
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbohydrate structure–activity relations of Au-catalysed base-free oxidations: gold displaying a platinum lustre†
Frits van der Klis ab,
Linda Gootjesb,
Noud Hendrik Verstijnenb,
Jacco van Haverenb,
Daniël Stephan van Es*b and
Johannes Hendrik Bitter*a
ab,
Linda Gootjesb,
Noud Hendrik Verstijnenb,
Jacco van Haverenb,
Daniël Stephan van Es*b and
Johannes Hendrik Bitter*a
aWageningen University Biobased Chemistry and Technology, Bornse Weilanden 9, 6708 WG, Wageningen, The Netherlands. E-mail: harry.bitter@wur.nl; Tel: +31 317 480 303
bWageningen Food & Biobased Research, Bornse Weilanden 9, 6708 WG, Wageningen, The Netherlands. E-mail: daan.vanes@wur.nl; Tel: +31 317 481 160
First published on 22nd March 2022
Abstract
This paper describes the base-free gold-catalysed oxidation of four different carbohydrates in a packed bed plug flow reactor. The influence of the carbohydrate structure on the catalytic activity and selectivity was investigated by comparing two neutral sugars (glucose (Glc) and galactose (Gal), both with primary alcohols at C6), with their sugar-acid analogues (glucuronic acid (GlcA) and galacturonic acid (GalA), both with carboxylic acids at C6). The orientation of OH-groups at the C4-position (equatorial in Glc/GlcA and axial in Glc/GlcA), and the C6-functionality (primary alcohols in Gal/Glc and carboxylic acids in GalA/GlcA) has a profound influence on the catalytic activity. When the OH-groups are in an axial position their reactivity was higher compared to the OH-groups in the equatorial position for both the sugars and the sugar acids. In addition the reactivity of carbohydrates over Au-catalysts under base-free conditions is different compared to alkaline conditions, and is more in line with a Pt-catalysed dehydrogenation mechanism.
1. Introduction
Agricultural residues with low nutritional value are attractive feedstocks for the production of chemicals and materials, while not competing with food or land-use. These residues are generally rich in carbohydrates, and can be converted into useful chemicals1 such as sugar acids and lactones via green catalytic oxidation reactions.2 These sugar acids and lactones have already shown many potential applications, ranging from platform molecules for polymer building blocks (including FDCA), sequestering agents, corrosion inhibitors, food additives, and many more.3–5Over the past decades, supported gold nanoparticles have proven to be excellent catalysts for carbohydrate oxidations. Extensive research on the influence of particle size, catalyst support, preparation methods, as well as catalyst deactivation mechanisms, has contributed to in-dept understanding of the influence of the Au-catalyst morphology on the oxidation performance.6–14 Furthermore, reaction mechanisms have been proposed for both alkaline- and base-free conditions. Despite this great knowledge on Au-catalyst structure–activity relationships, much less is known about the influence of carbohydrate structures on catalytic oxidation reactions at the gold surface.
To establish such a structure–activity relationship for carbohydrates, substituent variation of carbohydrates with otherwise identical conformations should be compared. Fig. 1 shows four (relevant and natural abundant) carbohydrates, organised in such a way that systematic comparison of only one substituent is possible over each axis. The horizontal axis shows sole variation in the orientation of the OH-group at the C4-position, while the vertical axis shows the variation in only the C6-position.
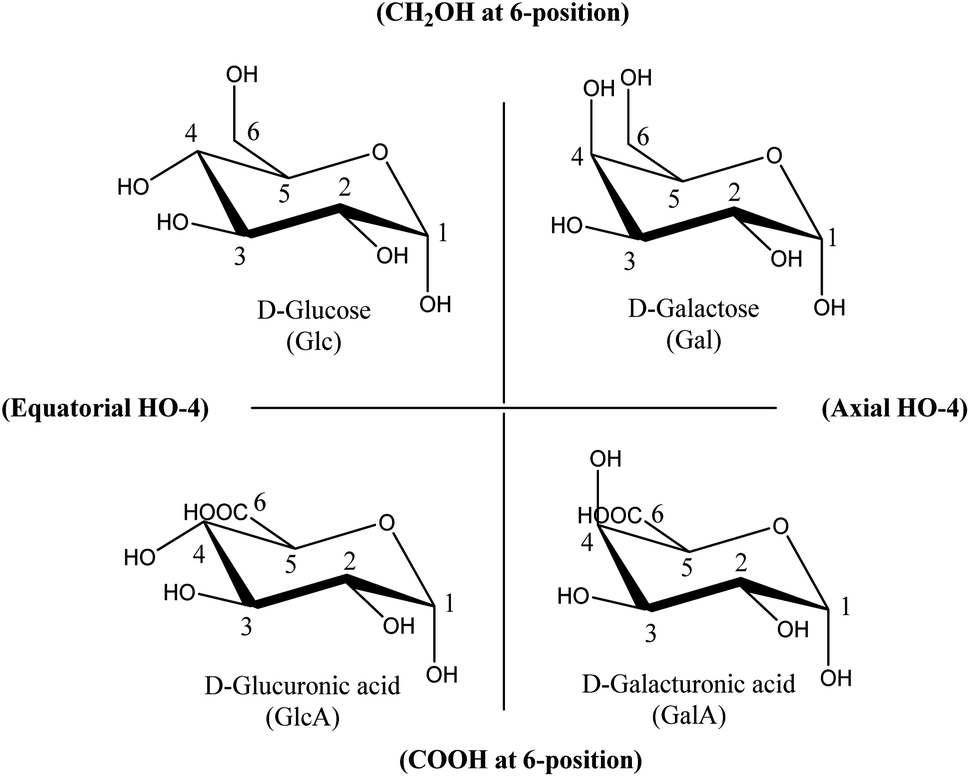 | ||
| Fig. 1 Carbohydrate structures investigated in this paper, represented as α-D-pyranosides. | ||
The influence of conformation of the four carbohydrate structures depicted in Fig. 1 on catalytic oxidations has been previously investigated for platinum catalysts: the pioneering work of De Wit et al.15 focussed on establishing carbohydrate oxidation rates (actually dehydrogenation rates) under strong alkaline conditions (1 M KOH) in the absence of oxygen over Pt- and Rh-catalysts.
The following influence on oxidation rate was found: for the HO-4 orientation Axial > Equatorial; and for the 6-position CH2OH > CO2−. (Thus for the carbohydrates in Fig. 1: Gal > Glc > GalA > GlcA.) The higher reactivity of Gal compared to Glc, only differing in their HO-4 orientation, was later confirmed by others for Pt- and Pd-catalysts under milder reaction conditions (pH 9) and in the presence of oxygen.16
For Au-catalysts, comparable research on carbohydrate structure–activity relations was solely performed under alkaline conditions in the presence of oxygen. Surprisingly, Au behaves completely different compared to Pt under these alkaline conditions: e.g. Glc oxidation is much faster compared to Gal16,17 (Equatorial HO-4 preferred over axial orientation for Au, contrary to Pt). Rautiainen et al.18 included variation at C6 (CH2OH and CO2−), by adding the uronic acid analogues GlcA and GalA, but did not find a large influence of C6-position on the reaction rate (Glc > Gal ≈ GlcA ≈ GalA), while De Wit et al.15 clearly found this influence for Pt-catalysts (Gal > Glc > GalA > GlcA). The ESI provides a complete overview of carbohydrate conformations (Table S1†) and previously reported oxidation rates (Table S2†).
The question what causes the observed differences in reactivity for gold compared to other metals like platinum and palladium was already raised in 2006 by Mirescu & Prüße:16
“The Au catalyst displayed the highest activity in glucose oxidation, whereas the same catalyst oxidized the galactose more slowly. It appears that the structure of the sugars influences the activity of the Au catalyst but not in the same manner as it does for Pt catalysts. A detailed mechanism of carbohydrates' oxidation on an Au catalyst has not yet been reported.”
As we will show later in this paper, Au-catalysts under base-free conditions do resemble the performance of Pt catalysts. Therefore we will focus on the mechanisms of Au oxidations in some more detail here as well. Despite today's knowledge on the oxidation mechanisms of Au-catalysts, to the best of our knowledge the question with regard to the reactivity differences between Pt and Au has not yet been answered. In the meantime, carbohydrate oxidations over Au-catalysts have evolved in two directions: besides the “classical” alkaline conditions, also base-free oxidations using Au have been developed.7,8,12,14,19–23 It is believed that Au-catalysed reactions under alkaline conditions follows a completely different reaction mechanism (Fig. 2A) compared to base-free conditions (Fig. 2B).
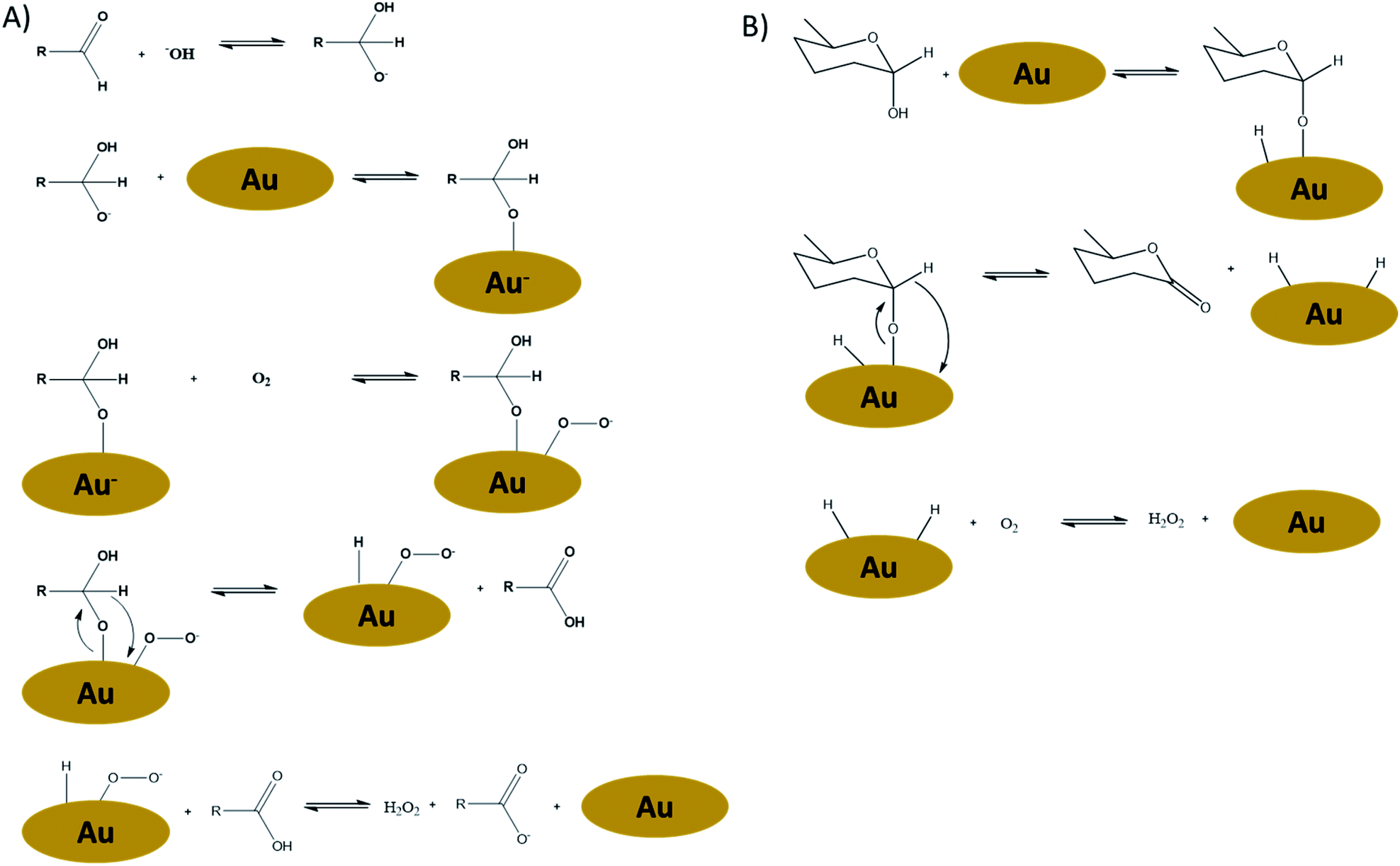 | ||
| Fig. 2 (A) Proposed reaction mechanism for Au-catalysed alkaline oxidation of carbohydrates in the presence of oxygen,24 and (B) proposed reaction mechanism for Au-catalysed base-free oxidation of carbohydrates in the presence of oxygen.22 | ||
The alkaline mechanism is believed to proceed via an open-ring intermediate, while the base-free mechanism occurs via the adsorption of the anomeric OH functionality, thus with the carbohydrate still in cyclic form. The base-free mechanism thereby closer resembles the Pt-dehydrogenation mechanism as proposed by De Wit et al.15 It is therefore to be expected that structural differences of the carbohydrates have a different impact, depending on the oxidation mechanism. In other words: for base-free oxidations a different influence of carbohydrate-structure on the reaction rates might be expected, than has been established for Au under alkaline conditions.
Our hypothesis is therefore that Au will behave like Pt under base-free conditions. Hence, the aim of this study is to establish a structure–activity relationship for Au-catalysed carbohydrate oxidations under base-free conditions by systematically comparing four structurally related carbohydrates under flow conditions in a fixed bed reactor over gold on titania.
2. Experimental
2.1 Materials and chemicals
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, silylation reagent, Supelco); Celite® (Alfa Aesar); D-(+)-gluconic acid δ-lactone (≥99.0%, Sigma Aldrich); D-galactose (≥99%, Sigma Aldrich); D-galacturonic acid monohydrate (batch C-9, gift by Royal Cosun); diethyl ether (≥99.5%, Actu-All chemicals); absolute ethanol (100%, VWR Chemicals); ethyl acetate (≥99.7%, Honeywell); hydrochloric acid (37%, VWR Chemicals); magnesium sulfate (Alfa Aesar); mucic acid (≥97%, Sigma Aldrich); potassium carbonate (≥99.5%, Merck); pyridine (≥99.5%, Merck); Sicapent® (Merck Millipore); sodium bicarbonate (≥99.5%, Merck); sodium bisulfite (Acros Organics); sodium hydroxide (2 M standard solution, Fluka analytical); sodium thiosulfate (≥98%, Sigma Aldrich); sulfuric acid (95–97%, Sigma Aldrich).
:1, silylation reagent, Supelco); Celite® (Alfa Aesar); D-(+)-gluconic acid δ-lactone (≥99.0%, Sigma Aldrich); D-galactose (≥99%, Sigma Aldrich); D-galacturonic acid monohydrate (batch C-9, gift by Royal Cosun); diethyl ether (≥99.5%, Actu-All chemicals); absolute ethanol (100%, VWR Chemicals); ethyl acetate (≥99.7%, Honeywell); hydrochloric acid (37%, VWR Chemicals); magnesium sulfate (Alfa Aesar); mucic acid (≥97%, Sigma Aldrich); potassium carbonate (≥99.5%, Merck); pyridine (≥99.5%, Merck); Sicapent® (Merck Millipore); sodium bicarbonate (≥99.5%, Merck); sodium bisulfite (Acros Organics); sodium hydroxide (2 M standard solution, Fluka analytical); sodium thiosulfate (≥98%, Sigma Aldrich); sulfuric acid (95–97%, Sigma Aldrich).2.2 Protocol for carbohydrate oxidation reactions in the packed bed plug flow reactor
All catalytic oxidation reactions were performed in a packed bed plug flow reactor set-up as described in detail previously.26 The catalyst bed of the packed bed reactor was filled with 1 wt% Au/TiO2 (7.7 g; crushed and sieved), and the catalyst particles were mixed with additional inert silicon carbide powder to fill the voids. For each reaction, the desired temperature was set (variation between 40–100 °C), and 0.1 M aqueous solutions of the carbohydrates were fed to the reactor at a flow rate of 10 mL min−1. Oxygen gas was applied at a flow rate of 50 mL min−1, and the total system pressure was kept constant at 10 bar. The contact time of the reactants with the catalyst bed was approximately 1.3 min.All oxidation reactions were performed using the above described set-up and conditions, and the following protocol to determine the conversion of the carbohydrates was applied: reactions were initiated by applying the fixed oxygen- and liquid flows at a system pressure of 10 bar, and the desired temperature was set. Next, the reactor was allowed to stabilize for 1 h. During this time no samples were collected, to allow the system to stabilise. After one hour, sample collection was started at time intervals of 10 min. For a period of 1 h (total reaction time including stabilisation is therefore 2 h per setting). As a sample was collected each 10 min, during 1 h collection time, for each condition a total of five samples was obtained.
The collected liquid samples were stored at 4 °C for analysis by HPLC, and/or colorimetric methods to determine the conversion and product composition. Additionally, some samples were subjected to crash freezing immediately after they left the reactor, followed by lyophilizing, to obtain dry samples for additional NMR analysis (Section 1 of ESI†).
Section 1 of the ESI† also provides a comparison to the appropriate reference compounds (see under 2.4) as well as an in-dept discussion on potential pathways and kinetics.
2.3 Analytical equipment
As the conversion of GlcA could not be determined by HPLC due to overlapping peaks, a colorimetric method described as the “TTC-Protocol” in Botelho da Silva et al.29 & Jaušovec et al.30 was used to determine the conversion for the GlcA reactions.
:1, silylation reagent, Supelco) followed by storing the sample at 70 °C for 20 min. prior to analysis.2.4 Synthesis of lactone references and detailed analysis of oxidation reaction mixtures
All potential lactone products, resulting from the catalytic oxidation reactions in Section 2.2, which were not commercially available were synthesized in-house. A detailed description of their synthesis and analysis can be found in Section 2 of the ESI.†3. Results and discussion
The structural influence of four common carbohydrates (Fig. 1) on their catalytic oxidation over Au/TiO2 under base-free conditions was investigated: D-glucose and D-galactose (differing only in HO-4 orientation) and their related uronic acid derivatives D-glucuronic acid and D-galacturonic acid (also differing in C-6 functionality).All conversions were performed without added base, so under initial neutral conditions for Glc and Gal, and under the natural pH (acidic) for GlcA and GalA. The conversions were investigated over a temperature range from 40–100 °C. The number of temperature variations performed with D-glucuronic acid was limited due to the relative high price of this substrate, and the high amounts needed in our experimental set-up.
Fig. 3 shows the conversion of the four carbohydrates as a function of reaction temperature. The diamond shapes represent the individual HPLC/colorimetric measurement points of the obtained samples (for each reaction temperature and carbohydrate, five individual measurements were obtained). The dotted lines are added as a visual reference to guide the eye over the conversion trend as a function of temperature. A similar graph, which expresses the same data as substrate conversion rates, is included in the ESI, Fig. S7.†
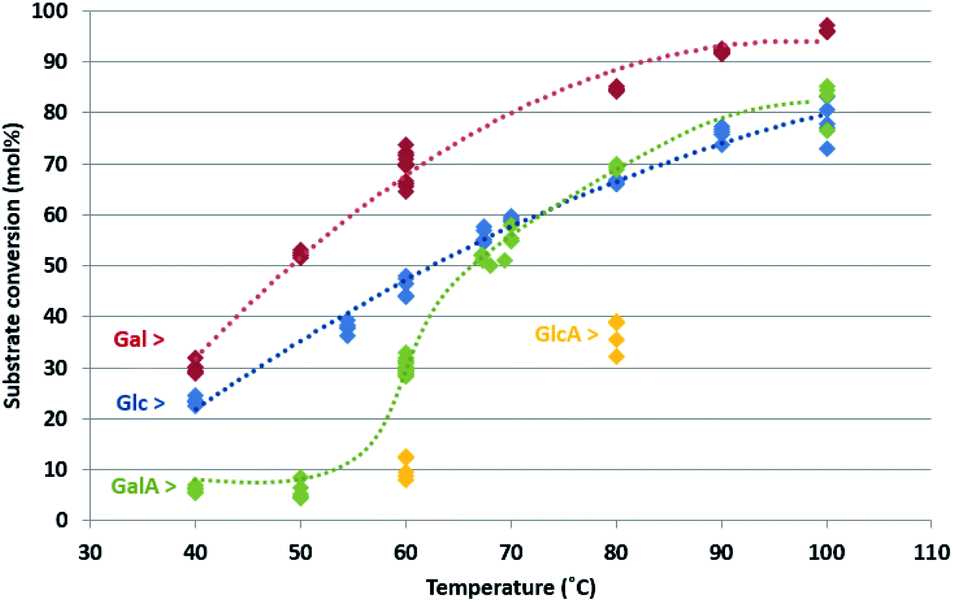 | ||
| Fig. 3 Substrate conversion as a function of reaction temperature; Gal (red), Glc (blue), GalA (green) and GlcA (yellow). Dots represent experimental data points, the dotted lines are added as a visual reference to guide the eye over the general conversion trends. Reaction conditions: Au/TiO2 catalyst, substrate concentration = 0.1 M (10 mL min−1), oxygen (10 bar pressure, 50 mL min−1). | ||
As can be seen from the graph in Fig. 3, the conversion of the carbohydrates was stable over the course of the reactions, resulting in a low spread (≤5% experimental error) in conversion.
The influence of the HO-4 orientation on the conversion can be found by comparing the two neutral sugars: over the whole temperature range, 40–100 °C, the conversion of Gal was higher compared to Glc. This indicates that for the neutral sugars (CH2OH at C6) the axial HO-4 orientation in Gal is preferred over the equatorial HO-4 orientation in Glc. A same preference was found for their acidic analogues with COOH at C6: the axial HO-4 orientation in GalA leads to higher reactivity compared to the equatorial HO-4 orientation in GlcA. The rate promoting effect of the axial HO-4 orientation is thus universal in both the neutral- and acidic sugars.
The influence of the C6 position is shown by the higher conversion of Glc (CH2OH at C6), compared to the lower conversion of its acidic analogue GlcA (COOH at C6). An idential effect is found for the galacto-series: Gal (CH2OH at C6) shows higher conversions compared to the sugar acid analogue GalA (COOH at C6) over the whole temperature range (40–100 °C). This result indicates a clear rate deteriorating effect of the carboxylic acid functionality compared to the primary OH at C6.
At temperatures between 40 and 60 °C, the reactivity of Glc is higher compared to GalA, while at temperatures above 60 °C the reactivity's are almost identical. This indicates that the extent of the reactivity promoting effect of axial HO-4 orientation, and the reactivity decreasing effect of a carboxylate group at C6, have different influence on the activation energy: a stronger effect of the carboxylic acid functionality compared to the effect of the HO-4 position. It is generally accepted that pH plays an important role during Au-catalysed oxidation reactions. The lowered reactivity of the acidic sugars compared to the neutral sugars might be caused by this effect. However, it should be realized that lactones and sugar acids are formed as products during reaction. We found that the pH of all reaction mixtures was between pH 1.5–2 when they left the reactor, indicating that there is not much variation in pH of the final product mixtures.
To summarize, the following effect of the carbohydrate structure on the reaction rate has been established: Gal > Glc > GalA > GlcA. These results, we have obtained in this study under base-free conditions, differs from the results previously observed by others for Au-catalysts under alkaline conditions: Glc > Gal ≈ GlcA ≈ GalA,18 but is an exact match with the previous findings for Pt-catalysts.15 This is a first confirmation of the hypothesis that Au under base-free conditions is acting in a similar way as Pt.
To gain more insight in the mechanism occurring during base-free carbohydrate oxidation, conformation of lactone formation during the reactions was needed. Therefore the presence of lactones in the reaction mixtures was investigated by taking samples of the reaction mixtures directly after they left the reactor, followed by immediate crash-freezing of the samples. Next, samples were lyophilized, and analysed by NMR in DMSO-D6. DMSO was chosen as the solvent, to avoid potential ring opening of lactones in D2O during the measurements. All obtained spectra can be found in Section 1 of the ESI.†
The samples of the reaction mixtures for Glc, Gal and GalA were compared to the NMR spectra of the starting materials, all potential lactone products (1,4- and 1,5-lactones), as well as the sugar acids in open form. In case reference samples were not commercially available, the lactones were synthesized via independent synthesis methods. An elaborate description of the synthesis of the reference lactones as well as their analysis is described in Section 2 of the ESI.†
For all investigated carbohydrate oxidation reactions, the presence of lactones in the freeze dried samples was confirmed: GalA oxidation resulted in a mixture of remaining starting material, galactaric acid-1,4-lactone, and galactaric acid (di-acid in open form); consult ESI, Sections 1.1 & 1.2.† For Gal, a very similar product mixture was found: Unreacted starting material, galactonic acid-1,4-lactone and galactonic acid in open form; consult ESI, Section 1.4.† For Glc, next to starting material and gluconic acid in open form, both the gluconic acid-1,4-lactone and gluconic acid-1,5-lactone were found; consult ESI, Section 1.3.† The synthesis of the reference compounds, the analysis results of the free-dried reaction mixtures, and an in-dept discussion on the obtained results can be found in the ESI.†
To summarize, the detailed product analysis which clearly indicated the formation of lactone products, is another indication that the mechanism of Au-catalysed oxidations under base-free conditions resembles a Pt-like oxidation (dehydrogenation) mechanism.
4 Conclusions
The base-free catalytic oxidation of four different carbohydrates over Au/TiO2 was investigated in a plug flow packed bed reactor set-up over a range of different temperatures, aiming to establish a structure–activity relationship.It was found that the reactivity of D-galactose (axial HO-group in 4-position) was higher compared to D-glucose (equatorial HO-group in 4-position) over the temperature range investigated (40–100 °C). The same influence of the HO-4 position was found by the higher reactivity of D-galacturonic acid (axial HO-group in 4-position) compared to D-glucuronic acid (equatorial HO-group in 4-position). The reactivity of the two uronic acids, galacturonic acid and glucuronic acid, which both have an carboxyl moiety at C6, is lower compared to their aldose analogues, both with primary CH2OH moieties at C6. This indicates a rate-deteriorating effect of carboxyl groups at C6 over primary alcohols at C6.
At temperatures between 40 and 60 °C, the reactivity of glucose is higher compared to galacturonic acid, while at temperatures above 60 °C the reactivity's are almost identical. This indicates that the extent of the reactivity promoting effect of axial HO-4 orientation, and the reactivity decreasing effect of a carboxylate group at C6, are both temperature dependant.
Our study has shown that the structure–activity relationship of carbohydrate oxidations over an Au-catalyst under base-free conditions is completely different from those under alkaline conditions. We tentatively attribute this to a difference between these two reaction mechanisms: under alkaline conditions the carbohydrates orientate in open form to the catalyst surface, while under acidic conditions the oxidation proceeds via adsorption of the carbohydrate in a closed ring conformation. This results in different interaction with the catalyst surface, thereby resulting in a different preference for the orientation of substituents interacting with this gold surface. Our hypothesis was that the Au-catalysed mechanism resembles a platinum-like hydrogen abstraction mechanism, and should therefore follow the same structure–activity relationship. This has now been confirmed for the first time, by showing that Au-catalysts under base-free conditions indeed resemble the results of Pt-catalysts. The results obtained in this study pave the way for further (computational) investigations, to elucidate the exact interaction of the carbohydrates with the catalyst surface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is part of a project that has received funding from the Bio Based Industries Joint Undertaking under the European Union's Horizon 2020 research and innovation programme under grant agreement No 669105. The authors would like to thank David Franciolus for assisting with the HPLC analysis, Guus Frissen for NMR measurements, and Royal Cosun for supplying galacturonic acid.Notes and references
- R. A. Sheldon, Curr. Opin. Green Sustain. Chem., 2018, 14, 89–95 CrossRef.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed.
- R. M. de Lederkremer and C. Marino, Adv. Carbohydr. Chem. Biochem., 2003, 58, 199–306 CrossRef CAS PubMed.
- T. Mehtiö, M. Toivari, M. G. Wiebe, A. Harlin, M. Penttilä and A. Koivula, Crit. Rev. Biotechnol., 2016, 36, 904–916 CrossRef PubMed.
- N. M. Xavier, A. P. Rauter and Y. Queneau, Top. Curr. Chem., 2010, 295, 19–62 CrossRef CAS PubMed.
- M. Sankar, Q. He, R. V. Engel, M. A. Sainna, A. J. Logsdail, A. Roldan, D. J. Willock, N. Agarwal, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2020, 120, 3890–3938 CrossRef CAS PubMed.
- C. Megías-Sayago, L. F. Bobadilla, S. Ivanova, A. Penkova, M. A. Centeno and J. A. Odriozola, Catal. Today, 2018, 301, 72–77 CrossRef.
- C. Megías-Sayago, J. L. Santos, F. Ammari, M. Chenouf, S. Ivanova, M. A. Centeno and J. A. Odriozola, Catal. Today, 2018, 306, 183–190 CrossRef.
- Y. Önal, S. Schimpf and P. Claus, J. Catal., 2004, 223, 122–133 CrossRef.
- S. Porsgaard, P. Jiang, F. Borondics, S. Wendt, Z. Liu, H. Bluhm, F. Besenbacher and M. Salmeron, Angew. Chem., Int. Ed., 2011, 50, 2266–2269 CrossRef CAS PubMed.
- U. Prüße, M. Herrmann, C. Baatz and N. Decker, Appl. Catal., A, 2011, 406, 89–93 CrossRef.
- P. Qi, S. Chen, J. Chen, J. Zheng, X. Zheng and Y. Yuan, ACS Catal., 2015, 5, 2659–2670 CrossRef CAS.
- F. Vigneron and V. Caps, C. R. Chim., 2016, 19, 192–198 CrossRef CAS.
- Y. Wang, S. Van de Vyver, K. K. Sharma and Y. Roman-Leshkov, Green Chem., 2014, 16, 719–726 RSC.
- G. de Wit, J. J. de Vlieger, A. C. Kock-van Dalen, R. Heus, R. Laroy, A. J. van Hengstum, A. P. G. Kieboom and H. van Bekkum, Carbohydr. Res., 1981, 91, 125–138 CrossRef CAS.
- A. Mirescu and U. Prüße, Appl. Catal., B, 2007, 70, 644–652 CrossRef CAS.
- U. Prüße, K. Heidkamp, N. Decker, M. Herrmann and K. D. Vorlop, Chem. Ing. Tech., 2010, 82, 1231–1237 CrossRef.
- S. Rautiainen, P. Lehtinen, J. Chen, M. Vehkamaki, K. Niemela, M. Leskela and T. Repo, RSC Adv., 2015, 5, 19502–19507 RSC.
- D. An, A. Ye, W. Deng, Q. Zhang and Y. Wang, Chem. - Eur. J., 2012, 18, 2938–2947 CrossRef CAS PubMed.
- Y. Cao, X. Liu, S. Iqbal, P. J. Miedziak, J. K. Edwards, R. D. Armstrong, D. J. Morgan, J. Wang and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 107–117 RSC.
- M. Comotti, C. D. Pina and M. Rossi, J. Mol. Catal. A: Chem., 2006, 251, 89–92 CrossRef CAS.
- B. T. Kusema, J.-P. Mikkola and D. Y. Murzin, Catal. Sci. Technol., 2012, 2, 423–431 RSC.
- P. J. Miedziak, H. Alshammari, S. A. Kondrat, T. J. Clarke, T. E. Davies, M. Morad, D. J. Morgan, D. J. Willock, D. W. Knight, S. H. Taylor and G. J. Hutchings, Green Chem., 2014, 16, 3132–3141 RSC.
- U. Prüße, S. Heidinger and C. Baatz, Agric. Res., 2011, 3, 261–272 Search PubMed.
- R. K. Pazhavelikkakath Purushothaman, F. van der Klis, A. E. Frissen, J. van Haveren, A. Mayoral, A. van der Bent and D. S. van Es, Green Chem., 2018, 20, 2763–2774 RSC.
- F. van der Klis, L. Gootjes, J. van Haveren, D. S. van Es and J. H. Bitter, React. Chem. Eng., 2018, 3, 540–549 RSC.
- M. Kosmulski, Adv. Colloid Interface Sci., 2002, 99, 255–264 CrossRef CAS PubMed.
- J. Sha, Bimetallic catalysts for oxidation of carbohydrates: looking for synergetic effects, Ecole Centrale de Lille, 2018 Search PubMed.
- S. Botelho da Silva, M. Krolicka, L. A. M. van den Broek, A. E. Frissen and C. G. Boeriu, Carbohydr. Polym., 2018, 186, 299–309 CrossRef CAS PubMed.
- D. Jaušovec, R. Vogrinčič and V. Kokol, Carbohydr. Polym., 2015, 116, 74–85 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00255h |
| This journal is © The Royal Society of Chemistry 2022 |