
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights into the chemistry of compound I formation in heme peroxidases: quantum chemical investigations of cytochrome c peroxidase†
Mohamed M. Aboelnga *
*
Chemistry Department, Faculty of Science, Damietta University, New Damietta, 34517, Egypt. E-mail: aboelng@uwindsor.ca
First published on 23rd May 2022
Abstract
Peroxidases are heme containing enzymes that catalyze peroxide-dependant oxidation of a variety of substrates through forming key ferryl intermediates, compounds I and II. Cytochrome c peroxidase (Ccp1) has served for decades as a chemical model toward understanding the chemical biology of this heme family of enzymes. It is known to feature a distinctive electronic behaviour for its compound I despite significant structural similarity to other peroxidases. A water-assisted mechanism has been proposed over a dry one for the formation of compound I in similar peroxidases. To better identify the viability of these mechanisms, we employed quantum chemistry calculations for the heme pocket of Ccp1 in three different spin states. We provided comparative energetic and structural results for the six possible pathways that suggest the preference of the dry mechanism energetically and structurally. The doublet state is found to be the most preferable spin state for the mechanism to proceed and for the formation of the Cpd I ferryl-intermediate irrespective of the considered dielectric constant used to represent the solvent environment. The nature of the spin state has negligible effects on the calculated structures but great impact on the energetics. Our analysis was also expanded to explain the major contribution of key residues to the peroxidase activity of Ccp1 through exploring the mechanism at various in silico generated Ccp1 variants. Overall, we provide valuable findings toward solving the current ambiguity of the exact mechanism in Ccp1, which could be applied to peroxidases with similar heme pockets.
Introduction
Heme enzymes are crucial in biology and exist in almost all living organisms.1 Exploiting the catalytic redox power of the iron-containing heme cofactor, they catalyze a wide range of biological redox reactions.2 Heme peroxidases are the most abundant heme enzymes and activate hydrogen peroxide to oxidize a variety of substrates.3 A defining feature across the whole heme enzyme family is the formation of key ferryl intermediates (compound I), involving a porphyrin π-cation radical, during their catalytic cycle.4–6 Identifying the chemical nature of the oxyferryl bond in compound I has been extensively studied but its precise chemical identity remains a matter of increasing debate.4,5,7–10The most widely accepted catalytic pathway for the formation of compound I (Cpd I) by peroxidases is the Poulos–Kraut acid–base mechanism.11 In this mechanism, the conserved His residue in the distal site acts as an acid–base catalyst that promotes heterolytic cleavage of the O–O bond of the peroxide group.12 The resting-state ferric iron binds H2O2 and the reaction is triggered by an electron transfer from the iron toward the substrate leading to the formation of compound I, a ferryl intermediate with an unprotonated iron–oxo double bond (FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).13 Previous computational studies explored this mechanism in various peroxidases and reported a two-step mechanism in which the heterolytic cleavage of the O–O bond is the rate-limiting step.12–15 According to these studies, the first step to form the ferric-hydroperoxide (FeIII–OOH) intermediate (Cpd 0) takes place through proton abstraction from the bound peroxide in FeIII–O2H2 by the nearby His acid–base catalyst (IC1), Scheme 1. This step is followed by a structural rearrangement of the imidazole side chain of the newly protonated His generating a second conformation of Cpd 0 (IC2), in which the His residue is hydrogen-bonded with the distal OH group of Fe–OOH, Scheme 1. Subsequently, the protonated His residue releases its proton to the distal OH, promoting heterolytic cleavage of the O–O bond together with formation of a water molecule.16 However, other theoretical studies support the proposal that a nearby water molecule may assist proton transfer from H2O2 to the His acid–base catalyst.17,18 This proposal is in consistent with the long distance reported between the catalytic His and H2O2 and the presence of crystalized water molecules around the ferryl oxygen.2,12 According to these studies, the incorporation of a bridged water molecule in the first step of the mechanism lowered the activation barrier.12,17 Thus, in contrast to the initial no-water (dry) pathway, an water-mediated (wet) mechanism has been proposed for peroxide activation by peroxidases.2,17,18 Indeed, water-assisted mechanism is well-known approach in facilitating the reaction in other fundamental enzymes.19,20
O).13 Previous computational studies explored this mechanism in various peroxidases and reported a two-step mechanism in which the heterolytic cleavage of the O–O bond is the rate-limiting step.12–15 According to these studies, the first step to form the ferric-hydroperoxide (FeIII–OOH) intermediate (Cpd 0) takes place through proton abstraction from the bound peroxide in FeIII–O2H2 by the nearby His acid–base catalyst (IC1), Scheme 1. This step is followed by a structural rearrangement of the imidazole side chain of the newly protonated His generating a second conformation of Cpd 0 (IC2), in which the His residue is hydrogen-bonded with the distal OH group of Fe–OOH, Scheme 1. Subsequently, the protonated His residue releases its proton to the distal OH, promoting heterolytic cleavage of the O–O bond together with formation of a water molecule.16 However, other theoretical studies support the proposal that a nearby water molecule may assist proton transfer from H2O2 to the His acid–base catalyst.17,18 This proposal is in consistent with the long distance reported between the catalytic His and H2O2 and the presence of crystalized water molecules around the ferryl oxygen.2,12 According to these studies, the incorporation of a bridged water molecule in the first step of the mechanism lowered the activation barrier.12,17 Thus, in contrast to the initial no-water (dry) pathway, an water-mediated (wet) mechanism has been proposed for peroxide activation by peroxidases.2,17,18 Indeed, water-assisted mechanism is well-known approach in facilitating the reaction in other fundamental enzymes.19,20
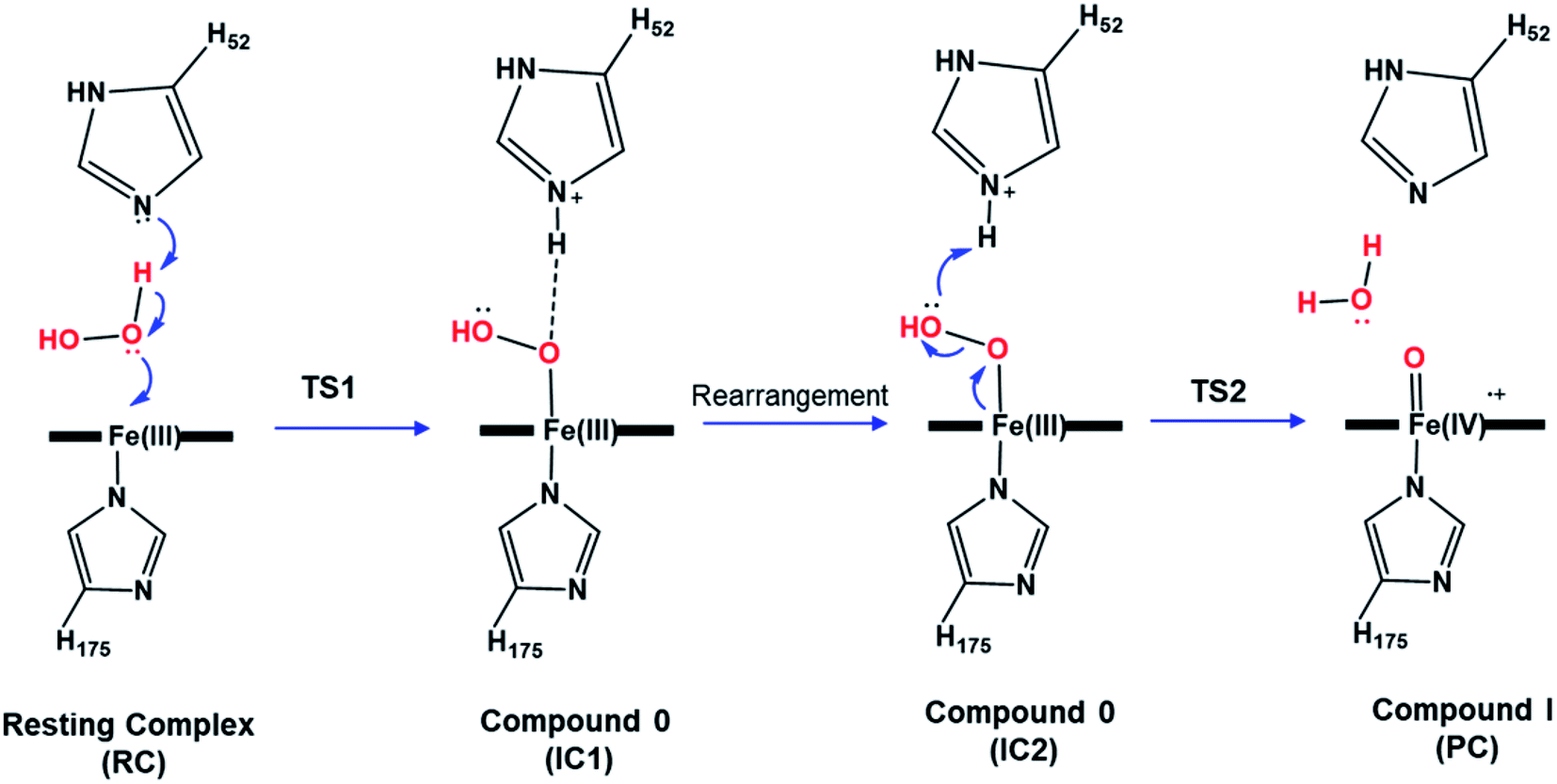 | ||
| Scheme 1 Proposed catalytic mechanism of Cpd I formation in heme peroxidases. | ||
Cytochrome c peroxidase (Ccp1) served for decades as a benchmark biochemical model for understanding the chemical and structural properties of other peroxidases.6 Its principal function is to eliminate the toxic H2O2 from the cell with the assistance of its redox partner cytochrome c (Cc).21 The ferrous heme in Cc is approximately 20 Å away from the ferric heme of peroxidase and the combined Ccp1–Cc complex has been considered as a paradigm for probing long-range electron transfer in biology.6,22 Despite the presence of a universal heme pocket amongst heme-peroxidases, Ccp1 known to stand out, with a well-characterized tryptophan (W191) π-cation radical compound I.23 This distinctive electronic feature of Ccp1's ferryl intermediate is attributed mainly to the electrostatic effect of the surrounding environment of the critical W191 residue.24 In fact, the nearby methionine residues (M230 and M231) are found to be vital for the stabilization of the W191 radical through S-aromatic interactions.25 The absence of these residues together with the presence of a cation binding site in ascorbate peroxidase (APX) resulted in a porphyrin π-cation radical instead as Cpd I.24,26 The W191 residue in Ccp1 together with the nearby redox-active (Trp, Met and Tyr) residues play an important role in the hole hopping pathway from the heme pocket to Ccp1's surface and eventually to Cc.27
The axial H175 is conserved amongst heme peroxidases and its ligation to the Fe centre is paramount to the redox activity of the system.28–30 Its strong H-bond interaction with the adjacent D235 residue is vital to induce imidazole character and thus lower the redox potential of the iron centre.31,32 Mutagenesis studies demonstrated the severe modulation of peroxidase activity upon changing the chemical identity of the axial ligand.32–34 Moreover, the proximal triad H-bond interaction between the conserved D232, W191 and H175 residues, Fig. 1, is found to play a fundamental role in correctly positioning W191 for the hole hoping pathway and thus efficient peroxidase activity of Ccp1.31,35–38
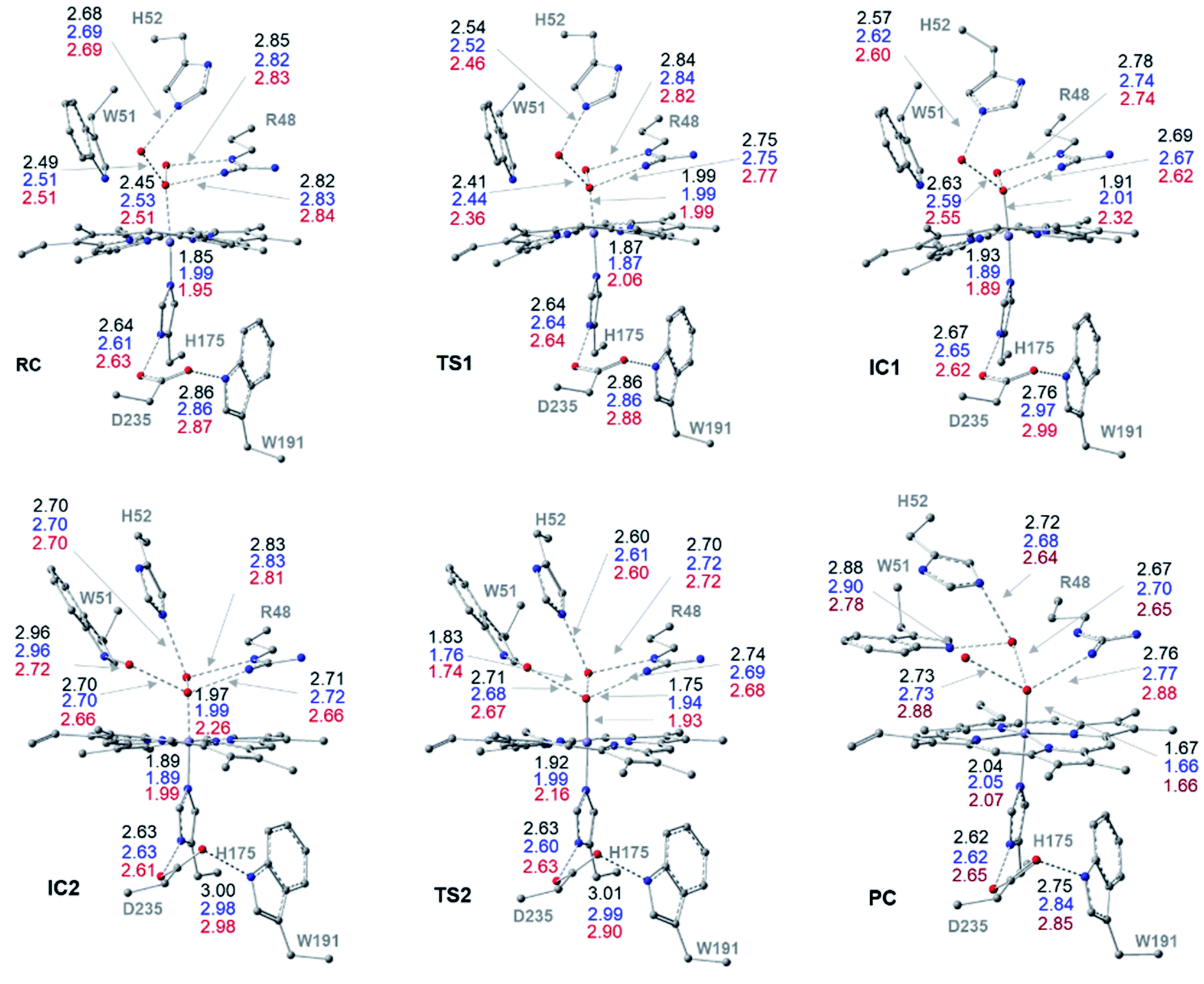 | ||
| Fig. 1 Cluster structures along the wet-mechanism pathway of Cpd I formation, with selected distances between heavy atoms (Å), for three different spin states: doublet (black), quartet (blue) and sextet (red). Geometries are optimized with UB3LYP-D3BJ/6-31G(d,p). | ||
Various key intramolecular interactions in both the proximal and distal sites around the heme are central for the system stability and reactivity.39–41 Three residues located in the distal site of the heme, namely W51, H52 and R48, are within close proximity to influence, via strong H-bond interactions, the proper positioning of H2O2 near the Fe(III) ion to trigger the peroxidase reaction.42 H52 residue is ideally oriented to act as an acid–base catalyst and its mutation leads to a significant drop in the catalytic rate of the reduction process.2,40,43,44 Both R48 and W51 residues feature side-chain geometries consistent with H-bond interaction with the oxyferryl oxygen in compound I and thus help stabilize it.5,45,46
Recent all-atom molecular dynamics (MD) simulations conducted by our group on Ccp1 with H2O2 bound to the ferric iron suggest that the catalytic H52 residue lies close to H2O2, with an average distance r(H52N⋯HO) = 2.32 Å consistent with strong H-bond interactions.47 This observation brings support for the dry mechanism of Cpd I formation in Ccp1, and not the water-assisted one proposed earlier.2,17 This is in agreement with previous MD investigations of horseradish peroxidase (HRP) that demonstrated the very low probability for a catalytic water molecules to bridge the catalytic His residue and H2O2.15,17,46 In addition to other quantum chemistry calculations (QM) on different heme peroxidases that suggested a dry mechanism for the formation of the Fe–hydroperoxide containing complex (Cpd 0).13,14 This warrants further investigation of the feasibility of the water-assisted vs. dry mechanism for the formation on Cpd I in heme peroxidases with Ccp1 as a case study. Accordingly, we report in this manuscript the results of QM calculations of the dry and wet mechanisms for Cpd I formation in Ccp1. Careful considerations were given to the possible spin states of each model which results in probing the catalytic mechanism for a total of 6-different models, our objective is to better characterize the preferable pathway and shed light onto the role of essential residues in both the formation and stability of the high-valent oxyferryl intermediate, Cpd I.
Detailed comparison between the structural and electronic features of Cpd I at different spin states has been also conducted for both wet and dry models. We obtained a kinetically preferred pathway in which we underlined various structural parameters that align with experimental ones. Moreover, the preferred mechanism was explored at various in silico generated variants for key residues to further clarify their precise roles in the mechanism, the geometry, and the electronic behaviour of Cpd I. Deep investigations were conducted to analyze all the obtained structures during the mechanism for each model. Our findings explain the impact of each point of mutation on the structures collected during the reaction mechanism. Due to the conserved nature of the heme pocket amongst heme-peroxidases, the obtained findings from our extensive investigation could be universal amongst them.
Computational methodology
QM cluster calculations have been successfully applied to model numerous catalytic reactions.48–52 The cluster model used herein is obtained from the crystal structure (PDB ID: 3M2I resolution: 1.40 Å) of the compound I ferryl intermediate.53 The resting-state ferric-containing heme structure was generated by morphing the bound oxygen and the nearby oxygen into H2O2 for the dry model. A single additional water molecule was placed in a bridging position between H52 and peroxide to build the water-assisted model. Our cluster models include the heme pocket residues that have been previously suggested to directly participate in the catalytic mechanism. These crucial residues participate in the mechanism through either activating H2O2 and/or neutralizing the developing charges in the transition states and intermediates, and therefore enhance the chemical reactivity of the heme. This QM model includes the distal R48, W51 and H52 residues in addition to the proximal H175, W191 and D235 residues. All the residues are truncated at their α-carbons and these latter carbons are kept frozen to their coordinates in the crystal structure (PDB ID: 3M2I). This resulted in dry and water-assisted models with a total of 170 and 173 atoms, respectively, and an overall charge of +1, as a starting point to investigate the mechanism of H2O2 activation, Scheme 1.54–56 The open d-shell of the heme iron allows for various possible electronic configurations and thus different multiplicity states, namely, doublet, quartet and sextet states, have been considered for each pathway.57,58 The spin multiplicity is indicated as a left-hand superscript for each structure calculated throughout.Density-functional theory (DFT) calculations are performed with the unrestricted B3LYP functional,59–61 which has been highly recommended for heme-system studies,62,63 in conjunction with the 6-31G(d,p) basis set, as implemented in Gaussian 16 (revision B.01).64 The D3 version of Grimme's dispersion with Becke–Johnson damping (GD3BJ) correction is used to improve the treatment of noncovalent and dispersion interactions.65 Spin contamination was investigated for all the optimized structures to ensure that it is not significant. Following geometry optimization, vibrational frequency analyses are performed to characterize the nature of the stationary points. Visualization of the atomic motion associated with the calculated imaginary-frequency mode was performed to connect every transition state (TS) to the corresponding intermediates on the potential energy surface. Electronic energies were further refined with a larger basis set, and the cluster environment was taken into account with the integrated effective fragment polarizable continuum model (IEFPCM) and a dielectric constant ε = 4 which is generally believed to represent the protein environment [IEFPCM-B3LYP-GD3BJ/6-311+G(2d,2p)].50,51,66 Another set of functions namely [IEFPCM-B3LYP-GD3BJ/6-311+G(2df,2p)] was conducted to calculate the energy for one full pathway to confirm the negligible impact of changing the basis set, Table S3.† The efficiency of using the current basis set for geometry optimization was confirmed to be accurate by comparison of structural results obtained for Cpd I in the three different spin states relative to more expensive basis sets (cf. ESI Fig. S1†). Environmental effects were further explored by repeating the calculations for Cpd 0 and Cpd I with dielectric constants ε = 4, 8, 16 and 78.4 (cf. ESI Tables S3 and S4†).
Results and discussion
The optimized structures of all intermediates and transition states along the wet and dry-mechanism pathways of Cpd I formation are shown in Fig. 1 and 2, respectively, while free energy profiles are depicted in Fig. 3 and the associated free energy values for all structures are collected in Table S1.†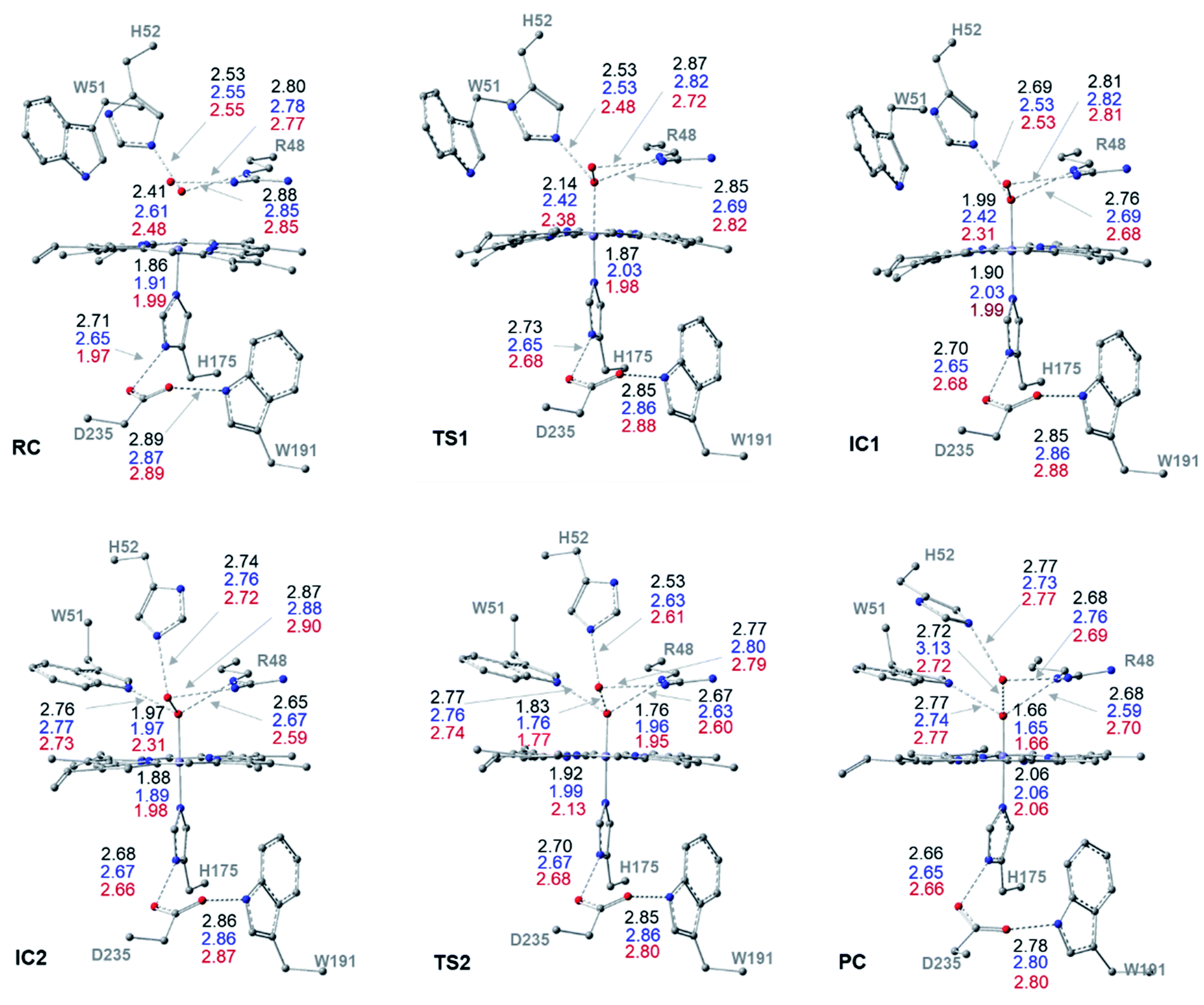 | ||
| Fig. 2 Cluster structures along the dry-mechanism pathway of Cpd I formation, with selected distances between heavy atoms (Å), for three different spin states: doublet (black), quartet (blue) and sextet (red). Geometries are optimized with UB3LYP-D3BJ/6-31G(d,p). | ||
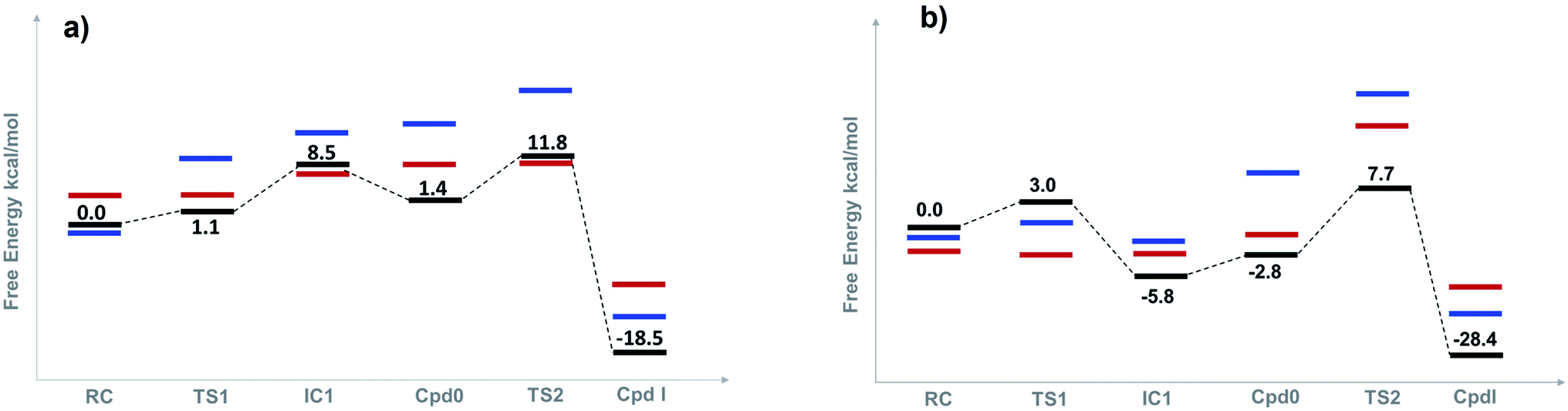 | ||
| Fig. 3 Free-energy profiles (kcal mol−1) for both the wet (a), and dry (b) mechanisms of Cpd I formation for three different spin states: doublet (black line), quartet (blue line) and sextet (red line). Free energies are estimated with UB3LYP-GD3BJ/6-311+G(2d,2p) with ε = 4 (see text for details). | ||
I. Wet mechanism of cpd I formation
The reaction mechanism starts from a fully optimized reactive complex (RC) in which the H2O2 is bound to the Fe(III) ion and close to the bridged water molecule (W). From the structures in Fig. 1, the latter water molecule is properly positioned to facilitate a proton transfer from H2O2 to the nearby catalytic H52 residue. The R48 residue geometry is consistent with H-bond interactions with the two oxygens of H2O2, demonstrating its role in positioning the ligand in the catalytic distal site. The H-bond interactions between the proximal H174, W191 and D235 residues appear to be maintained regardless of the system's spin state. The RC geometries obtained in the three spin states align structurally, with 6RC thermodynamically the least stable. The structures of 2RC and 4RC lie very close in energy, within 0.5 kcal mol−1, and 4RC is slightly more stable, Fig. 3a.The formation of the Cpd 0 intermediate IC1, in which the H52 residue becomes protonated, and a FeIII–OOH single bond is formed, takes place through the first transition state (TS1), Fig. 1. TS1 exhibits a water-assisted double proton transfer from H2O2 to the nearby H52 residue. The lowest free energy barrier for this concerted step is found to be 1.1 kcal mol−1 for 2TS1 relative to its 2RC, Fig. 3a. The second lowest free energy barrier is for 6TS1 (2.4 kcal mol−1 higher in energy than 2TS1) with has an activation energy of 2.1 kcal mol−1 relative to 6RC. It is also worth mentioning that the higher energy values of IC1 relative to its TS1 is noticed only upon adding the Gibbs energy correction. In fact, the relative energies for the obtained structures before adding the Gibbs correction are collected in Table S2† and TS1 is higher in energy than the following IC1. The structural parameters for TS1 in different spin states are very similar, and the most significant change is obtained for the length of Fe–O bond, which is 0.28 and 0.16 Å longer in both 4TS1 and 6TS1, respectively, than in 2TS1. Notably, the H52N⋯H-bond distance in 6TS1 is found to be 0.05 Å shorter than its value in both 2TS1 and 4TS1, Fig. 1.
The optimized geometries of 2IC1 and 6IC1 are found to be the most thermodynamically stable and pseudo-degenerate, with similar structures except for the elongation of the Fe⋯O bond by 0.41 Å in 6IC1 relative to 2IC1, Fig. 1. This intermediate encountered a structural rearrangement of the H52 imidazole side chain consistent with H-bond interactions with the distal OH of the ligated hydroperoxide group, Scheme 1 and Fig. 1, to form the other conformation of Cpd 0 intermediate (IC2). The structures of the IC2 intermediate are very similar in all three spin states but its most thermodynamically stable form is that of the doublet state. It is also worth noting that the axial Fe⋯O bond in 6IC2 remains the longest, with a difference of 0.29 Å relative to that for the doublet analogue (2IC2).
The second step of the mechanism proceeds through a proton transfer from the newly protonated H52 residue to the H-bonded distal –OH group, promoting heterolytic cleavage of the O–O bond. The lowest free energy barrier for this step is found to be 11.8 kcal mol−1 for 2TS2 relative to 2RC, Fig. 3a. Notably, 4TS2 and 6TS2 are found to be thermodynamically pseudo-degenerate with a single structural difference in the distance between the distal oxygen and the bound one (0.90 Å shorter in the doublet), Fig. 1 and 3a. The activation barrier for heterolytic cleavage of the O–O bond calculated here is in a good agreement with those obtained for similar peroxidases which were in the range of 9–14 kcal mol−1.12,13 This step is followed by formation of Cpd I (PC) which is found to be thermodynamically stable in all spin states, Fig. 3a. Geometries for PC in the three spin states exhibit minimal structural differences, and 2PC is found to be the most thermodynamically stable, Fig. 1 and 3. In these ferryl intermediates, the average Fe–O bond distance is 1.66 Å, in agreement with experimental observations,4,5 indicative of double-bond character (FeIVO), Fig. 1. The most considerable structural change is obtained at the distance between W molecule at the oxo-ferryl oxygen in 6PC that is only 0.15 Å longer than the corresponding distance in both 2PC and 4PC. While the second most significant change is at the distance of R48 with the oxo-ferryl group in 6PC which is 0.11 and 0.10 longer than the corresponding distances in 2PC and 4PC, respectively.
Inspection of the H175NH⋯−OOCD235 and D235COO−⋯HOW191 distances in the various structures, Fig. 1, indicates that the tight H-bond interactions between the proximal residues persist throughout the entire mechanism. Moreover, the geometry of the R48 residue suggests consistent H-bond interactions with the two oxygens of peroxide along the pathway, indicating its crucial role in the entire mechanism. Meanwhile, the W51 residue does not seem to be involved in any H-bond interaction with the distal oxygen up until IC2 formation, Fig. 1. Importantly, there are two water molecules, W molecule and the one liberated after the O–O bond cleavage, in the heme pocket of Cpd I upon Cpd I formation and both are H-bonded to the oxyferryl group, Fig. 1. This structure is consistent with the crystal structures of other ferryl intermediates which contain more than one single water molecule (PDB ID: 1ZBZ and 6P43),10,67 but unlike the crystal structure which was used to build the current model (PDB ID: 3M2I).53 In general, the free energy profile for Cpd I formation through the wet mechanism indicates the kinetic and thermodynamic preference of doublet spin state, Fig. 3a.
II. Dry mechanism of cpd I formation
The structures of the reactive complex (RC) for the dry mechanism are found to be very similar regardless of spin state but 6RC is found to be the most thermodynamically stable, Fig. 3b. In all spin states, H2O2 is orientated properly with respect to both the catalytic H52 residue and the Fe(III) centre with average H52NH⋯HOH2O2 and H2O2O⋯Fe distances of 2.50 and 2.55 Å, respectively, Fig. 2. This positioning of H2O2 illustrates the feasibility of a direct proton transfer toward the H52 residue in the first step of the reaction to access IC1 (Cpd 0), Scheme 1.The first transition state in the dry mechanism displayed low energy barrier relative to RC regardless of the spin states and 6TS1 suggests a barrierless reaction, Fig. 3b. The highest free energy barrier for this step is obtained for 2TS1, with a value of 3.0 kcal mol−1, Fig. 3b. Relative to the lowest barrier for the corresponding step in the water-assisted mechanism, the Gibbs free energy for this step is only 1.4 kcal mol−1 higher in energy than the corresponding step in the water-assisted mechanism, Fig. 3. Thereby, the inclusion of a water molecule to bridge the proton transfer from the peroxide to the catalytic H52 did not result in significant drop in the energy barriers unlike previous suggestions.17,18
The IC1 and IC2 (Cpd 0 intermediates) obtained in this mechanism differ only in the orientation of the H52's side chain relative to the distal hydroxyl of Fe–OOH, Scheme 1 and Fig. 2. Except for Fe–O distances (longer in 4IC1 and 6IC1 by 0.43 and 0.32 Å relative to 2IC1), the optimized IC1 geometries align structurally very well in all spin states, Fig. 2. Similar structural parameters are obtained for IC2 with the single geometrical difference lying in the Fe–O bond, which elongates by 0.34 Å in 6IC2. The determined 2IC1 and 2IC2 are found to be the most thermodynamically stable forms of the system after PC and lie 0.3 and 2.8 kcal mol−1 lower in energy than 2RC, respectively, Fig. 3b. Interestingly, 2IC1 is found to be more stable stationary point than 2RC. In fact, this latter observation is consistent with earlier studies on Ccp1 and APX that showed the better stability of the ferric hydroperoxide intermediate as a result of the high acidity of Fe–OOH2.12,14 Moreover, a barrier free process for the proton transfer from the distal Fe–OOH2 to the catalytic His has been obtained in these earlier studies.
The newly protonated H52 residue is within tight H-bond distance with the distal Fe–OOH group with r(H52N⋯O) = 2.74 Å in 2IC2 triggering the hydrolytic cleavage of the O–O bond as in TS2, Scheme 1 and Fig. 2. Despite structurally analogous TS2 for the three spin states, our obtained data demonstrate that 2TS2 is the most kinetically preferable with an activation energy of 7.7 (10.5) kcal mol−1 relative to the corresponding 2RC (2IC2), Fig. 3b. In comparison to the obtained energy barriers for the analogous step in the wet mechanism, the energy barrier for the current step relative to 2IC1 (a more stable intermediated the 2RC) is 1.3 kcal mol−1 lower and thus more kinetically preferable. Then, the collapse of latter TS resulted in the generation of a thermodynamically stable Cpd I by splitting of a water molecule associated with the formation of the unprotonated high-valent iron–oxo double bond (FeIVO) bond, Fig. 2. The average iron–oxo distance is 1.66 Å, indicative of double bond character in agreement with generally accepted bond-order to bond-distance values.5,68 The calculated Cpd I PC geometries are almost identical in the three spin states but 2PC is found to be the most thermodynamically stable, lying 28.4 (28.1) kcal mol−1 lower in free energy than 2RC (2IC1), in agreement with previous studies, Fig. 3b.14,69 The only observed structural difference between Cpd I in the three spin states is the position of the leaving water molecule relative to the proximal oxygen in FeIVO in 4PC, and thus affects its interactions with the nearby R48 residue, Fig. 2.
The role of the R48 residue is further clarified through positioning in conformation consistent with tight H-bond distances with the oxygen atoms of the H2O2 ligand in all the reaction steps regardless of the spin states, Fig. 2. This observation demonstrates its role in properly orienting the H2O2, neutralizing the developed charges in TSs and finally facilitating the formation of Cpd I. Meanwhile, W51 residue is within tight H-bond distance with the axial Fe–O group but only upon the IC2 formation and remained until the generation of Cpd I at all the spin states, Fig. 2. Notably, the distal H-bonded network in the proximal site (H175, W191, D235) and around the oxy-ferryl group in Cpd I is in a good agreement with the crystalized ferryl-intermediated Cpd I, Fig. S2.† In this structure, the R48 and W51 residues and the nearby water molecule is within tight H-bond distances with the oxyferryl group whereas H52 residue is H-bonded to the departed water molecule.
The spin density distributions for Cpd I in all three spin states of the dry model are shown in Fig. 4. The major contributing components in the spin density are found to be the oxyferryl group, and more importantly the H175 and W191 residues. It is observed from both the density surface that the spin density of the radical is localized on both W191 and H175 residues. The closer distance together with the π-stacking interaction between H175 and W191 residues resulted in mixing of their π-orbitals and thereby equal spin density distribution.70 Across all the three spin densities, the spin contamination on these two proximal residues is almost similar regardless of the spin states, Fig. 4. The major increase in the spin density is found to be located on the oxyferryl group of the sextet spin state. The antiferromagnetic coupling between the alpha radicals on oxyferryl group and the radical over the π orbitals of W191 and H175 residues generates doublet state, whereas the ferromagnetic coupling between them resulted in the quartet state, Fig. 4.
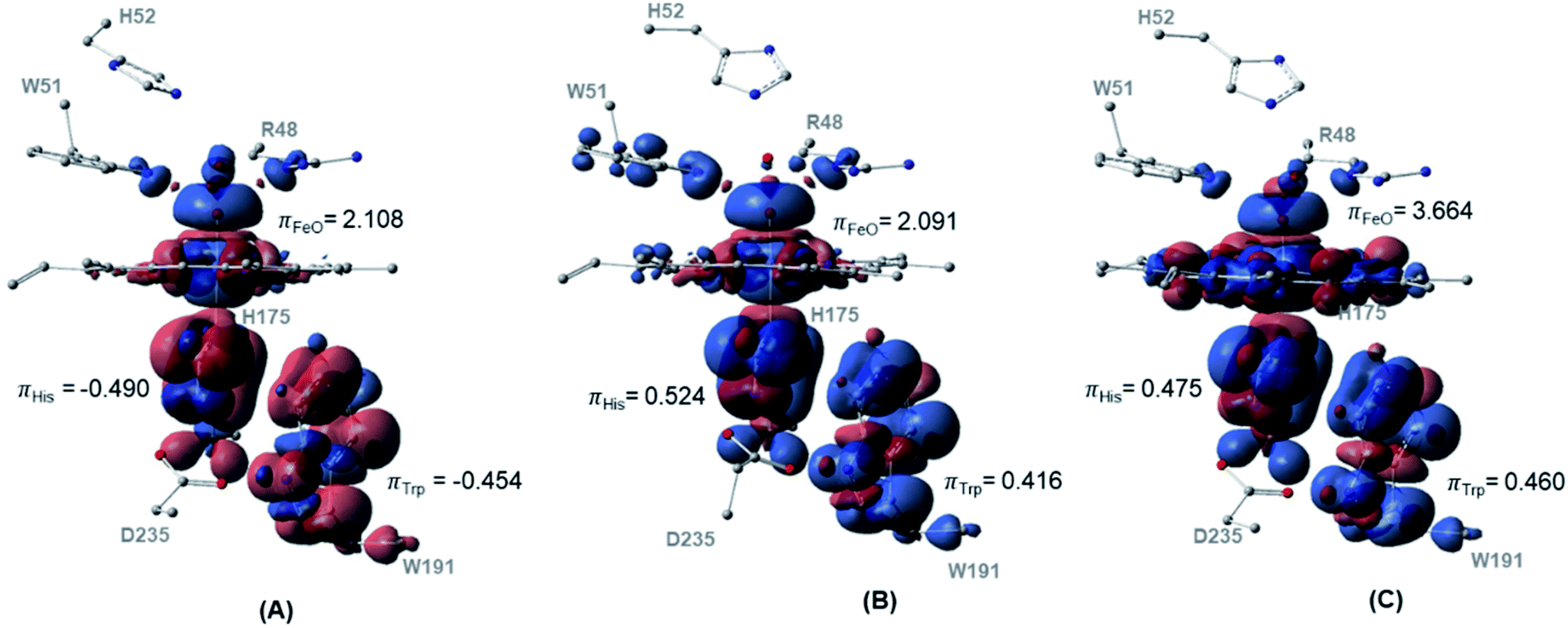 | ||
| Fig. 4 Spin density surfaces together with values for the three major spin contaminated components of (A) doublet (B) quartet (C) sextet. Calculated with UB3LYP-D3BJ/6-31G(d,p). | ||
Previous study concluded the dependency of the spin-energy splitting in peroxidases on the external perturbation.71 Thereby, to confirm the thermodynamic preference of Cpd 0 and Cpd I in the doublet spin states in various environments, we reoptimized the structures for both Cpd 0 and Cpd I for the three spin states at four different dielectric constants to represent four surrounding environments, Fig. S2.† The free energy values for Cpd 0 and Cpd I structures are collected in Table S2† while their corresponding geometries are depicted in Tables S3 and S4.† Our calculations demonstrate the negligible structural perturbation of the geometries in the studied environments. In addition, upon conducting energetic comparison between the Cpd 0 and Cpd I structures in different spin states, the doublet spin state for both intermediates dominate the other spin states supporting our initial findings. This observation also supports the proper representation of our current chemical cluster to explore the mechanism of Ccp1. In particular, the size of our chemical model aligns perfectly with the generally suggested size that is known to be less affected by the nature of the surrounding dielectric constants.50,72,73
III. Studying the mechanism of cpdI formation at different in silico generated QM variants
To further probe the impact of various key residues and their associated interactions on the mechanism of Cpd I formation, we studied the peroxidase mechanism for eight generated Ccp1 variants. Comparative structural, energetic, and electronic analysis of each individual point of mutation relative to the corresponding values in WT Ccp1 have been conducted. The generated in silico Ccp1 variants include the following: W191Y, W191A, W51A, R48A, W51/W191F (to model the active site of HRP)68 D235E, neutral D235, D235N, and D235A. For each one of these in silico generated variants, we explored the mechanism of Cpd I formation starting from the first conformation of Cpd 0 (IC1) in the doublet state through a dry model, the most preferred pathway obtained at WT Ccp1, Fig. 3b. Our attempts to locate RC for most of the studied Ccp1 variants have failed and the structure of the reactive complex fell apart toward more stable intermediate (IC1) and thus the latter has considered as the reaction starting complex. All the structures along the pathway for each of the variant models (starting from IC1) have been fully characterized and the distances for various key interactions are collected in Tables S7–S10,† while their associated Gibbs free energy for chemical reactions are collected in Table 1. Overall, none of the studied Ccp1 variants resulted in considerable geometrical change in the obtained key distances relative to WT Ccp1, except for a local change nearby the point of mutations, which is in a good agreement with former experimental observation.39,41,74–76| WT | R48A | W51A | W51F/W191F | W191Y | W191A | D235E | D235N | D235H | D235A | |
|---|---|---|---|---|---|---|---|---|---|---|
| a The energy values between brackets are calculated relative to IC2 in cases where it is thermodynamically more stable than IC1. | ||||||||||
| IC1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| IC2 | −2.5 | 3.3 | 0.8 | 1.6 | −2.6 | −3.8 | −3.1 | −3.1 | −4.1 | −5.6 |
| TS2a | 8.0 (10.5) | 13.0 | 14.3 | 11.8 | 8.3 (10.9) | 8.8 (12.6) | 7.4 (10.5) | 9.1 (12.2) | 7.4 (11.5) | 6.3 (11.9) |
| PC | −31.9 | −22.6 | −19.1 | −22.0 | −25.3 | −25.4 | −26.8 | −24.5 | −25.5 | −27.2 |
However, relative to the calculated Gibbs energy barrier obtained for WT Ccp1, the studied Ccp1 variants demonstrated higher energy barrier for the rate-limiting step, and thus lower reaction rate, that is the heterolytic cleavage of O–O bond. The maximum influence on the reaction barrier is obtained for W51A and R48A Ccp1 variants which gave energy barriers that are 3.8 and 2.5 kcal mol−1 higher than the barrier obtained for WT Ccp1, Table 1. This noticeable impact on the activation energies clearly indicates the essential roles of these residues in the overall Cpd I formation determined experimentally.34,39,41,67,76
The similar chemical electronic and chemical behaviour between the readily oxidizable tyrosine and tryptophan residues at 191 position has been shown experimentally.74 In agreement with that, the calculated barrier for W191Y Ccp1 is only 0.4 kcal mol−1 than the value of WT Ccp1. Greater impact on the energy barrier is found on the other proximal site substituted models except for D235E Ccp1. The Glu235 residue at the latter D235E Ccp1 model adopts a geometry consistent with forming H-bond interaction with both H175 and W191 residues similar to WT Ccp1 during its reaction coordinate, Table S7–S10.† Thereby, a similar reaction barrier to the WT Ccp1 is obtained for this variant. Except for variants related to the distal site residues, the 2nd conformation of Cpd 0 (IC2) is thermodynamically more stable than the corresponding 1st conformation (IC1). Furthermore, all the energy profiles possess a thermodynamically stable product complex (indicative of Cpd I) relative to their IC1, Table 1.
Analysis of the spin density distributions for Cpd I for all in silico Ccp1 variants were conducted and provided in Fig. 5 and their values are collected in Table S11.† Relative to WT Ccp1 which shows the major contribution of W191 in the overall spin contamination, W191Y, W51A and R48A Ccp1 variants display a similar electronic manner. However, W191A and all mutations at D235 position adopt porphyrin π-cation radical with minimal spin density over 191 positions but on W51 residue instead. W51F/W191F Ccp1 doublet mutant, that resembles the heme pocket of HRP, demonstrates a porphyrin π-cation radical in consistence with experimental observation.2 The geometry of E235 at D235E resulted in a spin contamination over W191 residue similar to WT Ccp1, whereas N235 residue at D235N, which affords only a single and weaker H-bond interaction with the nearby H175 residue, shows less spin density accumulated over W191. Overall, changing the net of interactions around the essential W191 display a great impact on the electronic behaviour of the system.
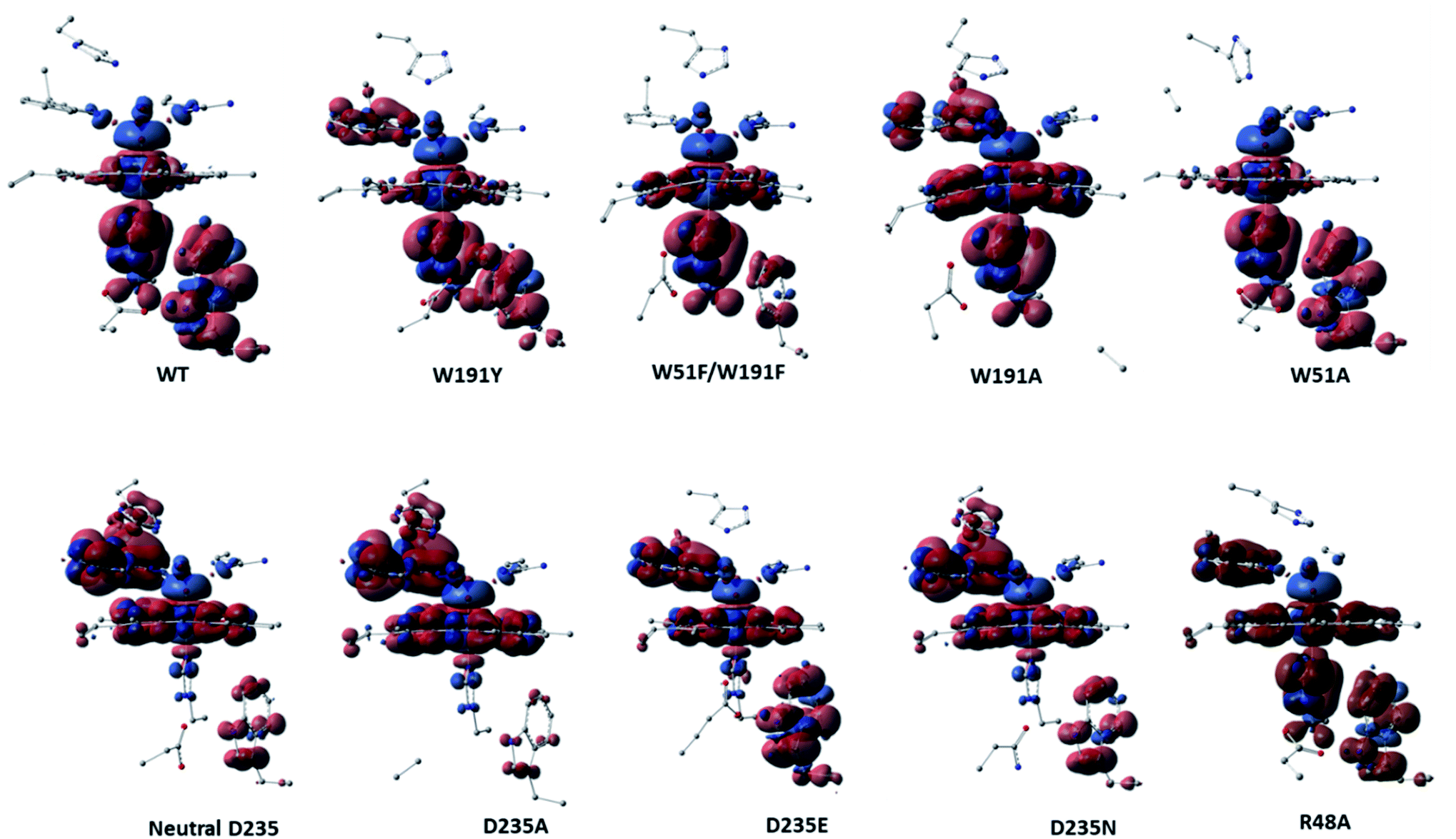 | ||
| Fig. 5 Spin density surfaces together with values for the three major spin contaminated components of WT Ccp1 and its in silico generated variants. Calculated with UB3LYP-D3BJ/6-31G(d,p). | ||
Conclusions
Extensive DFT calculations on a cluster model containing the heme pocket of Ccp1 were conducted to explore the mechanism of cpd I formation. Two different mechanisms, namely the water-assisted (wet) and no-water (dry) mechanisms previously proposed, in three possible spin states (doublet, quartet, and sextet) have been investigated. The overall mechanism proceeds through two main stages; the first is the formation of hydroperoxide containing intermediate (Cpd 0) which decomposes toward ferryl-intermediate (Cpd I) in the second stage. All the geometries for the studied six reactions have been fully characterized and analyzed from a structural and energetic perspectives.Our calculations demonstrated that the inclusion of a bridged water molecule to facilitate the reaction mechanism, did not result in significantly lowering the activation energy for the first step as suggested previously. The productive positioning of the H2O2 in RC of the dry model relative to both H52 residue and Fe(III) centre demonstrate the unnecessarily of a water molecule to assist this process. This observation is further supported by obtaining low free energy barrier for the associated step. The dry mechanism in the doublet spin sate resulted in low energy barriers and therefore, highlight its kinetic and thermodynamic preference.
Due to the thermodynamic stability of doublet Cpd 0 intermediates (IC1 and IC2) over its RC together with high kinetic feasibility of first step of the reaction, we suggest the Cpd 0 (2IC1) to be the initial reacting complex for the doublet state mechanism in the direct pathway. The geometry of the calculated Cpd I agrees structurally with available crystal structures and the doublet state is the most stable. Our data also support the notion that Cpd 0 and Cpd I exist in the doublet spin state and FeO bond has a double-bond character at all the considered models, spin states and in presence of various dielectric media. The profound roles of the W51 and R48 distal residues in the chemical reactivity of Ccp1 have been also clarified. The geometries of the two residues are within tight H-bond distances to H2O2 and the oxo-ferly group and thus contribute to the charge neutralization developed over the transition states. Moreover, the R48 residue dictates the proper positioning of the H2O2 for the reaction to proceed.
Our investigation was further expanded to reveal the impact of the hem pocket residues in the mechanism of Cpd I formation by exploring the mechanistic, structural, and electronic behaviour of various in silico generated Ccp1 variants. Relative to the Gibbs energy barrier of WT Ccp1, the calculated barriers of the Ccp1 mutants are lower indicating the significance of the substituted residues in the overall mechanism. The role of these residues in obtaining a distinctive Trp π-cation radical was also explored by displaying the radical distributions for the Cpd I of the studied variants. Globally, our findings should pave the road toward further investigation of the feasibility of the proposed dry mechanism in other peroxidases due to conserved structure of the heme pocket amongst a wide range of peroxidases.
Conflicts of interest
I declare that there is no conflict of interest.Abbreviations
| Ccp1 | Cytochrome c peroxidase |
| Cpd0 | Compound 0 intermediate |
| Cpd1 | Compound I intermediate |
| RC | Reactive complex |
| IC | Intermediate complex |
| TS | Transition state |
| PC | Product complex |
Acknowledgements
I am thankful for Concordia University for the Horizon Post-doctoral Fellowship at the beginning of this study and Compute Canada for the Computational resources.References
- C. J. Reedy and B. R. Gibney, Heme Protein Assemblies, Chem. Rev., 2004, 104, 617–650 CrossRef CAS PubMed.
- T. L. Poulos, Heme Enzyme Structure and Function, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- G. Smulevich, A. Feis and B. D. Howes, Fifteen Years of Raman Spectroscopy of Engineered Heme Containing Peroxidases: What Have We Learned?, Acc. Chem. Res., 2005, 38, 433–440 CrossRef CAS PubMed.
- P. C. E. Moody and E. L. Raven, The Nature and Reactivity of Ferryl Heme in Compounds I and II, Acc. Chem. Res., 2018, 51, 427–435 CrossRef CAS PubMed.
- A. Gumiero, C. L. Metcalfe, A. R. Pearson, E. L. Raven and P. C. Moody, Nature of the ferryl heme in compounds I and II, J. Biol. Chem., 2011, 14, 1260–1268 CrossRef PubMed.
- T. L. Poulos, Thirty years of heme peroxidase structural biology, Arch. Biochem. Biophys., 2010, 500, 3–12 CrossRef CAS PubMed.
- H. Kwon, J. Basran, C. M. Casadei, A. J. Fielding, T. E. Schrader, A. Ostermann, J. M. Devos, P. Aller, M. P. Blakeley, P. C. E. Moody and E. L. Raven, Direct visualization of a Fe(IV)–OH intermediate in a heme enzyme, Nat. Commun., 2016, 7, 13445 CrossRef CAS PubMed.
- C. M. Casadei, A. Gumiero, C. L. Metcalfe, E. J. Murphy, J. Basran, M. G. Concilio, S. C. Teixeira, T. E. Schrader, A. J. Fielding, A. Ostermann, M. P. Blakeley, E. L. Raven and P. C. Moody, Heme enzymes. Neutron cryo-crystallography captures the protonation state of ferryl heme in a peroxidase, Science, 2014, 345, 193–197 CrossRef CAS PubMed.
- R. K. Behan and M. T. Green, On the status of ferryl protonation, J. Inorg. Biochem., 2006, 100, 448–459 CrossRef CAS PubMed.
- C. A. Bonagura, B. Bhaskar, H. Shimizu, H. Li, M. Sundaramoorthy, D. E. McRee, D. B. Goodin and T. L. Poulos, High-Resolution Crystal Structures and Spectroscopy of Native and Compound I Cytochrome c Peroxidase, Biochemistry, 2003, 42, 5600–5608 CrossRef CAS PubMed.
- T. L. K. J. Poulos, The stereochemistry of peroxidase catalysis, J. Biol. Chem., 1980, 255, 8199–8205 CrossRef CAS PubMed.
- E. Derat and S. Shaik, The Poulos-Kraut Mechanism of Compound I Formation in Horseradish Peroxidase: A QM/MM Study, J. Phys. Chem. B, 2006, 110, 10526–10533 CrossRef CAS PubMed.
- H. Chen, H. Hirao, E. Derat, I. Schlichting and S. Shaik, Quantum Mechanical/Molecular Mechanical Study on the Mechanisms of Compound I Formation in the Catalytic Cycle of Chloroperoxidase: An Overview on Heme Enzymes, J. Phys. Chem. B, 2008, 112, 9490–9500 CrossRef CAS PubMed.
- M. Wirstam, M. R. A. Blomberg and P. E. M. Siegbahn, Reaction Mechanism of Compound I Formation in Heme Peroxidases: A Density Functional Theory Stud, J. Am. Chem. Soc., 1999, 121, 10178–10185 CrossRef CAS.
- C. Zazza, A. Amadei, A. Palma, N. Sanna, S. Tatoli and M. Aschi, Theoretical Modeling of Enzyme Reactions: The Thermodynamics of Formation of Compound 0 in Horseradish Peroxidase, J. Phys. Chem. B, 2008, 112, 3184–3192 CrossRef CAS PubMed.
- T. L. Poulos, Heme Proteins, Heme Peroxidases, Encyclopedia of Metalloproteins, ed. R. H. Kretsinger, V.N. Uversky and E. A. Permyakov, Springer, New York, NY. 2013 Search PubMed.
- P. Vidossich, G. Fiorin, M. Alfonso-Prieto, E. Derat, S. Shaik and C. Rovira, On the Role of Water in Peroxidase Catalysis: A Theoretical Investigation of HRP Compound I Formation, J. Phys. Chem. B, 2010, 114, 5161–5169 CrossRef CAS PubMed.
- E. Derat, S. Shaik, C. Rovira, P. Vidossich and M. Alfonso-Prieto, The Effect of a Water Molecule on the Mechanism of Formation of Compound 0 in Horseradish Peroxidase, J. Am. Chem. Sci., 2007, 129, 6346–6347 CrossRef CAS PubMed.
- M. M. Aboelnga, J. J. Hayward and J. W. Gauld, Unraveling the Critical Role Played by Ado762′OH in the Post-Transfer Editing by Archaeal Threonyl-tRNA Synthetase, J. Phys. Chem. B, 2018, 122, 1092–1101 CrossRef CAS PubMed.
- M. M. Aboelnga, J. J. Hayward and J. W. Gauld, A water-mediated and substrate-assisted aminoacylation mechanism in the discriminating aminoacyl-tRNA synthetase GlnRS and non-discriminating GluRS, Phys. Chem. Chem. Phys., 2017, 19, 25598–25609 RSC.
- T. L. Poulos, S. T. Freer, R. A. Alden, S. L. Edwards, U. Skogland, K. Takio, B. Eriksson, X. Nguyen-huu, T. Yonetani and J. Kraut, The crystal structure of cytochrome c peroxidase, J. Biol. Chem., 1980, 255, 575–580 CrossRef CAS PubMed.
- K. Van de Water, Y. G. J. Sterckx and A. N. Volkov, The low-affinity complex of cytochrome c and its peroxidase, Nat. Commun., 2015, 6, 7073 CrossRef PubMed.
- M. Sivaraja, D. B. Goodin, M. Smith and B. M. Hoffman, Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES, Science, 1989, 245, 738–740 CrossRef CAS PubMed.
- C. A. Bonagura, M. Sundaramoorthy, H. S. Pappa, W. R. Patterson and T. L. Poulos, An engineered cation site in cytochrome c peroxidase alters the reactivity of the redox active tryptophan, Biochemistry, 1996, 35, 6107–6115 CrossRef CAS PubMed.
- Y. T. Meharenna, P. Oertel, B. Bhaskar and T. L. Poulos, Engineering Ascorbate Peroxidase Activity into Cytochrome c Peroxidase, Biochemistry, 2008, 47, 10324–10332 CrossRef CAS PubMed.
- T. P. Barrows and T. L. Poulos, Role of electrostatics and salt bridges in stabilizing the compound I radical in ascorbate peroxidase, Biochemistry, 2005, 44, 14062–14068 CrossRef CAS PubMed.
- T. P. Barrows, B. Bhaskar and T. L. Poulos, Electrostatic control of the tryptophan radical in cytochrome c peroxidase, Biochemistry, 2004, 43, 8826–8834 CrossRef CAS PubMed.
- T. L. Poulos, The role of the proximal ligand in heme enzymes, J. Biol. Inorg Chem., 1996, 1, 356–359 CrossRef CAS.
- P. Rydberg, E. Sigfridsson and U. Ryde, On the role of the axial ligand in heme proteins: a theoretical study, J. Biol. Inorg Chem., 2004, 9, 203–223 CrossRef CAS PubMed.
- T. H. Yosca, R. K. Behan, C. M. Krest, E. L. Onderko, M. C. Langston and M. T. Green, Setting an Upper Limit on the Myoglobin Iron(IV)Hydroxide pKa: Insight into Axial Ligand Tuning in Heme Protein Catalysis, J. Am. Chem. Soc., 2014, 136, 9124–9131 CrossRef CAS PubMed.
- J. Heimdal, P. Rydberg and U. Ryde, Protonation of the Proximal Histidine Ligand in Heme Peroxidases, J. Phys. Chem. B, 2008, 112, 2501–2510 CrossRef CAS PubMed.
- A. P. Green, T. Hayashi, P. R. E. Mittl and D. Hilvert, A Chemically Programmed Proximal Ligand Enhances the Catalytic Properties of a Heme Enzyme, J. Am. Chem. Soc., 2016, 138, 11344–11352 CrossRef CAS PubMed.
- J. Hirst, S. K. Wilcox, J. Ai, P. Moënne-Loccoz, T. M. Loehr and D. B. Goodin, Replacement of the Axial Histidine Ligand with Imidazole in Cytochrome c Peroxidase. 2. Effects on Heme Coordination and Function, Biochemistry, 2001, 40, 1274–1283 CrossRef CAS PubMed.
- M. Ortmayer, K. Fisher, J. Basran, E. M. Wolde-Michael, D. J. Heyes, C. Levy, S. L. Lovelock, J. L. R. Anderson, E. L. Raven, S. Hay, S. E. J. Rigby and A. P. Green, Rewiring the “Push-Pull” Catalytic Machinery of a Heme Enzyme Using an Expanded Genetic Code, ACS Catal., 2020, 10, 2735–2746 CrossRef CAS PubMed.
- A. P. Green, T. Hayashi, P. R. E. Mittl and D. Hilvert, A Chemically Programmed Proximal Ligand Enhances the Catalytic Properties of a Heme Enzyme, J. Am. Chem. Sci., 2016, 138, 11344–11352 CrossRef CAS PubMed.
- K. Mittra, K. Sengupta, A. Singha, S. Bandyopadhyay, S. Chatterjee, A. Rana, S. Samanta and A. Dey, Second sphere control of spin state: Differential tuning of axial ligand bonds in ferric porphyrin complexes by hydrogen bonding, J. Inorg. Biochem., 2016, 155, 82–91 CrossRef CAS PubMed.
- A. D. Pardillo, A. N. Morozov and D. C. Chatfield, Proximal Pocket Hydrogen Bonds Significantly Influence the Mechanism of Chloroperoxidase Compound I Formation, J. Phys. Chem. B, 2015, 119, 12590–12602 CrossRef CAS PubMed.
- J. Hirst, S. K. Wilcox, P. A. Williams, J. Blankenship, D. E. McRee and D. B. Goodin, Replacement of the axial histidine ligand with imidazole in cytochrome c, Biochemistry, 2001, 40, 1265–1273 CrossRef CAS.
- L. B. Vitello, J. E. Erman, M. A. Miller, J. M. Mauro and J. Kraut, Effect of Asp-235 to Asn substitution on the absorption spectrum and hydrogen peroxide reactivity of cytochrome c peroxidase, Biochemistry, 1992, 31, 11524–11535 CrossRef CAS.
- J. E. Erman, L. B. Vitello, M. A. Miller, A. Shaw, K. A. Brown and J. Kraut, Histidine 52 is a critical residue for rapid formation of cytochrome c peroxidase compound I, Biochemistry, 1993, 32, 9798–9806 CrossRef CAS PubMed.
- D. B. Goodin and D. E. McRee, The Asp-His-iron triad of cytochrome c peroxidase controls the reduction potential electronic structure, and coupling of the tryptophan free radical to the heme, Biochemistry, 1993, 32, 3313–3324 CrossRef CAS.
- M. C. Foshay, L. B. Vitello and J. E. Erman, Effect of alternative distal residues on the reactivity of cytochrome c peroxidase, Biochim. Biophys. Acta, 2011, 1814, 525–535 CrossRef CAS PubMed.
- L. Bateman, C. Léger, D. B. Goodin and F. A. Armstrong, A Distal Histidine Mutant (H52Q) of Yeast Cytochrome c Peroxidase Catalyzes the Oxidation of H2O2 Instead of Its Reduction, J. Am. Chem. Soc., 2001, 123, 9260–9263 CrossRef CAS PubMed.
- M. C. Foshay, L. B. Vitello and J. E. Erman, Relocation of the Distal Histidine in Cytochrome c Peroxidase: Properties of CcP(W51H), CcP(W51H/H52W), and CcP(W51H/H52L), Biochemistry, 2009, 48, 5417–5425 CrossRef CAS PubMed.
- A. Iffland, P. Tafelmeyer, C. Saudan and K. Johnsson, Directed Molecular Evolution of Cytochrome c Peroxidase, Biochemistry, 2000, 39, 10790–10798 CrossRef CAS PubMed.
- S. Tatoli, C. Zazza, N. Sanna, A. Palma and M. Aschi, The role of arginine 38 in horseradish peroxidase enzyme revisited: a computational investigation, Biophys. Chem., 2009, 141, 87–93 CrossRef CAS PubMed.
- M. M. Aboelnga, Exploring the structure function relationship of heme peroxidases: Molecular dynamics study on cytochrome c peroxidase variants, Comput. Biol. Chem., 2022, 146, 105544 CAS.
- A. D. Pardillo, A. N. Morozov and D. C. Chatfield, Proximal Pocket Hydrogen Bonds Significantly Influence the Mechanism of Chloroperoxidase Compound I Formation, J. Phys. Chem. B, 2015, 119, 12590–12602 CrossRef CAS PubMed.
- D. C. Chatfield and A. N. Morozov, Proximal Pocket Controls Alkene Oxidation Selectivity of Cytochrome P450 and Chloroperoxidase toward Small, Nonpolar Substrates, J. Phys. Chem. B, 2018, 122, 7828–7838 CrossRef CAS PubMed.
- F. Himo, Recent Trends in Quantum Chemical Modeling of Enzymatic Reactions, J. Am. Chem. Soc., 2017, 139, 6780–6786 CrossRef CAS PubMed.
- M. R. A. Blomberg, T. Borowski, F. Himo, R.-Z. Liao and P. E. M. Siegbahn, Quantum Chemical Studies of Mechanisms for Metalloenzymes, Chem. Rev., 2014, 114, 3601–3658 CrossRef CAS PubMed.
- M. Aboelnga and S. D. Wetmore, Unveiling a Single-Metal-Mediated Phosphodiester Bond Cleavage Mechanism for Nucleic Acids: A Multiscale Computational Investigation of a Human DNA Repair Enzyme, J. Am. Chem. Sci., 2019, 141, 8646–8656 CrossRef CAS PubMed.
- Y. T. Meharenna, T. Doukov, H. Li, S. M. Soltis and T. L. Poulos, Crystallographic and Single-Crystal Spectral Analysis of the Peroxidase Ferryl Intermediate, Biochemistry, 2010, 49, 2984–2986 CrossRef CAS PubMed.
- E. Derat and S. Shaik, The Poulos−Kraut Mechanism of Compound I Formation in Horseradish Peroxidase: A QM/MM Study, J. Phys. Chem. B, 2006, 110, 10526–10533 CrossRef CAS PubMed.
- P. Vidossich, G. Fiorin, M. Alfonso-Prieto, E. Derat, S. Shaik and C. Rovira, On the Role of Water in Peroxidase Catalysis: A Theoretical Investigation of HRP Compound I Formation, J. Phys. Chem. B, 2010, 114, 5161–5169 CrossRef CAS PubMed.
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, The catalytic pathway of horseradish peroxidase at high resolution, Nature, 2002, 417, 463–468 CrossRef CAS PubMed.
- H. Chen, H. Hirao, E. Derat, I. Schlichting and S. Shaik, Quantum Mechanical/Molecular Mechanical Study on the Mechanisms of Compound I Formation in the Catalytic Cycle of Chloroperoxidase: An Overview on Heme Enzyme, J. Phys. Chem. B, 2008, 112, 9490–9500 CrossRef CAS PubMed.
- H. Chen, H. Hirao, E. Derat, I. Schlichting and S. Shaik, QM/MM modeling of compound I active species in cytochrome P450, cytochrome C peroxidase, and ascorbate peroxidase, J. Comput. Chem., 2008, 112, 2006–9500 Search PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Phys. Chem., 1993, 98, 5648–5652 CrossRef CAS.
- A. Altun, J. Breidung, F. Neese and W. Thiel, Correlated Ab Initio and Density Functional Studies on H2 Activation by FeO+, J. Chem. Theory Comput., 2014, 10, 3807–3820 CrossRef CAS PubMed.
- H. Chen, W. Lai and S. Shaik, Exchange-Enhanced H-Abstraction Reactivity of High-Valent Nonheme Iron(IV)-Oxo from Coupled Cluster and Density Functional Theories, J. Phys. Chem. Lett., 2010, 1, 1533–1540 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. Ehudin, L. B. Gee, S. Sabuncu, A. Braun, P. Moënne-Loccoz, B. Hedman, K. O. Hodgson, E. I. Solomon and K. D. Karlin, Tuning the Geometric and Electronic Structure of Synthetic High-Valent Heme Iron(IV)-Oxo Models in the Presence of a Lewis Acid and Various Axial Ligands, J. Am. Chem. Soc., 2019, 141, 5960 Search PubMed.
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, The catalytic pathway of horseradish peroxidase at high resolution, Nature, 2002, 417, 468 CrossRef PubMed.
- E. Derat, S. Cohen, S. Shaik, A. Altun and W. Thiel, Principal Active Species of Horseradish Peroxidase, Compound I: A Hybrid Quantum Mechanical/Molecular Mechanical Study, J. Am. Chem. Soc., 2005, 127, 13621 CrossRef PubMed.
- Y. I. A. Reyesa and F. C. Franco Jr, DFT study on the effect of proximal residues on the Mycobacterium tuberculosis catalase-peroxidase (katG) heme compound I intermediate and its bonding interaction with isoniazid, Phys. Chem. Chem. Phys., 2019, 21, 16515–16525 RSC.
- S. P. de Visser, What Affects the Quartet−Doublet Energy Splitting in Peroxidase Enzymes?, J. Phys. Chem. A, 2005, 109, 11050–11057 CrossRef CAS PubMed.
- K. H. Hopmann and F. Himo, Quantum Chemical Modeling of the Dehalogenation Reaction of Haloalcohol Dehalogenase, J. Chem. Theory Comput., 2008, 4, 1137 CrossRef PubMed.
- P. Georgieva and F. Himo, Quantum chemical modeling of enzymatic reactions: The case of histone lysine methyltransferase, J. Comput. Chem., 2010, 31, 1707–1714 CAS.
- T. M. Payne, E. F. Yee, B. Dzikovski and B. R. Crane, Constraints on the Radical Cation Center of Cytochrome c Peroxidase for Electron Transfer from Cytochrome c, Biochemistry, 2016, 55, 4807–4822 CrossRef CAS PubMed.
- J. Wang, J. M. Mauro, S. L. Edwards, S. J. Oatley, L. A. Fishel, V. A. Ashford, X. Nguyen Huu and J. Kraut, X-ray structures of recombinant yeast cytochrome c peroxidase and three heme-cleft mutants prepared by site-directed mutagenesis, Biochemistry, 1990, 29, 7160–7173 CrossRef CAS PubMed.
- D. Nonaka, H. Wariishi, K. G. Welinder and H. Fujii, Paramagnetic 13C and 15N NMR Analyses of the Push and Pull Effects in Cytochrome c Peroxidase and Coprinus cinereus Peroxidase Variants: Functional Roles of Highly Conserved Amino Acids around Heme, Biochemistry, 2010, 49, 49–57 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01073a |
| This journal is © The Royal Society of Chemistry 2022 |