
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of hierarchical nanocrystalline β zeolite as efficient catalyst for alkylation of benzene with benzyl alcohol
Pan Zhoua,
Meng-Nan Liua,
Qun-Xing Luo abc,
Jianbo Zhangabc,
Huiyong Chenabc,
Xiaoxun Maabc and
Qing-Qing Hao*abc
abc,
Jianbo Zhangabc,
Huiyong Chenabc,
Xiaoxun Maabc and
Qing-Qing Hao*abc
aSchool of Chemical Engineering, Northwest University, Xi'an, Shaanxi 710069, China. E-mail: haoqq@nwu.edu.cn
bChemical Engineering Research Center of the Ministry of Education for Advanced Use Technology of Shanbei Energy, Xi'an, Shaanxi 710069, China
cInternational Science & Technology Cooperation Base of MOST for Clean Utilization of Hydrocarbon Resources, Collaborative Innovation Center for Development of Energy and Chemical Industry in Northern Shaanxi, Xi'an, Shaanxi 710069, China
First published on 9th February 2022
Abstract
To develop an efficient solid acid catalysts for the Friedel–Crafts alkylation reaction, especially for involving bulky molecules, the direct synthesis of hierarchical nanocrystalline β zeolites were achieved by using amphiphilic organosilane ([(CH3O)3SiC3H6N(CH3)2C18H37]Cl, TPOAC) as collaborative structure-directing agent (SDA). The growth evolution of β crystals and the influence of TPOAC/SiO2 molar ratio on the mesoporous structure, crystal size, and acidic properties of β zeolites were investigated and discussed in detail. The characterization results reveal that intracrystalline mesopores and intercrystalline mesopores/macropores via the stacking of β nanocrystals were generated over the hierarchical β zeolites. Moreover, most of the strong acid sites were well remained compared with the conventional microporous β zeolite. Consequently, the hierarchical nanocrystalline β zeolite synthesized under the optimized synthesis conditions shows improved specific catalytic activity of acid sites (turnover number, TON) in alkylation of benzene with benzyl alcohol, which can be attributed to the integrated balance of considerable mesoporosity, accessibility of the acid sites, and well-remained strong acid sites in the hierarchical β zeolite.
1. Introduction
Friedel–Crafts (F–C) alkylation is one of the most important reactions in the field of fine chemical industry for the production of various valuable chemicals.1 Among these chemicals, benzyl aromatic compounds and their derivates are important compounds in the pharmaceutical, cosmetic, petrochemical, and dyestuff industries.2–4 However, homogeneous catalysts (such as AlCl3, FeCl3, BF3, HF, H2SO4, etc.) are commonly used in the liquid phase alkylation of aromatic compounds, which is contrary to the concept of green chemistry5 because of the excessive pollution, severe equipment corrosion and separation disadvantages.In order to overcome these problems, heterogeneous solid acid catalysts are highly desired to substitute the conventional homogenous catalysts in F–C alkylation reactions.6 Among the solid acid catalysts, zeolites have been received increasingly broad attention in both industrial and academic domains due to their tunable acidity and porous structure, large surface area, and shape-selectivity.7 Mordenite, β, and Y zeolites with large pore size have been widely studied in the alkylation of benzene with benzyl alcohol.8–11 According to previous research, it was found that the porous structure and acidity of zeolites play a crucial role in the catalytic performance in aromatics alkylation reactions.12 Specially, β zeolite is an excellent catalyst for the alkylation of benzene and benzyl alcohol. However, acidic sites inside the micropores of beta zeolite are often deactivated rapidly in the alkylation of benzene with benzyl alcohol, which can be attributed to the internal pore blockage by the bulky products and side products due to their lower turnover rate in the longer micropore channel.13,14 Therefore, introducing mesopores into the conventional sole microporous β zeolite to form hierarchical β zeolite is an effective method to solve the above problems.15–17 The hierarchical β zeolite can not only increase the accessibility of acid sites inside the micropore channel of zeolite, but also reduce the diffusion limitation of the bulky products in alkylation reaction.18,19
In recent years, a variety of methods for preparing hierarchical zeolites have been developed, such as post-treatment methods (esilication20,21 and dealumination22) and direct synthesis methods (hard template method23,24 and soft template method25,26). Among them, the mesoporous formed by the post-treatment method is likely to cause crystal defects of zeolite, to decrease the amount of strong acid sites heavily, and hardly to control the mesoporous size distribution.27 On the contrary, it has been widely reported that the soft template-directing method is an effective method for the synthesis of different zeolites with uniform mesopore size distribution, such as ZSM-5,28 β, and mordenite zeolites.29,30 The mesopores formed are mainly two different manners, which are the intracrystalline mesopores formed after template removal, and the intercrystalline mesopores formed by nanocrystalline aggregations.
Different type of reagent, such as surfactant,31–33 cationic polymer,34 and organosilane,35 have been used as mesopore-generating agents for the synthesis of hierarchical zeolites. Xiao et al.36 synthesized hierarchical β zeolite with large amount of mesoporous volume (0.17 cm3 g−1) by using cationic polymer [poly(diallyldimethyl ammonium chloride)] as mseopores directing agent. Choi et al.37 reported an efficient strategy for the synthesis of hierarchical MFI and LTA zeolites by using the amphiphilic organosilane surfactants as combinational SDA. Following this method, the nanosized or mesoporous MCM-22,38 SAPO-34,39 FAU-X,40 AlPO4-5,41 and MOR11,42 zeolites were also successfully synthesized by using an amphiphilic organosilane surfactant as an assistant SDA.
However, it should be noted that the effectivity of organosilane surfactants for the synthesis of hierarchical zeolites can be significantly influenced by the topological structure of the zeolites. Specifically, it has been reported that simply addition of organosilane surfactants in the synthesis gel of the MOR zeolite is not effective at all and it will produce impurity crystals. To resolve this limitation, a seed-assisted method was developed to synthesize hierarchical MOR zeolite using organosilane surfactants as a mesoporous SDA. It was found that the impurity crystal (GIS) will be formed in the absences the bulk MOR seed.43 In our previous work,42 we reported organosilane surfactant-directed synthesis of mesoporous MOR zeolite in the absence of the bulk seed by decreasing the alkalinity of the synthesis gel. Therefore, it is necessary to investigate the effectivity of organosilane surfactants for the synthesis of hierarchical β zeolites with BEA topological structure.
Based on these understandings, in this work, the β zeolites with tunable hierarchical meso-/microporosity structure were synthesized through one-pot hydrothermal treatment under dynamic condition, in which the commercial organosilane surfactant TPOAC and tetraethylammonium hydroxide (TEAOH) were used as the mesopore-generating and micropore SDA, respectively. The effects of TPOAC/SiO2 molar ratio on the crystallinity, porous structure, and acidity of β zeolite were systematically investigated and discussed in detail. The catalytic performances of the obtained hierarchical β zeolites with different mesoporous structures and acid properties in the alkylation of benzene with benzyl alcohol, including activity, product selectivity and TON value, were systematically evaluated and compared with those of the β zeolite with sole microporous structure.
2. Experimental
2.1 Chemicals
Colloidal silica (Ludox, AS-40, 40%), tetraethylammonium hydroxide (TEAOH, 20 wt% in water), and dimethyloctadecyl[3-(trimethoxysilyl)propyl]ammonium chloride (TPOAC, 42 wt% in methanol) were purchased from Sigma-Aldrich. Sodium aluminate (NaAlO2, 41% Al2O3), sodium hydroxide (NaOH, 96%), ammonium nitrate (NH4NO3, 99%), benzene (99.5%), and benzyl alcohol (99.5%) were purchased from Sinopharm Chemical Reagent Co. Ltd (China). All reagents were used as received without further purification.2.2 Synthesis of β zeolites
The hierarchical β zeolites were synthesized by using TEAOH and commercially available TPOAC as the micropore and mesopores SDA, respectively. In a typical experiment, 0.40 g sodium aluminate are dissolved in 13.30 g deionized water at room temperature, and then 6.01 g colloidal silica was added to the solution. After stirring the mixture at room temperature for 1 h, 5.89 g TEAOH and desired amount of TPOAC were added to give a gel molar composition of 0.06 Na2O/1 SiO2/0.04 Al2O3/0.2 TEAOH/x TPOAC/30 H2O (x = 0, 0.02, 0.04, 0.06). The gel was allowed to age under stirring for 1 h at room temperature. Finally, the reaction mixture was transferred into a 50 mL Teflon-lined stainless steel autoclave, which was rotated at 60 rpm and heated at 150 °C for needed crystallization times. After that, the autoclaves were quenched in cold water and the product was filtered and washed with deionized water to reduce the pH to 7. The product was obtained after drying at 120 °C for 12 h, and then calcined in air at 550 °C for 6 h. The calcined products were ion-exchanged with 1 M NH4NO3 three times at 80 °C for 2 h. Finally, the ion-exchanged products dried at 120 °C and calcined at 550 °C for 6 h in air to obtain Hβ. The obtained samples were designed as Hβ-x–y, in which x and y referred to the molar ratio of TPOAC/SiO2 and crystallization times, respectively.2.3 Characterizations
The crystal structure of β zeolites were measured by powder X-ray diffraction (XRD) using (SmartLab SE, Rigaku) diffractometer with Cu Kα radiation (operation at 40 kV, 40 mA, λ = 0.15406 nm). The scanning range was set between 5° and 50° in 2θ with a step size of 0.02° and the scanning rate of 4° per minute. Scanning electron microscopy (SEM) images were recorded on a Zeiss Sigma instrument with a field-emission gun operated at 5.0 kV, and all samples were coated with Pt (approximately 1 nm). Transmission electron microscopy (TEM) images were obtained using a FEI Tecnai G2F20S-TWIN electron microscope operated at 200 kV. Nitrogen adsorption/desorption isotherms were measured at −196 °C with a physisorption analyser of Micromeritics ASAP 2460 instrument. Approximately 10 mg of powder samples were outgassed at 300 °C for 12 h before the measurements. The Brunauer–Emmett–Teller (BET) equation was employed to calculate the total surface area, and the pore size distribution was determined based on the Barrett–Joyner–Halenda (BJH) method by using the data of adsorption branches. The mole ratio of SiO2/Al2O3 was measured with inductively coupled plasma optical emission spectroscopy (ICP-OES) on an Optima 7000DV (PerkinElmer) spectrometer. The NH3-TPD measurements were performed on a BELCAT II (Microtrac BEL) instruments. Prior to the NH3-TPD measurements, the catalyst (50 mg) was loaded and degassed at 550 °C for 1 h under He flow to completely remove molecules adsorbed on the samples. Then the sample was cooled to 120 °C and saturated in the gas mixture of NH3 gas (5 vol% in He) for 30 min, and flushed with He flow for 1 h at 120 °C. Ammonia desorption was carried out with increasing temperature from 120 °C to 600 °C at a constant heating rate of 10 °C min−1, and the concentration of desorbed ammonia was obtained by a thermal conductivity detector connected to the TPD system. 27Al MAS NMR experiments were performed on Bruker AVANCE III 600 spectrometer at a resonance frequency of 119.2 MHz. 27Al MAS NMR spectra were recorded on a 4 mm probe by small-flip angle technique with a pulse length of 0.5 μs (<π/12) and a 1 s recycle delay and a spinning rate of 14 kHz. The chemical shifts of 27Al MAS were referenced to 1 mol L−1 aqueous Al(NO3)3.2.4 Catalytic tests
The alkylation of benzene with benzyl alcohol (BA) were carried out in septum-sealed thick-walled glass tube with magnetic stirring. Prior to reaction, the β zeolites calcined at 550 °C in muffle furnace for 2 h to remove the adsorbed water, and the benzene and BA are dried by 5A zeolite. Typically, 7.81 g of benzene and 0.54 g of benzyl alcohol were added to a 35 mL thick-walled glass tube and mixed, and then 100 mg of the catalyst was added into the reactants. During the reaction, the oil bath temperature was controlled at 85 °C and the stirring rate was 630 rpm. Moreover, the catalytic performance of β zeolites for the alkylation of mesitylene with BA was also evaluated using same reaction conditions with that of alkylation of benzene with BA. The reaction products were analyzed by using GC with a KB-1 column (30 m × 0.25 mm × 0.5 nm) and a flame ionization detector FID (GC-9790 II, Fuli Chromatographic Analysis Co., Ltd.). Since the benzene/mesitylene was excessive in the reaction with BA, the conversion was calculated based on BA. The conversion of BA and selectivity of diphenylmethane (DP) and 2-benzyl-1,3,5-trimethylbenzene (BTMB) were calculated as follows: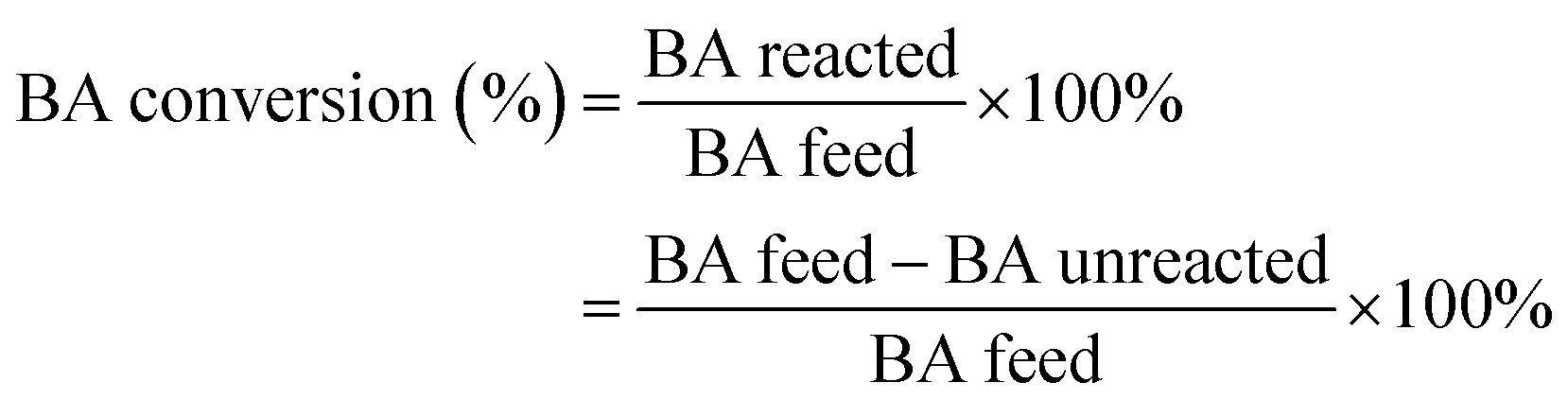
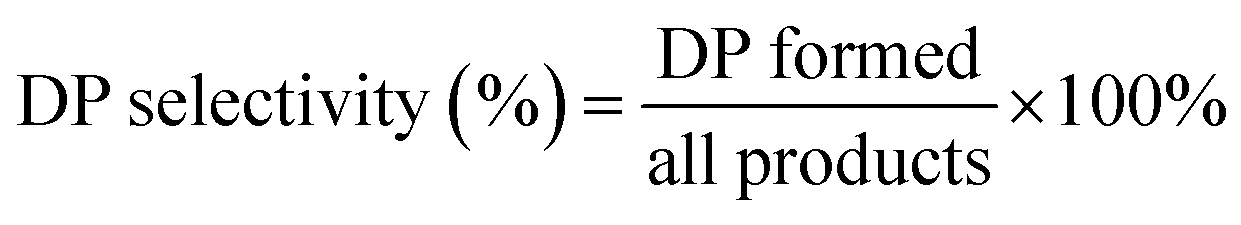
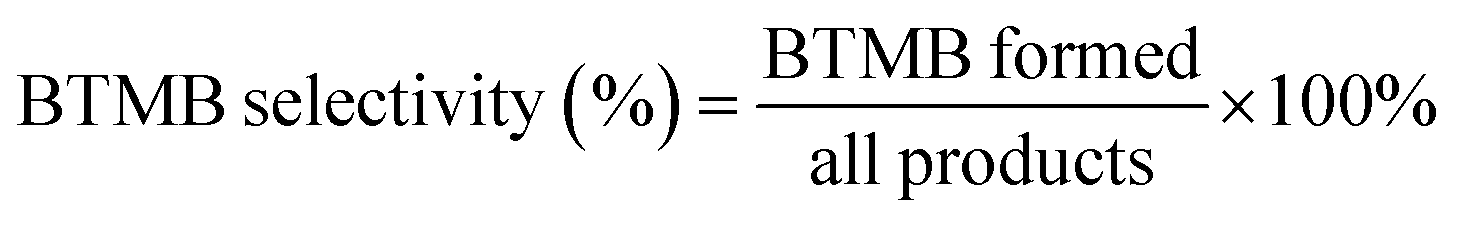
3. Results and discussion
3.1 Crystalline structure of β zeolites
The influence of TPOAC/SiO2 molar ratio in the synthesis gel and crystallization time on the crystalline structure of β zeolites were investigated. Fig. 1A shows the samples synthesized at crystallization time of 6 days. The Hβ-0–6d presents the typical diffraction pattern of BEA topology with diffraction peaks at 7.8°, 22.4°, 27.0°, 29.3°, and 43.7°, corresponding to (101), (302), (008), (306), and (526) lattice planes, respectively. Moreover, there is no impurity crystal phase in the Hβ-0.02–6d, suggesting the introduction of TPOAC into the synthesis gel cannot result in the formation of other impurity crystals. However, as the TPOAC/SiO2 molar ratio in the gel mixture is increased from 0.02 to 0.04, the intensity of the diffraction peaks for Hβ-0.04–6d are clearly decreased. Meanwhile, there is obvious amorphous silica phase (2θ range of 15–30°) in the Hβ-0.04–6d, indicating the crystallinity is significantly decreased. Furthermore, when the molar ratio of TPOAC/SiO2 increases to 0.06, there is almost no diffraction peak of β zeolite in the XRD patterns. In order to obtain the growth evolution of crystalline structure of β zeolites during the crystallization process, the crystallization time is prolonged and the XRD results of the samples are shown in Fig. 1B. As the crystallization time is increased from 6 days to 8 days, the crystallinity of Hβ-0.04–8d is obviously increased in compared with that of Hβ-0.04–6d. However, for the TPOAC/SiO2 molar ratio is 0.06, there is obvious amorphous silica phase in the Hβ-0.06–8d even though the crystallization time was prolonged to 8 days, suggesting the lower crystallinity of the synthesized sample. It should be noted that the crystallinity of Hβ-0.06–10d synthesized at crystallization time of 10 days is clearly increased and the amorphous silica phase can hardly be seen. Thus, the pure Hβ zeolites with high crystallinity can be synthesized through prolong the crystallization time when using the TPOAC as mesopores SDA. These results indicate that the crystallization rate of β zeolite is gradually decreased as increasing the content of TPOAC in the synthesis gel, but the introduction of TPOAC will not result in the generation of impurity crystals phase for the synthesis system of β zeolites.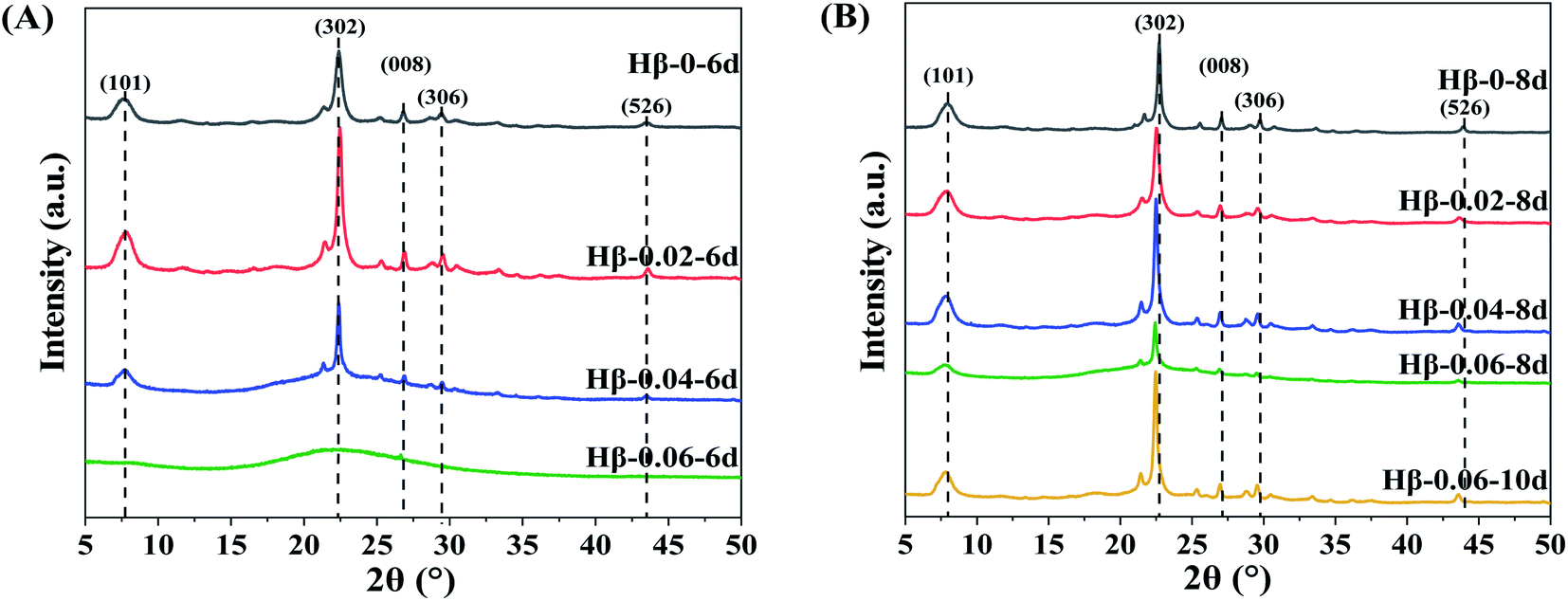 | ||
| Fig. 1 XRD patterns of Hβ zeolites synthesized with crystallization time of 6 days (A) and 8 days (B). (Hβ-0.06–10d is synthesized with 10 days). | ||
3.2 Textural properties of β zeolites
The existence and evolution of the mesoporous structure of β zeolites as the increasing of TPOAC content can be revealed by N2 adsorption–desorption isotherms. As shown in Fig. 2A, the Hβ-0–8d synthesized in the absence of TPOAC presents the type I isotherm with most of the adsorbed N2 at low relative pressures (P/P0 < 0.05), indicating the microporous structure of Hβ-0–8d. In compares with the Hβ-0–8d, the H −0.02–8d, Hβ-0.04–8d, and Hβ-0.06–10d show a type IV isotherm, together with the obvious increase of N2 adsorption in the range of P/P0 0.2 to 0.6, suggesting the formation of intracrystalline mesopores inside the β crystals due to the directing effect of TPOAC. Furthermore, the N2 adsorption in the range of relative pressure P/P0 > 0.9 for hierarchical β zeolites are increased sharply, indicating the generation of intercrystalline mesopores or macropores via the stacking of the nanocrystals of β zeolites. This can be clearly revealed by the BJH pore size distribution in Fig. 2B. The Hβ-0.02–8d present broad mesopore size distribution in the range of 2–10 nm, which indicates the hierarchical structure of β zeolites with intracrystalline and intercrystalline mesopores. We speculated that partial of the TPOAC tends to incorporate onto the external surface of the β nanocrystals with the lower concentration of TPOAC in the gel mixture. In this condition, the TPOAC acts as the mesopores-directing agent and crystal growth inhibitor to suppress the growth of the nanocrystals of β zeolites. It should be noted that as the molar ratio of TPOAC/SiO2 in the gel mixture is increased from 0.02 to 0.06, the mesopore size distribution of Hβ-0.04–8d and Hβ-0.06–10d become more uniform and narrower, which indicates that the micelles will be generated as increasing the concentration of TPOAC in the synthesis gel. The textural properties of various β zeolites are summarized in Table 1. The β zeolites synthesized in the presence TPOAC exhibits not only a considerable microporosity, but also an obviously increased mesopore volume, indicating that the Hβ-0.02–8d, Hβ-0.04–8d, and Hβ-0.06–10d are the hierarchical zeolites with micro- and mesoporous porosity. Moreover, the Hβ-0.02–8d shows the largest BET surface area and external surface area, which may be related to its optimal content of TPOAC in the gel mixture.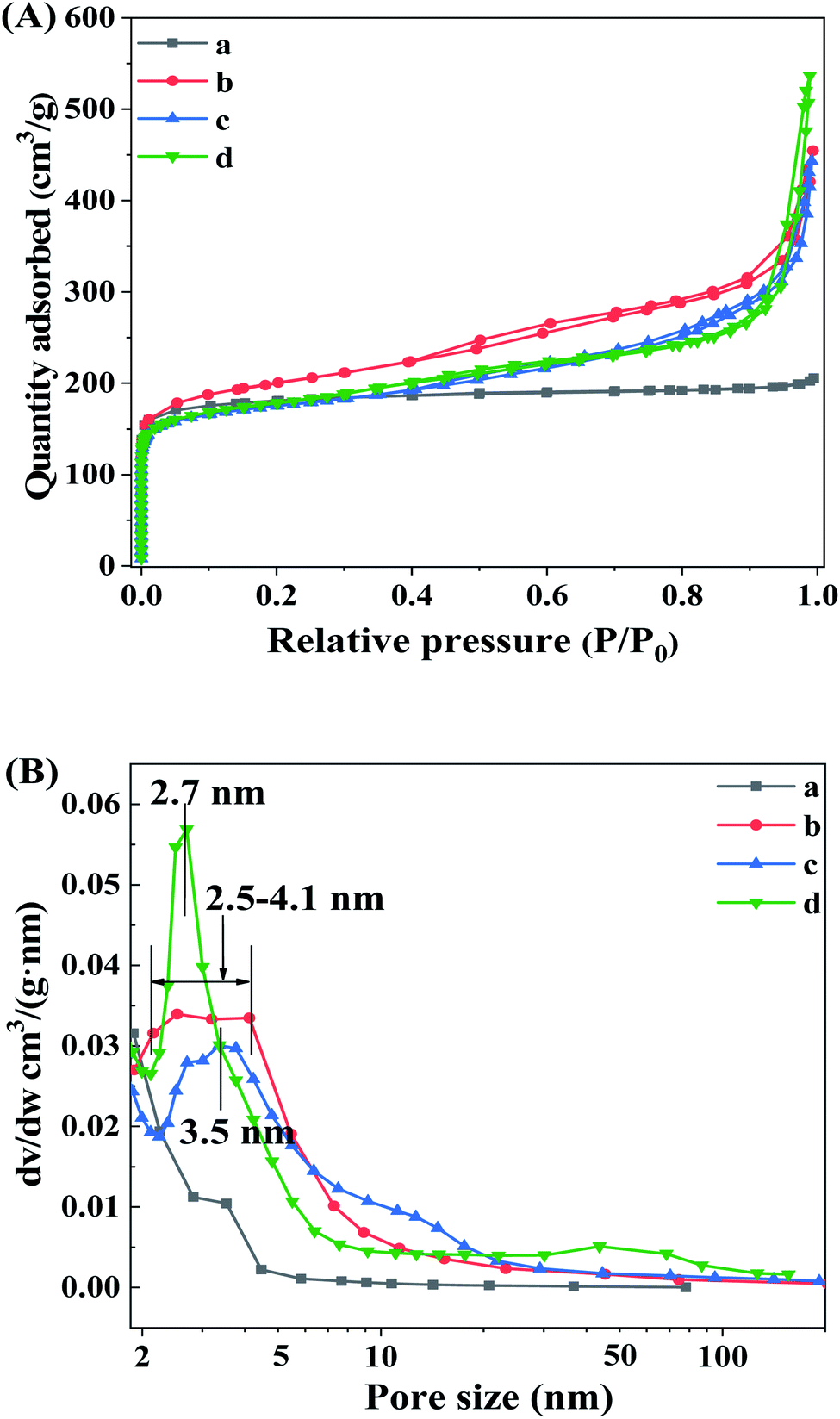 | ||
| Fig. 2 N2 adsorption–desorption isotherms (A) and BJH pore size distribution (B) of Hβ-0–8d (a), Hβ-0.02–8d (b), Hβ-0.04–8d (c), and Hβ-0.06–10d (d). | ||
3.3 Crystal morphology of β zeolites
In order to verify the morphology of β zeolites, the scanning electron microscopy (SEM) images are shown in Fig. 3. The Hβ-0–8d exhibits aggregates with particle size about 1 μm, which is formed by irregular β crystals. In contrast, when TPOAC was added into the synthesis gel, the particle size of synthesized β zeolites were clearly decreased. These observations indicate that the TPOAC not only act as a mesopores SDA, but also the long alkyl hydrophobic tail of TPOAC can restrict the agglomeration of the β crystals via incorporate onto the external surface of β crystals. | ||
| Fig. 3 SEM images of Hβ-0–8d (a), Hβ-0.02–8d (b), Hβ-0.04–8d (c), and Hβ-0.06–10d (d). | ||
TEM images were further carried out to illustrate the crystal structure and the crystal size of β zeolites. As shown in Fig. 4, micron-sized crystals can be clearly observed for Hβ-0–8d synthesized in the absence of TPOAC. In contrast, the crystal size of Hβ-0.02–8d, Hβ-0.04–8d, and Hβ-0.06–10d is decreased to 30–70 nm, which can be reasonably attributed to the inhibiting effect of TPOAC for the crystal growth. Moreover, the intercrystalline mesopores constructed by the stacking of nanocrystals can be clearly observed. However, the intracrystalline mesopores inside the nanocrystals cannot be observed in the TEM images due to the lower magnification times.
 | ||
| Fig. 4 TEM images of Hβ-0–8d (a), Hβ-0.02–8d (b), Hβ-0.04–8d (c), and Hβ-0.06–10d (d). | ||
3.4 Acidic properties of β zeolites
The amount of the acid sites and the distributions of weak and strong acid sites was evaluated by NH3-TPD method. As shown in Fig. 5, two clear NH3 desorption peaks can be observed for all the β zeolites synthesized in this work, which are assigned to the weak and strong acidic sites, respectively. From the NH3-TPD profiles, we can see that the shape and peak area of desorption peaks can be significantly influenced by the content of TPOAC in the synthesis gel. It is clear that the temperature of desorption peaks, especially the high-temperature desorption peaks, are decreased as increasing the content of TPOAC in the synthesis gel. This result indicates that the strength of the acid sites of hierarchical β zeolites is decreased significantly due to the incorporation of TPOAC into the framework of β zeolites. To clearly illustrate the amount and distribution of weak and strong acid sites for the β zeolites, the NH3-TPD profiles were deconvoluted into two peaks, and the calculated results are summarized in Table 2. The amount of total acid sites of hierarchical β zeolites is decreased compared to the Hβ-0–8d. This result can be attributed to the incorporation additional Si source derived from TPOAC, which result in the increasing of SiO2/Al2O3 ratio in the framework of hierarchical β zeolites (Table 2). Moreover, it should be noted that the amount of strong acid sites is gradually decreased as increasing the molar ratio of TPOAC/SiO2. These observations indicate that the TPOAC have clear influence on the coordination environment of Al species in the framework of β zeolites. Therefore, the 27Al MAS NMR experiments were carried out to detect the coordination state of aluminum species in the β zeolites (Fig. 6). It can be clearly observed that the spectra of Hβ-0–8d exhibits a strong peak centered at around 54 ppm corresponding to tetrahedral coordination aluminum, but the peak at around 0 ppm corresponding to octahedral coordination aluminum is almost invisible. This result indicates that there is almost no extra-framework Al in the Hβ-0–8d zeolite. On the contrary, all the hierarchical β zeolites synthesized in the presence of TPOAC shows a clear peak at around 0 ppm except the peak at around 54 ppm, indicating Al coordination in the hierarchical β zeolites mainly consisted of tetrahedral coordination with a certain amount of octahedral coordination. This observation indicates that partial of the Al atoms are found in octahedrally coordinated extra-framework positions due to the introduction of TPOAC in the synthesis gel. Thus, the decreasing of strong acid sites over hierarchical β zeolites can be reasonably attributed to the formation of extra-framework Al.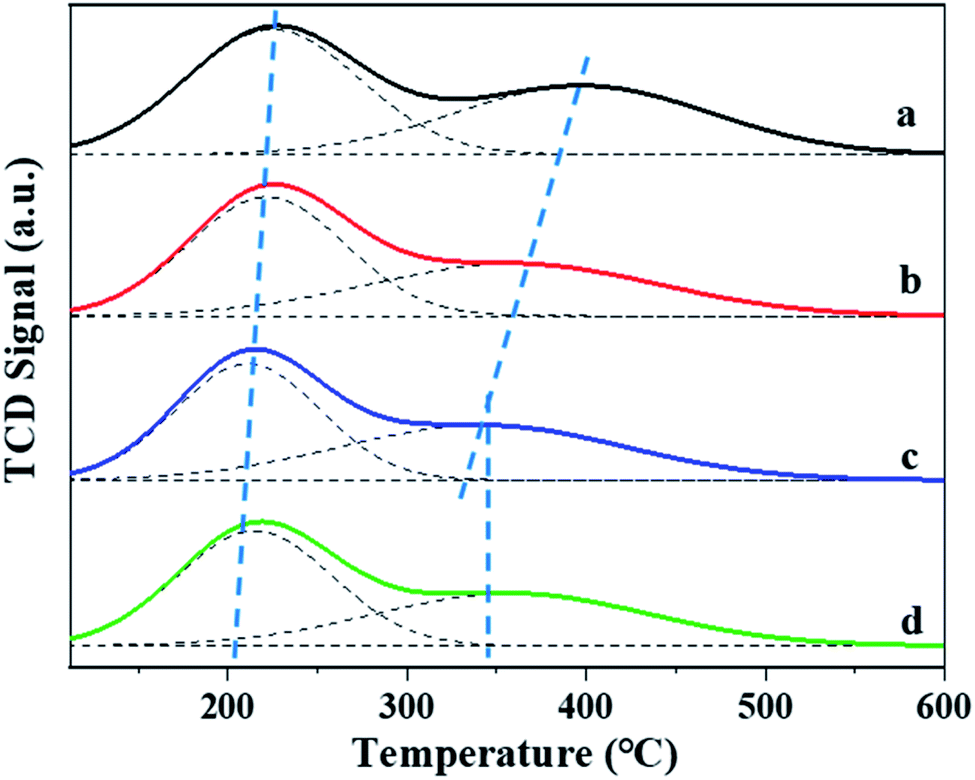 | ||
| Fig. 5 NH3-TPD profiles of Hβ-0–8d (a), Hβ-0.02–8d (b), Hβ-0.04–8d (c), and Hβ-0.06–10d (d). | ||
| Samples | SiO2/Al2O3 | Total acid sites (mmol g−1) | Weak acid sites (mmol g−1) | Strong acid sites (mmol g−1) |
|---|---|---|---|---|
| Hβ-0–8d | 27 | 1.166 | 0.642 | 0.524 |
| Hβ-0.02–8d | 30 | 1.104 | 0.608 | 0.496 |
| Hβ-0.04–8d | 34 | 0.975 | 0.505 | 0.470 |
| Hβ-0.06–10d | 32 | 0.980 | 0.541 | 0.439 |
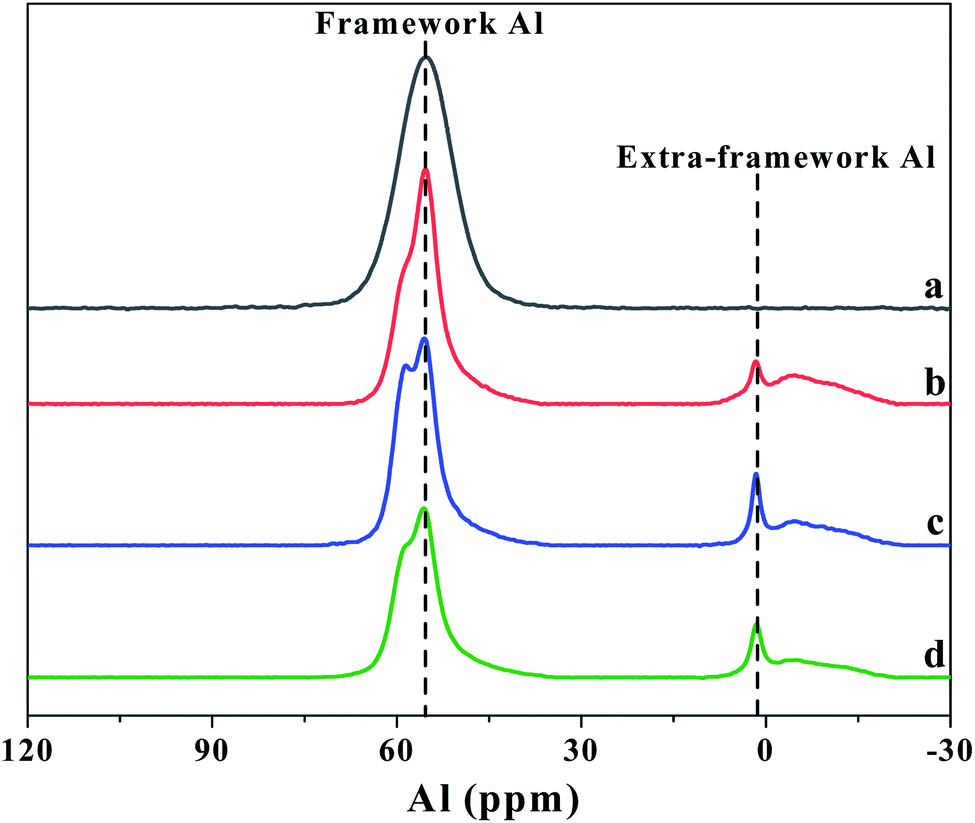 | ||
| Fig. 6 27Al MAS NMR spectra of Hβ-0–8d (a), Hβ-0.02–8d (b), Hβ-0.04–8d (c), and Hβ-0.06–10d (d). | ||
3.5 Catalytic performance of β zeolites
The catalytic performances of β zeolites synthesized in this work were evaluated using the Friedel–Crafts alkylation of benzene with BA and alkylation of mesitylene with BA in the thick-walled glass tube, respectively. As shown in Scheme 1A, the products of alkylation of benzene with BA are composed of diphenylmethane (DP) and dibenzyl ether (DE) according to two different reaction routes. As shown in Fig. 7A, the BA conversion is gradually increased with reaction time over all β zeolites. Significantly, the initial BA conversion over Hβ-0.02–8d, Hβ-0.04–8d, and Hβ-0.06–10d are slightly higher than that of Hβ-0–8d. However, as reaction time was prolonged to 3 h, the Hβ-0.02–8d exhibits a highest BA conversion, and the BA conversion decreased in the order of Hβ-0.02–8d > Hβ-0.04–8d > Hβ-0.06–10d > Hβ-0–8d. This observation reveals that the porous structure has obvious influence on the utilization efficiency of acid sites over β zeolites. Generally speaking, the catalytic performance of zeolites is affected by many factors, such as the porous structure, crystal size, and strength of acid sites. It is well known that the strong acid sites are required for alkylation of benzene with BA.44–46 Thus, to further obtain the catalytic efficiency of per active sites over β zeolites with different porous structure, the turnover number (TON) normalized by the number of strong acid sites was calculated (Fig. 7B). It is obviously that the TON values over hierarchical β zeolites, i.e., Hβ-0.02–8d, Hβ-0.04–8d, and Hβ-0.06–10d, are significantly higher than that of Hβ-0–8d with sole microporous structure. This result indicates that the reaction rates over per active site for hierarchical β zeolites are increased due to the decreased diffusional limitation. Specifically, the Hβ-0.02–8d present highest TON value, which can be reasonably attributed to the highest external surface area (277 m2 g−1) and the easily accessible of acid sites. As revealed from pore-size distribution, Hβ-0.02–8d exhibits a broad mesopore-size distribution (2–10 nm), which can accelerate the diffusion rate of the products (DP) with large molecular size over the acidic sites. Moreover, the TON values over Hβ-0.04–8d and Hβ-0.06–10d are slightly lower than that of Hβ-0.02–8d. It should be noted that the mesopore-size distribution of Hβ-0.04–8d is very similar to that of Hβ-0.02–8d. Therefore, we speculate that the lower external surface area, mesoporous volume, and strength of acid sites are the main reason for the decreased TON value of Hβ-0.04–8d and Hβ-0.06–10d.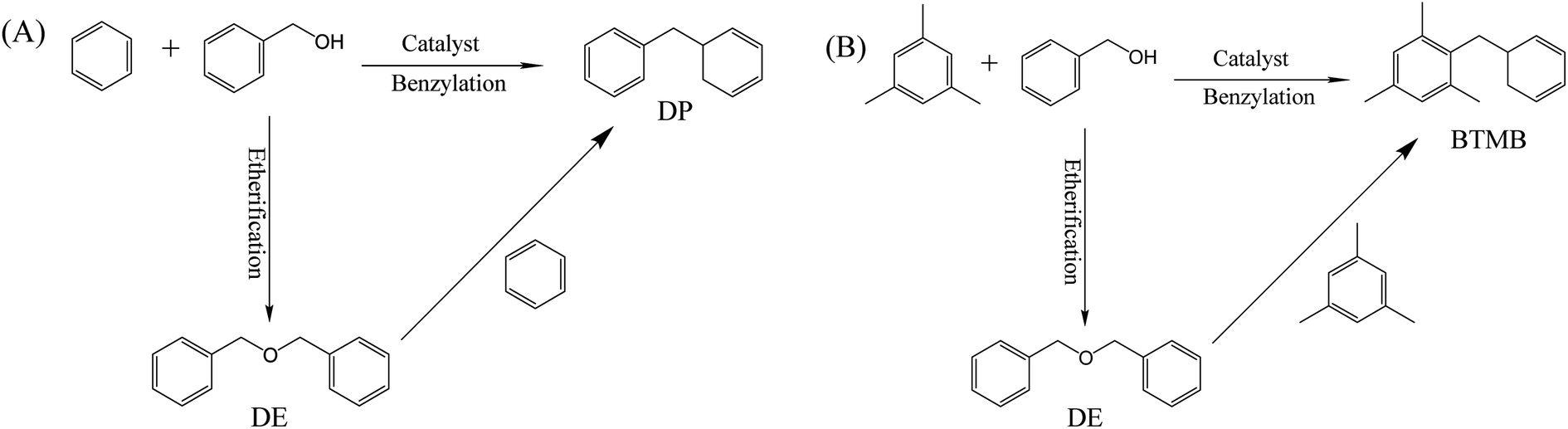 | ||
| Scheme 1 Alkylation reaction pathway of benzene with BA (A) and mesitylene with BA (B). | ||
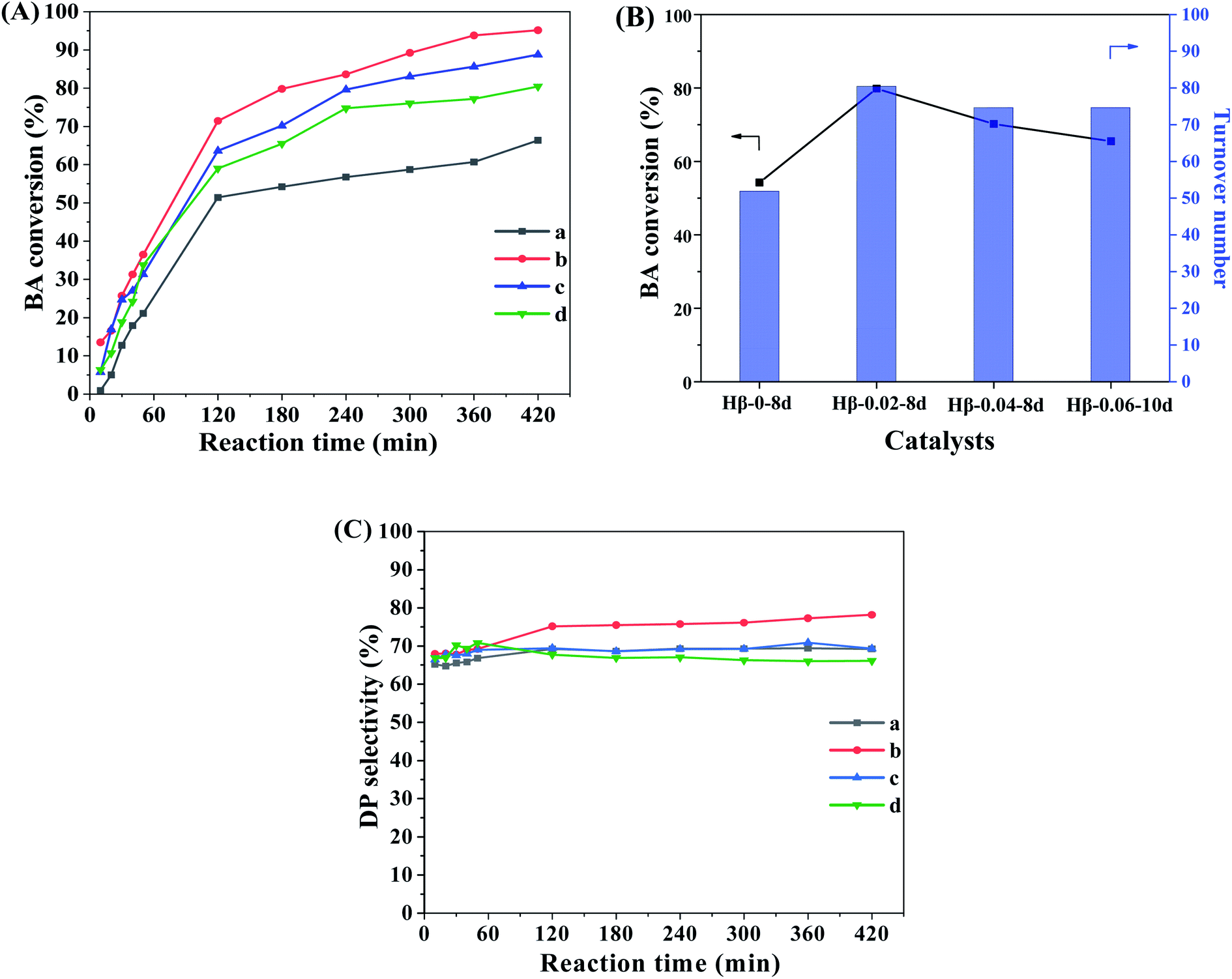 | ||
| Fig. 7 BA Conversion versus reaction time (A), TON normalized by the number of strong acid sites (3 h) (B), and DP selectivity (C) over Hβ-0–8d (a), Hβ-0.02–8d (b), Hβ-0.04–8d (c), and Hβ-0.06–10d (d). Reaction conditions: molar ratio of benzene/benzyl alcohol = 20, 85 °C. | ||
Fig. 7C shows the product selectivity for the alkylation of benzene with BA over β zeolites. It can be observed that the selectivity of target product DP (C-alkylation) over hierarchical β zeolites is very similar to that over sole microporous β zeolite, although the DP selectivity of Hβ-0.02–8d is slightly higher than that of other three samples. It is well known that the C-alkylation of benzene with BA and the self-etherification of BA can take place inside the 12-ring micropores of β zeolite, and there is almost no shape selectivity for the target product DP. Therefore, the selectivity of the target product DP cannot be significantly improved over hierarchical β zeolites, although the multilevel pore structures can effectively enhance the accessibility of acid sites, turnover of reactants, and diffusion rate of bulky products in the alkylation of benzene with BA.
To better understand the maximum diffusion enhancement offered by multilevel pore structures in hierarchical β zeolites, a more diffusion-constrained probe molecule mesitylene (∼0.87 nm) was used to evaluate the catalytic performance in the alkylation of mesitylene with BA. As shown in Scheme 1B, the reaction between mesitylene and BA involves parallel reactions, alkylation and self-etherification of BA, which produce 1,3,5-trimethyl-2-benzylbenzene (BTMB) and dibenzyl ether (DE) products, respectively. It has been reported that the alkylation occurred solely on the external surface of zeolites owing to the diffusion limitation of bulk mesitylene, while the self-etherification of BA occurs over external surface acid sites, acid sites in pore mouth and acid sites in micropore of zeolites.47 As shown in Fig. 8, the hierarchical β zeolite (Hβ-0.02–8d) exhibits excellent catalytic activity compared to β zeolite with sole micropores (Hβ-0–8d). Notably, the Hβ-0–8d exhibits almost constant selectivity of BTMB (∼30%) with the increase of reaction time, indicating the products reached at the state of thermodynamic equilibrium and that the balance composition was not changed. On the contrary, the BTMB selectivity over Hβ-0.02–8d is gradually increased with the reaction time, which is obviously higher than that of Hβ-0–8d after reaction time of 420 min. This result indicates that the mesopore in β zeolite can effectively reduce the diffusional limitation of bulky mesitylene, which significantly improve the reaction rate of mesitylene to BTMB. Moreover, the selectivity of BTMB is gradually increased to ∼67%, indicating the DE is reversibly converted back to BA and reacted with mesitylene to generate BTMB over the external acid sites. These obversions can be reasonably attributed to the integrated balance of considerable mesoporosity and well-remained strong acid sites in the hierarchical β zeolites.
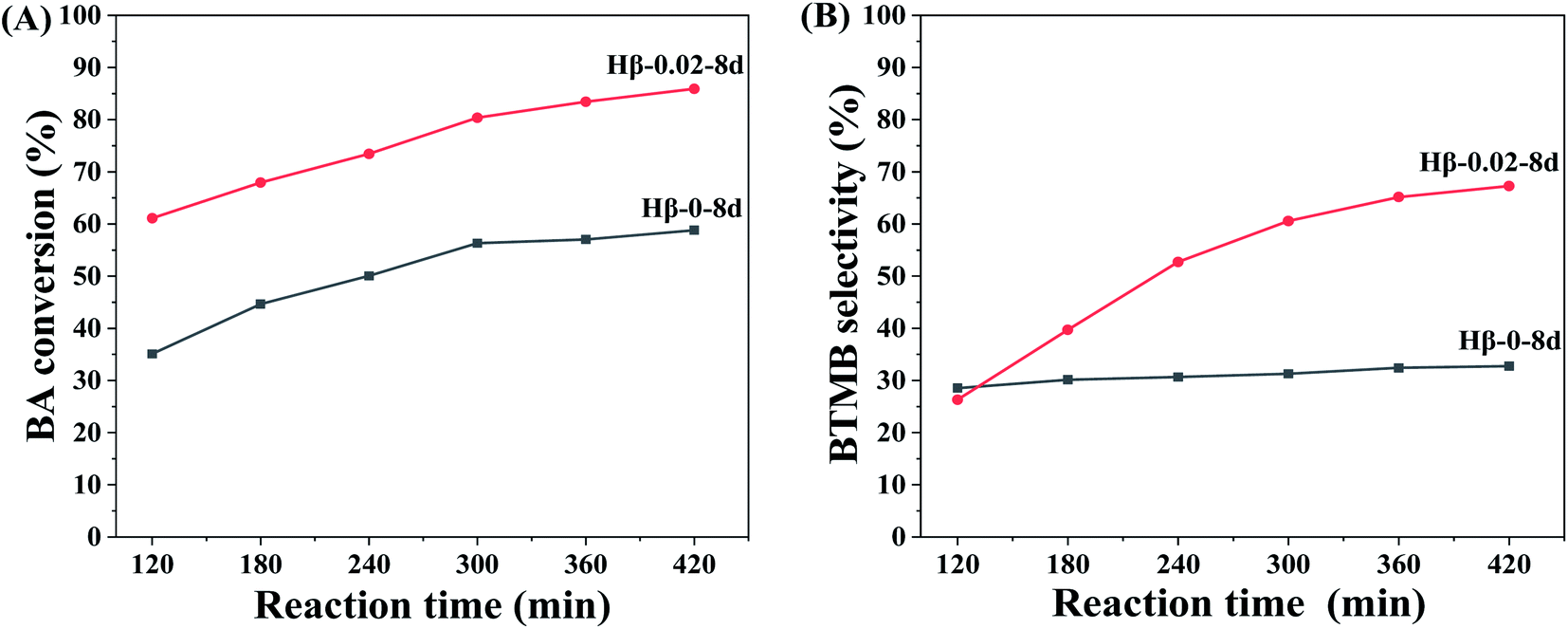 | ||
| Fig. 8 BA conversion (A) and BTMB selectivity (B) versus reaction time over Hβ-0–8d and Hβ-0.02–8d. Reaction conditions: molar ratio of mesitylene/benzyl alcohol = 20, 85 °C. | ||
4. Conclusion
In summary, hierarchical β zeolites were successfully synthesized by using the commercially available TEAOH and TPOAC as co-SDAs. Our findings revealed that the crystallization rate of β zeolites was decreased as increasing the content of TPOAC in the synthesis gel. Moreover, the mesopore size distribution, external surface area, crystal size, and strength of acid sites can be significantly influenced by the molar ratio of TPOAC/SiO2 in the mixture gel. More importantly, the TPOAC not only act as a mesopores SDA to generate the intracrystalline mesopores inside the β crystals, but also act as crystal growth inhibitor to decrease the crystal size and generate intercrystalline mesopores or macropores via the stacking of the β nanocrystals. The hierarchical β zeolites synthesized in this work exhibits enhanced catalytic activity in alkylation of benzene with BA and alkylation of mesitylene with BA compared with the conventional sole microporous β zeolite, which can be attributed to the integrated balance of considerable mesoporosity and well-remained strong acid sites in the hierarchical β zeolites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial supports from the National Natural Science Foundation of China (No. 29178233), China Postdoctoral Science Foundation (2016M600810 and 2017T100767), Natural Science Basic Research Program of Shaanxi (No. 2020JQ-572), Scientific Research Program of Shaanxi Provincial Education Department (No. 19JC039), and Scientific Research Project of Xi'an Science and Technology Bureau (2020KJRC0113) are highly appreciated.References
- Y. Wang, Y. Sun, C. Lancelot, C. Lamonier, J.-C. Morin, B. Revel, L. Delevoye and A. Rives, Microporous Mesoporous Mater., 2015, 206, 42–51 CrossRef CAS.
- B. Liu, Z. Liao, X. Xiao, S. Gan, R. Luo, Y. Wu, H. Chen, Y. Fang and J. Dong, Ind. Eng. Chem. Res., 2020, 59, 13932–13939 CrossRef CAS.
- B. Viswanathan and B. Jacob, Catal. Rev.: Sci. Eng., 2005, 47, 1–82 CrossRef CAS.
- S. Al-Khattaf, S. Ali, A. Aitani, N. Žilková, D. Kubička and J. Cejka, Catal. Rev.: Sci. Eng., 2014, 56, 333–402 CrossRef CAS.
- I. Iovel, K. Mertins, J. Kischel, A. Zapf and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 3913–3917 CrossRef CAS PubMed.
- N. Narender, K. Mohan, S. Kulkarni and I. Reddy, Catal. Commun., 2006, 7, 583–588 CrossRef CAS.
- P. Gupta and S. Paul, Catal. Today, 2014, 236, 153–170 CrossRef CAS.
- G. Winé, M.-J. Ledoux and C. Pham-Huu, Top. Catal., 2007, 45, 111–116 CrossRef.
- N. Candu, M. Florea, S. Coman and V. Parvulescu, Appl. Catal., A, 2011, 393, 206–214 CrossRef CAS.
- S. Jun and R. Ryoo, J. Catal., 2000, 195, 237–243 CrossRef CAS.
- M.-N. Liu, Z.-X. Xie, Q.-X. Luo, J. Zhang, H. Chen, L. Xu, M. Sun, J. Zhang, H. Chen, X. Ma and Q.-Q. Hao, New J. Chem., 2022, 61, 1078–1088 CAS.
- K. Zheng, B. Liu, J. Huang, K. Zhang, F. Li and H. Xi, Inorg. Chem. Commun., 2019, 107, 107468–107475 CrossRef CAS.
- B. Liu, J. Huang, Z. Liao, C. Zhu, Q. Chen, G. Sheng, Y. Zhu, Y. Huang and J. Dong, AIChE J., 2021, 67, 17177–17190 Search PubMed.
- K. Mantri, K. Komura, Y. Kubota and Y. Sugi, J. Mol. Catal. A: Chem., 2005, 236, 168–175 CrossRef CAS.
- S. Fernandez, M. Ostraat, J. Lawrence III and K. Zhang, Microporous Mesoporous Mater., 2018, 263, 201–209 CrossRef CAS.
- V. Valtchev and L. Tosheva, Chem. Rev., 2022, 25, 6734–6760 Search PubMed.
- A. Feliczak-Guzik, Microporous Mesoporous Mater., 2018, 259, 33–45 CrossRef CAS.
- J.-C. Kim, K. Cho, S. Lee and R. Ryoo, Catal. Today, 2015, 243, 103–108 CrossRef CAS.
- J.-C. Kim, K. Cho and R. Ryoo, Appl. Catal., A, 2014, 470, 420–426 CrossRef CAS.
- M. Ogura, S.-Y. Shinomiya, J. Tateno, Y. Nara, M. Nomura, E. Kikuchi and M. Matsukata, Appl. Catal., A, 2001, 219, 33–43 CrossRef CAS.
- J. Groen, J. Jansen, J. Moulijn and J. Pérez-Ramírez, J. Phys. Chem. B, 2004, 108, 13062–13065 CrossRef CAS.
- G. Kerr, J. Phys. Chem., 1967, 71, 4155–4156 CrossRef CAS.
- C. Jacobsen, C. Madsen, J. Houzvicka, I. Schmidt and A. Carlsson, J. Am. Chem. Soc., 2000, 122, 7116–7117 CrossRef CAS.
- I. Schmidt, A. Krogh, K. Wienberg, A. Carlsson, M. Brorson and C. Jacobsen, Chem. Commun., 2000, 21, 2157–2158 RSC.
- D. Xu, S. Che and O. Terasaki, New J. Chem., 2016, 40, 3982–3992 RSC.
- H. Luo, V. Michaelis, S. Hodges, R. Griffin and Y. Roman-Leshkov, Chem. Sci., 2015, 6, 6320–6324 RSC.
- K. Leng, Y. Wang, C. Hou, C. Lancelot, C. Lamonier, A. Rives and Y. Sun, J. Catal., 2013, 306, 100–108 CrossRef CAS.
- Y. Sun and R. Prins, Appl. Catal., A, 2008, 336, 11–16 CrossRef CAS.
- B. Singh, Y. Kim, S. Kwon and K. Na, Catalysts, 2021, 11, 1541–1571 CrossRef CAS.
- L.-H. Chen, M.-H. Sun, Z. Wang, W. Yang, Z. Xie and B.-L. Su, Chem. Rev., 2020, 120, 11194–11294 CrossRef CAS PubMed.
- A. Sachse and J. García-Martínez, Chem. Mater., 2017, 29, 3827–3853 CrossRef CAS.
- Y. Zhu, Z. Hua, J. Zhou, L. Wang, J. Zhao, Y. Gong, W. Wu, M. Ruan and J. Shi, Chem.–Eur. J., 2011, 17, 14618–14627 CrossRef CAS PubMed.
- J. Zhou, Z. Hua, Z. Liu, W. Wu, Y. Zhu and J. Shi, ACS Catal., 2011, 1, 287–291 CrossRef CAS.
- Y. Yuan, P. Tian, M. Yang, D. Fan, L. Wang, S. Xu, C. Wang, D. Wang, Y. Yang and Z. Liu, RSC Adv., 2015, 5, 9852–9860 RSC.
- D. Serrano, J. Aguado, J. Escola, J. Rodríguez and Á. Peral, Chem. Mater., 2006, 18, 2462–2464 CrossRef CAS.
- F.-S. Xiao, L. Wang, C. Yin, K. Lin, Y. Di, J. Li, R. Xu, D. Su, R. Schlögl, T. Yokoi and T. Tatsumi, Angew. Chem., Int. Ed., 2006, 45, 3090–3093 CrossRef CAS PubMed.
- M. Choi, H. Cho, R. Srivastava, C. Venkatesan, D.-H. Choi and R. Ryoo, Nature, 2006, 5, 718–723 CrossRef CAS PubMed.
- J.-Q. Chen, Y.-Z. Li, Q.-Q. Hao, H. Chen, Z.-T. Liu, C. Dai, J. Zhang, X. Ma and Z.-W. Liu, Natl. Sci. Rev., 2021, 8, nwaa236 CrossRef CAS PubMed.
- H. Chen, M. Wang, M. Yang, W. Shang, C. Yang, B. Liu, Q. Hao, J. Zhang and X. Ma, J. Mater. Sci., 2019, 54, 8202–8215 CrossRef CAS.
- T. Yutthalekha, C. Wattanakit, C. Warakulwit, W. Wannapakdee, K. Rodponthukwaji, T. Witoon and J. Limtrakul, J. Cleaner Prod., 2016, 142, 1244–1251 CrossRef.
- M. Choi, R. Srivastava and R. Ryoo, Chem. Commun., 2006, 42, 4380–4382 RSC.
- M.-N. Liu, Y.-Z. Li, Z.-X. Xie, Q.-Q. Hao, Q.-X. Luo, J. Zhang, H. Chen, C. Dai and X. Ma, New J. Chem., 2020, 44, 16638–16644 RSC.
- J. Kim, C. Jo, S. Lee and R. Ryoo, J. Mater. Chem. A, 2014, 2, 11905–11912 RSC.
- M.-T. Yuan, D.-Y. Zhao, Q.-Q. Hao, Q.-X. Luo, J. Zhang, H. Chen, M. Sun, L. Xu and X. Ma, Ind. Eng. Chem. Res., 2020, 59, 16312–16320 CrossRef CAS.
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, R. Messinger, B. Chmelka and R. Ryoo, Science, 2011, 333, 328–332 CrossRef CAS.
- H. Luo, V. Michaelis, S. Hodges, R. Griffin and Y. Roman-Leshkov, Chem. Sci., 2015, 6, 6320–6324 RSC.
- L. Emdadi, S. Oh, Y. Wu, S. Oliaee, Y. Diao, G. Zhu and D. Liu, J. Catal., 2016, 335, 165–174 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |