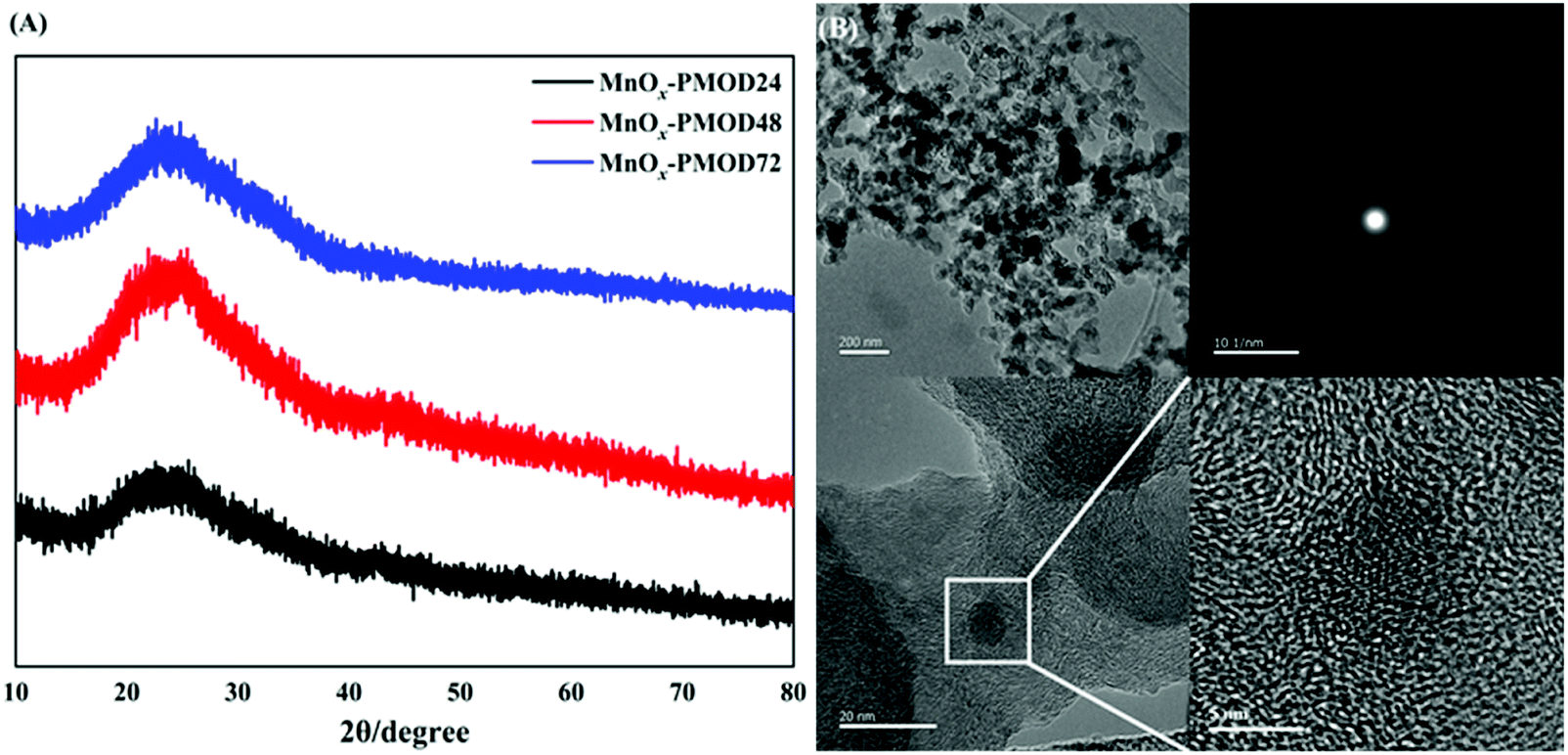 | ||
| Fig. 1 (A) XRD patterns of MnOx-PMOD24 (black), MnOx-PMOD48 (red), MnOx-PMOD72 (blue). (B) TEM and HRTEM images of MnOx-PMOD48 after ultrasonic dispersion. | ||
The SEM images in Fig. S3† show the microstructures of MnOx-PMOD24, MnOx-PMOD48, and MnOx-PMOD72. The amorphous catalysts prepared by PMOD showed an irregular 2D morphology different from the 1D structure of crystalline manganese oxide. These surfaces had different degrees of folds and cracks. With different photolysis times, the morphologies of the catalysts showed similar film structures. For example, the energy dispersive spectrum (EDS) of MnOx-PMOD72 (Fig. S4†) shows that manganese and oxygen mainly existed in the irregular 2D lamellar morphology, while carbon mainly existed in the form of XC-72 carbon particles in the substrate. The irregular 2D structure has the advantages of mechanical peeling control, large surface area, and abundant edge active sites, which will improve the catalyst performance.40–43
In the electrochemical tests, the catalysts were modified on the surface of the GC electrode to affect the real morphology. We dispersed MnOx-PMOD48 in a Nafion–ethanol solution and performed SEM and EDS tests after natural drying. A SEM test was performed on an XC-72 sample subjected to the same treatment for comparison. The SEM images of MnOx-PMOD48 and XC-72 show that the carbon nanoparticles were stacked in the same way, as shown in Fig. S5.† The MnOx-PMOD48 nanoparticles appeared closer than those of XC-72. In other words, amorphous manganese oxide had no obvious shape characteristics after being dispersed in ethanol, and the original 2D structure disappeared. The EDS image of MnOx-PMOD48 in Fig. S6† shows that manganese and oxygen were enriched in the carbon particles. In actual electrode materials, manganese oxides can be combined with XC-72 carbon particles. XC-72 can provide support for manganese oxides, which is helpful for improving the stability of catalysts during the catalytic process and reducing the crystallisation of manganese oxides. Amorphous manganese oxides were evenly dispersed in XC-72, which contributed to its better catalytic activity.
TEM characterisation was used to further explore the microstructure and the relationship between the amorphous manganese oxides and XC-72 carbon particles, as shown in Fig. 1B. In the TEM images, the morphology of crystalline manganese oxides was not observed, and only the accumulation of particles was observed. As shown in the HRTEM images in Fig. 1B, fuzzy lattice fringes appear within a narrow range of less than 10 nm, but it is difficult to match the crystal planes of crystalline manganese oxides. We believe that this nanoregion was formed by the initial crystallisation of manganese oxides. Amorphous manganese oxides began to show an ordered arrangement, but because of their high defect density, a stable and visible lattice structure could not be formed. In addition, the corresponding selective area electron diffraction image shows an obvious diffuse halo, and no regular spot group or diffraction ring is observed, which proves that the region existed in an amorphous form. The above characterisations confirm that catalysts composed of amorphous manganese oxide were successfully prepared.
3.2 Electrochemical properties and analysis
The ORR and OER performances of the amorphous catalysts MnOx-PMOD24, MnOx-PMOD48, and MnOx-PMOD72 were compared using CV and LSV measurements, as shown in Fig. S7 and S8.† MnOx-PMOD24 showed the worst catalytic activity for the ORR, as confirmed by the CV and ORR results. The ORR onset potentials of MnOx-PMOD48 and MnOx-PMOD72 were similar, and the ORR-limiting diffusion current of MnOx-PMOD72 was the highest among the three catalysts. For the OER performance, MnOx-PMOD48 showed the lowest overpotential at 10 mA cm−2 and the highest current density in the measurement potential range. MnOx-PMOD48 showed better catalytic activity for the OER than MnOx-PMOD24 and MnOx-PMOD72. MnOx-PMOD48 was found to be the best ORR/OER catalyst among the three tested catalysts. The electrochemical active surface area of each catalyst was estimated from the electrochemical double-layer capacitance (CDL) of the catalytic surface and the specific capacitance, as shown in Fig. S9.† The CDL was calculated from the CV measurements at different scan rates, and the scan range was ±60 mV around the open circuit potential of each catalyst. The CDL calculation result for MnOx-48 was 6.135 mF, compared with 2.785 and 4.355 mF for MnOx-PMOD24 and MnOx-PMOD72, respectively. MnOx-PMOD48 had a larger electrochemically active surface area than the other catalysts, leading to better catalytic performance. Therefore, we chose MnOx-PMOD48 as the amorphous catalyst for subsequent tests.According to Zhu et al., using manganese(II) 2-ethylhexanoate as the raw material for UV photolysis should produce a mixture of MnO and Mn2O3.32 The Mn2+ in MnO is unstable at room temperature and can continue to oxidise to MnO2. Therefore, Mn2+, Mn3+, and Mn4+ may coexist on the surfaces of the as-prepared amorphous MnOx samples. Because all the generated products exist in amorphous form, the molecular formula shown in this work is convenient for characterising the valence state of manganese and the number of Mn–O bonds. The obtained valence state is not consistent with the actual form of manganese oxides.
XPS was used to determine the valence state of the MnOx-PMOD24, MnOx-PMOD48, and MnOx-PMOD72 sample surfaces. The narrow sweep of manganese was limited to a binding energy of 632–660 eV, as shown in Fig. S10.† The Mn 2p spectrum includes two main regions, which can be divided into two peaks of Mn 2p1/2 and Mn 2p3/2. Based on the NIST XPS database and the previous XRD patterns, the valence states of the MnOx-PMOD samples are identified as Mn2+(2p3/2 for 641.1 eV and 2p1/2 for 653.0 eV), Mn3+(2p3/2 for 641.6 eV and 2p1/2 for 653.3 eV), Mn4+(2p3/2 for 642.2 eV and 2p1/2 for 653.8 eV). The peak areas and ratios of the samples at different calcination temperatures are listed in Table S1† for comparing the areas of Mn2+, Mn3+, and Mn4+.
The Mn2+ ratio in the three catalysts was the highest at 24 h and decreased continuously on increasing the photolysis time to 72 h. In contrast, the Mn4+ ratio increased gradually. This result is consistent with the mechanism by which Mn2+ is oxidised to Mn4+ at room temperature. The Mn3+ ratio is unique in that the highest ratio was observed for MnOx-PMOD24, i.e. after 24 h of UV photolysis. These results, in combination with the FTIR results in Fig. S1,† confirm that the oxidation of amorphous metal continued because of incomplete photolysis of the manganese source, and the ratio of Mn3+ increased slightly from MnOx-PMOD24 to MnOx-PMOD48. Because the manganese source had completely reacted from MnOx-PMOD48 to MnOx-PMOD72, the formation of Mn3+ is ended. Because of the high stability of Mn3+, only a small amount of Mn3+ was further oxidised to Mn4+; thus, the Mn3+ ratio was only slightly reduced. In our previous work, we found that the Mn3+ ratio obviously impacted the catalytic performance.27 The electrochemical tests have proved that MnOx-PMOD48 is the best catalyst among the three tested herein.
The ORR and OER performances of the amorphous catalyst MnOx-PMOD48 and crystalline catalysts MnOx and MnOx/Ti4O7 were evaluated by LSV measurements, as shown in Fig. 2A. MnOx and MnOx/Ti4O7 are crystalline manganese oxide catalysts obtained in a previous work. The MnOx/Ti4O7 catalyst includes Ti4O7 as a catalyst support. The ORR onset potential of MnOx-PMOD48 was higher than those of MnOx and MnOx/Ti4O7, and the ORR-limiting diffusion current density of MnOx-PMOD48 was the highest. In terms of OER performance, MnOx-PMOD48 showed the lowest overpotential at 10 mA cm−2 for the OER and the highest current in the measurement potential range, whereas the OER currents of MnOx and MnOx/Ti4O7 did not reach 10 mA cm−2. This result is consistent with the fact that amorphous manganese oxides exhibit better ORR/OER performance than crystalline manganese oxides. Chronoamperometry tests were performed to compare the stability of the catalysts, as shown in Fig. 2B. After a 10000 s test at 0.68 V vs. RHE, the current of MnOx-PMOD48 decreased by 35.4% compared with the initial current. The current declined by 50.1% and 39.4% for MnOx and MnOx/Ti4O7, respectively. Therefore, amorphous manganese oxide showed improved stability, performing even better than the Ti4O7-supported crystalline catalyst MnOx/Ti4O7.
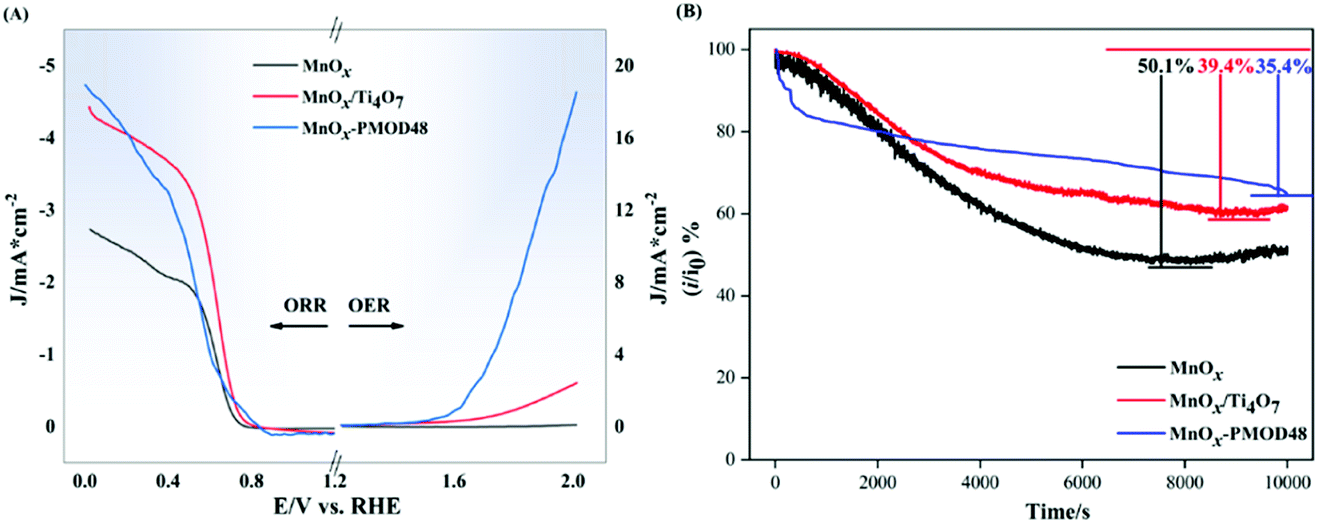 | ||
| Fig. 2 (A) OER/ORR performance of amorphous catalyst MnOx-PMOD48 and crystalline catalysts MnOx and MnOx/Ti4O7 at a rate of 10 mV s−1 after saturated with O2. (B) Chronoamperometry tests of MnOx-PMOD48, MnOx, and MnOx/Ti4O7. | ||
3.3 Influence of Ti4O7 on manganese oxides
In our previous work, adding Ti4O7 particles into a crystalline catalyst led to better performance.27 This is mainly because Ti4O7 induces the growth of manganese oxides and increases the number of active sites. The Ti4O7 particles also had a supporting effect on the MnOx catalyst. We tested the effect of Ti4O7 on the amorphous manganese oxides. The XRD, SEM, TEM, and EDS mapping results in Fig. 3 show the microstructure and elemental distribution of the amorphous catalyst MnOx/Ti4O7-PMOD. In Fig. 3A, the peaks at 2θ = 26.38° and 31.76° can be assigned to Ti4O7 (PDF#71-1428) and no peaks corresponding to the crystal planes of manganese oxide were observed. The introduction of Ti4O7 did not affect the formation of amorphous manganese oxides. The distribution of titanium was different from those of manganese and oxygen in the SEM image, as shown in Fig. 3B. The manganese oxides showed 2D growth on the Ti4O7 particles. Therefore, the surface of MnOx/Ti4O7-PMOD is more concave and convex than that of MnOx-PMOD48, which leads to more folds and introduces more marginal active sites. This result is consistent with that of our previous work. In Fig. 3C, the TEM image of MnOx/Ti4O7-PMOD shows that the Ti4O7 particle size was approximately 100 nm, and no lattice characteristics of manganese oxide were observed. According to the EDS mapping results in Fig. 3C, the distribution of titanium, manganese, and oxygen was identical, which confirms the above speculation about the growth of amorphous manganese oxide on Ti4O7 particles. Amorphous manganese oxides have low stability and are easy to crystallise. Because of the support and stabilisation of Ti4O7 particles, amorphous manganese oxide can be maintained well, which may further improve the catalytic performance and stability of MnOx/Ti4O7-PMOD.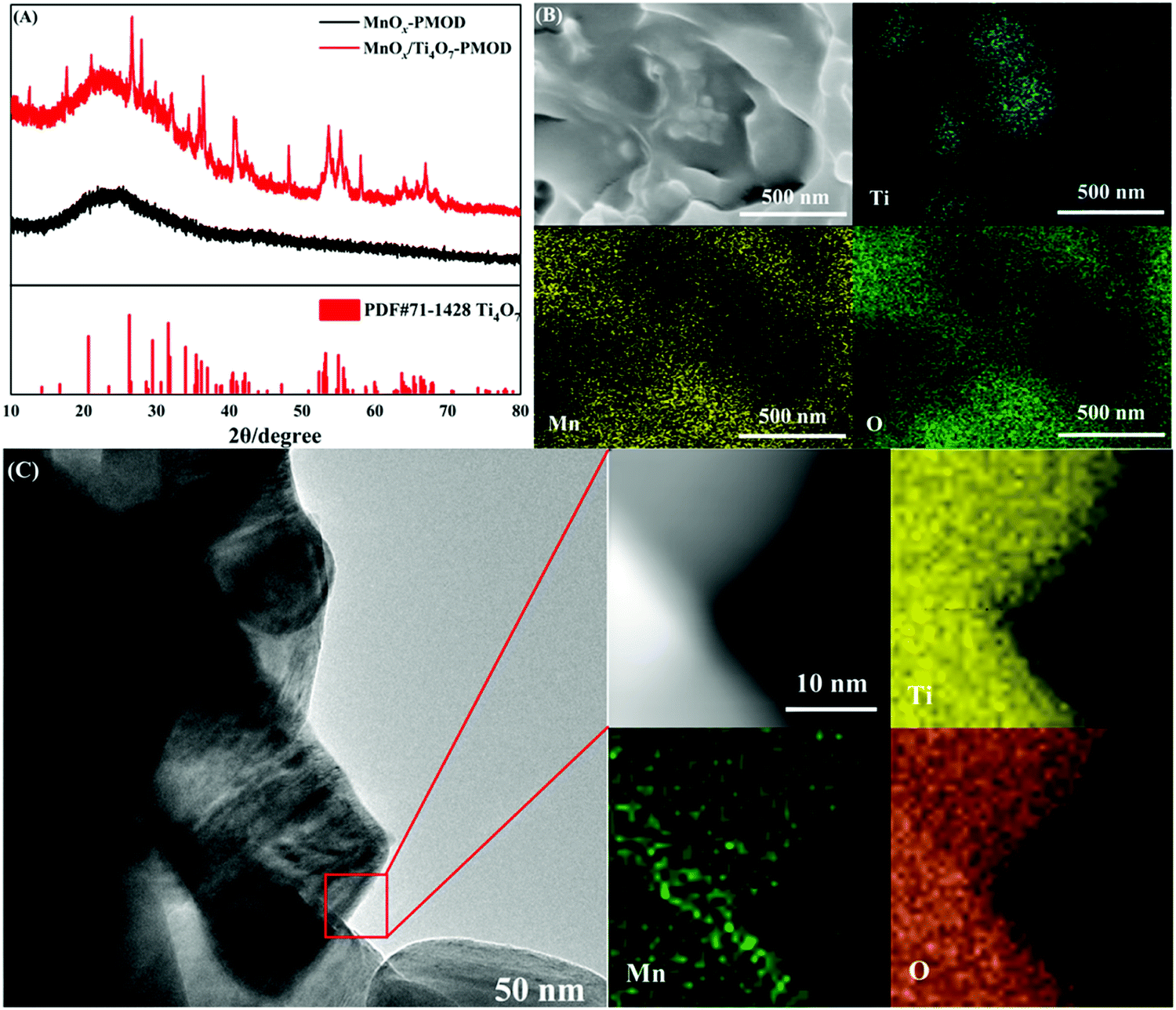 | ||
| Fig. 3 (A) XRD of amorphous catalysts MnOx/Ti4O7-PMOD and MnOx-PMOD, (B) SEM of MnOx/Ti4O7-PMOD and corresponding EDS mapping element distribution of Ti, Mn and O, (C) TEM of MnOx/Ti4O7-PMOD and corresponding EDS mapping element distribution of Ti, Mn and O. | ||
We chose MnOx/Ti4O7-PMOD as the best amorphous manganese oxide catalyst to compare with crystalline catalyst MnOx/Ti4O7-300, the crystalline manganese oxide catalyst from our previous work. As shown in Fig. 4A, the ORR halfwave potential and ORR-limiting diffusion current of MnOx/Ti4O7-PMOD were 0.75 V VS RHE and −5.21 mA cm−2, respectively, better than those of MnOx/Ti4O7-300 at 0.72 V vs. RHE and −4.34 mA cm−2, respectively. As shown in Fig. 4B, the OER performance of MnOx/Ti4O7-PMOD was also better than that of MnOx/Ti4O7-300, with a potential of 1.60 V vs. RHE at 1 mA cm−2 and an overpotential of 530 mV at 10 mA cm−2; for comparison, MnOx/Ti4O7-300 had a potential of 1.71 V vs. RHE and could not reach 10 mA cm−2. Amorphous materials exhibit better ORR/OER performance because they show a high defect state with more abundant edge active sites and more surface-exposed catalytic active sites. A comparison of the ORR/OER performance of the MnOx/Ti4O7-PMOD synthesised in this work and those from other studies is presented in Table S2.† MnOx/Ti4O7-PMOD showed ORR/OER bifunctional catalytic performance, which led us to assemble a lithium–oxygen battery with it for further comparison with the crystalline catalyst MnOx/Ti4O7-300.
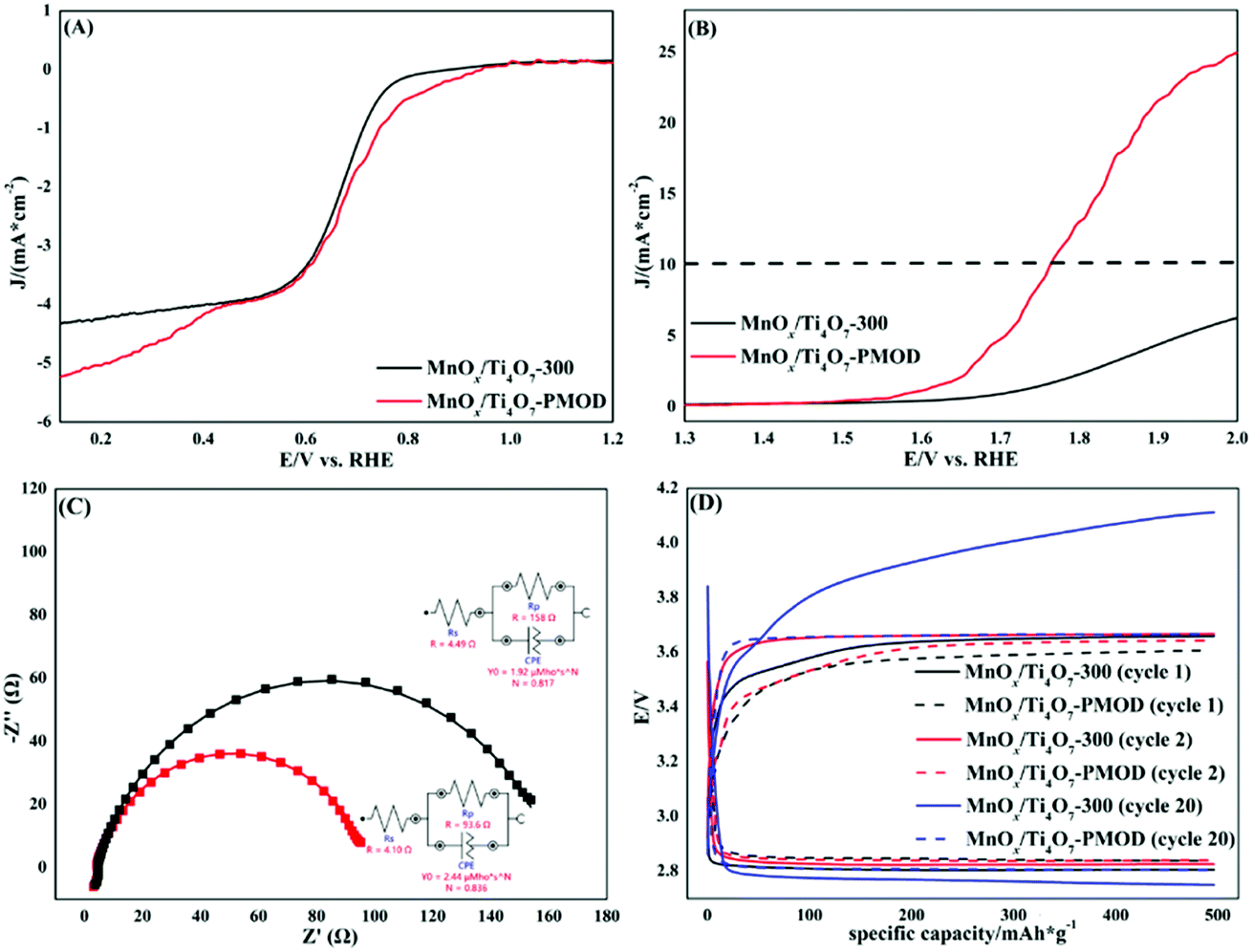 | ||
| Fig. 4 Comparison of crystalline catalyst MnOx/Ti4O7-300 (black) and amorphous catalyst MnOx/Ti4O7-PMOD (red): (A) ORR, (B) OER, (C) EIS tests and corresponding equivalent circuit of Li–O2 battery, and (D) constant current and capacity performance of Li–O2 battery. | ||
The K–L equation and RRDE curves could also be used to further confirm the catalytic performance based on the electron transfer numbers, as shown in Fig. S11–S14.† The electron transfer numbers of MnOx/Ti4O7-PMOD and MnOx/Ti4O7-300, calculated from the K–L equation, were 3.89 and 3.87, respectively. The electron transfer numbers of MnOx/Ti4O7-PMOD calculated from the RRDE curves were 3.86, 3.89, and 3.86 at 400, 900, and 1600 rpm, respectively. The average electron transfer number of MnOx/Ti4O7-PMOD, calculated from the RRDE curves, was 3.87, which is consistent with the K–L result. In contrast, the RRDE-calculated electron transfer numbers of MnOx/Ti4O7-300 were 3.39, 3.39, and 3.42 at 400, 900, and 1600 rpm, respectively. The average electron transfer number of MnOx/Ti4O7-300 from the RRDE curves was 3.40. The electron transfer number of MnOx/Ti4O7-PMOD is closer to the theoretical value of 4 than MnOx/Ti4O7-300. This means that MnOx/Ti4O7-PMOD has more efficient O2 utilisation. The production rate of H2O2 (%H2O2), calculated from the RRDE curves, also confirms this. The average %H2O2 of MnOx/Ti4O7-PMOD was 7.09% compared to 29.90% for MnOx/Ti4O7-300. More oxygen is directly reduced to OH− than to OH2−.
According to the battery performance comparison shown in Fig. 4C and D, the lithium–oxygen battery assembled with the amorphous catalyst MnOx/Ti4O7-PMOD is better than that assembled with the crystalline catalyst MnOx/Ti4O7-300, which is consistent with the ORR/OER performance results. As shown by the EIS tests in Fig. 4C, the charge transfer resistance of MnOx/Ti4O7-PMOD was 93.6 Ω, smaller than that of MnOx/Ti4O7-300 at 158 Ω. In the constant-current and capacity-performance tests, shown in Fig. 4D, the initial charge–discharge voltage platforms of the two batteries were similar. After 20 cycles, the discharge voltage, charging voltage, and platform voltage difference of MnOx/Ti4O7-PMOD were 2.81, 3.67, and 0.86 V, respectively, while the corresponding values of MnOx/Ti4O7-300 were 2.78, 4.10, and 1.32 V, respectively. After a long period of operation, the polarisation of the MnOx/Ti4O7-300 battery was more obvious than that of the MnOx/Ti4O7-PMOD battery, and the degradation of battery performance was more severe.
A comparison of the MnOx/Ti4O7-PMOD and MnOx/Ti4O7-300 catalysts is shown in Table S3.† Overall, the ORR/OER bifunctional catalytic activity of the amorphous catalyst MnOx/Ti4O7-PMOD was better than that of the crystalline catalyst MnOx/Ti4O7-300, especially in terms of OER catalysis performance. The performance of the lithium–oxygen battery assembled with MnOx/Ti4O7-PMOD was superior to that of the battery assembled with MnOx/Ti4O7-300. The amorphous catalyst prepared in this study not only has excellent ORR/OER bifunctional catalytic performance but also has good prospects for further development and potential for application in metal–air batteries.
4. Conclusions
Amorphous materials exhibit a high defect state with more abundant edge active sites and more surface-exposed catalytic active sites, which improves the catalytic performance. Amorphous materials also tend to form crystals, thereby losing active sites and reducing the catalytic activity. By adding a carrier like Ti4O7, the durability and conductivity of amorphous materials can be effectively increased. In this work, we prepared amorphous manganese oxide catalysts that show comparable ORR/OER catalytic performance to catalysts from other studies. Several conclusions can be drawn from this study.(1) Amorphous manganese oxides were prepared by PMOD and their composition, phase and morphology were proved using SEM, XPS, TEM, Raman spectroscopy, and FTIR spectroscopy. The ratio of Mn3+ was different with different UV light irradiation times, which is a key point in the performance of the amorphous manganese oxide catalyst. Among the catalysts prepared with different irradiation times, MnOx-PMOD48 with 48 h irradiation had the highest Mn3+ ratio and showed the best ORR/OER performance.
(2) MnOx-PMOD48 also showed better ORR/OER performance than the crystalline manganese oxide catalyst obtained in a previous work because of the high defect state with more abundant edge active sites and more surface-exposed catalytic active sites.
(3) Following our previous experience with the use of Ti4O7 as a catalyst support for crystalline manganese oxide, we added Ti4O7 during the synthesis of MnOx-PMOD48 and obtained amorphous manganese oxides supported with Ti4O7, denoted as MnOx/Ti4O7-PMOD. Ti4O7 particles induced the formation of manganese oxides on themselves and supported the catalyst, leading to better stability. Comparing the amorphous catalyst MnOx/Ti4O7-PMOD and crystalline catalyst MnOx/Ti4O7-300, both of which are supported by Ti4O7, we can conclude that the amorphous catalyst has better ORR/OER performance.
In the future, we may perform more work to further stabilise the amorphous catalyst for applications like batteries. The PMOD method can also be used for the synthesis of other amorphous materials. This will help us to understand nanoparticles and find more choices for the wider application of metal-air batteries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to the National Natural Science Foundation of China (NSFC 51964041, 52074016) for their financial support.References
- M. F. Sanad, A. E. Shalan, S. O. Abdellatif, E. S. A. Serea, M. S. Adly and M. A. Ahsan, Top. Curr. Chem., 2020, 378, 1–43 CrossRef PubMed.
- K. Kannan, D. Radhika, K. K. Sadasivuni, K. R. Reddy and A. V. Raghu, Adv. Colloid Interface Sci., 2020, 281, 102178 CrossRef CAS PubMed.
- K. Kannan, D. Radhika, K. R. Reddy, A. V Raghu, K. K. Sadasivuni, G. Palani and K. Gurushankar, Nano Express, 2021, 2, 010014 Search PubMed.
- D. P. Suhas, A. V. Raghu, H. M. Jeong and T. M. Aminabhavi, RSC Adv., 2013, 3, 17120–17130 RSC.
- D. P. Suhas, T. M. Aminabhavi, H. M. Jeong and A. V. Raghu, RSC Adv., 2015, 5, 100984–100995 RSC.
- F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- Y. T. Sun, X. R. Liu, Y. M. Jiang, J. Li, J. Ding, W. B. Hu and C. Zhong, J. Mater. Chem. A, 2019, 7, 18183–18208 RSC.
- D. Banham, S. Ye, K. Pei, J. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS.
- X. M. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. L. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- Y. H. Bing, H. S. Liu, L. Zhang, D. Ghosh and J. J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- K. A. Stoerzinger, L. Qiao, M. D. Biegalski and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 1636–1641 CrossRef CAS PubMed.
- M. Sevim, T. Sener and O. Metin, Int. J. Hydrogen Energy, 2015, 40, 10876–10882 CrossRef CAS.
- K. Kannan, D. Radhika, A. S. Nesaraj, K. K. Sadasivuni, K. R. Reddy, D. Kasai and A. V. Raghu, Mater. Sci. Energy Technol., 2020, 3, 853–861 CAS.
- W. Xia, A. Mahmood, Z. B. Liang, R. Q. Zou and S. J. Guo, Angew. Chem., Int. Ed., 2016, 55, 2650–2676 CrossRef CAS PubMed.
- M. Srinivas, C. V. Reddy, K. R. Reddy, P. S. Nagaraj, M. S. Reddy and A. V. Raghu, Mater. Res. Express, 2019, 6, 125502 CrossRef CAS.
- M. I. A. A. Maksoud, R. A. Fahim, A. E. Shalan, M. A. Elkodous, S. O. Olojede, A. I. Osman, C. Farrell, A. H. Al-Muhtaseb, A. S. Awed, A. H. Ashour and D. W. Rooney, Environ. Chem. Lett., 2021, 19, 375–439 CrossRef.
- K. Zhang, X. P. Han, Z. Hu, X. L. Zhang, Z. L. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- J. E. Post, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447–3454 CrossRef CAS PubMed.
- M. Pourbaix, Atlas of electrochemical equilibria in aqueous solutions pergamon press, Pergamon Press, New York, 1966 Search PubMed.
- Y. J. Xue, S. S. Sun, Q. Wang, Z. H. Dong and Z. P. Liu, J. Mater. Chem. A, 2018, 6, 10595–10626 RSC.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Norskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- J. L. Hodala, D. J. Moon, K. R. Reddy, C. V. Reddy, T. N. Kumar, M. I. Ahamed and A. V. Raghu, Int. J. Hydrogen Energy, 2021, 46, 3289–3301 CrossRef CAS.
- F. Bai, L. C. Xu, D. D. Wang, L. An, Z. Z. Hao and F. Li, RSC Adv., 2021, 11, 1524–1530 RSC.
- C. T. Campbell and C. H. F. Peden, Science, 2005, 309, 713–714 CrossRef CAS PubMed.
- H. B. Huang, S. H. Luo, C. L. Liu, Q. Wang, Z. Y. Wang, Y. H. Zhang, A. M. Hao, Y. G. Liu, J. Z. Li, Y. C. Zhai and Y. N. Dai, J. Alloys Compd., 2017, 726, 939–946 CrossRef CAS.
- R. Shwetharani, H. R. Chandan, M. Sakar, G. R. Balakrishna, K. R. Reddy and A. V. Raghu, Int. J. Hydrogen Energy, 2020, 45, 18289–18309 CrossRef CAS.
- A. A. Avey and R. H. Hill, J. Am. Chem. Soc., 1996, 118, 237–238 CrossRef CAS.
- H. J. Zhu and R. H. Hill, J. Non-Cryst. Solids, 2002, 311, 174–184 CrossRef CAS.
- L. S. Andronic and R. H. Hill, J. Photochem. Photobiol., A, 2002, 152, 259–265 CrossRef CAS.
- S. Trudel, E. D. Crozier, R. A. Gordon, P. S. Budnik and R. H. Hill, J. Solid State Chem., 2011, 184, 1025–1035 CrossRef CAS.
- R. D. L. Smith, M. S. Prevot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prevot, R. D. Fagan, Z. P. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- C. J. Zhang, S. Trudel and C. P. Berlinguette, Eur. J. Inorg. Chem., 2014, 2014, 660–664 CAS.
- K. R. Reddy, B. Hemavathi, G. R. Balakrishna, A. V. Raghu, S. Naveen and M. V. Shankar, Polymer Composites with Functionalized Nanoparticles, 2019, pp. 357–379 Search PubMed.
- M. F. Sanad, A. E. Shalan, M. A. Ahmeda and M. F. A. Messih, RSC Adv., 2021, 11, 22352–22364 RSC.
- H. Zhu, F. L. Lyu, M. L. Du, M. Zhang, Q. F. Wane, J. M. Yao and B. C. Guo, ACS Appl. Mater. Interfaces, 2014, 6, 22126–22137 CrossRef CAS PubMed.
- L. M. Wang, W. L. Chen, D. D. Zhang, Y. P. Du, R. Amal, S. Z. Qiao, J. W. Bf and Z. Y. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- M. P. Browne, Z. Sofer and M. Pumera, Energy Environ. Sci., 2019, 12, 41–58 RSC.
- K. M. Zhao, W. W. Zhu, S. Q. Liu, X. L. Wei, G. Y. Ye, Y. K. Su and Z. He, Nanoscale Adv., 2020, 2, 536–562 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08618a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |