
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and characterization of photopolymerized poly(L-lactide-co-ε-caprolactone-co-N-vinyl-2-pyrrolidone) network as anti-biofouling materials
Shuyin Zuoab,
Xiaorong Lanb,
Yong Wang ab,
Sai Li*a,
Zhonglan Tang*bc and
Yunbing Wangb
ab,
Sai Li*a,
Zhonglan Tang*bc and
Yunbing Wangb
aSchool of Chemical Engineering, Sichuan University, China
bNational Engineering Research Center for Biomaterials, Sichuan University, China. E-mail: tang.zhonglan@scu.edu.cn; Tel: +86-28-6423-2936
cInstitute of Regulatory Science for Medical Device, Sichuan University, China
First published on 21st March 2022
Abstract
The anti-biofouling properties have important applications in the medical field. In this study, cross-linked networks were prepared by photopolymerizing two synthetic macromonomers, including fumaric acid monoethyl ester (FAME) functionalized, three-armed poly(L-lactide) prepolymers (3-PLLA-F) and poly(ε-caprolactone) prepolymers (2-PCL-F), with N-vinyl-2-pyrrolidone (NVP) as the diluent. The prepared networks were characterized by their thermal properties, mechanical properties, cytotoxicity experiments and anti-biofouling properties. The Young's modulus and tensile strength of networks decreased by increasing PCL content. In contrast, the elongation of networks significantly increased. Moreover, no obvious cytotoxicity was observed, and the adhesion of L929 fibroblasts and platelets was resisted. Combined with Digital Light Processing technology (DLP) in the future, the designed polymer network could potentially be commercial in the field of biological anti-fouling materials.
1. Introduction
Anti-biofouling properties play critical roles in many medical applications such as medical implants, biosensors and separation membranes.1 Research and development of anti-biofouling materials for reducing the side effects of implant materials in the healing process, such as inflammation, thrombosis and blood coagulation, has important value in clinical applications. So far, a variety of biocompatible polymers have been synthesized and developed, including polyvinyl alcohol, poly(N-vinyl-2-pyrrolidone), poly(2-oxazoline), polyethylene glycol and zwitterionic polymers.2These polymers possess strong anti-biofouling capability and good biocompatibility. However, most of the molecular chains were polyacrylate or polyacrylamide, which are not biodegradable. These materials could not be metabolized in the body, then remain in the body to form foreign bodies, which would disturb the metabolism of normal cells. One of the future development directions of anti-biofouling materials is to develop biodegradable antifouling materials to expand their research and application in the biomedical field. Biodegradable antifouling materials could be obtained by introducing antifouling functional molecules into biodegradable matrix molecules (such as aliphatic polyesters, polycarbonates). I. Dueramae et al. synthesized a novel polyurethane with high mechanical properties and non-toxic degradation products. Poly(carboxymethyl betaine) was interpenetrated in novel polyurethane materials, to achieve additionally anti-biofouling property. The novel materials would be expected to be used in aneurysm and postoperative adhesion prevention.3 Guoxiong Dai et al. prepared biodegradable copolymer with good antifouling efficacy for more than 6 months.4 The improved antifouling efficacy was attributed by the optimal main-chain degradation as well as the side-group hydrolysis.
The current medical products applied in the market are not suitable for personalized medical needs, because most of them were manufactured by the technology of assembly-line production and then put into use. However, for the implant material in vivo, personalized product could reduce the additional adverse effects owing to a high-precision size at the implant site and the most matched mechanical properties. Recently, 3D printing technologies have been applied in many biomedical material productions. Among 3D printing technology, Digital Light Processing technology (DLP), which possesses the advantages of higher printing accuracy and faster printing speed rather than Fused Deposition Modeling (FDM), is based on polymerizing liquid photosensitive resin layer by layer. Robert van Lith et al. developed a bioresorbable biomaterial ink. Due to the polymerization by exposure to ultraviolet light, this ink could be used for fast 3D printing. With the customizable, compressed and self-expanded properties within a clinically relevant time frame, this ink could be potential to print a customizable, patient-specific stent for the patients with an arterial blockage.5 Le Hoang Sinh et al. demonstrated a novel photo-curable polyurethane resin for stereolithography, which could accurately and precisely simulate artificial skin blood vessel model.6
There are few reports on the application of resins combined with UV-curing 3D molding technology to obtain degradable materials for anti-biofouling. In our previous work, a cross-linked network of functionalized poly(L-lactic) (PLLA) with NVP was developed for application in the field of antifouling.7 PLLA was confirmed to be non-toxic, low cost in use, good mechanical properties, biocompatible and biodegradable, thus has attracted much attention in the biomedical polymer field.8–10 Poly(N-vinyl-2-pyrrolidone) (PVP) possesses strong anti-biofouling capability and good biocompatibility, which has been used as a blood plasma substitute.11–13 However, PLA is generally considered to be a brittle polymer.14 When copolymerization with poly(ε-caprolactone) (PCL), which exhibits high flexibility and low glass transition temperature, the toughness of PLA could increase.15,16 At present, poly(ε-caprolactone) derivatives prepared by introducing antifouling molecules, as antifouling materials, have been described in some literatures.17,18
In this study, to increase the toughness of the network, PCL chains was introduced in PLLA/NVP cross-linked networks. 2-PCL was end-functionalized with fumaric acid monoethyl ester (FAME). Since fumaric acid is a compound naturally found in the body, fumaric acid derivatives are attractive compounds for end-functionalization reactions without toxicity upon implantation.19 The photosensitive resin has achieved by mixing photosensitive FAME-functionalized 3-PLLA prepolymers with photosensitive FAME-functionalized 2-PCL prepolymers, reactive diluent of NVP and photoinitiator of Irgacure 2959. A complex PLLA/PCL/NVP cross-linked network was formed by UV curing. Therefore, a poly(L-lactide) material modified by poly(ε-caprolactone) was prepared, and its thermal properties, hydrophilicity, mechanical properties, biocompatibility and antifouling properties were studied.
2. Materials and methods
2.1. Materials
L-Lactide was obtained from Jinan Daigang Biomaterial Co., Ltd. (Shandong, China), which was purified by recrystallization from anhydrous ethyl acetate twice before used. Poly(ε-caprolactone) diol (2-PCL) with a nominal number-average molecular weight (Mn) of 2000 g mol−1 and Tin 2-ethylhexanoate (Sn(Oct)2) were purchased from Sigma Aldrich (Darmstadt, Germany). NVP was obtained from Chengdu Best Reagent Co., Ltd. (Chengdu, China). 4-Dimethylaminopyridine (DMAP), fumaric acid monoethyl ester (FAME), 1,3-dicyclohexylcar-bodiimide (DCC) and Irgacure 2959 (2-hydroxy-1-[4-(hydroxyethoxy)phenyl]-2-methyl-1-propanone) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Toluene (b.p. 111 °C) and dichloromethane (b.p. 40 °C) were obtained from Chengdu Kelong Chemical Reagent Factory (Chengdu, China), and distilled from calcium hydride before used.Dulbecco's modified eagle medium (DMEM), phosphate buffer solution (PBS), fetal bovine serum (FBS), and penicillin–streptomycin mixture were purchased from Thermo Fisher Scientific Co., Ltd (Waltham, MA). 4′,6-Diamidino-2-phenylindole (DAPI) and tetramethylrhodamine–phalloidin (TRITC–phalloidin) were received from Solarbio Life Sciences Co., Ltd (Beijing, China). Water used in this study was purified by a water purification system (PCDX-WJ) (Pincheng Science and Technology Ltd., Sichuan, China) unless otherwise mentioned.
All animal procedures were performed in accordance with the animal use regulations of the Animal Protection Commission of China and Sichuan University, and experiments were approved by the Medical Ethics Committee of Sichuan University (SN: K2019014).
2.2. Preparation of three-armed PLLA prepolymers
Three-armed poly(L-lactiede) prepolymer (3-PLLA) was prepared according to a previous reported.7 Briefly, 3-PLLA was synthesized by using glycerol as an initiator to ring-open and polymerize L-lactide (LLA) (Fig. 1A). Glycerol, LLA, and Sn(Oct)2 as a catalyst were dissolved in dry toluene, which was performed degassing by three freeze–pump–thaw cycles. Under argon protection, the radical polymerization was conducted at 80 °C. After 24 h, the reaction mixture was then poured into an excess amount of diethyl ether to precipitate 3-PLLA. The precipitated compound was obtained through a filter and dried under vacuum.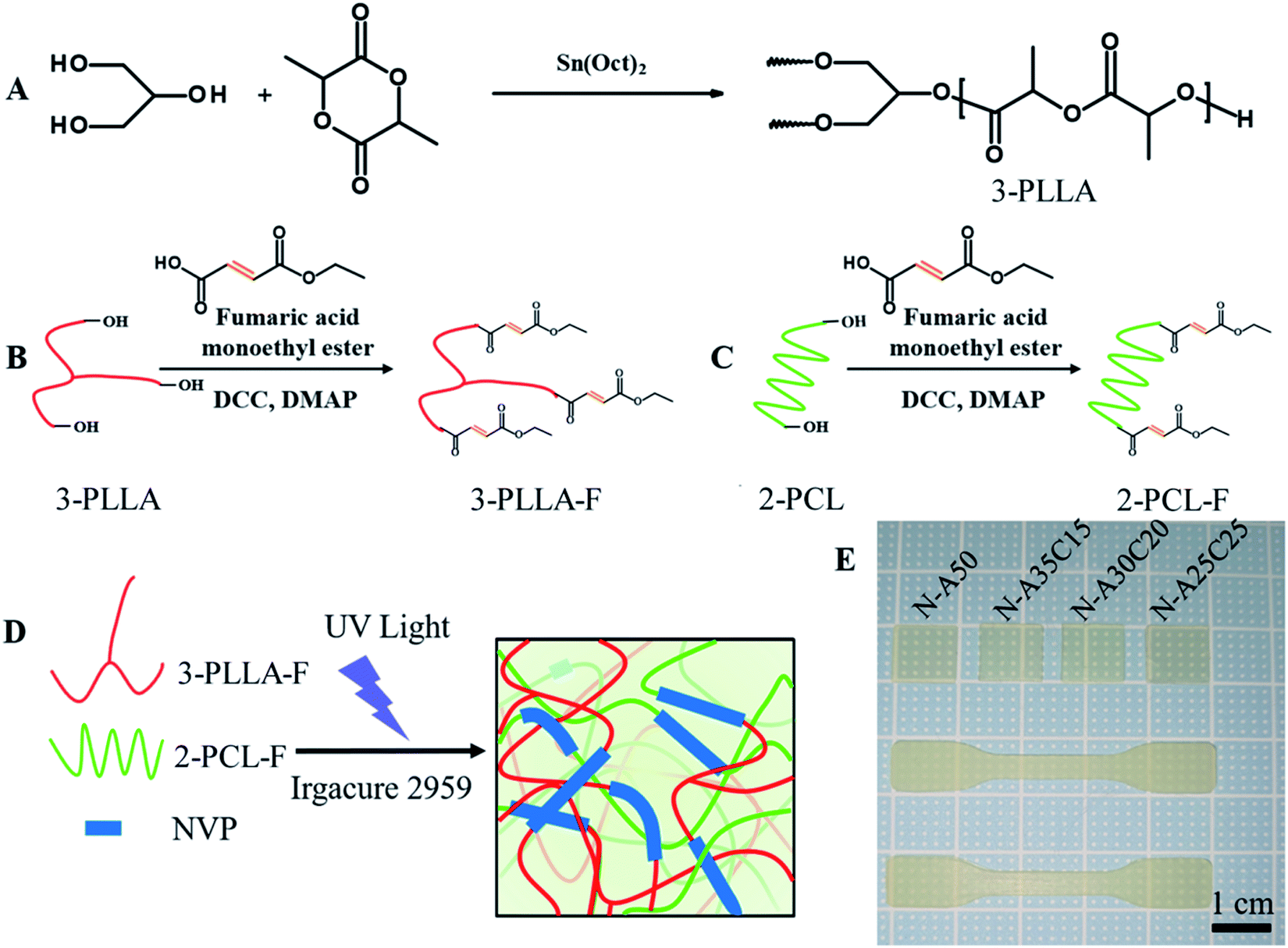 | ||
| Fig. 1 Synthetic schemes of 3-PLLA (A), FAME-functionalized 3-PLLA prepolymer (3-PLLA-F) (B) and FAME-functionalized 2-PCL prepolymer (2-PCL-F) (C). A schematic to describe the formation of cross-linked networks (D). 3-PLLA-F, 2-PCL-F and NVP polymers are covalently cross-linked upon exposure to UV light in the presence of Irgacure 2959 photoinitiator to form a highly elastic network. The specimen sheets (10 × 10 × 0.5 mm) and the dumbbell-shaped test specimen (50 × 10 × 1 mm) from prepared resins were obtained (E). | ||
2.3. Preparation of FAME-functionalized 3-PLLA and 2-PCL prepolymers
FAME-functionalized 3-PLLA (3-PLLA-F) and 2-PCL (2-PCL-F) prepolymers were prepared via esterification reaction between hydroxyl groups at the terminal of oligomer chains and carboxyl group of FAME, respectively (Fig. 1B and C). The 2-PCL or 3-PLLA were dissolved in dry DCM with a three-necked round-bottom flask, which was connected with two-way stopcocks and a drooping funnel. The reaction mixture was stirred at 0 °C for 30 min under argon atmosphere. Then, an exceeded amount of FAME was added into the mixture. After complete dissolution, a DCM solution containing DCC as a coupling agent and DMAP as a catalyst was added dropwise to the former stirred solution at 0 °C. After being stirring under argon protection for 48 h, the solution was filtered to remove dicyclohexylurea and subsequently purified by precipitation in cool ether, filtration and vacuum drying.2.4. Resin formulation and network preparation
3-PLLA-F and 2-PCL-F were diluted with NVP (50 wt%) in different weight ratios. Irgacure 2959 (5% of total mass) was added to the resin as a photoinitiator. After 30 min vacuum degassing, the resultant resin was carefully spread on a Teflon® mould. A cross-linked network was formed (Fig. 1D) by irradiating specimens cast with 365 nm UV light (INTELLIRAY 600, intensity 50 mW cm−2) for 180 seconds. After exposing, the samples were extracted with alcohol three times for 10 minutes each to strip soluble impurities and dried at 60 °C overnight. In this study, the dumbbell-shaped test specimen (50 × 10 × 1 mm) and the specimen sheets (10 × 10 × 0.5 mm) were prepared. Throughout this paper, all networks are labelled as N-AXCY, in which X stands for the content of 3-PLLA-F of networks and Y for 2-PCL-F. For example, N-A25C25 stands for the network obtained after photo-polymerisation of PLLA-F (25 wt%), PCLD-F (25 wt%) and NVP (50 wt%).2.5. Chemical characterization of prepolymers
The Mn of 3-PLLA and 3-PLLA-F, and their polydispersity (Mw/Mn) were measured by a gel permeation chromatography system (GPC, Agilent 1260 Infinity). The powder of 3-PLLA and 3-PLLA-F were dissolved in chloroform solution. The calibration curve was prepared with a series of monodisperse polystyrene standards, while the chloroform (HPLC grade) was used as the eluent. The GPC analysis was performed with 30 μL polymer solution at a flow rate of 1 ml min−1 at 35 °C.Proton nuclear magnetic resonance (1H NMR) spectra of 3-PLLA, 3-PLLA-F, 2-PCL and 2-PCL-F were monitored by an AV III HD 400 MHz NMR spectrometer (Bruker, Germany). All samples were dissolved in deuterated chloroform. The degrees of functionalization of products were measured from the spectra. A total of 32 scans were acquired for each 1H NMR spectra. Chemical shifts (δ) were given in ppm with respect to tetramethylsilane. Unless otherwise indicated, all were run at room temperature (24 ± 1 °C).
To assess further whether FAME was integrated in both ends of the polymer chain, Fourier Transform Infrared Spectrometer (FTIR spectrometer) with attenuated total reflection mode was used on 3-PLLA, 3-PLLA-F, 2-PCL and 2-PCL-F. Each sample of polymer power mixing with 10 mg of KBr was compressed to prepare pellet. The pellets were placed into the holder of FTIR spectrometer (INVENIO R Bruker, Germany), and 16 scans with a resolution of 4 cm−1 were performed. The data was analyzed using OriginPro 8 software.
2.6. Thermal characterization of prepolymer
Differential scanning calorimetry (DSC) was performed to study the thermal properties of the prepared networks with the TGA/DSC 3 + (Mettler Toledo, Switzerland) in a nitrogen atmosphere. In order to eliminate the thermal history, all of samples (10–15 mg) were heated from 30 to 180 °C at a heating rate of 10 °C min−1, and then kept isothermal for 5 min, cooled to 30 °C at 10 °C min−1. After 5 min, a second heating scan was recorded under nitrogen gas flow rate of 50 ml min−1.2.7. Characterization of polymer networks
| Mass loss (%) = (W0 − W)/W0 × 100 |
MFCs were seeded by adding 100 μL cell suspension in 96-well plates (3000 cells per well) and cultured for 6 h. After MFCs were confirmed to adhere on the surface of well, the culture medium was replaced by the sample extract with FBS. MFCs were continued to culture for 1 d, 3 d and 5 d. MFCs on the plate were counted at each time point by using the Cell Counting Kit-8 (CCK-8) assay. To study the morphology of MFCs under the sample extract, MFCs were cultured in 24-well plates with a similar procedure. Namely, MFCs were seeded in 24-well plates with same cell density, and cultured for 6 h. Then the sample extract with FBS was used to culture the cells instead of original medium. Cell morphology was monitored by using an inverted light microscope. High molecular weight polylactide (HW-PLA) sheets and tissue culture polystyrene (TCPS) were used as a control sample.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well and incubated for 24 h and 72 h in a cell incubator, respectively. After this time period, cells were fixed by formalin (4% paraformaldehyde) and permeabilized using 0.5% Tiriton X-100. The specimens were stained using DAPI and TRITC–phalloidin. Fluorescent images were recorded by using a confocal laser scanning microscope (CLSM) (Nikon A1R MP+). Then, the specimens were observed by SEM (JEOL, JSM-7500F, Japan) followed by fixation, dehydration, freeze-drying treatment and gold spray treatment.
000 cells per well and incubated for 24 h and 72 h in a cell incubator, respectively. After this time period, cells were fixed by formalin (4% paraformaldehyde) and permeabilized using 0.5% Tiriton X-100. The specimens were stained using DAPI and TRITC–phalloidin. Fluorescent images were recorded by using a confocal laser scanning microscope (CLSM) (Nikon A1R MP+). Then, the specimens were observed by SEM (JEOL, JSM-7500F, Japan) followed by fixation, dehydration, freeze-drying treatment and gold spray treatment.3. Results and discussion
3.1. Preparation of 3-PLLA
3-PLLA was prepared by ring-opening polymerization of L-lactide with glycerol as a trifunctional initiator. The chemical structure of the synthesized polymer was characterized by 1H-NMR. As shown in Fig. 2A, two sharp proton peaks at δ = 5.16 ppm and δ = 1.58 ppm belonged to the methylene (3) and methyl (4) of the L-lactide repeat unit, respectively. The weak peak at δ = 4.37 ppm was attributed to the terminal hydroxyl group of 3-PLLA (5′). These results indicated that PLLA oligomers were successfully synthesized. The degree of polymerization of 3-PLLA in each arm has been calculated as 7.65 by 1H-NMR according to the previous reported method.7 As a result, the Mw of 3-PLLA was about 3400 g mol−1. By a GPC system the Mn and polydispersity of 3-PLLA were determined as 4500 g mol−1 and 1.17, respectively. Such narrow molecular weight distribution has also been reported in other literatures,20–22 which has been explained that alcohol-induced ring-opening polymerization of lactones could yield star copolymers.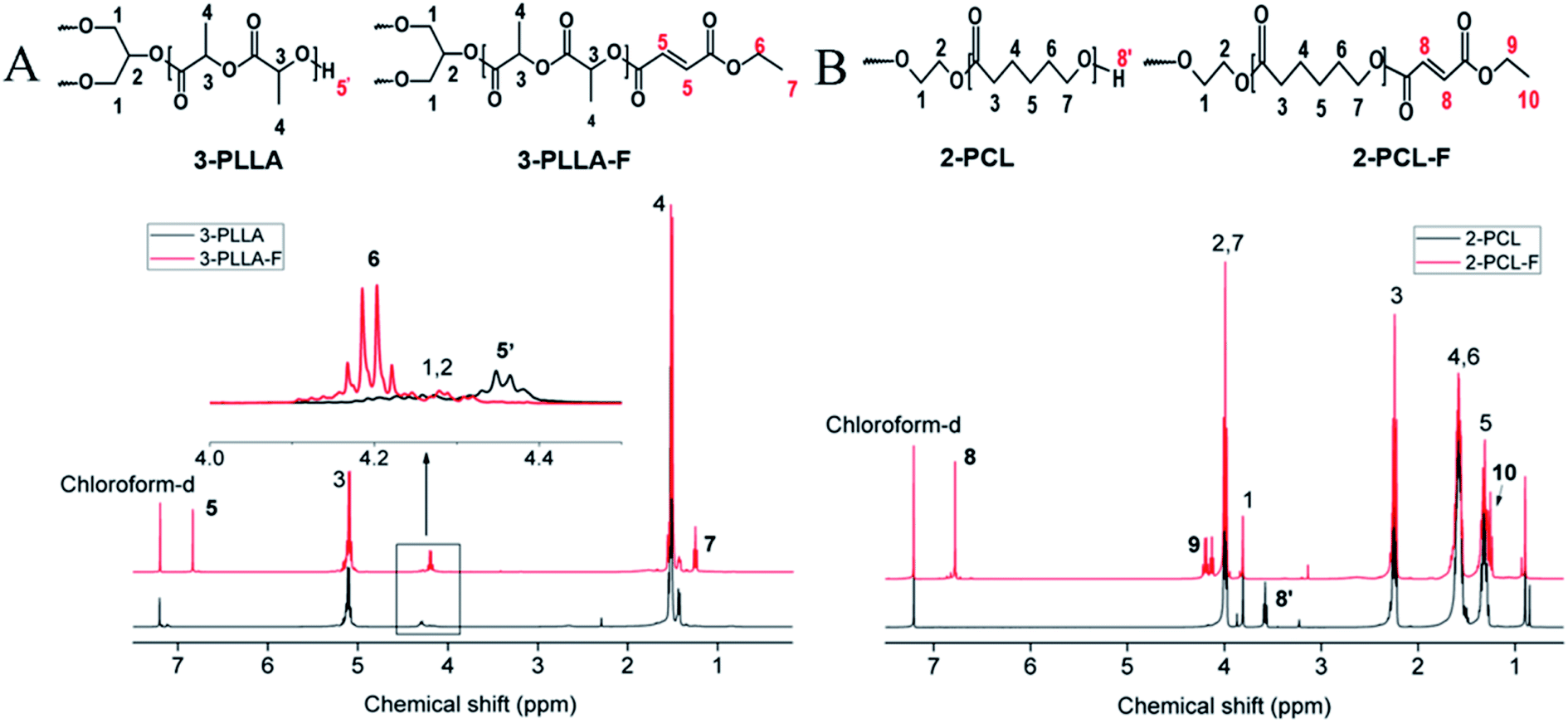 | ||
| Fig. 2 1H NMR spectra dissolved in chloroform-d. (A) The spectra of 3-PLLA and 3-PLLA-F. (B) The spectra of 2-PCL and 2-PCL-F. | ||
3.2. Preparation and characterization of prepolymer
The prepolymers of 3-PLLA-F and 2-PCL-F were prepared by coupling of 3-PLLA and 2-PCL with FAME. The 1H NMR spectra were measured to confirm the esterification reaction. As shown in Fig. 2A, the proton peak of hydroxyl (5′, δ = 4.37 ppm) belonged to 3-PLLA disappeared, and the proton peaks of double bond (5, δ = 6.83 ppm) and ethyl group (–CH2–, 6, δ = 4.25 ppm; –CH3, 7, δ = 1.32 ppm) appeared. Similarly, as shown in Fig. 2B, the proton peak of hydroxyl (8′, δ = 3.58 ppm) belonged to 2-PCL disappeared, and the proton peaks of double bond hydrogen (8, δ = 6.83 ppm) and ethyl group (–CH2–, 9, δ = 4.25 ppm; –CH3, 10, δ = 1.32 ppm) appeared. These results indicated that active double bond has been introduced at the end of polymer chain resulting in the photosensitivity prepolymer of 3-PLLA-F and 2-PCL-F. The degrees of substitution assigned to the ratio of fumarate groups substituted for hydroxyl groups was calculated to be about 97% and 91% for 3-PLLA-F and 2-PCL-F, respectively, based on the integral of the peak intensity of double bond at δ = 6.83 from the NMR spectrum. The high degree of substitution indicated the high functionalization of double bond, which could provide enough photosensitive sites in the following polymerization.To further confirm the functionalization of 3-PLLA and 2-PCL, the chemical structures of 3-PLLA-F and 2-PCL-F were characterized by FTIR spectroscopy. FTIR analysis of resultant 3-PLLA-F showed the presence of small peak at 1645 cm−1, which stands for the characteristic peak of –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– bonds of 3-PLLA-F (Fig. 3A). 3-PLLA and 3-PLLA-F had a strong peak around 1745 cm−1 due to –CO moieties. FTIR spectrum for 3-PLLA and 3-PLLA-F exhibited characteristic peaks at 2989 cm−1 (–CH2–, asymmetric and stretching) and 2946 cm−1 (–CH2–, symmetric stretching). These absorption peaks indicated that FAME was successfully introduced into 3-PLLA, which is consistent with the results of NMR.
CH– bonds of 3-PLLA-F (Fig. 3A). 3-PLLA and 3-PLLA-F had a strong peak around 1745 cm−1 due to –CO moieties. FTIR spectrum for 3-PLLA and 3-PLLA-F exhibited characteristic peaks at 2989 cm−1 (–CH2–, asymmetric and stretching) and 2946 cm−1 (–CH2–, symmetric stretching). These absorption peaks indicated that FAME was successfully introduced into 3-PLLA, which is consistent with the results of NMR.
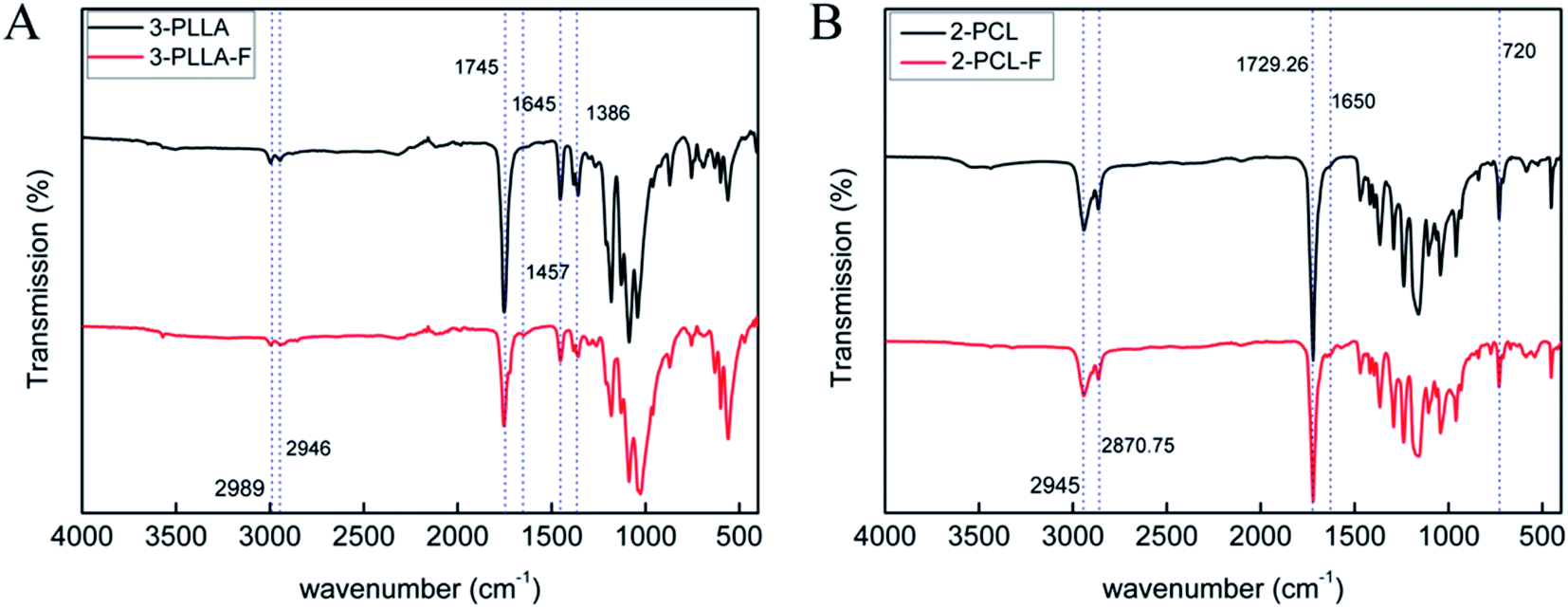 | ||
| Fig. 3 FTIR spectra. (A) The spectra of 3-PLLA and 3-PLLA-F. (B) The spectra of 2-PCL and 2-PCL-F. | ||
Similarly, FTIR spectrum of 2-PCL and 2-PCL-F was shown in Fig. 3B. FTIR spectrum of the 2-PCL-F exhibited characteristic peaks at 1650 cm−1 (–CHCH–, stretching), 2945 cm−1 (–CH2–, asymmetric and stretching), 2870 cm−1 (–CH2–, symmetric stretching) and 1729 cm−1 (–CO, stretching), indicating that 2-PCL-F was successfully synthesized, which is consistent with the results of NMR.
3.3. Preparation and characterization of polymer networks
Photosensitive resin was prepared by mixing 3-PLLA-F, 2-PCL-F, NVP as active solvent and Irgacure 2959 as a biocompatible initiator. Upon irradiation of UV-light, Irgacure 2959 molecules dissociate into radicals, which initiate the addition photopolymerization of 3-PLLA-F, 2-PCL-F and NVP, resulting in a cross-linked network (Fig. 1D and E). Three kinds of polymer networks were prepared with different ratio of polymers, which were labeled as N-A35C15, N-A30C20, N-A25C25, and N-A50 as control.| 2-PCL-F content (wt%) | Tg (°C) | |
|---|---|---|
| a N-A35C15: the network obtained after photo-polymerisation of PLLA-F (35 wt%), PCLD-F (15 wt%) and NVP (50 wt%). N-A30C20: the network obtained after photo-polymerisation of PLLA-F (30 wt%), PCLD-F (20 wt%) and NVP (50 wt%). N-A25C25: the network obtained after photo-polymerisation of PLLA-F (25 wt%), PCLD-F (25 wt%) and NVP (50 wt%). | ||
| N-A35C15 | 15 | 68.23 |
| N-A30C20 | 20 | 68.33 |
| N-A25C25 | 25 | 66.98 |
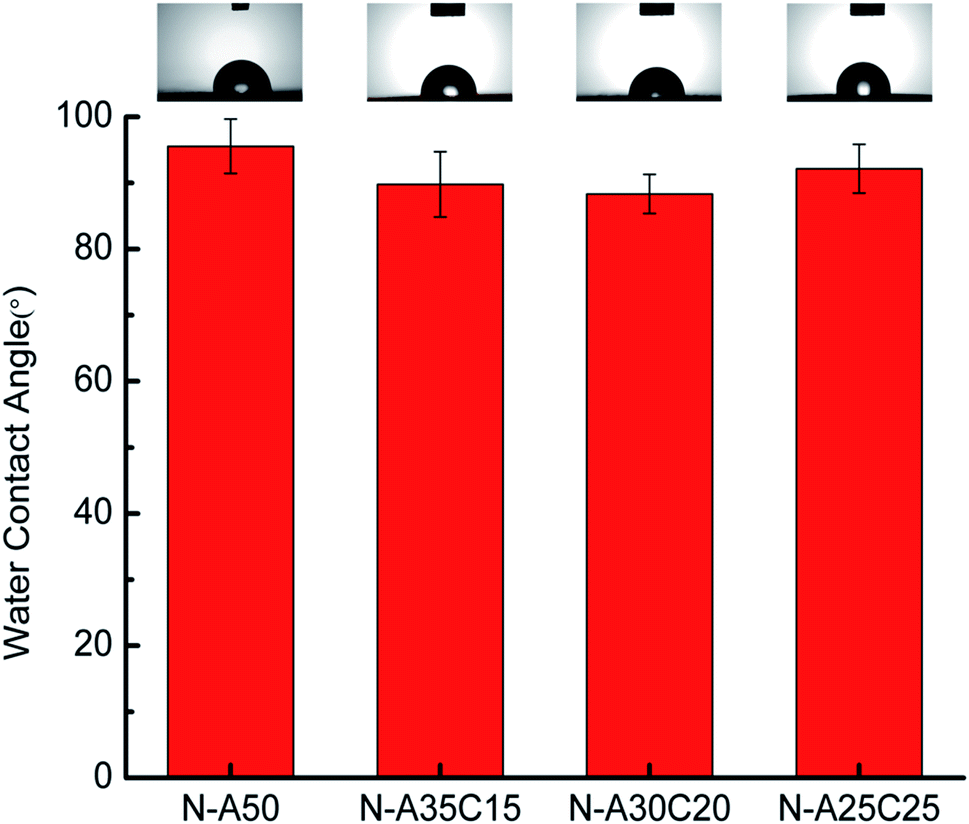 | ||
| Fig. 4 Static water-in-air contact angle measurements for N-A50, N-A35C15, N-A30C20 and N-A25C25. | ||
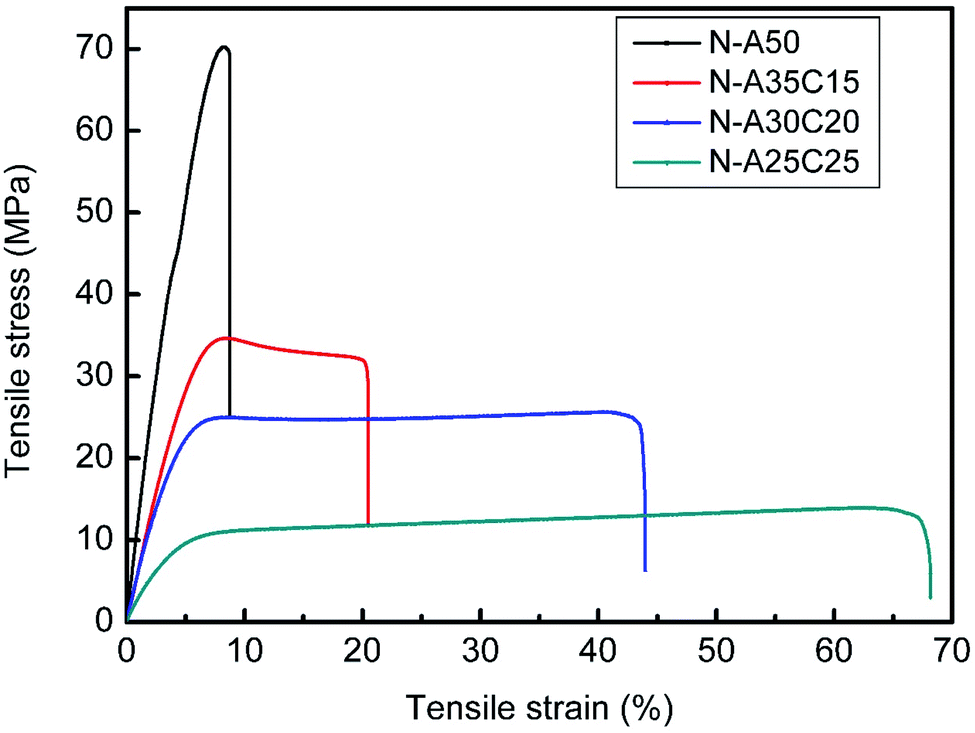 | ||
| Fig. 5 The stress–strain curve of N-A50, N-A35C15, N-A30C20, and N-A25C25 with a 1000 N load cell. | ||
| Young's modulus (Mpa) | Tensile strength (Mpa) | Elongation (%) | Fracture energy (MJ m−3) | |
|---|---|---|---|---|
| a The data in the table were expressed as mean ± standard deviation; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||||
| N-A50 | 1238 ± 22 | 71.8 ± 3.2 | 8.8 ± 1.0 | 373 ± 8 |
| N-A35C15 | 749 ± 93*** | 37.0 ± 3.9**** | 19.6 ± 4.4*** | 579 ± 9**** |
| N-A30C20 | 601 ± 6**** | 25.5 ± 0.8**** | 39.2 ± 6.7*** | 1194 ± 12**** |
| N-A25C25 | 94 ± 1**** | 13.9 ± 0.1**** | 65.4 ± 3.9**** | 738 ± 1**** |
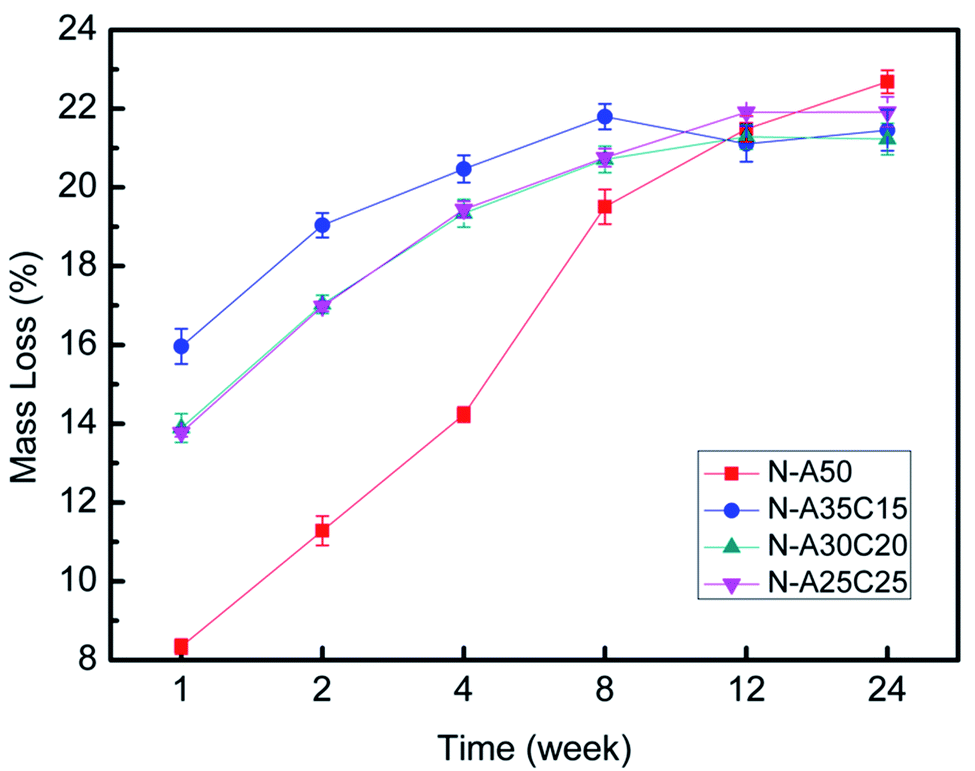 | ||
| Fig. 6 The mass loss of N-A50, N-A35C15, N-A30C20, and N-A25C25 with the degradation over time. | ||
MFCs were co-cultured with each sample extract for 24 h and 72 h, respectively. As shown in Fig. 7, the cell survival rates of all samples were higher than 90%, which indicated that the cytotoxicity of the samples was low. The growth and viability of cells were no significant difference among various extracts. Moreover, the number of cells cultured with sample extracts was gradually increasing as the time went by, as well as that on TCPS, indicating that there was no leached cytotoxic compound from the resultant networks. The morphology of MFCs cultured in the sample extracts for 1D, 3D and 5D was shown in Fig. 8. The MFCs adhered on each surface of PLLA/NVP network and PLLA/PCL/NVP network well, and showed a spread morphology as well as that on TCPS and HW-PLLA, indicating that no cytotoxicity could be found in the resultant networks.
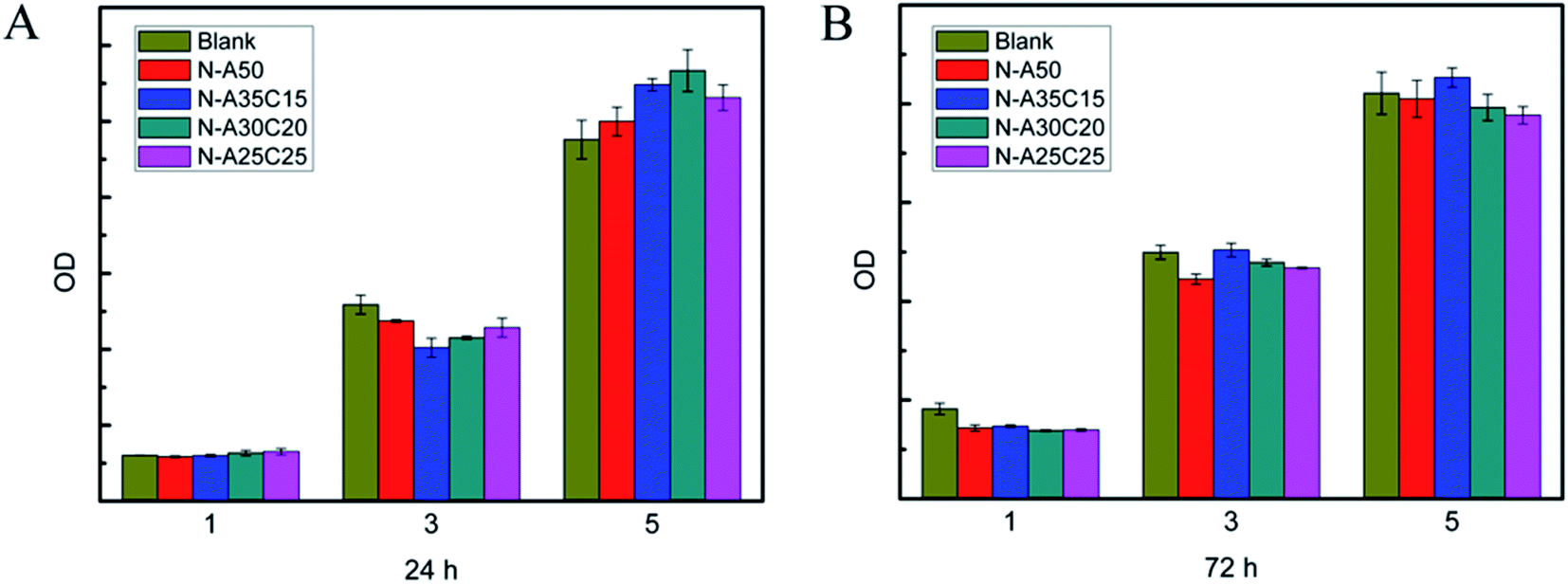 | ||
| Fig. 7 Cell viability of fibroblast MFCs cultured in the samples' leaching liquor for 1, 3 and 5 days. The samples' leaching liquor has been prepared for 24 h (A) or 72 h (B). | ||
 | ||
| Fig. 8 The morphology of MFCs cultured in the samples' leaching liquor for 1, 3 and 5 days. The samples' leaching liquor has been prepared for 24 h (A) or 72 h (B). | ||
To evaluate the cell adhesion and proliferation on the surface of resultant networks, MFCs were cultured on specimen sheets directly. Fig. 9 shows fluorescence microscopy images of DAPI and TRITC–phalloidin-stained MFCs growing on resultant networks and HW-PLA after 24 h and 72 h of incubation. After seeding on networks for 24 hours, MFCs barely adhered on the PLLA/PCL/NVP network and PLLA/NVP network. However, a few cells were observed on the surface of HW-PLA. After 72 h culture, the number of cells on the surface of HW-PLA increased significantly. In contrast, few cells were observed on the surface of N-A35C15, N-A30C20 and N-A25C25 polymer networks. The morphology of MFCs adhered on each surface has been visualized by SEM (Fig. 10). After 72 h incubation, MFCs were observed with a spindle shape on the surface of HW-PLA, which suggested that MFCs could adhere and spread on the surface of HW-PLA, and proliferate. Though a few of MFCs were found on the surface of N-A50 in Fig. 8, cells attached on the surface with a spherical shape by SEM study, which suggested a weak adhesion between cells and the surface. Moreover, MFCs not only barely adhered on the surface of PLLA/PCL/NVP network, but presented a spherical shape. These results indicated that N-A35C15, N-A30C20 and N-A25C25 polymer networks had better anti-cell adhesion performance than that of N-A50.
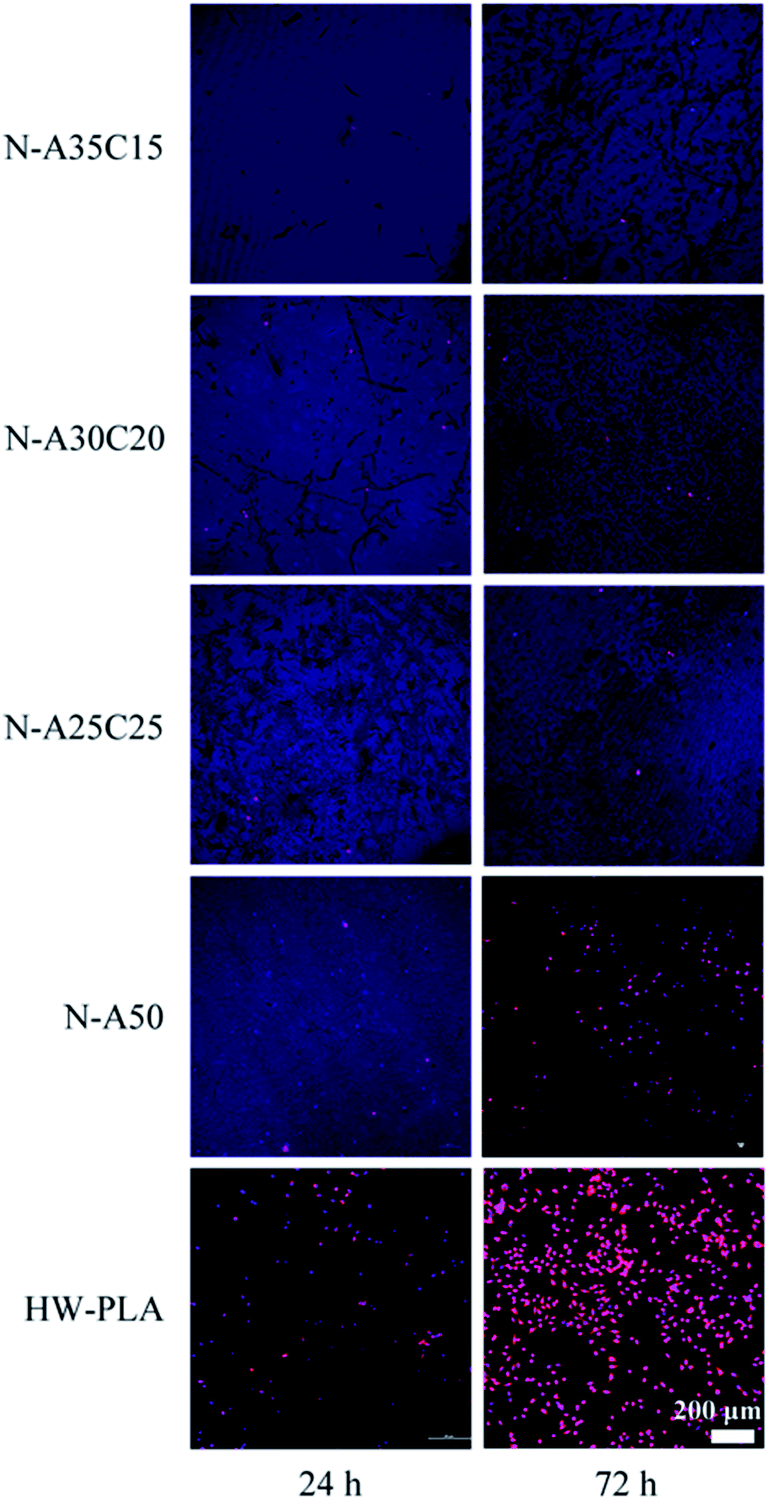 | ||
| Fig. 9 Fluorescein stain images of MFCs seeding on polymer networks for 24 h or 72 h. The cells were labeled by DAPI and TRITC–phalloidin, and images were taken by a laser-scanning confocal microscope. | ||
 | ||
| Fig. 10 SEM images of MFCs on polymer networks after 72 h. | ||
Platelet-rich plasma contains clotting factors present in the plasma as well as proteins such as fibrinogen, contained within the α-granules of platelets, which are capable promoting clot formation. Adherence and activation of platelets are key steps of coagulation. Therefore, to evaluate the platelet adhesion and coagulation properties of the resultant specimens, in vitro experiments of platelet adsorption were used as assessment criteria. N-A35C15, N-A30C20, N-A25C25, N-A50 and HW-PLA samples were co-cultured with platelets for 1 h in vitro to study the adhesion and activation of platelets. Hardly any platelets could be observed on the surface of N-A35C15, N-A30C20, N-A25C25 and N-A50 samples as visualized by SEM (Fig. 11). However, a significant number of platelets were adhered on HW-PLA surfaces. Moreover, the platelets on the surfaces of HW-PLA were observed in the severely activated and completely spread state. Obviously, the platelets produced pseudopods on smooth surfaces and tended to adhere to the surface. These results indicated that PLLA/PCL/NVP network could resist platelet absorption and activation as well as PLLA/NVP network.
 | ||
| Fig. 11 SEM images of platelets adhesion in vitro. SEM micrographs of samples after incubation in platelet-rich plasma. | ||
The statistical results were given in Fig. 12A and B showed adhesion of E. coli to each sample after 4 h exposure to a bacterial suspension. The CFUs on LB agar plate of PLLA/NVP or PLLA/PCL/NVP networks were significantly lower than that of HW-PLA, indicating that PLLA/NVP or PLLA/PCL/NVP networks exhibited better anti-bacterial adhesion. Compared with N-A50, other specimens containing 2-PCL-F exhibited slightly fewer CFUs.
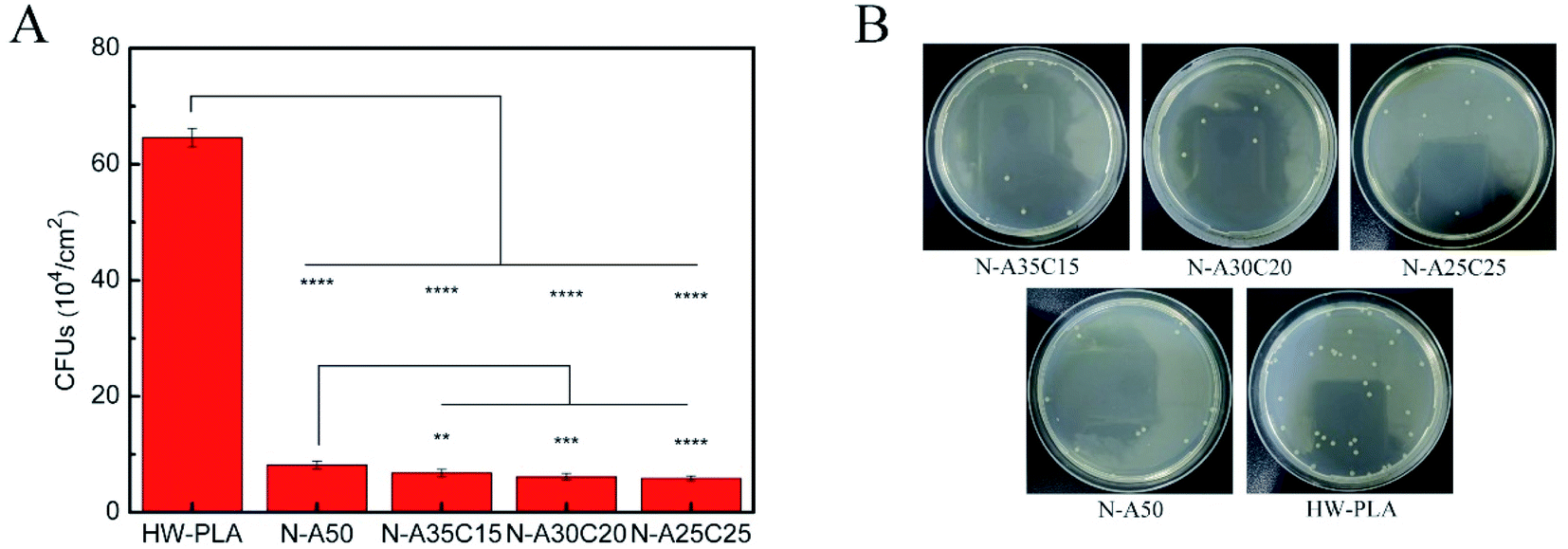 | ||
| Fig. 12 The amount of CFUs that E. coli adheres to different networks was cultured for 1 d (A). Images of E. coli colonies on the HW-PLA, N-A50, N-A35C15, N-A30C20 and N-A25C25 surfaces after spread on agar plate (B). ns **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
Based on the molecular level inside the cell, anti-cell adhesion is that the molecules on the surface of biomaterials could not be recognized with the receptor (protein) on the surface of cell membrane, so it cannot produce adhesion. Improving the hydrophilicity of surface could effectively resist the non-specific adsorption of proteins. Because the hydration layer would prevent direct contact between proteins and the surface, and strongly repel proteins close to the surface, which could avoid the protein adsorbing to the surface of material. PVP has been reported to form a stable and dense water barrier to avoid the adhesion of proteins and cells. Similarly, in this study, the hydrophilic segment of PVP content in N-A35C15, N-A30C20, and N-A25C25 as well as that in N-A50 could form a hydration layer on the surface of the material resulting in anti-biofouling surface. Moreover, the rest of hydroxyl groups surface in PLLA/PCL/NVP network could increase the hydrophily on the material as mentioned above, resulting in better anti-biofouling performance.
4. Conclusions
Photosensitive resin has been prepared by mixing 3-PLLA-F, 2-PCL-F, NVP as reactive diluent and Irgacure 2959 as photoinitiator, which could form PLLA/PCL/NVP cross-linked networks by UV irradiation. By increasing the segment of PCL content, the Young's modulus and tensile strength of resultant polymer networks decreased, but the elongation of networks increased significantly. No obvious cytotoxicity has been found in PLLA/PCL/NVP networks. Moreover, the PLLA/PCL/NVP networks could resist platelets and cells adhesion better than that of PLLA/NVP networks, because the rest of hydroxyl groups could enhance the hydrophilicity of material surface. Combined with UV-curing 3D Printing technology in the future, it is potential to develop an anti-biofouling material with the custom-fit requirements for medical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Key Research and Development Program of China (Grant No. 2017YFE0102600).References
- C. A. Del Grosso, C. Leng, K. Zhang, H.-C. Hung, S. Jiang, Z. Chen and J. J. Wilker, Chem. Sci., 2020, 11, 10367–10377 RSC.
- J. Ma, C. Ma and G. Zhang, Langmuir, 2015, 31, 6471–6478 CrossRef CAS PubMed.
- I. Dueramae, M. Nishida, T. Nakaji-Hirabayashi, K. Matsumura and H. Kitano, J. Mater. Chem. B, 2016, 4, 5394–5404 RSC.
- G. Dai, Q. Xie, C. Ma and G. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 11947–11953 CrossRef CAS PubMed.
- R. van Lith, E. Baker, H. Ware, J. Yang, A. C. Farsheed, C. Sun and G. Ameer, Adv. Mater. Technol., 2016, 1, 1600138 CrossRef.
- L. H. Sinh, K. Harri, L. Marjo, M. Minna, N. D. Luong, W. Jürgen, W. Torsten, S. Matthias and S. Jukka, RSC Adv., 2016, 6, 50706–50709 RSC.
- Y. Wang, X. Lan, S. Zuo, Y. Zou, S. Li, Z. Tang and Y. Wang, RSC Adv., 2021, 11, 20997–21005 RSC.
- L. G. Griffith, Acta Mater., 2000, 48, 263–277 CrossRef CAS.
- X. Chen, X. Wu, Z. Fan, Q. Zhao and Q. Liu, Polym. Adv. Technol., 2018, 29, 1684–1696 CrossRef CAS.
- K. Madhavan Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef CAS PubMed.
- T. Mao, G. Lu, C. Xu, H. Yu and J. Yu, Prog. Org. Coat., 2020, 141, 105317 CrossRef CAS.
- J. Jiang, L. Zhu, L. Zhu, H. Zhang, B. Zhu and Y. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 12895–12904 CrossRef CAS PubMed.
- Z. Wu, W. Tong, W. Jiang, X. Liu, Y. Wang and H. Chen, Colloids Surf. B, 2012, 96, 37–43 CrossRef CAS PubMed.
- C. Zhang, T. Zhai, L.-S. Turng and Y. Dan, Ind. Eng. Chem. Res., 2015, 54, 9505–9511 CrossRef CAS.
- G. Maglio, A. Migliozzi, R. Palumbo, B. Immirzi and M. G. Volpe, Macromol. Rapid Commun., 1999, 20, 236–238 CrossRef CAS.
- L. Wang, W. Ma, R. A. Gross and S. P. McCarthy, Polym. Degrad. Stab., 1998, 59, 161–168 CrossRef CAS.
- C. Ma, W. Zhang, G. Zhang and P.-Y. Qian, ACS Sustainable Chem. Eng., 2017, 5, 6304–6309 CrossRef CAS.
- J. Yao, S. Chen, C. Ma and G. Zhang, J Mater Chem B, 2014, 2, 5100–5106 RSC.
- J. Jansen, M. P. Tibbe, G. Mihov, J. Feijen and D. W. Grijpma, Acta Biomater., 2012, 8, 3652–3659 CrossRef CAS PubMed.
- R. F. Storey, S. C. Warren, C. J. Allison and A. D. Puckett, Polymer, 1997, 38, 6295–6301 CrossRef CAS.
- B. Sandner, S. Steurich and U. Gopp, Polymer, 1997, 38, 2515–2522 CrossRef CAS.
- M. P. K. Turunen, H. Korhonen, J. Tuominen and J. V. Seppälä, Polym. Int., 2001, 51, 92–100 CrossRef.
- J. F. Mano, Y. Wang, J. C. Viana, Z. Denchev and M. J. Oliveira, Macromol. Mater. Eng., 2004, 289, 910–915 CrossRef CAS.
- S. Jin, Y. Pu, Z. Guo, W. Zhu, S. Li, X. Zhou, W. Gao and B. He, J Mater Chem B, 2021, 9, 3863–3873 RSC.
- J. Teng, S. Bates, D. A. Engers, K. Leach, P. Schields and Y. Yang, J. Pharm. Sci., 2010, 99, 3815–3825 CrossRef CAS PubMed.
- T.-T. Li, H. Zhang, S.-Y. Huang, X. Pei, Q. Lin, S. Tian, Z. Ma and J.-H. Lin, J. Polym. Res., 2021, 28, 156 CrossRef CAS.
- H. Tsuji and Y. Ikada, J. Appl. Polym. Sci., 1998, 67, 405–415 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |