
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidation of the reaction mechanism on dry reforming of methane in an electric field by in situ DRIFTs†
Naoya Nakanoa,
Maki Torimotoa,
Hiroshi Sampei a,
Reiji Yamashitaa,
Ryota Yamanoa,
Koki Saegusaa,
Ayaka Motomuraa,
Kaho Nagakawaa,
Hideaki Tsunekia,
Shuhei Ogobc and
Yasushi Sekine*a
a,
Reiji Yamashitaa,
Ryota Yamanoa,
Koki Saegusaa,
Ayaka Motomuraa,
Kaho Nagakawaa,
Hideaki Tsunekia,
Shuhei Ogobc and
Yasushi Sekine*a
aDepartment of Applied Chemistry, Waseda University, 3-4-1, Okubo, Shinjuku, Tokyo, 169-8555, Japan. E-mail: ysekine@waseda.jp
bDepartment of Marine Resources Science, Faculty of Agriculture and Marine Science, Kochi University, Nankoku, 783-8502, Japan
cCenter for Advanced Marine Core Research, Kochi University, Nankoku, 783-8502, Japan
First published on 23rd March 2022
Abstract
With increasing expectations for carbon neutrality, dry reforming is anticipated for direct conversion of methane and carbon dioxide: the main components of biogas. We have found that dry reforming of methane in an electric field using a Pt/CeO2 catalyst proceeds with sufficient rapidity even at a low temperature of about 473 K. The effect of the electric field (EF) on dry reforming was investigated using kinetic analysis, in situ DRIFTs, XPS, and DFT calculation. In situ DRIFTs and XPS measurements indicated that the amount of carbonate, which is an adsorbed species of CO2, increased with the application of EF. XPS measurements also confirmed the reduction of CeO2 by the reaction of surface oxygen and CH4. The reaction between CH4 molecules and surface oxygen was promoted at the interface between Pt and CeO2.
1. Introduction
As expectations for the achievement of carbon neutrality continue to rise worldwide, the effective use of biomass resources is attracting attention as one means of achieving that goal. Biogas obtained from anaerobic fermentation is composed mainly of methane and carbon dioxide, both of which are known to be greenhouse gases. A promising technology is one that could directly convert methane and carbon dioxide into high value-added substances such as synthesis gas by dry reforming (DRM: eqn (1)).1,2 However, because of thermodynamic and kinetic constraints, the conventional catalytic process for dry reforming requires temperatures higher than 1000 K. Moreover, the reaction is known to be prone to carbon deposition.| DRM: CH4 + CO2 → 2CO + 2H2, ΔH0298 = 247 kJ mol−1 | (1) |
The reaction mechanism of CO2 in these reactions has been well-studied through in situ DRIFTs measurements. CO2 dissociates on the active metal via carbonyl species3–6 and supports oxides using oxygen vacancies via intermediates such as carbonate and formate.7–10 Chen et al.11 reported that CO was formed via carbonate, which is adsorbed species of CO2 using CeO2 lattice oxygen defects, in DRM using Pt–Ni supported CeO2. Bobadilla et al.12 revealed that the reaction proceeds via formate species as the key intermediate in RWGS (reverse water gas shift) of Pt-loaded CeO2.
In addition, various unconventional studies have been conducted in recent years to promote the conversion of CO2 by application of external stimuli such as electricity, magnetic fields, and light.13–17 Wang et al. reported that Xe lamp irradiation to Cu-loaded CeO2 increases the amount of lattice defects of CeO2 and shows high CO2 reduction performance.18 Zhu et al.19 reported that plasma application accelerates the formation and decomposition of the intermediate formate in DRM using Au-loaded CeO2. Consequently, a process that accelerates the reaction by providing an external stimulus is becoming a more attractive CO2 conversion technology.20,21
Under these circumstances, our group has proposed a catalytic reaction in an electric field (EF) in which direct current is applied by inserting electrodes at the top and bottom edges of the catalyst layer to promote the reaction.22–25 We earlier reported various effects of enhanced activity and lowered apparent activation energy at low temperatures in DRM,26 RWGS,27 and CO2 methanation.28
However, details of the reactivity of CO2 and its adsorption ability in the EF have not been clarified. Therefore, in situ DRIFTs measurements were taken for this study to confirm EF effects on the CO2 conversion reaction by examining changes of intermediates when the EF is applied during a DRM reaction.
2. Experimental
2.1 Catalyst preparation
The catalyst of 1 wt% Pt-supported ceria (1wt%Pt/CeO2) was prepared using an impregnation method. We used ceria (CeO2; JRC-CEO-1) supplied by the Catalysis Society of Japan as a support. Pt was loaded on the support using the following procedure. First, Pt(NH2)4(NO3) (Sigma-Aldrich Corp.) was dissolved in distilled water. The powder of CeO2 support was added to the solution and was stirred for 2 h. Subsequently, the solution was dried at 393 K for 24 h. The obtained powder was calcined at 773 K for 3 h. The obtained sample was pressed and sieved using 355–500 μm mesh. Subsequently it was reduced with 20% H2 diluted with Ar at a 100 SCCM flow rate at 723 K for 30 min.2.2 In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTs)
In situ DRIFTs measurements were taken with an FT-IR spectrophotometer (FT/IR-6200; Jasco Corp.) equipped with a ZnSe window and MCT-M detector cooled with liquid nitrogen. Spectra were recorded with 4 cm−1 resolution and 50–200 times scans. The DRIFT cell was made of PTFE, which is an insulator for inserting two electrodes as described in an earlier report.29 After a pelletized catalyst of 50 mg (without EF) or 110 mg (with EF) was set in the DRIFT cell, the EF was applied with 3 mA of direct current. A schematic image of the DRIFT cell was described in an earlier report.29 The background spectrum was recorded under Ar flow at 90 SCCM at 473 K without EF. During the experiment, CH4 and CO2 were supplied respectively at 10 SCCM. They were diluted with Ar at 90 SCCM at 473 K.2.3 Activity test
Catalytic activity tests were conducted with a fixed bed flow type reactor as presented in Fig. S1.† A 6.0 mm-diameter quartz tube was used, with 300 mg of the catalyst charged in it. A thermocouple was inserted into the catalyst bed at the bottom. The reaction temperature was fixed at 473 K. The temperature was controlled with the thermocouple of the catalyst bed, which includes the Joule heat with EF. For that reason, electrical heating of the catalyst bed is negligible in this case. The feed gas (Ar![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH4:CO2 = 2:1:1) was supplied with the total flow rate of 80 SCCM. A direct current voltage power supply was used to apply the EF. The current and applied voltage were monitored using a digital phosphor oscilloscope (TDS2100; Tektronics Corp.). After passing a cold trap, the reacted gas was analyzed using GC-FID, TCD (GC-2014; Shimadzu Corp.) with a Porapak Q column and a methanizer. The conversion, yield, and input power were calculated by some equations (see ESI†).
:CH4:CO2 = 2:1:1) was supplied with the total flow rate of 80 SCCM. A direct current voltage power supply was used to apply the EF. The current and applied voltage were monitored using a digital phosphor oscilloscope (TDS2100; Tektronics Corp.). After passing a cold trap, the reacted gas was analyzed using GC-FID, TCD (GC-2014; Shimadzu Corp.) with a Porapak Q column and a methanizer. The conversion, yield, and input power were calculated by some equations (see ESI†).
2.4 Characterization
Powder X-ray diffractometry (XRD, RINT-Ultima III; Rigaku Corp.) was used to characterize the crystalline structure of catalysts before and after the activity test. The anode voltage and current were 40 kV, 40 mA, respectively, with Cu-Kα radiation. Fig. S2† shows XRD patterns of catalysts before and after the activity test. The diffraction peaks at 28.6, 33.0, 47.4, 56.4, 69.3, and 76.7° were assigned respectively to the CeO2 crystal facets of (111), (200), (220), (311), (222), and (400). No significant change in the crystalline structure occurred during the reaction.The nitrogen physical ad-/desorption isotherm of catalysts was measured using a surface area analyser (Gemini VII; Micromeritics Instrument Corp.) at 77 K. The specific surface area was obtained using the BET method. The specific surface area of the catalyst before the activity test was 158 m2 g−1.
The Pt particle size was measured using a field-emission electron microscope (FE-TEM, JEM-2100F; Jasco Corp.) with 200 kV accelerating voltage and 0.1 nm resolution. Fig. S3† shows HAADF-STEM images and EDX mappings of 1wt%Pt/CeO2. The image of elemental mapping shows that Pt was highly dispersed on CeO2 and that the particle size could not be estimated. The Pt loading amount on the catalyst was confirmed by ICP-OES. The X-ray photoelectron spectroscopy (XPS) measurements were taken (PHI 5000 Versa Probe II; ULVAC-PHI Corp.) with a monochromatic Al-Kα X-ray source. Before the measurement, samples were pretreated with H2 (H2:Ar = 1:4; 100 SCCM total flow rate) and O2 (O2:Ar = 1:4; 100 SCCM total flow rate) at 693 K for 30 min. Subsequently they were exposed to CH4, CO2, and Ar flow, respectively, for 20 min. In each flow, the EF was applied for 10 min at 3 mA. The samples were transported to the instrument using a transfer vessel while avoiding exposure to air. The obtained binding energies were calibrated using C 1s assigned to C–H or C–C (284.8 eV). The peak fittings were conducted using the Shirley method.30 The Ce3+ fraction was calculated using the equation shown in ESI.†
2.5 DFT calculation
All Density Functional Theory (DFT) calculations were performed using the Vienna Ab initio Simulation Package (VASP) 5.4.4 with the PAW (Projector Augmented Wave) potentials.31–33 The Perdew–Burke–Ernzerhof (PBE) of the generalized gradient approximation (GGA) was used as the functional, and the van der Waals force was corrected by DFT-D3.34,35 The plane wave cutoff was set to 400 eV, and the on-site Coulomb interaction of Ce 4f orbitals was taken into account using the DFT+U method with U = 5.0 eV.36–41 As reciprocal space sampling, (5 × 5 × 5) and (1 × 1 × 1) k-points were sampled using Gaussian smearing for CeO2 bulk and slab, respectively, and 0.08 Å−1 k-points were sampled using the first order Methfessel–Paxton method for Pt bulk.42 All sampling points were decided with Monkhorst–Pack method.43The calculated lattice constant of CeO2 bulk was 5.437 Å with an error of 0.4% from the experimental value.44 The CeO2(111) slab was represented by the three repeated layers of (3 × 6) supercells with 20 Å vacuum layers. The lowest layer was fixed during the structural optimization. The calculated lattice constant of Pt bulk was 3.909 Å with an error of 0.4% from the experimental value.45 A Pt rod were prepared from the (111) and (100) planes of the Pt bulk as the side and top, respectively, and placed on a CeO2 slab to express Pt/CeO2 surface. The O2 molecules in the gas phase were placed in a 10 × 10 × 10 Å box and calculated at Γ point. All calculation models were depicted using VESTA.46 (Fig. S13 and S14†) In addition, the oxygen vacancy formation energies were calculated according to the following equation.
| EVox = E(Pt/CeO2 without O) + (½E(O2) − E(Pt/CeO2)) | (2) |
3. Results and discussion
3.1 Catalytic activity
First, activity tests were performed to evaluate the activity with EF at low temperatures. Yabe et al.26 reported DRM activity with EF: the reaction proceeds at a temperature as low as 473 K for Ni-supported La-doped ZrO2 with EF. As the catalyst, Pt/CeO2 was selected because CeO2 has been studied for other reactions with EF, such as methane steam reforming29 and dehydrogenation of methylcyclohexane.47 Then, Pt/CeO2 catalysts with various supported amounts of Pt were investigated (Fig. S4†), and 1wt%Pt/CeO2 showed highest activity and was selected for suitable catalyst. Table 1 presents the catalytic activity of 1wt%Pt/CeO2 at 473 K. The activities at different temperatures are presented in Table S1.† Fig. S5† shows the relation between reaction temperature and conversion in DRM (left), the Arrhenius plot (middle), and H2/CO ratio (right). The conversion of CH4 and CO2 at 473 K was more than 15% (it is conducted in a kinetic region), whereas the reaction did not proceed without EF at this temperature. The cause of catalyst deactivation, the amount of carbon deposition, was negligible because the carbon balance was almost 100%. No carbon formation on the catalyst was apparent after the reaction. The apparent activation energy calculated from the Arrhenius plot (Fig. S5†) without EF was 104 kJ mol−1, but it was markedly lower with EF: only 4.82 kJ mol−1. As reported from an earlier study, the H2/CO ratio with EF was high in the low-temperature range.26 In addition, the power efficiency, the value obtained by dividing the gained enthalpy during the reaction by the applied power, was calculated as around 20%. As described above, the 1wt%Pt/CeO2 showed high activity for DRM at 473 K in the electric field. Therefore, we investigated the reaction pathway related to this process using various methods including DRIFTs.| Temp./K | CH4 conv./% | CO2 conv./% | H2/CO/— | Power/W | Energy efficiency/— | |
|---|---|---|---|---|---|---|
| With EF | 453 | 15.1 | 16.4 | 0.763 | 2.81 | 0.186 |
| 473 | 15.0 | 16.5 | 0.754 | 2.91 | 0.179 | |
| 523 | 15.9 | 17.9 | 0.728 | 2.74 | 0.203 | |
| 573 | 16.4 | 19.2 | 0.696 | 2.61 | 0.223 | |
| 623 | 17.4 | 20.9 | 0.667 | 2.54 | 0.244 | |
| 672 | 19.1 | 24.0 | 0.625 | 2.63 | 0.262 | |
| 723 | 27.5 | 34.6 | 0.623 | 2.87 | 0.347 | |
| Without EF | 472 | 0 | 0 | — | — | — |
| 523 | 0.00 | 0.01 | 0.00 | — | — | |
| 573 | 0.03 | 0.07 | 0.00 | — | — | |
| 623 | 0.15 | 0.42 | 0.00 | — | — | |
| 673 | 0.66 | 1.60 | 0.135 | — | — | |
| 723 | 2.21 | 4.75 | 0.237 | — | — |
3.2 Effects of EF on CO2 adsorption
To elucidate reasons why DRM proceeds effectively even at such low temperatures, in situ DRIFTs measurements were performed to observe the changes in CO2 adsorption state before and after applying the EF. The adsorption of CO2 on CeO2-based catalysts has been well-studied. Many reports have assigned the peaks of carbonaceous species. The assignment of carbonaceous species used for this study and their values in the literature are shown in ESI.†5,11,48–73 We assigned peaks in the range of 1800–2100 cm−1 to carbonyl species adsorbed onto Pt, but they are not shown in the table. In this measurement, we applied the EF to the catalyst in the CO2 flow and compared the spectra before and after the application of EF. Fig. 1 shows (a) DRIFT spectra before and after the application of EF and (b) the difference spectrum obtained by subtracting the spectrum without EF from that with EF. As shown in Fig. 1(b), the peak area at around 1160 cm−1 was increased by the application of EF. The peaks at 1160, 1131, 1074, and 1031 cm−1 are assigned respectively as monodentate, bridged, polydentate, and bidentate carbonate peaks (see ESI†). The increase of the peak area at around 1160 cm−1 signifies that the number of carbonate species adsorbed onto CeO2 increased because of application of the EF.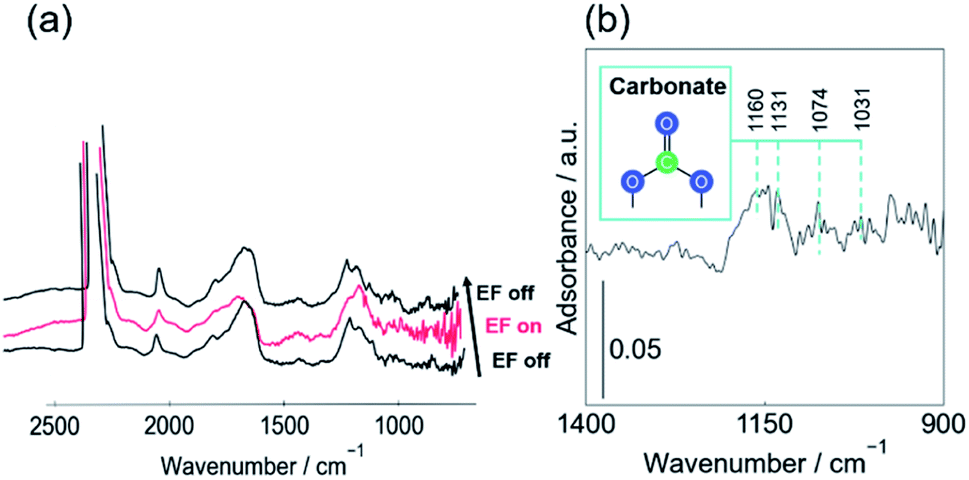 | ||
| Fig. 1 (a) In situ DRIFT spectra with, without EF under CO2 flow over 1wt%Pt/CeO2, (b) difference spectra in 900–1400 cm−1 region (with EF − without EF); CO2:Ar = 1:8; 90 SCCM total flow rate; 3.0 mA input current; at 473 K. | ||
In general, CO2 adsorbs on metal oxides and forms adsorbed species such as carbonate and bicarbonate.71,74 DFT calculations and various experimentally obtained results revealed that lattice oxygen defects enhance the adsorption of gas-phase CO2.75–77 Lattice oxygen in the catalyst support dissociates CH4 in the DRM reaction, leading to the formation of oxygen vacancies.75,78,79
Consequently, the reaction of CH4 with lattice oxygen is expected to be an important factor supporting oxygen vacancy formation. Therefore, to investigate the effects of CH4 on the formation of lattice oxygen defects with or without EF, we took XPS measurements and evaluated the reducibility of CeO2. We then compared the fractions of Ce3+ and Ce4+ of 1wt%Pt/CeO2 after exposing catalysts to CH4, CO2, and Ar flow, respectively with and without EF at 473 K. Table 2 shows the calculated Ce3+ fraction of 1wt%Pt/CeO2 spent in each condition. Fig. S7† presents the obtained XPS spectra. In the Ce 3d spectrum, three doublet peaks at (u′′′: 916.6, v′′′: 898.2), (u′′: 907.5, v′′: 888.8), and (u: 900.7, v: 882.3) eV are assigned to Ce4+; two doublet peaks at (u′: 903.8, v′: 885.2), (u0: 899.3, v0: 880.7) eV are assigned to Ce3+.80 Table 2 shows that the fraction of Ce3+, after being kept in the CH4 flow with EF, was larger than that without EF. The increase in the Ce3+ fraction in the CH4 flow suggests that the oxygen species were removed easily by reaction with CH4 in the EF, resulting in the formation of more lattice oxygen defects. Fig. S8† depicts changes in DRIFT spectra before and after application of the EF under the CH4 flow. Fig. S8† (b) shows that the peak area of carbonate species increased. The increase of the carbonate species signifies that CH4 consumed lattice oxygen, which is the only oxygen source, resulting in the formation of lattice oxygen defects.
| Condition | Surface content of Ce3+/% |
|---|---|
| CH4 flow without EF | 16.8 |
| CH4 flow with EF | 19.4 |
| Ar flow without EF | 16.2 |
| Ar flow with EF | 16.7 |
| CO2 flow without EF | 17.5 |
| CO2 flow with EF | 18.3 |
The increase of Ce3+ fraction by application of the EF in Ar flow was 0.5%, which is much less than that in the CH4 flow. Therefore, the oxygen species are not released simply by application of the EF, but are released by the reaction with CH4 in the EF. Therefore, the oxygen of CeO2 is regarded as activated by the EF. Thus the reaction of lattice oxygen with CH4 is promoted, thereby forming the oxygen defect.
The fraction of Ce3+ increased from 17.5% to 18.3% after applying the EF in the CO2 flow, indicating an increase of carbonate species by CO2 adsorption. According to Yi et al.,81 Ce4+ decreases along with the carbonate species formation because the carbonate species are formed by oxidization of the adsorbed CO2 by the surface oxygen of the CeO2 support. Therefore, the reduction of Ce in the EF in the CO2 flow also supports enhancement of the CO2 adsorption.
The enhanced reactivity of oxygen in the EF was confirmed using oxygen isotope exchange test. Fig. S9† portrays results of the oxygen isotope exchange test. For this measurement, the EF was applied to the catalyst in the 18O2 flow after the surface oxygen species in CZO were filled with 16O. Then a mass spectrometer was used to analyze the produced gas. As shown in Fig. S9,† after the application of EF, the amount of 18O2 decreased immediately. Simultaneously, 16O18O appeared. However, the appearance of 16O2 was slight. These results indicate that lattice oxygen is not removed merely by applying EF. Rather, it is removed by reaction with the gas species. This result also supports the increased reactivity of the oxygen in the EF.
3.3 Reactivity of carbonate species
Next, an alternating feed test between CO2 and CH4 was conducted to elucidate the behaviour of CO2 and CH4 in the DRM reaction with and without EF. In the alternating feed test, CO2 and CH4 were switched; GC was used to analyze the produced gas. Fig. 2 shows the calculated formation rate of CO after switching the gas with EF. Actually, CO was not observed without EF, although it was formed with EF. The CO formed after either the CO2 or CH4 supply. This CO formation signifies that oxidation of CH4 and reduction of CO2 occur on Pt/CeO2 with EF. In both gas feeds, the amount of the produced CO decreased with the time course. The oxidation and reduction performance of the catalyst decreased with time because of the continuous supply or consumption of oxygen. From earlier results and research, we assumed the role of lattice oxygen in this test. In the CH4 flow, lattice oxygen was removed by CH4. Then depletion of the activated oxygen caused the decrease of the produced CO amount. However, CO2 filled the lattice oxygen defect in the CO2 flow. The amount of produced CO decreased along with the decrease of the lattice oxygen defects.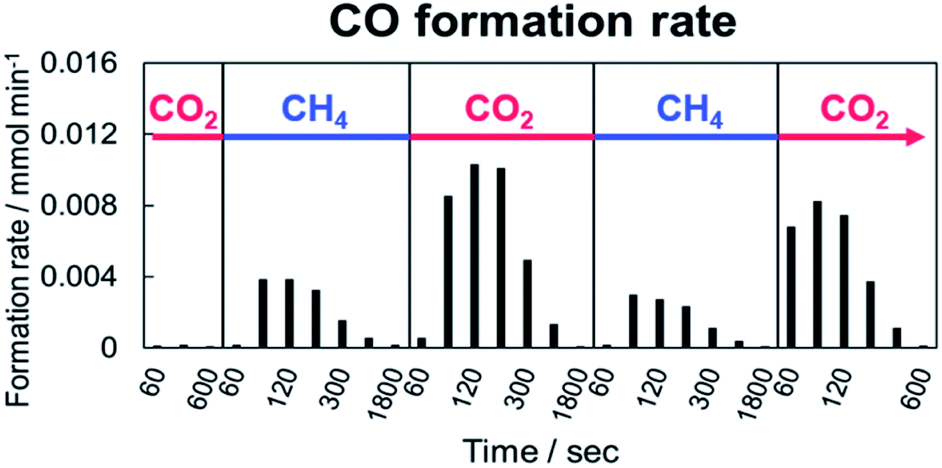 | ||
| Fig. 2 CO formation rate after switching between CO2 and CH4 over 1wt%Pt/CeO2 catalyst with EF; 80 SCCM total flow rate (CH4:Ar = 1:3 under CH4 flow, CO2:Ar = 1:3 under CO2 flow) at 473 K. | ||
In situ DRIFTs measurements were conducted in the CO2, CH4 alternating feed test to observe the reactivity of the carbonate species. Fig. 3 portrays the change in spectra in the test between CH4 and CO2 (a) with EF, (b) without EF, and the difference in the spectrum before and after CH4 supply (c) with EF, (d) without EF. The peaks assigned to carbonate species decreased by stopping the CO2 supply, irrespective of the presence of EF. A similar decrease in the carbonate peak was observed when the supply gas was switched from CO2 to Ar without EF (Fig. S10 and S11†). Without EF, CO was not formed when the CO2 gas feed was switched to CH4 or Ar flow, suggesting that carbonate species desorbed as CO2. However, when the EF was applied, CO was formed, suggesting that some of the carbonate species reduced and dissociated as CO.
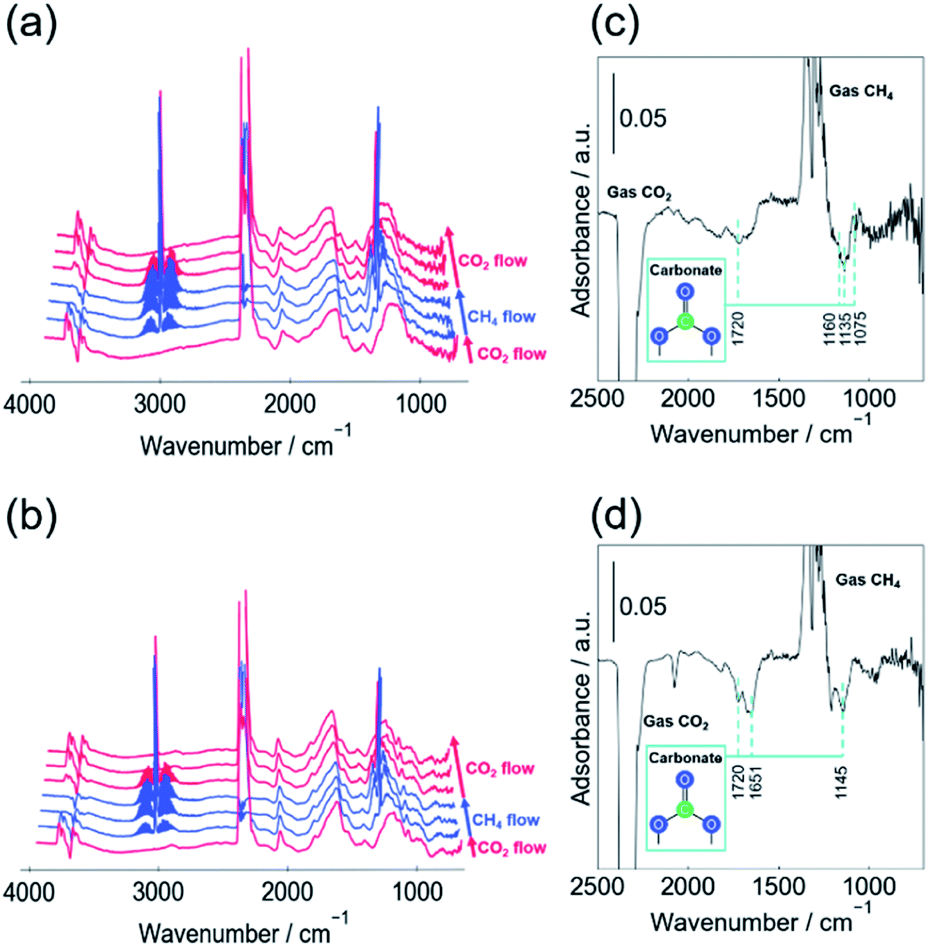 | ||
| Fig. 3 In situ DRIFT spectra over 1wt%Pt/CeO2 catalyst after switching between CO2 and CH4 feeds at 473 K. 120, 600, 1200 seconds after switching to CH4, 120, 600, 1200 seconds after switching to CO2, (a) with (b) without EF; difference spectrum in 700–2500 cm−1 (CH4 at steady state – CO2 at steady state) (c) with (d) without EF; 90 SCCM total flow rate (CH4:Ar = 1:8 under CH4 flow, CO2:Ar = 1:8 under CO2 flow at 473 K); 3.0 mA input current. | ||
3.4 Oxygen release ability of CeO2
DFT calculations were performed to investigate the tendency of oxygen defect formation on Pt-loaded CeO2. Fig. S15† shows the calculated oxygen defect locations, and Table S3† shows the oxygen defect formation energy at each location. The formation energy of the oxygen defects became higher as the distance from Pt increased. The results show that the oxygen defects are more easily generated at the interface of Pt and CeO2 than other locations. This result supports the reaction of CO2 at the interface of Pt and CeO2 because the oxygen defects are more easily generated near the interface, and the generated oxygen defects facilitate the adsorption of CO2 near the interface.75 Note that the sign of the value does not mean whether it is spontaneous or not, because the energy of half of the oxygen molecule is used as the energy of the oxygen atom and the term of pressure, and the temperature is not taken into account in the calculation of the oxygen defect formation energy.3.5 Discussion
From these results, we consider the effect of EF on DRM and the reaction mechanism.It is reported that the enhancement in CO2 adsorption is related to the oxygen activity of the oxide support. Chen et al.82 investigated the adsorption and oxidation, redox behaviour of CO and CO2 on CeO2 using DFT calculations. They reported that highly reactive surface oxygen, which Ce weakly bonds, lowers CO2 adsorption and carbonate species formation barriers. In addition, results of several studies suggest that EF promoted the activation of the oxygen in the oxide support.83–85 High catalytic activity due to the activated oxygen on supports by applying EF was reported in the OCM reaction using CeO2,85 and in the TWC catalytic reaction of exhaust gases using cerium zirconium oxides (Ce0.7Zr0.3O2) as support.84 Therefore, we considered that lattice oxygen was activated by the EF, and promoted CO2 adsorption.
The XPS measurements taken for this study revealed that CH4 molecules react with surface oxygen, consume them, and thereby reduce CeO2 more with EF than without EF (Table 2). This improved reactivity of CH4 with surface oxygen might be attributed to the enhanced reactivity of the oxygen by the EF.
Next, the CO2, CH4 alternating test shows the CO2 dissociation ability of CeO2. When the gas was switched from CO2 to Ar without EF, the carbonate species desorbed as CO2 (Fig. S9 and S10†). When the gas was switched to CH4 without EF, the carbonate species also desorbed as CO2 and CO did not form. However, CO was formed when the supply gas was switched to CH4 with EF, indicating that a part of the carbonate species reacted and desorbed as CO (Fig. 2 and 3). These results indicate that the EF promotes dissociation of CO2 adsorbed on CeO2.
On the other hand, CH4 dissociates on Pt at the interface of metal (Pt) and support (CeO2) with EF75 and without EF.86,87 CH4 and CO2 with high reactivity at the interface of Pt and CeO2 suggests that active site of this reaction is the interface of Pt and CeO2.
First, with the EF, the reaction of CO2 is considered to proceed by the Mars–van Krevelen (MvK) mechanism using the lattice oxygen of the support. Yabe et al.75 have shown that the dissociation of CO2 proceeds via the lattice oxygen in the EF by 18O isotope test. This indicates that the dissociation of CO2 by the MvK mechanism proceeds at low temperatures with EF.
Contrarily, CH4 dissociation proceeds on Pt. Previously, many reports have suggested the rate determining step (RDS) of the reaction of CH4. Otsuka et al.88 showed that CH4 dissociation and recombination proceed much faster than the oxidation of CH4 on CeO2 and Pt/CeO2 by the CH4, CD4 exchange test. Wei and Iglesia89 also suggested C–H bond activation on Pt is reversible, using the CH4, CD4 exchange test in DRM reaction. Thus, the oxidation of H species formed after CH4 dissociation is necessary for the reaction to proceed. Das et al.86 have also reported that reaction of CH4 and lattice oxygen is the RDS in the DRM reaction. Therefore, we considered that the RDS in this reaction is oxidation of the H species. The EF promotes the reaction to activate lattice oxygen.
From these results, we considered the possible DRM mechanism as follows.
(1) Methane dissociation on Pt90
| CH4(g) → CH4(Pt) | (3) |
| CH4(Pt) → CH4−x(Pt) + xH(Pt) | (4) |
(2) Diffusion of H species86,90
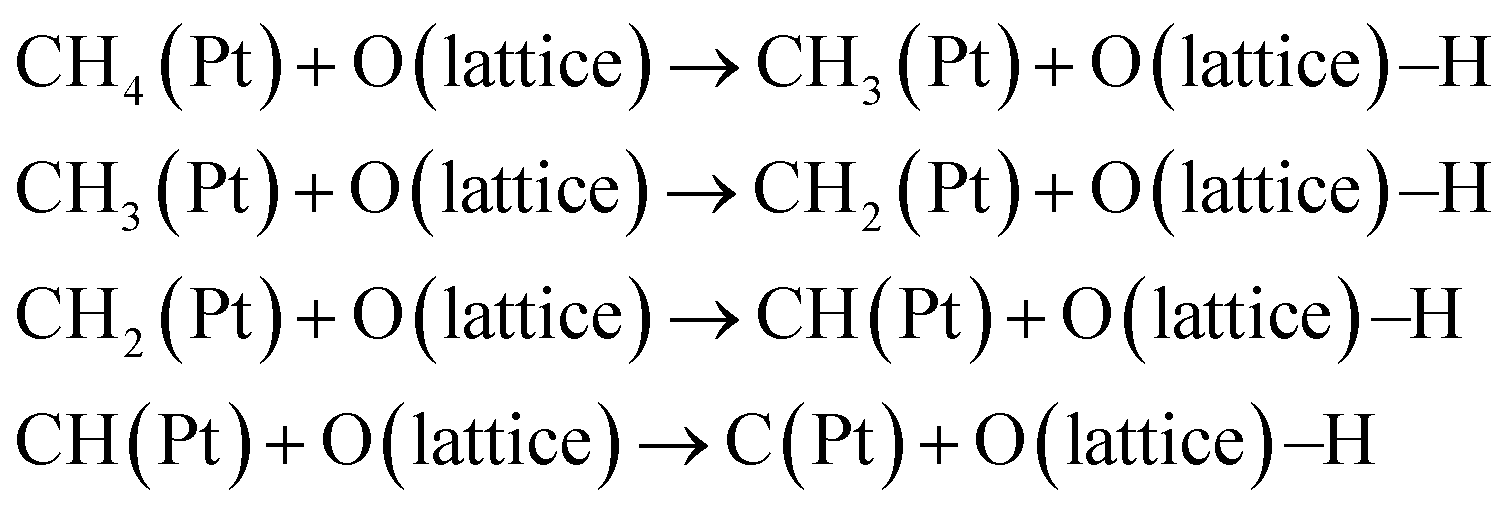 | (5) |
| H(Pt) + O(lattice) → O(lattice)–H | (6) |
(3) Methane desorption as CO86
| C(Pt) + O(lattice) → CO + Vox | (7) |
(4) CO2 adsorption
| CO2(g) + O(lattice) + Vox → CO3(CeO2) | (8) |
(5) Hydrogenation of CO2 species
| CO2(g) + O(lattice)–H → HCO3(CeO2) | (9) |
| CO3(CeO2) + H(Pt, CeO2) → HCO3(CeO2) | (10) |
(6) Desorption of carbonaceous species
| HCO3(CeO2) + H(Pt, CeO2) → CO(g) + H2O(g) + O(lattice) + Vox | (11) |
| HCO3(CeO2) → CO(CeO2) + O(lattice)–H + O(lattice) | (12) |
(7) Refilling of oxygen vacancy
| Vox + H2O → O(lattice) + 2H(CeO2) | (13) |
(8) Formation of H2
| 2H(CeO2) → H2(g) | (14) |
| 2H(Pt) → H2(g) | (15) |
First, the EF effect is to enhance the oxygen activity of oxide support because the adsorption of CO2 was promoted significantly by EF application (eqn (8)). The activated surface oxygen reacts easily with C and H species dissociated from CH4 and forms oxygen defects (eqn (5)–(7)).
The activated oxygen and the oxygen defects of oxide support both contribute to enhancement of the CO2 adsorption. Finally, the adsorbed CO2 forms carbonate species, reacts, and desorbs as CO. The remaining oxygen species again work as the oxidant of CH4. Consequently, the DRM reaction in the EF proceeds via a redox cycle of CeO2 support.
4. Conclusion
The effect of the electric field (EF) on DRM reaction was investigated using 1wt%Pt/CeO2 by activity tests, in situ DRIFTs, XPS, and DFT calculation. 1wt%Pt/CeO2 showed high activity for DRM with EF, even at 473 K, at which thermal catalysis proceeds only slightly. In situ DRIFTs measurements in CO2 flow revealed that the adsorbed species of CO2 increases when the EF is applied. This increase signifies that the adsorption of CO2 is promoted by the EF. XPS measurements of the catalysts after the treatment in CO2 atmosphere with EF suggest the enhancement of CO2 adsorption on CeO2. In addition, XPS measurements of catalysts after exposure to CH4 flow with EF confirmed that the Ce3+ fraction of CeO2 increased, indicating that CeO2 support was reduced after the reaction of surface oxygens and CH4 molecules in EF, even at 473 K. These results suggest that the oxygen in the oxide support was activated in the EF. Furthermore, the reaction of CH4 and the surface oxygen was promoted, thereby forming lattice oxygen defects and CO. The activated oxygen in the EF and the resulting lattice oxygen defects by the reaction with CH4 molecules both promote CO2 adsorption. The reaction is considered to proceed at the interface of Pt and CeO2. DFT calculations also support the reaction mechanism of DRM at the interface of Pt and CeO2.In summary, the reaction mechanism of DRM in the EF was inferred as presented below. The EF can improve the oxygen activity and can therefore promote the reaction of CH4 molecules and surface oxygens at the Pt–CeO2 interface, forming the oxygen defects. Both the activated oxygen and the oxygen defects of oxide support enhance the adsorption of CO2, forming more carbonate species on CeO2 in the EF. Finally, CO2 molecules dissociate to form CO via the carbonate species.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly achieved through the use of the supercomputer system at the information initiative center, Hokkaido University, Sapporo, Japan. This work was the result of using characterization equipments (JEM-2100F and Versa Probe II: Material Characterization Central Laboratory in Waseda University) shared in MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting construction of core facilities) Grant Number JPMXS0440500021 and JPMXS0440500022.References
- J. R. Rostrup-Nielsen and J. H. B. Hansen, J. Catal., 1993, 144, 38–49 CrossRef CAS
.
- A. Sternberg and A. Bardow, ACS Sustainable Chem. Eng., 2016, 4, 4156–4165 CrossRef CAS
- V. T. Sagar and A. Pintar, Appl. Catal., A, 2020, 599, 117603 CrossRef
- M. A. Álvarez, L. F. Bobadilla, V. Garcilaso, M. A. Centeno and J. A. Odriozola, J. CO2 Util., 2018, 24, 509–515 CrossRef
- Z. Liu, F. Zhang, N. Rui, X. Li, L. Lin, L. E. Betancourt, D. Su, W. Xu, J. Cen, K. Attenkofer, H. Idriss, J. A. Rodriguez and S. D. Senanayake, ACS Catal., 2019, 9, 3349–3359 CrossRef CAS
- M. García-Diéguez, E. Finocchio, M. Á. Larrubia, L. J. Alemany and G. Busca, J. Catal., 2010, 274, 11–20 CrossRef
- B. Bachiller-Baeza, C. Mateos-Pedrero, M. A. Soria, A. Guerrero-Ruiz, U. Rodemerck and I. Rodríguez-Ramos, Appl. Catal., B, 2013, 129, 450–459 CrossRef CAS
- M. Németh, Z. Schay, D. Srankó, J. Károlyi, G. Sáfrán, I. Sajó and A. Horváth, Appl. Catal., A, 2015, 504, 608–620 CrossRef
- C. C. Chong, Y. W. Cheng, H. D. Setiabudi, N. Ainirazali, D. V. N. Vo and B. Abdullah, Int. J. Hydrogen Energy, 2020, 45, 8507–8525 CrossRef CAS
- S. Das, M. Sengupta, A. Bag, M. Shah and A. Bordoloi, Nanoscale, 2018, 10, 6409–6425 RSC
- H. Chen, S. Chansai, S. Xu, S. Xu, Y. Mu, C. Hardacre and X. Fan, Catal. Sci. Technol., 2021, 11, 5260–5272 RSC
- L. F. Bobadilla, J. L. Santos, S. Ivanova, J. A. Odriozola and A. Urakawa, ACS Catal., 2018, 8, 7455–7467 CrossRef CAS
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed
- W. C. Chung, K. L. Pan, H. M. Lee and M. B. Chang, Energy Fuels, 2014, 28, 7621–7631 CrossRef CAS
- X. Gao, Z. Lin, T. Li, L. Huang, J. Zhang, S. Askari, N. Dewangan, A. Jangam and S. Kawi, Catalysts, 2021, 11, 455 CrossRef CAS
- X. Zhang, C. S. M. Lee, D. M. P. Mingos and D. O. Hayward, Catal. Lett., 2003, 88, 129–139 CrossRef CAS
- M. Wang, M. Shen, X. Jin, J. Tian, M. Li, Y. Zhou, L. Zhang, Y. Li and J. Shi, ACS Catal., 2019, 9, 4573–4581 CrossRef CAS
- X. Zhu, J. H. Liu, X. S. Li, J. L. Liu, X. Qu and A. M. Zhu, J. Energy Chem., 2017, 26, 488–493 CrossRef
- D. Mei, X. Zhu, Y. L. He, J. D. Yan and X. Tu, Plasma Sources Sci. Technol., 2015, 24, 015011 CrossRef CAS
- X. Tu and J. C. Whitehead, Appl. Catal., B, 2012, 125, 439–448 CrossRef CAS
- M. Torimoto, S. Ogo, Y. Hisai, N. Nakano, A. Takahashi, Q. Ma, J. G. Seo, H. Tsuneki, T. Norby and Y. Sekine, RSC Adv., 2020, 10, 26418–26424 RSC
- S. Okada, R. Manabe, R. Inagaki, S. Ogo and Y. Sekine, Catal. Today, 2018, 307, 272–276 CrossRef CAS
- S. Ogo and Y. Sekine, Chem. Rec., 2017, 17, 726–738 CrossRef CAS PubMed
- M. Torimoto, K. Murakami and Y. Sekine, Bull. Chem. Soc. Jpn., 2019, 92, 1785–1792 CrossRef CAS
- T. Yabe, K. Mitarai, K. Oshima, S. Ogo and Y. Sekine, Fuel Process. Technol., 2017, 158, 96–103 CrossRef CAS
- K. Oshima, T. Shinagawa, Y. Nogami, R. Manabe, S. Ogo and Y. Sekine, Catal. Today, 2014, 232, 27–32 CrossRef CAS
- K. Yamada, S. Ogo, R. Yamano, T. Higo and Y. Sekine, Chem. Lett., 2020, 49, 303–306 CrossRef CAS
- R. Manabe, S. Okada, R. Inagaki, K. Oshima, S. Ogo and Y. Sekine, Sci. Rep., 2016, 6, 38007 CrossRef CAS
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef PubMed
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- J. J. Carey and M. Nolan, Catal. Sci. Technol., 2016, 6, 3544–3558 RSC
- K. Murakami, Y. Mizutani, H. Sampei, A. Ishikawa and Y. Sekine, J. Chem. Phys., 2021, 154, 164705 CrossRef CAS PubMed
- M. D. Krcha, A. D. Mayernick and M. J. Janik, J. Catal., 2012, 293, 103–115 CrossRef CAS
- H. T. Chen and J. G. Chang, J. Chem. Phys., 2010, 132, 214702 CrossRef PubMed
- M. Nolan, J. Mater. Chem., 2011, 21, 9160–9168 RSC
- A. D. Mayernick and M. J. Janik, J. Phys. Chem. C, 2008, 112, 14955–14964 CrossRef CAS
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS PubMed
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef
- A. Sundaresan, R. Bhargavi, N. Rangarajan, U. Siddesh and C. N. R. Rao, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 161306 CrossRef
- I. N. Leontyev, A. B. Kuriganova, N. G. Leontyev, L. Hennet, A. Rakhmatullin, N. V. Smirnova and V. Dmitriev, RSC Adv., 2014, 4, 35959–35965 RSC
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS
- K. Takise, A. Sato, K. Murakami, S. Ogo, J. G. Seo, K. I. Imagawa, S. Kado and Y. Sekine, RSC Adv., 2019, 9, 5918–5924 RSC
- M. Daturi, C. Binet, J. C. Lavalley and G. Blanchard, Surf. Interface Anal., 2000, 30, 273–277 CrossRef CAS
- L. Fan, J. Zhang, K. Ma, Y. Zhang, Y. M. Hu, L. Kong, A. ping Jia, Z. Zhang, W. Huang and J. Q. Lu, J. Catal., 2021, 397, 116–127 CrossRef CAS
- L. Atzori, M. G. Cutrufello, D. Meloni, B. Onida, D. Gazzoli, A. Ardu, R. Monaci, M. F. Sini and E. Rombi, Front. Chem. Sci. Eng., 2021, 15, 251–268 CrossRef
- Z. Wu, A. K. P. Mann, M. Li and S. H. Overbury, J. Phys. Chem. C, 2015, 119, 7340–7350 CrossRef CAS
- Z. Wu, M. Li and S. H. Overbury, J. Catal., 2012, 285, 61–73 CrossRef CAS
- T. Shido and Y. Iwasawa, J. Catal., 1993, 141, 71–81 CrossRef CAS
- S. Y. Zhao, S. P. Wang, Y. J. Zhao and X. Bin Ma, Chin. Chem. Lett., 2017, 28, 65–69 CrossRef CAS
- Z. Wu, M. Li and S. H. Overbury, J. Catal., 2012, 285, 61–73 CrossRef CAS
- K. Yoshikawa, H. Sato, M. Kaneeda and J. N. Kondo, J. CO2 Util., 2014, 8, 34–38 CrossRef CAS
- Y. Wang, Z. Liu, M. P. Confer, J. Li and R. Wang, Mol. Catal., 2021, 509, 111629 CrossRef CAS
- O. Pozdnyakova, D. Teschner, A. Wootsch, J. Kröhnert, B. Steinhauer, H. Sauer, L. Toth, F. C. Jentoft, A. Knop-Gericke, Z. Paál and R. Schlögl, J. Catal., 2006, 237, 1–16 CrossRef CAS
- P. A. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catal. Today, 2013, 215, 201–207 CrossRef CAS
- P. Bea, A. L. Camera, A. Hornes and A. Martınez-Arias, J. Phys. Chem. C, 2009, 113, 10689–10695 CrossRef
- S. M. Lee, Y. H. Lee, D. H. Moon, J. Y. Ahn, D. D. Nguyen, S. W. Chang and S. S. Kim, Ind. Eng. Chem. Res., 2019, 58, 8656–8662 CrossRef CAS
- F. C. Meunier, D. Tibiletti, A. Goguet, D. Reid and R. Burch, Appl. Catal., A, 2005, 289, 104–112 CrossRef CAS
- G. Busca and V. Lorenzelli, Mater. Chem., 1982, 7, 89–126 CrossRef CAS
- S. Hilaire, X. Wang, T. Luo, R. J. Gorte and J. Wagner, Appl. Catal., A, 2004, 258, 271–276 CrossRef CAS
- T. Tabakova, F. Boccuzzi, M. Manzoli and D. Andreeva, Appl. Catal., A, 2003, 252, 385–397 CrossRef CAS
- C. Li, Y. Sakata, T. Arai, K. Domen, K. I. Maruya and T. Onishi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 1451–1461 RSC
- L. Chen, S. Wang, J. Zhou, Y. Shen, Y. Zhao and X. Ma, RSC Adv., 2014, 4, 30968–30975 RSC
- M. Wang, M. Shen, X. Jin, J. Tian, Y. Shao, L. Zhang, Y. Li and J. Shi, Chem. Eng. J., 2022, 427, 130987 CrossRef CAS
- Q. Pan, J. Peng, S. Wang and S. Wang, Catal. Sci. Technol., 2014, 4, 502–509 RSC
- G. Finos, S. Collins, G. Blanco, E. Del Rio, J. M. Cíes, S. Bernal and A. Bonivardi, Catal. Today, 2012, 180, 9–18 CrossRef CAS
- C. Li, Y. Sakata, T. Arai, K. Domen, K. I. Maruya and T. Onishi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 929–943 RSC
- A. Yee, S. J. Morrison and H. Idriss, J. Catal., 1999, 186, 279–295 CrossRef CAS
- C. Binet, M. Daturi and J. C. Lavalley, Catal. Today, 1999, 50, 207–225 CrossRef CAS
- G. Ramis, G. Busca and V. Lorenzelli, Mater. Chem. Phys., 1991, 29, 425–435 CrossRef CAS
- T. Yabe, K. Yamada, K. Murakami, K. Toko, K. Ito, T. Higo, S. Ogo and Y. Sekine, ACS Sustainable Chem. Eng., 2019, 7, 5690–5697 CrossRef CAS
- B. Liu, C. Li, G. Zhang, X. Yao, S. S. C. Chuang and Z. Li, ACS Catal., 2018, 8, 10446–10456 CrossRef CAS
- Z. Ma, P. Li, L. Ye, Y. Zhou, F. Su, C. Ding, H. Xie, Y. Bai and P. K. Wong, J. Mater. Chem. A, 2017, 5, 24995–25004 RSC
- K. Kousi, D. Neagu, L. Bekris, E. I. Papaioannou and I. S. Metcalfe, Angew. Chem., 2020, 132, 2531–2540 CrossRef
- K. Sutthiumporn, T. Maneerung, Y. Kathiraser and S. Kawi, Int. J. Hydrogen Energy, 2012, 37, 11195–11207 CrossRef CAS
- L. Óvári, S. Krick Calderon, Y. Lykhach, J. Libuda, A. Erdohelyi, C. Papp, J. Kiss and H. P. Steinrück, J. Catal., 2013, 307, 132–139 CrossRef
- Y. Yu, B. Mao, A. Geller, R. Chang, K. Gaskell, Z. Liu and B. W. Eichhorn, Phys. Chem. Chem. Phys., 2014, 16, 11633–11639 RSC
- F. Chen, D. Liu, J. Zhang, P. Hu, X. Q. Gong and G. Lu, Phys. Chem. Chem. Phys., 2012, 14, 16573–16580 RSC
- A. Sato, S. Ogo, Y. Takeno, K. Takise, J. G. Seo and Y. Sekine, ACS Omega, 2019, 4, 10438–10443 CrossRef CAS PubMed
- Y. Omori, A. Shigemoto, K. Sugihara, T. Higo, T. Uenishi and Y. Sekine, Catal. Sci. Technol., 2021, 11, 4008–4011 RSC
- K. Sugiura, S. Ogo, K. Iwasaki, T. Yabe and Y. Sekine, Sci. Rep., 2016, 6, 25154 CrossRef CAS PubMed
- S. Das, A. Jangam, S. Jayaprakash, S. Xi, K. Hidajat, K. Tomishige and S. Kawi, Appl. Catal., B, 2021, 290, 119998 CrossRef CAS
- J. H. Bitter, K. Seshan and J. A. Lercher, Top. Catal., 2000, 10, 295–305 CrossRef CAS
- K. Otsuka, Y. Wang, E. Sunada and I. Yamanaka, J. Catal., 1998, 175, 152–160 CrossRef CAS
- J. Wei and E. Iglesia, J. Phys. Chem. B, 2004, 108, 4094–4103 CrossRef CAS
- A. P. Ferreira, I. Rodriguez-Ramos, J. A. Anderson and A. Guerrero-Ruiz, Appl. Catal., A, 2000, 202, 183–196 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00402j |
| This journal is © The Royal Society of Chemistry 2022 |