
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymerization kinetics of bicyclic olefins and mechanism with symmetrical ansa-metallocene catalysts associated with active center count: relationship between their activities and structure and activation path†
Amjad Ali ai,
Ahmad Naveeda,
Khurram Shehzad*bcd,
Tariq Azize,
Tahir Rasheedf,
Jamile Mohammadi Moradiang,
Mobashar Hassana,
Abdul Rahmanbh,
Fan Zhiqiangi and
Li Guo*a
ai,
Ahmad Naveeda,
Khurram Shehzad*bcd,
Tariq Azize,
Tahir Rasheedf,
Jamile Mohammadi Moradiang,
Mobashar Hassana,
Abdul Rahmanbh,
Fan Zhiqiangi and
Li Guo*a
aResearch School of Polymeric Materials, School of Materials Science & Engineering, Jiangsu University, Zhenjiang, 212013, P. R. China. E-mail: liguo@ujs.edu.cn
bSchool of Micro-Nano Electronics, Hangzhou Global Scientific and Technological Innovation Center (HIC), Zhejiang University, Xiaoshan 311200, China. E-mail: khurrams@zju.edu.cn
cState Key Laboratory of Silicon Materials, Zhejiang University, Hangzhou 310027, China
dZJU-UIUC Joint Institute, Zhejiang University, Jiaxing 314400, China
eWestlake University, School of Engineering Yunqi Campus, Hangzhou, Zhejiang 310024, PR China
fInterdisciplinary Research Center for Advanced Materials, King Fahd University of Petroleum and Minerals (KFUPM), Dhahran 31261, Saudi Arabia
gBiofuels Institute, School of Envinoment, Jiangsu University, Zhenjiang, 212013, P. R. China
hStoddart Institute of Molecular Science, Department of Chemistry, Zhejiang University, Hangzhou 310027, China
iMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, 310027, P. R. China
First published on 19th May 2022
Abstract
Copolymerization of ethylene (E) with 5-vinyl-2-norbornene (VNB) catalyzed by ansa-metallocenes allows the precise control of essential polymeric properties such as comonomer incorporation, molecular weight (Mw), and polydispersity (Đ). Significant efforts have been devoted to synthesizing and developing novel catalysts, cocatalysts, and activators, although the fundamental elements of catalytic processes remain unclear. For example, it is questionable how polymeric catalysts are divided across dormant and active sites and how this distribution affects the order of monomers for the propagation rate, which widely vary in the literature. Furthermore, although the empirical correlation between the monomers and average Mw has been established in many systems, the fundamental processes of chain termination remain unknown. Furthermore, the involvement of ion-pairing in metallocene-catalyzed polymerization and the termination mechanisms are also contentious issues. In this study, we describe the use of a quenched-labeling technique based on acyl chloride to selectively quench the zirconium metal–polymeric bond, which can be used to study the kinetics, active site [Zr][C*] counting, copolymer microstructure, and molecular weight distribution (MWD) to determine the rate laws for chain initiation, chain propagation rate (Rp), propagation rate constant (kp) and chain termination. In addition, we also predict previously unknown chemical characteristics of E/bicyclic copolymerization processes, where either a cis-endocyclic double bond with steric properties or a vinyl exocyclic double bond affects the activity, i.e., [Zr]/[*C], (Rp) and (kp). All these properties require the implementation of a particular kinetic mechanism that assumes the low activity of the building copolymer chains incorporating a single ethylene/VNB unit, i.e., the Cp2Zr–C2H5 group, in the ethylene addition process in the Cp2Zr–C bond. Due to β-agostic stabilization, the Cp2Zr–C2H5 group exhibits a distinct feature. These effects were confirmed experimentally, such as the E/VNB co-polymer activity and VNB mol%, propagation rate decrease in the polymerization time (tp) of 120 s to 1800 s, crystalline properties, and significant increase in molecular weight. The active center [Zr]/[*C] fraction considerably increased in the initial (tp) 840 s, and subsequently tended to the steady stage of 33%, which is lower than previously reported E homo- and E/P copolymerization. The lower [C*]/[Zr] in both the early and stable stages, decrease in VNB mol%, and Rp with tp can be associated with the more significant fraction of Cp2Zr–CH2CH3-type dormant site by the β-agostic hydrogen interaction with the Cp2Zr metal. The tp versus RpE, RpVNB, kpE, kpVNB, and [Zr]/[C*] count could be fitted to a model that invokes deactivation of the growing polymer chains. In the case of the thermal behavior of the copolymers (melting temperature (Tm) and crystalline temperature (ΔHm)), Tm varied from 101 °C to 121 °C, while ΔHm varied from 9 to 16 (J g−1).
Introduction
Homogeneous catalysts can be used to precisely regulate the properties of polymer such as their microstructure, molecular weight (Mw), and dispersity index (Đ).1–6 The modern homogeneous metallocenes can covert simple alkenes into polymers with the regio-stereospecificity and rate accelerations surpassing that of enzymatic catalysts.7–9 Catalysis is purely a kinetic phenomenon.10–13 Given the previous impact of metallocene catalysts on polyolefin research and manufacturing, it was surprising that there are no well-defined rate laws for the fundamental steps of polymerization such as initiation (ki) by the addition of monomers to the metal–polymeric bond, chain propagation (kp), re-initiation from a metal–hydrogen bond (M–H) bond and chain termination for a logical and practical metallocene-catalyzed polymerization reaction with applicable propagation rate (Rp).14–18 The absence of well-defined rate laws is partially attributed to the problems posed by metallocene catalysts with high catalytic activity and great sensitivity to contaminants.19–21 In addition, it is difficult to identify the percentage of metallocenes dynamically constructing a polymer in any instant, such as the active site count. Thus, it is challenging to accept empirical rate laws with molecular pathways without information on the concentration and speciation of the catalytically dynamic species.22,23 In the literature, many theoretical investigations have been reported on monomer insertion, the role of cocatalyst, activity, and polymerization mechanism of metallocene systems. In this contribution, a methylalumoxane (MAO) cocatalyst, which includes various activating species, complicates the study given that it could further speciate the active catalytic sites.24,25 As shown in Scheme S1,† the critical features of the model are as follows: (1) ion-pair solvent separation occurs before alkene insertion; simple bimolecular mechanisms accomplish (2) initiation and propagation; and (3) β-hydride elimination is the predominant chain termination step. However, it is difficult to explain many facts with this straightforward explanation.26–28 Thus, this basic model cannot explain a large amount of data.The commercial production of EPE EPDM olefins by polymerization exceeds 100 million tons per year. Some kinetic polymerization investigations assume that 100% of the polymeric catalyst added to the system is active during the reactions.29,30 However, numerous comprehensive kinetic investigations showed wide variations in the active sites amongst the different catalysts used in olefin polymerization. In addition, identical polymeric catalysts can have variable active site concentrations, depending on the reaction parameters, for example, monomer pressure, temperature, solubility of monomers, additives, and activators. In fact, in homogenous metallocenes, their active center concentration is less than 5% and even 1%. Therefore, it is essential to determine why some metallocenes have higher active site counts and others do not. This is critical for understanding the chemistry of catalysts and the polymerization procedure more generally. Also, it is challenging but essential to identify the origin of 100% polymerization catalyst activity, which can lead to significant improvements in catalyst productivity due to the optimization of the reaction conditions or catalyst design.6,31,32
Generally, polymerization reactions are seriously impacted by either the low efficiency of the selected comonomers or the excessive amount of comonomers chosen, despite being achievable with some late transition metal-based catalysts and cocatalysts.18,33,34 Determining the source of 100% catalyst activity may lead to a considerable increase in catalytic efficiency based on changes in the catalyst design or reaction parameters.35–37 Nonetheless, no kinetic and mechanistic study of the [rac-Me2Et(Ind)2ZrCl2]/[(Ph3C)B(C6F5)4]-catalyzed copolymerization processes of ethylene with cyclic olefins (E/VNB) with quench flow observation has been published to date. The study of the kinetics of E/VNB copolymerization with the involvement of [rac-Me2Et(Ind)2ZrCl2]/[(Ph3C)B(C6F5)4]/TIBA using the TPCC technique perfectly demonstrates that the metal–polymeric species provides an efficient method for the real-time measurement of the commoner consumption and catalyst species diversity as a function of time. TPCC works by selectively quenching the metal–polymer bonds through acyl chloride, which has been proven in studies using nickel–diimine, metallocene/MMAO, metallocene/TEA, metallocene/TIBA, and Ziegler–Natta heterogeneous catalyst polymerization reactions.19,35,38–40 Furthermore, all the obtained E/VNB were characterized via 1HNMR, GPC and DSC.
Overall procedures and materials
Standard glovebox and Schlenk-line procedures were used for all reactions under an Ar or pure N2 environment. A gas purification column and molecular sieves were employed to purify ethylene. Cyclic comonomer 5-vinyl-2-norbornene (VNB) was manufactured by Aldrich, which was dehydrated by stirring with CaH2 overnight. The catalyst (rac-Et(Ind)2ZrCl2) was purchased from Sigma-Aldrich and its solution prepared in toluene. Triisobutylaluminum (TIBA) was purchased from Albemarle Co. and diluted in heptane. Borate (Ph3C)B(C6F5)4 was synthesized according to the literature and dissolved in toluene for the reaction.40,41 TPCC with 98% purity was bought from J&K Scientific and prepared as a 2 M solution in dry heptane.Polymerization
All the E/VNB copolymers reactions were carried out in a 100 mL round-bottom Schlenk flask to ensure uniform polymerization. Thirty minutes before starting the polymerization process, the reactor was heated to 90–100 °C under vacuum before being backfilled with N2 gas. An appropriate amount of toluene was added to the Schlenk flask reactor, and then saturated with E. After introducing VNB in the reactor, 2 M TIBA was added, operated for 5 min, and then the appropriate amount of zirconocene catalyst and borate in toluene solutions was added. TPPC (TPCC/Al = 2) was added after the polymerization reaction was complete. In addition, to decompose the borate, metallocene, TPCC, and TIBA, 2% HCl was used. The obtained copolymers were washed several time with 95% ethanol, and subsequently dried under vacuum.40–42Characterizations
A YHTS-2000 fluorescence sulfur detector was employed to measure the sulfur (S) content in each copolymer sample. Using the average of three sulfur (S) content measurements for each sample, the percentage of sulfur was calculated.The S content in the blank E/VNB copolymers prepared under similar conditions, without TPPC quenching was 0. Then, it was compared with the quench-labelled E/VNB copolymer, which contained an S content ranging from 5 to 35 ppm. The mechanistic model presented in Scheme S2† briefly explains the TPCC quenching phenomena. How the combination of E and VNB produces non-active sites, how the VNB exocyclic double bonds interact with the Zr metal and interact with the catalyst ligand, and how VNB activates the inactive sites is discussed in the later sections herein.
1HNMR
The content of the commoner (VNB) was calculated via 1HNMR spectroscopy. 1HNMR spectra were recorded using a Varian Mercury Plus 300 spectrometer. The measurements were performed in o-dichlorobenzene-d4 at 120 °C. Hexamethyldisiloxane was used as the internal reference. Previously reported literature was employed to conform to the E/VNB 1HNMR peak assignment.41 The 1HNMR spectrum of the E/VNB copolymers used for the kinetic and mechanistic study is presented in the ESI, Fig. S4.† The peaks at 4.5 and 5.4 ppm confirmed the insertion of the endocyclic π bond in the polymer backbone.43–45The thermal properties of the E/VNB copolymers were performed using differential scanning calorimetry (DSC) analysis. The TA Q200 (DSC) instrument was calibrated with water and indium. An appropriate amount of polymer sample was sealed in an aluminum pan. To eliminate the thermal history of the sealed sample, it was firstly heated to 150 °C for 5 min. Secondly, it was cooled at a rate of 10 °C min−1 to 20 °C. Finally, it was gradually heated to 180 °C at a rate of 10 °C min−1 to produce the melting curve.46,47
The polydispersity and molecular weight of E/VNB were calculated via high-temperature PL 220 gel permeation chromatography (GPC). The eluent 1,2,4-trichlorobenzene was used in the PL-gel 10 m MIXED-B column with a flow rate of 1.0 mL min−1 at 150 °C. It was envisioned that thin polystyrene standards would be used for universal calibration.
Results and discussion
E/VNB copolymerization with zirconocenes/borate/TIBA was firstly studied. For the kinetic and mechanistic studies of these catalyst systems, a series of E/VNB copolymerization tests was performed. The subsequent protocol was followed to maintain the performance of the catalyst, where after injecting VNB, a suitable amount of TIBA was added, which reacted with VNB and the solvent impurities. Our group reported the synthesis of ethylene–propylene co- and E/P/diene terpolymers with a symmetrical metallocene catalyst activated by MMAO and TIBA/borate cocatalyst system.17,36,39 We observed that the addition of diene decreased the polymerization activity.19,20,41,48 Herein, we performed our experiments with a stoichiometric amount of VNB 0.06 mol L−1. The time-dependent variation of RpE and RpVNB was determined from the differentiation of the curve of copolymer yield versus polymerization time (tp). The sulfur (S) content of the copolymers was calculated in the purified samples to measure [C*]/[Zr] according to the equation [S] = [C*], as shown in ESI Tables S1 and S2.† The propagation rate constants kpE and kpVNB were calculated according to the equation Rp = kp[C*][M], which was developed for homogenous metallocenes, α-diamine nickel complexes, and heterogeneous Ziegler–Natta catalysts.38–40 The results of the E/VNB copolymerization, including activity, Mw, MWD, time-dependent [C*]/[Zr] fraction, RpE, kpVNB, kpE, kpVNB, and thermal properties are summarized in Tables 1 and 2.| RUN | Time (s) | VNB in polymerb | Activity (107 gpoly Mt−1 h−1) | Mwc (g mol−1) | PDc | Tmd (°C) | ΔHmd (J g−1) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: toluene = 50 mL, E = 0.1 MPa, VNB = 0.06 mol L−1, TIBA 1000 μmol, borate = 2.50 μmol, catalyst = 1.25 μmol and TPCC = 2000 μmol.b Determined by high-temperature 1HNMR.c Determined by high-temperature GPC.d Determined by DSC. | |||||||
| 1.1 | 120 | 10.92 | 0.36 | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
2.08 | 122.14 | 7.78 |
| 1.2 | 240 | 9.33 | 0.20 | 15100 |
2.25 | 120.43 | 9.77 |
| 1.3 | 360 | 8.46 | 0.15 | 16000 |
2.45 | 120.44 | 9.48 |
| 1.4 | 480 | 8.05 | 0.13 | 17800 |
2.69 | 120.12 | 15.01 |
| 1.5 | 840 | 7.85 | 0.12 | 21000 |
3.20 | 120.28 | 14.92 |
| 1.6 | 1200 | 7.22 | 0.11 | 25500 |
3.15 | 120.62 | 3.66 |
| 1.7 | 1500 | 7.09 | 0.10 | 27300 |
3.27 | 120.49 | 12.76 |
| 1.8 | 1800 | 5.73 | 0.09 | 29300 |
3.70 | 121.46 | 6.41 |
| RUN | Time (s) | VNB in polymer | *Cb (%) | RpVNBc (molVNB molcat−1 s−1) | RpEc (molE molcat−1 s−1) | kpEd (L mol−1 s−1) | kpVNBd (L mol−1 s−1) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: toluene = 50 mL, E = 0.1 MPa, VNB = 0.06 mol L−1, TIBA 1000 μmol, borate = 2.50 μmol, catalyst = 1.25 μmol and TPCC = 2000 μmol.b *C determined through the sulfur analyzer (see ESI).c RpVNB and RpE in units of mmolpoly molMt−1 s−1.d Propagation constant of VNB and E. | |||||||
| 1.1 | 120 | 10.92 | 5.71 | 0.7 | 10.23 | 4266 | 233 |
| 1.2 | 240 | 9.33 | 7.55 | 0.67 | 9.87 | 3113 | 173 |
| 1.3 | 360 | 8.46 | 10.1 | 0.58 | 9.52 | 2244 | 116 |
| 1.4 | 480 | 8.05 | 14.08 | 0.52 | 9.18 | 1552 | 78.9 |
| 1.5 | 840 | 7.85 | 19.68 | 0.36 | 8.25 | 998 | 42.04 |
| 1.6 | 1200 | 7.22 | 26.17 | 0.25 | 7.41 | 674 | 23.395 |
| 1.7 | 1500 | 7.09 | 29.93 | 0.18 | 6.77 | 539 | 15.727 |
| 1.8 | 1800 | 5.73 | 31.67 | 0.14 | 6.19 | 465 | 11.5397 |
According to the literature, the amount of linear conjugated, non-conjugated, and cyclic dienes increases in the reaction system, and as predicted, the activities of metallocene, Ziegler–Natta, and α-diamine nickel polymeric catalysts decrease.20,33,39 For example, VNB was used as a commoner, similar to ENB used in EPDM manufacturing, but the response of ENB against the metallocene is much more complicated than VNB.20,48 In E/VNB copolymerization, the activity decreased in the time of 120–1800 s, and [Zr]/[*C] considerably increased, and consequently tended to the stable level of 31.67%. This is similar to polypropylene (PP) but lower than E homo, E/P and E/ENB copolymerization.17,39,49 The addition of 0.06 mol L−1 VNB resulted in the maximum activity of 0.36 × 107 g molMt−1 h−1 at 120 s, as shown in Fig. 1. However, when the reaction time increased, the activity declined until it reached 0.09 × 107 g molMt−1 h−1. The obtained copolymers containing higher than 5 mol% VNB were soluble in 1,2,4-trichlorobenzene at temperatures greater than 100 °C. Swaminathan Sivaram et al. demonstrated that increasing the mol% of dienes in the feed reduces the intrinsic viscosity of the polymer and catalytic activity.50
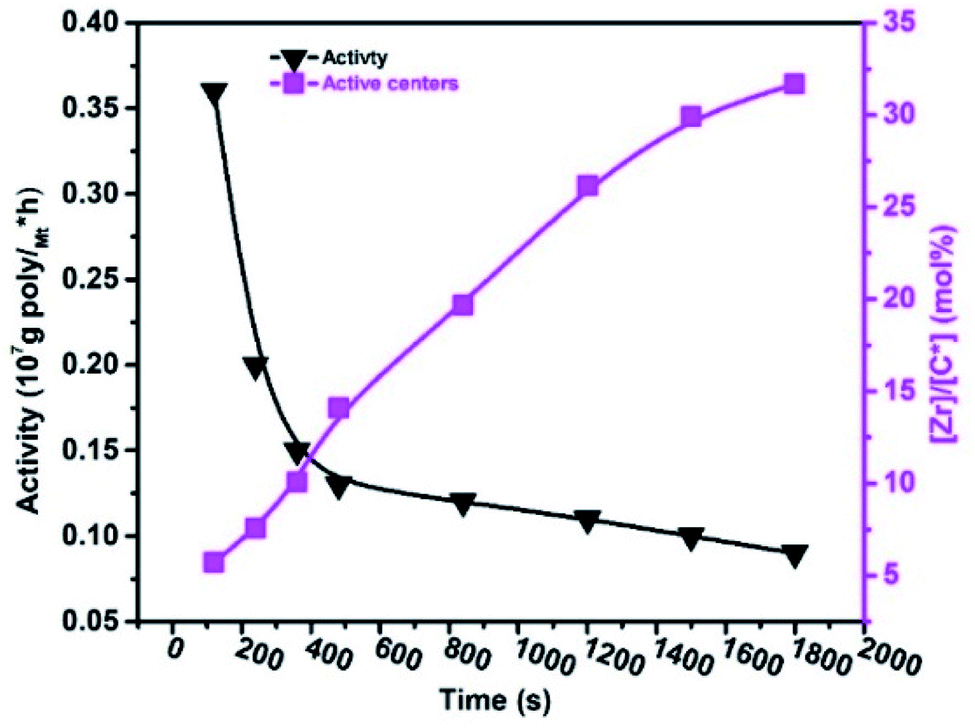 | ||
| Fig. 1 Change in the active centre [Zr]/[C*] fraction and catalytic activity with polymerization time (tp) during E/VNB copolymerization. | ||
We observed that the effect of VNB on metallocene activity was lower in the initial stage of the reaction than the later stage. It is recommended that metallocene/borate presented a high E/VNB copolymerization rate at 120 s, which can be used to confirm the quick initiation of the active catalytic sites. The plot of RpE and kpVNB with tp is shown in Fig. S3.†
In contrast, the rapid increase in active centres in the initial polymerization level also defined the immediate commencement of active sites located on the surface of the metallocene catalyst molecules. These active sites are the molecules that are accessible to incoming monomers and activators and can be activated during the pre-contact process. Guo et al. well-defined in their study the increase in the amount of the deactivated or dormant sites in the metallocene/MAO system by the interaction of the metallocene catalyst with TMA, and they recommended the incomplete activation of the catalyst.36 In our previous study, we explained that chain transfer with alkylaluminum led to polyethene (PE) chains ending with saturation.17,19 During the E/VNB copolymerization, a significant change in the solvent reaction viscosity was observed, and the growing polymer fibres entangled on the reaction stirrer were noted. The developing catalytic active sites are compressed in the precipitated copolymers through this phase, inhibiting the incoming monomers from diffusing into the active sites, which subsequently reduces the polymerization rate.
The Mw of the E/VNB copolymerization decreased in the initial polymerization time and slightly increased with an increase in the reaction time. When correlated with the Mw of PE, E/P, and E/ENB copolymers produced with rac-Et(Ind)2ZrCl2/MAO, rac-Et(Ind)2ZrCl2/MMAO, and rac-Et(Ind)2ZrCl2/TIBA, that of the E/P copolymers is comparable. However, PE and E/ENB were produced with a higher molecular weight. This suggests that the chain transfer of the active sites with the TIBA cocatalyst in the E/VNB copolymers is much faster than that in the PE and E/VNB systems. The MWD of the E/VNB copolymers was broader and became even broader as the reaction time reached 1800 s, which was significantly greater than 2. According to the literature, a theoretical polydispersity index (Đ) more than 2 of a true single-site metallocene suggests the presence of numerous active sites in the system (see Fig. S1†).
1HNMR is one of the main methodologies to elucidate the microstructure of polymers and calculate the co-monomer content. It can be observed that the 1HNMR spectra of all the E/VNB copolymer samples demonstrated identical peaks, which are consistent with that reported by Randall et al.51 VNB exhibits two different double bonds, with the peaks for the distinct exocyclic vinyl pattern between 4.5 and 5.9 ppm in the copolymer backbone, and the absence of a strong triplet peak at 6.1 ppm for the endocyclic π bonds, as shown in Fig. S5.† The exocyclic π bonds are nearly nonreactive with the Z–N catalyst. VNB, being a commoner with bulky alkyl groups, has more considerable ring strain, leaving the exocyclic bond unreacted. The exocyclic unreacted bonds were also used to quantify the mol% of VNB comonomers. The mol% of VNB is more significant in the initial reaction time than later. The [Zr]/[C*] rapidly increased initially, presenting evidence of the quick activation of the active sites at the start of the reaction, which was dormant for ethylene polymerizations.
It is well known that 5–10% of dienes are the essential requirement of applicable industrial elastomers. The properties of E/VNB copolymers depend on the VNB content, and the resulting polymers have a melting temperature (Tm) of 100–120 °C or are amorphous. The effect of time and VNB mol% on the thermal properties of the E/VNB copolymers is illustrated in Table 1. Commonly, the cyclic olefins NB, ENB, and VNB are bulkier structures than E or P, and cyclic olefins constrain the rotational movement of the polymers, resulting in a direct effect on their thermal properties such as crystalline (ΔHm) and melting temperature (Tm).
The E/VNB copolymer ΔHm temperature varies from 9 to 16 (J g−1) and Tm ranges from 100 °C to 120 °C. In addition, these results are strongly based on endocyclic π and exocyclic π insertion and the VNB composition. According to the analysis of the HNMR spectra, that endocyclic π-inserted VNB and exocyclic π composition is more than 5 mol%. E/VNB copolymers with VNB-rich segments were obtained in the earlier stage if the reaction melted at a lower temperature, and later due to the higher solubility of E, the incorporation of VNB decreased and it melted at a higher temperature. Interestingly, we discovered that the crystallinity of the E/VNB copolymer was lower and the initial polymerization rates (RpE and RpVNB) were higher due to the more significant insertion of VNB into the growing polymer chain. As the reaction time increased, the mol% of VNB and RpE and RpVNB in the E/VNB copolymers decreased. This is because the chain termination response in E/VNB is more rapid in the latter stage than the early stage.
The results of the E/VNB copolymerization, including time-dependent [C*]/[Zr], are summarized in Table 2, together with the kinetic data including RpVNB, RpE, kpVNB, and kpE. As shown in Fig. 2 and 3, all the kinetic properties studied, such as RpVNB, RpE, kpVNB, and kpE with tp, showed a moderate decrease.
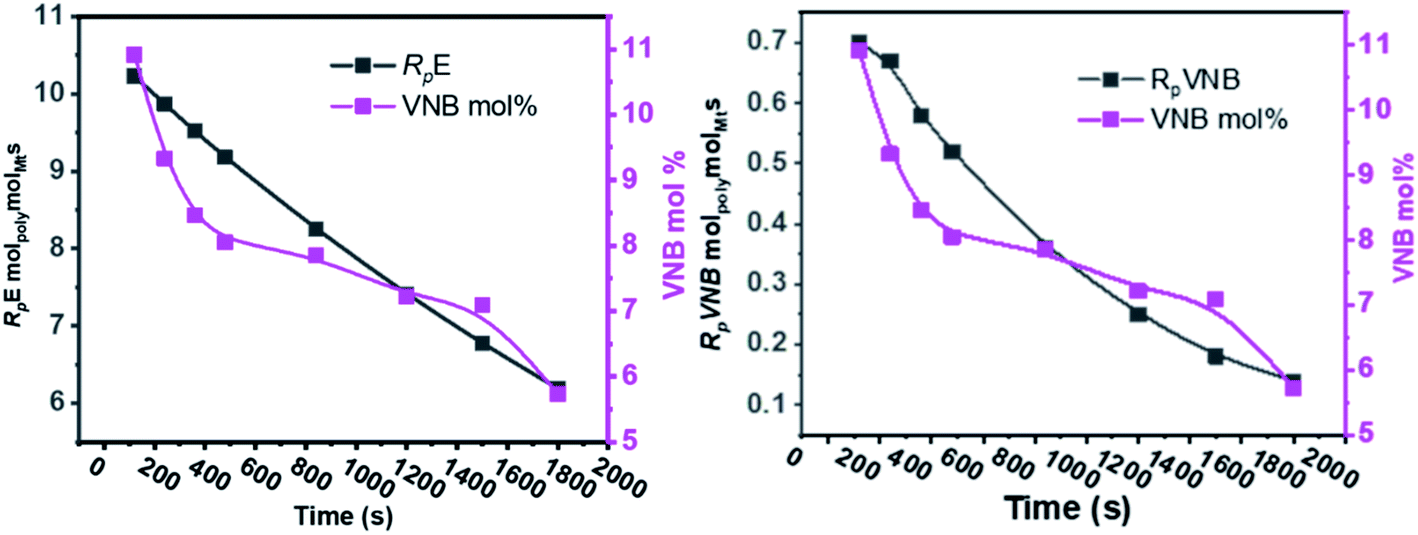 | ||
| Fig. 2 As the polymerization time progressed, the amount of VNB in mol% and the ratios of RpE and RpVNB decreased. | ||
 | ||
| Fig. 3 Fluctuation in the active center fractions RpE and RpVNB in the E/VNB copolymers as a function of polymerization time. | ||
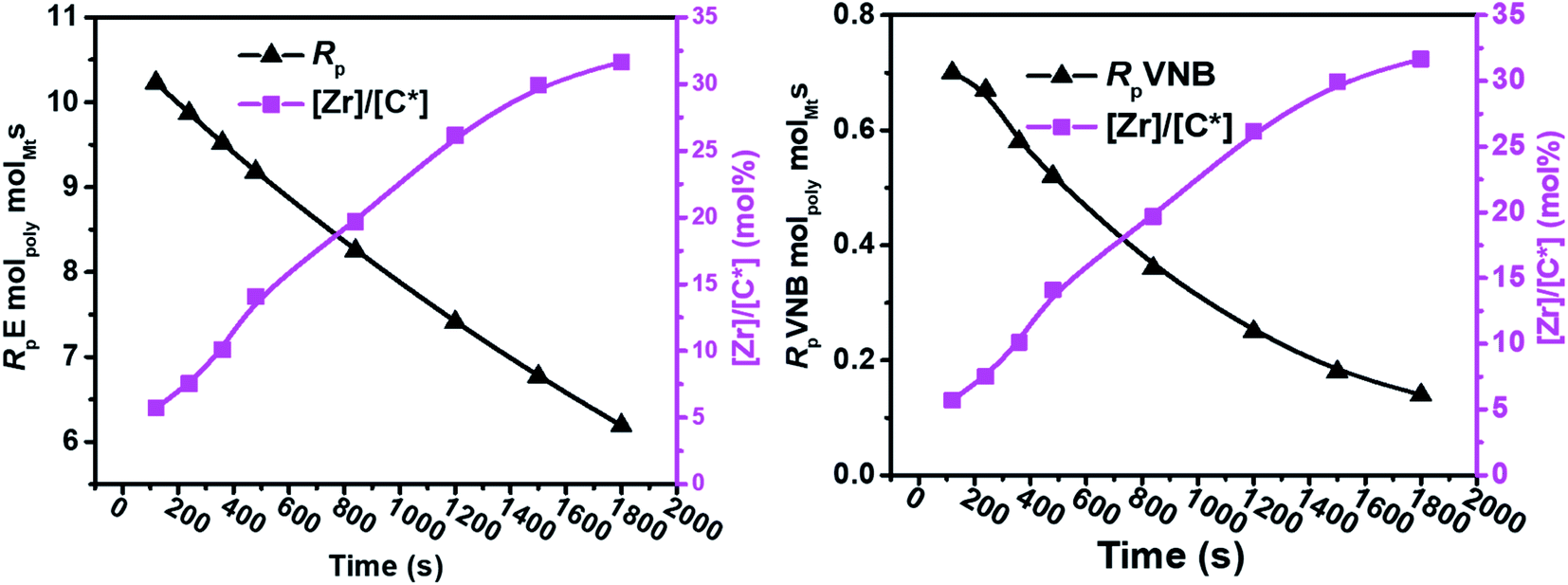
In contrast, the active sites continuously increased with time tp. The E/VNB copolymers had a lower [C*]/[Zr] fraction (3–8% at 240 s), but later, the system reached 33.7% in 1800 s. In previous studies, the same catalyst system-catalyzed E/ENB copolymers showed a much greater initial [C*]/[Zr] fraction (16–30% at 240 s) and relativity quicker increase in the active centers up to stable stage. During the E homo polymerization after 1800 s, the reaction time [C*]/[Zr] fraction reached 73%, which is higher than that of this system. Thus, this means that ethylene copolymerized with ENB and VNB has different characteristics. Alternatively, kpE, kpVNB, and kpVNB are much lower than kpE, which suggests that the minor diffusion barriers in the early stages are close to the actual propagation rate constant. Similarly, at 120 s, the kpE and kpVNB values were 4466 and 233 L mol−1 s−1, respectively, which are lower than that of the PE and E/ENB polymers, indicating that the diffusion barriers in E/VNB are higher than that in the PE and E/ENB systems.
When considering the continuous decrease in kpE and kpVNB with the polymerization time, this may be due to the drop in the local-monomer concentration around the active site as a result of the diffusion barrier, given that the rate of monomer diffusion from the bulk of solution to the position of active sites quickly decreased once the first set of polymerization chains was formed and aggregated in and around the active sites of the catalyst.
As shown in Fig. 4, in the early 500 s, an increase in the [Zr]/[C*] percentage by more than threefold was observed. The potential reactivity variation across the active sites triggered at various reaction phases may also have caused a significant variation in the kpE and kpVNB values. This is because the active catalytic sites in the time range of 120–480 s are 30% more active than the active catalytic sites in the later time range. The former account for 40% of the total [Zr]/[C*], and the average kpE and kpVNB values at the polymerization time of 1000 s were only 50% of that at the polymerization time of 480 s. Thus, it is reasonable to assume that there is no diffusion barrier in the system. In contrast, as reaction time increased, both kpE and kpVNB and the mol% of VNB decreased, as shown in Fig. 5.
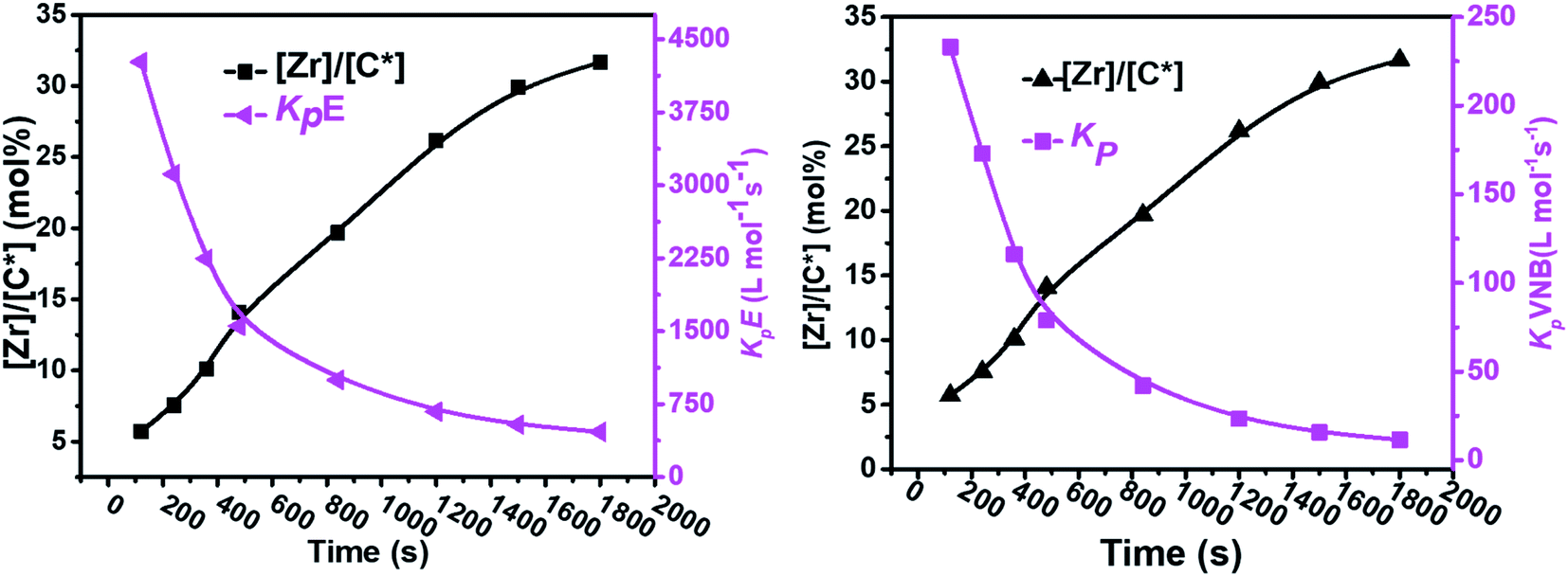 | ||
| Fig. 4 Variation in the propagation rate constants kpE and kpVNB and active center fraction in the E/VNB copolymers with polymerization time. | ||
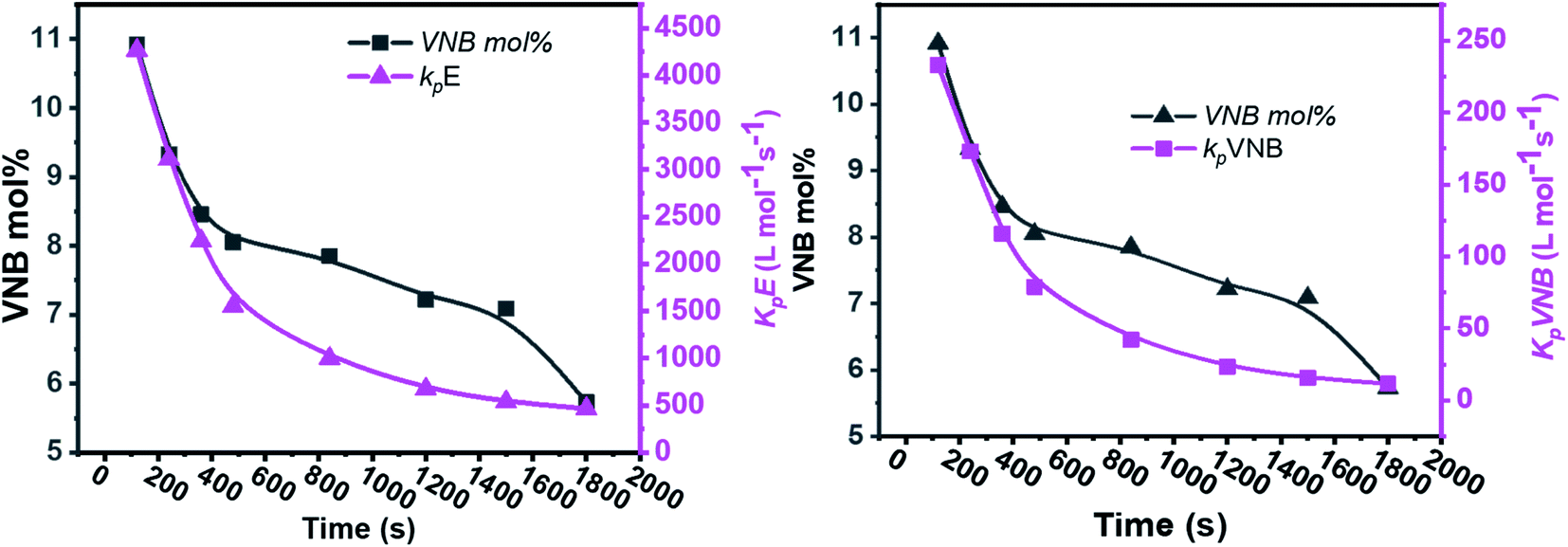 | ||
| Fig. 5 Relationship between polymerization time and alteration in the propagation rate constants kpE and kpVNB and the mole% of VNB in the E/VNB copolymers. | ||
The formerly investigated initial kpPE of polyethene and E/ENB copolymerization kpE values differed, whereas the kpPE of polyethene and kpE of the E/VNB copolymer were the same at 120 s (4110, 9115, and 4266, respectively). This is because the [C*]/[Zr] fraction of E/VNB copolymerization at the initial level is the same with polyethene but lower than that of the E/ENB copolymers, indicating that PE and E/VNB have the same type of active centers, whereas that if E/ENB is different. The active centers in the polyethylene and E/VNB copolymerization that produce E/VNB copolymers may be made up of contact ion pairs with lower kpPE and kpE values. In contrast, the active center in the E/ENB copolymers could be made up of loosely associated ion pairs with higher kpE values. Thus, to make the comparisons among the kp of PE, E/ENB, and E/VNB polymerization more understandable, the curves of polymerization time vs. kp are illustrated in Fig. 6 and 7.
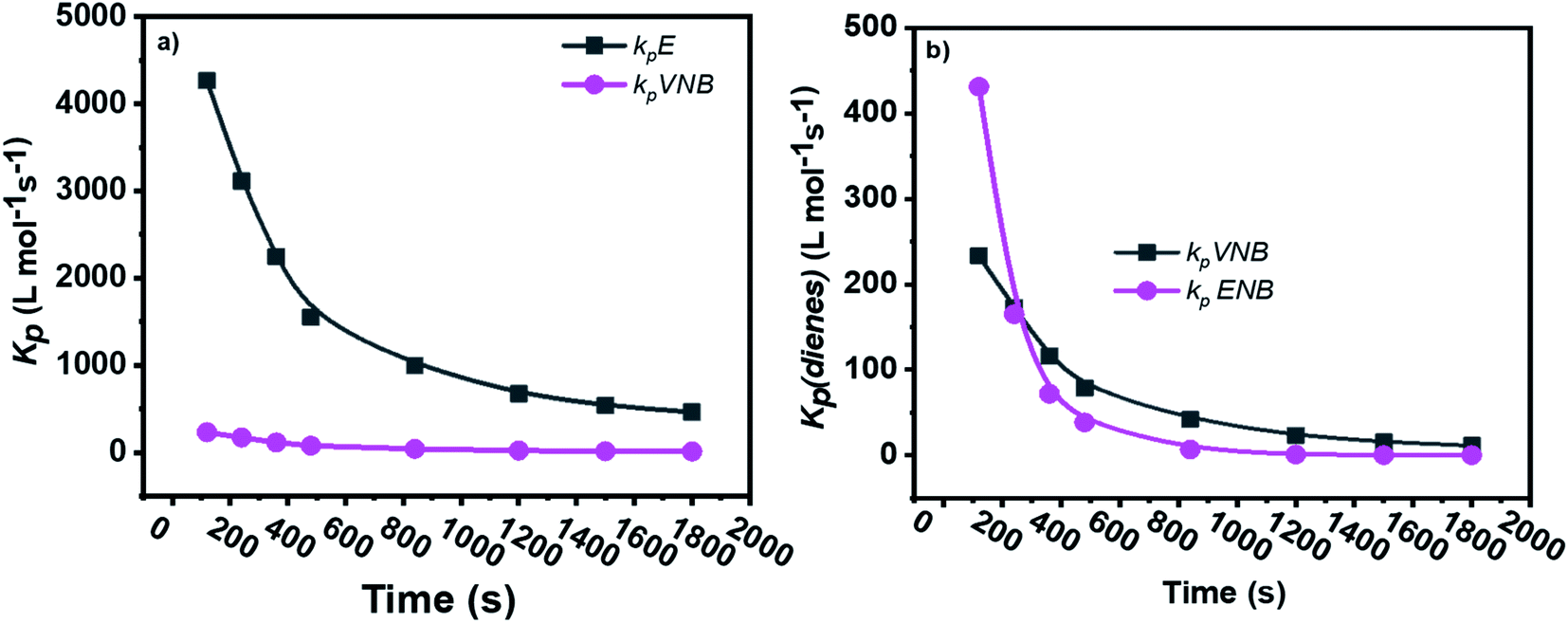 | ||
| Fig. 6 Comparisons between the propagation rate constants of (a) kpE and kpVNB of E/VNB copolymers and (b) kpVNB and kpENB with the same reaction parameters. | ||
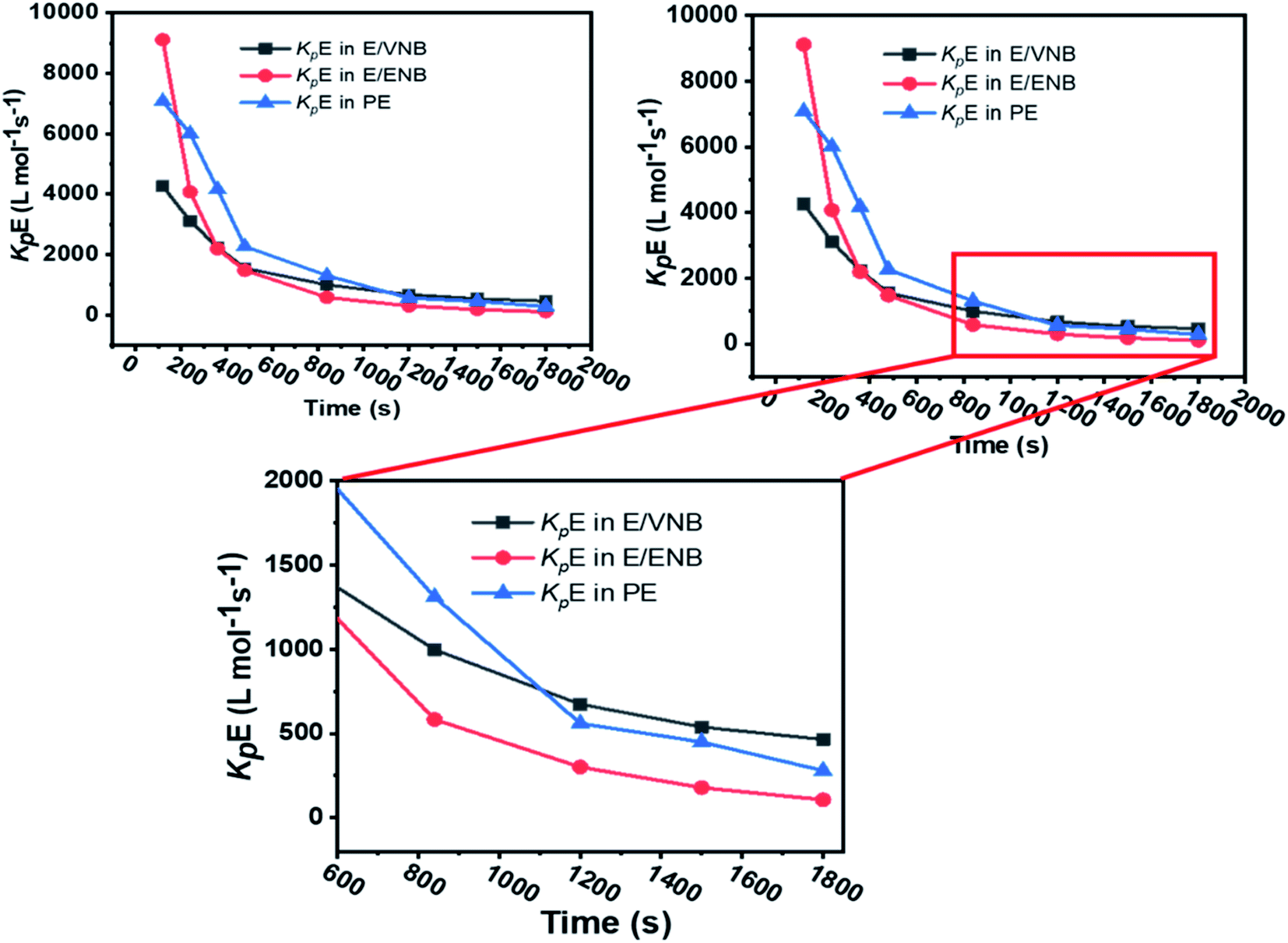 | ||
| Fig. 7 Comparison of the propagation rate constants kp of polyethylene (PE), E/ENB, and E/VNB polymerization. | ||
The initial values of kpE and kpVNB of E/VNB are similar with that of E/P but lower than that of the E/ENB polymers because E/ENB are less crystalline. Simultaneously, E/P and E/ENB are nearly amorphous, have low thermal values, and are comparatively easily dissolved in toluene. This difference may be due to the lower diffusion barrier in the E/VNB system and higher propagation rate content.
The diffusion barrier described the reasonable decay of kpE and kpVNB with tp in the above discussion. However, the influence of [C*]/[Zr] on kpE and kpVNB must be considered. The [C*]/[Zr] fraction in the E/VNB copolymers is much lower than that in the polyethylene and E/ENB copolymers, particularly in the stable reaction stage. The [C*]/[Zr] generated in the later stage of the E/VNB copolymers has lower kpE and kpVNB values than kpPE, kpE, and kpENB. It is well known that ENB, VNB, and P have a bulkier structure than the PE part of a polymeric catalyst such as metallocene in the shape of contact ion pairs, which may be unable to accept the coming VNB monomer for further coordination. Subsequently, it behaves as inactive or dormant sites and has a lower [C*]/[Zr] fraction. Consistent with earlier studies, the polymerization conditions employed in this study were the same. It is considered that the reaction parameters adopted in the given polymerizations are identical to that used in the previous work,19,35,40,52 and thus it can be said that changing the type of comonomers can influence the efficiency of the metallocene activation kinetic behaviors.
Discussion on mechanism
Our previous work investigated the kinetics of PE and PP.19,36 The significate increase in Mw and MWD with reaction time was briefly explained through the earlier activation of the active centers with a longer cation–anion distance, and later with a shorter cation–anion distance.17,19,53 The loose ion pairs found in the initial reaction time showed a higher polymerization rate, activity, and E/VNB yield with lower molecular weight. Subsequently, the interference from the counter-anion on the E and VNB monomers coordinated to the cationic active sites is poor and the conformational conversion of the propagation chain for β-hydrogen transfer becomes easier in the relatively open ion pair. According to Fan et al., if the ion-pairs exhibit a shorter cation–anion distance, they will be less active for E coordination due to the strong interference from the counter anion for monomer coordination.17,36 Given that we observed a similar increase in the Mw and Đ of E/VNB with the reaction time, we can describe the phenomena with identical models, as shown in Scheme 1. The MWD curves of E/VNB were obtained at different reaction times (see ESI, Fig. S1†), which slowly increased with an increase in the reaction time tp. The time-dependent change in the MWD for E/VNB was lower than that for PE. The time-dependent slow variations of MWD in tp of 120–1800 s may be attributed to the slower increase in [C*]/[Zr] in the same period, and thus we can assume that the later-stage reaction activated active center has a longer cation distance than PE and produced E/VNB with a lower Mw.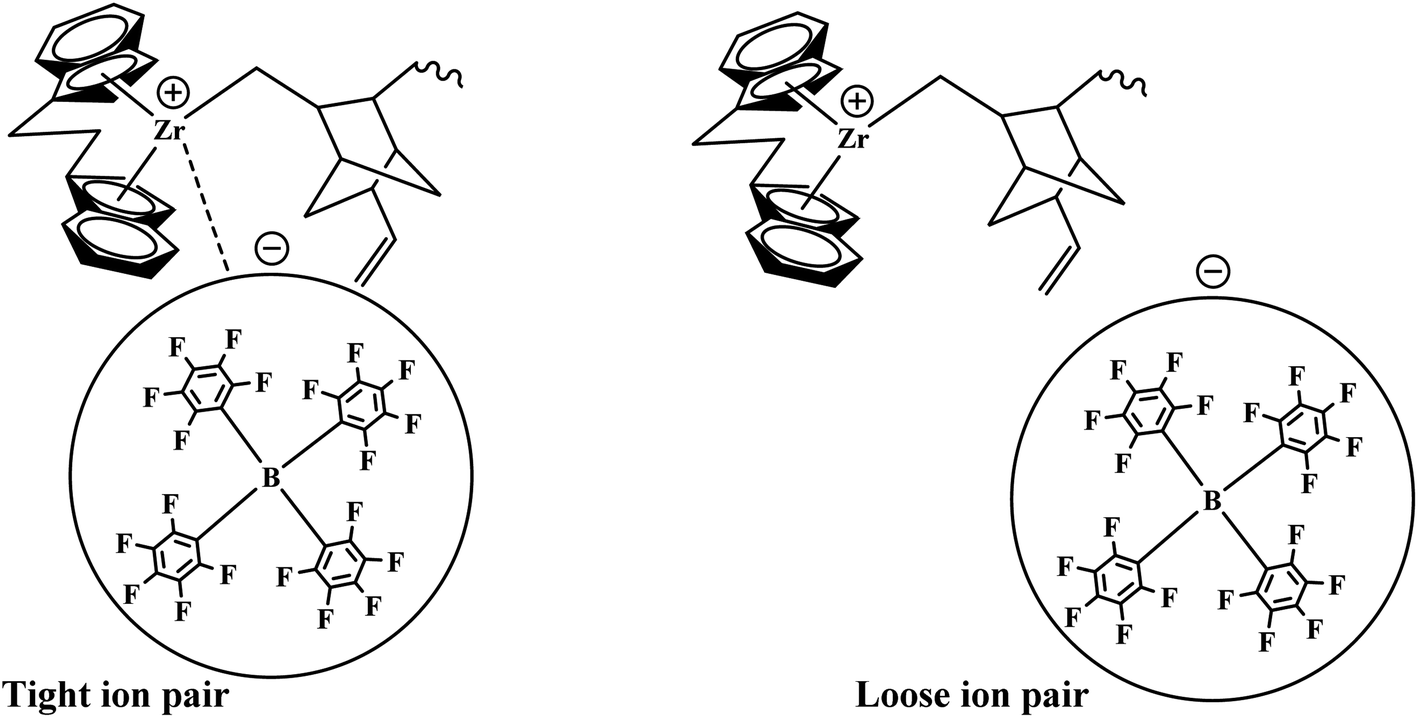 | ||
| Scheme 1 Zirconocene/borate catalyst active centers with varying cation and anion distances. | ||
The kinetic behaviors of the same catalyst for different monomers (PE, ENB, and VNB) can be described through this model. We compared the kpE and kpVNB with the kpPE and kpENB values in the first part of this paper, and we noticed that kpE and kpVNB were lower than kpPE and kpENB in most of the reaction times; meanwhile, the same catalyst system produced E/VNB with a lower Mw. This shows that the active site of the catalyst in the E/VNB copolymers has longer cation–anion distances than that for PE.
In the initial stage of E/VNB polymerization, the amount of active centers is slightly higher than that of PE, indicating that VNB sterically activated the active site in the metallocene precursor, which was dormant in the initial phase of PE. Despite the fact that the bicyclic VNB has an exocyclic and sterically endocyclic double bond, it is more difficult to synthesize than α-olefins such as propylene and 1-hexene. However, the lower [Zr]/[C*] fraction of the E/VNB copolymers than that of the PE and E/ENB copolymers at the stable and later stages can be attributed to the higher fraction of dormant sites in this system. This means that VNB as a comonomer deactivated the active sites in the same catalyst system, which was active in the PE and E/ENB polymerizations.
Scheme 2 shows the insertion of E into Zr–C and the insertion of VNB into Zr–E. VNB is sterically more interesting than P and 1-hexene, and the insertion barriers are more significant for Zr–C than P. The insertion of VNB as a comonomer in the system leads to a considerable decay inactivity. The steric impact of VNB, insertion in Zr–E, β-hydrogen transfer, repulsion between the catalyst ligand and VNB are explained by the mechanism presented in Scheme 2.
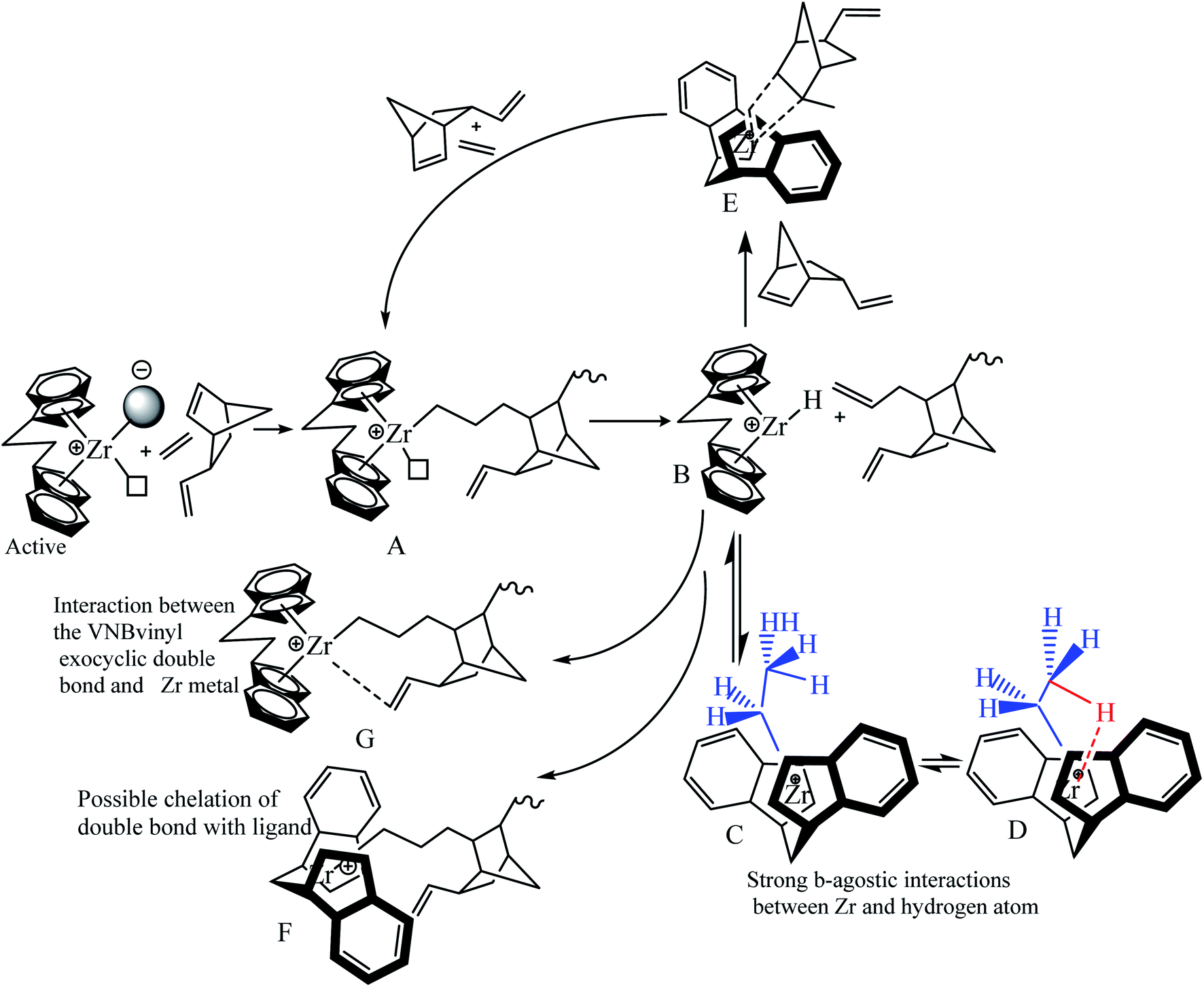 | ||
| Scheme 2 Mechanistic model for the E/VNB copolymerization reactions, explaining how the inactive sites are formed by the steric impact of VNB, β-hydrogen transfer, and repulsion between the catalyst ligand VNB exocyclic π bonds, and how VNB reactivate them. | ||
In the proposed mechanism (Scheme 2), the active catalytic sites L2Zr–CH2–CH2–VNB bearing propagation chain (A) possibly undergo β-H transfer reaction, and subsequently generate species (B) with a metal–hydrogen bond in the formation of L2Zr–H, which is further converted into L2Zr–CH2H3 (C) by inserting E into the L2Zr–H bond between the center of Zr and methyl hydrogen-produced Zr–CH2CH3 (D) species, which makes them unapproachable to the incoming VNB monomers. These species are involved in reducing the activity of the system. Many reports have been published in the literature on reactivating this type of dormant or inactive species. For example, Y. V. Kissin and colleagues investigated the co-co-monomer activation mechanism in heterogeneous Z–N catalyst systems through their model. They explained the E/P copolymerization catalyzed with Z–N and showed how propylene activates the dormant ethylene sites. Similarly, Fan et al. employed this model in E/P copolymerization catalyzed with metallocene/MMAO and metallocene/TIBA/borate, and they analyzed that the insertion of the P comonomers in the zirconium–hydrogen bond (Zr–H) can bypass the reaction and significantly increase [Zr]/[C*], which was higher than that of their homopolymers. However, in the case of E/VMB polymerization, the insertion of the VNB zirconium–hydrogen bond (Zr–H) is very slow compared to ENB and P and hardly bypasses the reaction and converts to (E), leading to a slight increase in [Zr]/[C*], which is slightly higher than that for the homopolymerization of E. In addition, ENB and VNB are the nearly same family of olefins, but there are more robust β-agnostic contacts between the Zr metal and methyl hydrogen in VNB compared to ENB, resulting in a higher content of these dormant or inactive sites in the E/VNB copolymerization. This is the main reason for the lower [Zr]/[C*] fraction in the earlier and later stages than that previously reported for E/ENB copolymerization.
VNB polymerizes as cyclic diolefins with an alkyl substituent using an endocyclic π (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) bond and has a significant ring strain. The endocyclic π bond insertion of VNB in H–Zr increases the reaction rate and active centers through (E). This reaction, as mentioned above, was elucidated by the presence of peaks of exocyclic bonds in the polymer backbone, which can be utilized for additional post-polymerization investigation. Furthermore, the formation of L2Zr–H and L2Zr–CH2H3 can be achieved via the suitable adjustment of alkyl and zirconium due to the sufficient space between the L2Zr+–CH2H2–VNB cation and X− anion. This prediction was supported by the significant reduction in the rate of VNB incorporation during the initial stages of the reaction. As illustrated in Table 2 and Fig. 2, 3, the active center fraction is 10–14% lower in the time of 120–480 s. With a further increase in the reaction time, it reached 33.7%, and the incorporation rate of VNB in the initial period was found to be higher than later. This indicates that the catalytic species L2Zr+–R.X.− activated initially can insert more VNB than the later activated catalytic species.
CH–) bond and has a significant ring strain. The endocyclic π bond insertion of VNB in H–Zr increases the reaction rate and active centers through (E). This reaction, as mentioned above, was elucidated by the presence of peaks of exocyclic bonds in the polymer backbone, which can be utilized for additional post-polymerization investigation. Furthermore, the formation of L2Zr–H and L2Zr–CH2H3 can be achieved via the suitable adjustment of alkyl and zirconium due to the sufficient space between the L2Zr+–CH2H2–VNB cation and X− anion. This prediction was supported by the significant reduction in the rate of VNB incorporation during the initial stages of the reaction. As illustrated in Table 2 and Fig. 2, 3, the active center fraction is 10–14% lower in the time of 120–480 s. With a further increase in the reaction time, it reached 33.7%, and the incorporation rate of VNB in the initial period was found to be higher than later. This indicates that the catalytic species L2Zr+–R.X.− activated initially can insert more VNB than the later activated catalytic species.
Conclusions
In this work, we investigated the kinetic behavior of ethylene/VNB copolymers catalyzed with rac-Et(Ind)2ZrCl2/TIBA/borate. The highest activity of 0.36 × 107 g molMt−1 h−1 was achieved at 120 s, which decreased with a further increase in reaction time to 0.09 × 107 g molMt−1 h−1, and [Zr]/[*C] considerably increased, and consequently tended to the stable level of 31.67%. The E/VNB copolymers containing higher than 5 mol% VNB were soluble in 1,2,4-trichlorobenzene at 120 °C. The MWD was broader and significantly greater than 2, indicating the presence of multiple active sites in the system. The molecular weight decreased in the initial polymerization time and slightly increased with an increase in the reaction time. The crystalline (ΔHm) temperature varied from 9 to 16 (J g−1) and melting temperature (Tm) ranged from 100 °C to 120 °C. The crystallinity of the E/VNB copolymer was lower and its initial polymerization rates (RpE and RpVNB) were higher due to the more significant insertion of VNB in the growing polymer chain. The propagation rate constants kpE and kpVNB were much higher at the initial level (4266 and 233 L mol−1 s−1) than later with values of 465 and 11.53 L mol−1 s−1, respectively, which indicate that the minor diffusion barriers in the early stages are close to the actual propagation rate constant. The continual decrease in kpE, and kpVNB with tp may be partially attributed to the reduction in the local-monomer concentration around the active site. In addition, the lower kpE and kpVNB in the later stage compared to earlier could be due to the lower mol% of VNB, which reduced the amorphous properties and hardly liquefied in the reaction solvent, subsequently leading to a higher diffusion barrier in the later system. The lower [C*]/[Zr] ratio of the E/VNB copolymers in both the early and stable stages and poor amorphous properties with lower VNB mol% at a later stage require the use of a specific kinetic mechanism that assumes the low activity of developing polymer chains containing one of ZrCH2CH3-type dormant site by β-agostic contact between the methyl hydrogens and the cationic Zr center and the interaction between the Zr metal and VNB endocyclic π bonds.Conflicts of interest
The representative authors have no conflict of interest to disclose in any capacity, either competing or financial.Acknowledgements
The authors are thankful to the National Natural Foundation of China (51803081) for their financial support.References
- D. L. Nelsen, B. J. Anding, J. L. Sawicki, M. D. Christianson, D. J. Arriola and C. R. Landis, Chromophore quench-labeling: an approach to quantifying catalyst speciation as demonstrated for (EBI)ZrMe2/B(C6F5)3-catalyzed polymerization of 1-hexene, ACS Catal., 2016, 6(11), 7398–7408 CrossRef CAS.
- A. Shamiri, M. H. Chakrabarti, S. Jahan, M. A. Hussain, W. Kaminsky, P. V. Aravind and W. A. Yehye, The influence of Ziegler-Natta and metallocene catalysts on polyolefin structure, properties, and processing ability, Materials, 2014, 7(7), 5069–5108 CrossRef PubMed.
- W. Kaminsky and M. Arndt, Metallocenes for polymer catalysis, in Polymer Synthesis/Polymer Catalysis, Springer, 1997, pp. 143–187 Search PubMed.
- W. Kaminsky, Highly active metallocene catalysts for olefin polymerization, J. Chem. Soc., Dalton Trans., 1998,(9), 1413–1418 RSC.
- P. S. Kulyabin, G. P. Goryunov, M. I. Sharikov, V. V. Izmer, A. Vittoria, P. H. Budzelaar, V. Busico, A. Z. Voskoboynikov, C. Ehm and R. Cipullo, ansa-Zirconocene Catalysts for Isotactic-Selective Propene Polymerization at High Temperature: A Long Story Finds a Happy Ending, J. Am. Chem. Soc., 2021, 143(20), 7641–7647 CrossRef CAS PubMed.
- A. Ali, A. Naveed, T. Rasheed, T. Aziz, M. Imran, Z.-K. Zhang, M. W. Ullah, A. A. Kubar, A. U. Rehman and Z. Fan, Methods for Predicting Ethylene/Cyclic Olefin Copolymerization Rates Promoted by Single-Site Metallocene: Kinetics Is the Key, Polymers, 2022, 14(3), 459 CrossRef CAS PubMed.
- K. Angermund, G. Fink, V. R. Jensen and R. Kleinschmidt, Toward quantitative prediction of stereospecificity of metallocene-based catalysts for α-olefin polymerization, Chem. Rev., 2000, 100(4), 1457–1470 CrossRef CAS.
- V. Busico, L. Caporaso, R. Cipullo, L. Landriani, G. Angelini, A. Margonelli and A. Segre, Propene Polymerization Promoted by C2-Symmetric Metallocene Catalysts: From Atactic to Isotactic Polypropene in Consequence of an Isotope Effect, J. Am. Chem. Soc., 1996, 118(8), 2105–2106 CrossRef CAS.
- M. D. Bruce, G. W. Coates, E. Hauptman, R. M. Waymouth and J. W. Ziller, Effect of metal on the stereospecificity of 2-arylindene catalysts for elastomeric polypropylene, J. Am. Chem. Soc., 1997, 119(46), 11174–11182 CrossRef CAS.
- E. S. Cueny, H. C. Johnson, B. J. Anding and C. R. Landis, Mechanistic studies of hafnium-pyridyl amido-catalyzed 1-octene polymerization and chain transfer using quench-labeling methods, J. Am. Chem. Soc., 2017, 139(34), 11903–11912 CrossRef CAS.
- A. C. Brezny and C. R. Landis, Recent developments in the scope, practicality, and mechanistic understanding of enantioselective hydroformylation, Acc. Chem. Res., 2018, 51(9), 2344–2354 CrossRef CAS PubMed.
- M. A. Naseer, M. K. Tufail, A. Ali, S. Hussain, U. Khan and H.-B. Jin, Review on Computational-Assisted to Experimental Synthesis, Interfacial Perspectives of Garnet-Solid Electrolytes for All-Solid-State Lithium Batteries, J. Electrochem. Soc., 2021, 168, 060529 CrossRef CAS.
- A. Ali, T. Aziz, J. Zheng, F. Hong, M. F. Awad, S. Manan, F. Haq, A. Ullah, M. N. Shah, Q. Javed, A. A. Kubar and L. Guo, Modification of Cellulose Nanocrystals with 2-Carboxyethyl Acrylate in the Presence of Epoxy Resin for Enhancing Its Adhesive Properties, Front. Bioeng. Biotechnol., 2022, 9, 797672 CrossRef.
- R. Leino, F. J. Gómez, A. P. Cole and R. M. Waymouth, Syndiospecific Propylene Polymerization with C1 Symmetric Group 4 ansa-Metallocene Catalysts, Macromolecules, 2001, 34(7), 2072–2082 CrossRef CAS.
- J. Huang and G. L. Rempel, Kinetic study of propylene polymerization using Et(H4Ind)2ZrCl2/methylalumoxane catalysts, Ind. Eng. Chem. Res., 1997, 36(4), 1151–1157 CrossRef CAS.
- A. Vittoria, V. Busico, F. D. Cannavacciuolo and R. Cipullo, Molecular kinetic study of “chain shuttling” olefin copolymerization, ACS Catal., 2018, 8(6), 5051–5061 CrossRef CAS.
- A. Ali, N. Muhammad, S. Hussain, M. I. Jamil, A. Uddin, T. Aziz, M. K. Tufail, Y. Guo, T. Wei and G. Rasool, Kinetic and thermal study of ethylene and propylene homo polymerization catalyzed by ansa-zirconocene activated with alkylaluminum/borate: effects of alkylaluminum on polymerization kinetics and polymer structure, Polymers, 2021, 13(2), 268 CrossRef CAS.
- F. Song, R. D. Cannon and M. Bochmann, Zirconocene-catalyzed propene polymerization: a quenched-flow kinetic study, J. Am. Chem. Soc., 2003, 125(25), 7641–7653 CrossRef CAS PubMed.
- A. Ali, M. A. Akram, Y. Guo, H. Wu, W. Liu, A. Khan, X. Liu, Z. Fu and Z. Fan, Ethylene–propylene copolymerization and their terpolymerization with dienes using ansa-zirconocene catalysts activated by borate/alkylaluminum, J. Macromol. Sci., Part A: Pure Appl.Chem., 2020, 57(2), 156–164 CrossRef CAS.
- A. Ali, M. K. Tufail, M. I. Jamil, W. Yaseen, N. Iqbal, M. Hussain, A. Ali, T. Aziz, Z. Fan and L. Guo, Comparative Analysis of Ethylene/Diene Copolymerization and Ethylene/Propylene/Diene Terpolymerization Using Ansa-Zirconocene Catalyst with Alkylaluminum/Borate Activator: The Effect of Conjugated and Nonconjugated Dienes on Catalytic Behavior and Polymer Microstructure, Molecules, 2021, 26(7), 2037 CrossRef CAS PubMed.
- C. R. Landis and M. D. Christianson, Metallocene-catalyzed alkene polymerization and the observation of Zr-allyls, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(42), 15349–15354 CrossRef CAS PubMed.
- Z. Liu, E. Somsook and C. R. Landis, A 2H-labeling scheme for active-site counts in metallocene-catalyzed alkene polymerization, J. Am. Chem. Soc., 2001, 123(12), 2915–2916 CrossRef CAS PubMed.
- C. R. Landis, D. R. Sillars and J. M. Batterton, Reactivity of secondary metallocene alkyls and the question of dormant sites in catalytic alkene polymerization, J. Am. Chem. Soc., 2004, 126(29), 8890–8891 CrossRef CAS PubMed.
- E. S. Cueny, H. C. Johnson and C. R. Landis, Selective quench-labeling of the hafnium-pyridyl amido-catalyzed polymerization of 1-octene in the presence of trialkyl-aluminum chain-transfer reagents, ACS Catal., 2018, 8(12), 11605–11614 CrossRef CAS.
- H. C. Johnson, E. S. Cueny and C. R. Landis, Chain transfer with dialkyl zinc during hafnium–pyridyl amido-catalyzed polymerization of 1-octene: relative rates, reversibility, and kinetic models, ACS Catal., 2018, 8(5), 4178–4188 CrossRef CAS.
- M. F. Ryan, A. R. Siedle, M. J. Burk and D. E. Richardson, Parameter scale for substituent effects in cyclopentadienyl complexes based on gas-phase electron-transfer equilibrium studies of ruthenocene derivatives, Organometallics, 1992, 11(12), 4231–4237 CrossRef CAS.
- A. Siedle, W. Lamanna, R. Newmark, J. Stevens, D. Richardson and M. Ryan, The role of non-coordinating anions in homogeneous olefin polymerization, in Makromolekulare Chemie Macromolecular Symposia, Wiley Online Library, 1993, pp. 215–224 Search PubMed.
- B. M. Moscato, B. Zhu and C. R. Landis, Mechanistic investigations into the behavior of a labeled zirconocene polymerization catalyst, Organometallics, 2012, 31(5), 2097–2107 CrossRef CAS.
- E. S. Cueny and C. R. Landis, The hafnium-pyridyl amido-catalyzed copolymerization of ethene and 1-octene: how small amounts of ethene impact catalysis, ACS Catal., 2019, 9(4), 3338–3348 CrossRef CAS.
- A. Uddin, R. Khatoon, D. Estevez, M. Salem, A. Ali, S. Attique, J. Lu and F. Qin, Waste Paper Cellulose Based-Mos2 Hybrid Polymer Composites: Towards Sustainable Green Shielding, Available at SSRN 4044245.
- A. Naveed, A. Ali, T. Rasheed, X. Wang, P. Ye, X. Li, Y. Zhou, S. Mingru and Y. Liu, Revisiting recent and traditional strategies for surface protection of Zn metal anode, J. Power Sources, 2022, 525, 231122 CrossRef CAS.
- E. S. Cueny, L. R. Sita and C. R. Landis, Quantitative validation of the living coordinative chain-transfer polymerization of 1-hexene using chromophore quench labeling, Macromolecules, 2020, 53(14), 5816–5825 CrossRef CAS.
- J. C. Chien and W. M. Tsai, Zirconocenium cation catalysis of propene polymerization, in Makromolekulare Chemie Macromolecular Symposia, Hüthig & Wepf Verlag, Basel, 1993, vol. 66, pp. 141–156 Search PubMed.
- W. Yaseen, M. Xie, B. A. Yusuf, Y. Xu, N. Ullah, M. Rafiq, A. Ali and J. Xie, Synergistically coupling of Co/Mo2C/Co6Mo6C2@C electrocatalyst for overall water splitting: the role of carbon precursors in structural engineering and catalytic activity, Appl. Surf. Sci., 2022, 579, 152148 CrossRef CAS.
- A. Ali, M. I. Jamil, A. Uddin, M. Hussain, T. Aziz, M. K. Tufail, Y. Guo, B. Jiang, Z. Fan and L. Guo, Kinetic and thermal study of ethylene-propylene copolymerization catalyzed by ansa-zirconocene activated with alkylaluminium/borate: effects of linear and branched alkylaluminium compounds as cocatalyst, J. Polym. Res., 2021, 28(5), 1–15 Search PubMed.
- Y. Guo, Z. Zhang, W. Guo, A. Khan, Z. Fu, J. Xu and Z. Fan, Kinetics and mechanism of metallocene-catalyzed olefin polymerization: comparison of ethylene, propylene homopolymerizations, and their copolymerization, J. Polym. Sci., Part A: Polym. Chem., 2017, 55(5), 867–875 CrossRef CAS.
- A. Khan, Y. Guo, Z. Zhang, A. Ali, Z. Fu and Z. Fan, Kinetics of short-duration ethylene–propylene copolymerization with MgCl2-supported Ziegler–Natta catalyst: differentiation of active centers on the external and internal surfaces of the catalyst particles, J. Appl. Polym. Sci., 2018, 135(12), 46030 CrossRef.
- M. A. Akram, X. Liu, B. Jiang, B. Zhang, A. Ali, Z. Fu and Z. Fan, Effect of alkylaluminum cocatalyst on ethylene/1-hexene copolymerization and active center distribution of MgCl2-supported Ziegler-Natta catalyst, J. Macromol. Sci., Part A: Pure Appl.Chem., 2021, 1–11 CAS.
- A. Ali, M. Nadeem, J. Lu, J. M. Moradian, T. Rasheed, T. Aziz, C. Maouche, Y. Guo, M. Awais and F. Zhiqiang, Rapid kinetic evaluation of homogeneous single-site metallocene catalysts and cyclic diene: how do the catalytic activity, molecular weight, and diene incorporation rate of olefins affect each other?, RSC Adv., 2021, 11(50), 31817–31826 RSC.
- B. Jiang, B. Zhang, Y. Guo, A. Ali, W. Guo, Z. Fu and Z. Fan, Effects of titanium dispersion state on distribution and reactivity of active centers in propylene polymerization with MgCl2-supported Ziegler-Natta catalysts: a kinetic study based on active center counting, ChemCatChem, 2020, 12(20), 5140–5148 CrossRef CAS.
- R. Fang, J. Pi, T. Wei, A. Ali and L. Guo, Stimulus-Responsive Polymers Based on Polypeptoid Skeletons, Polymers, 2021, 13(13), 2089 CrossRef CAS PubMed.
- B. Jiang, B. Zhang, Y. Guo, A. Ali, W. Guo, Z. Fu and Z. Fan, Effects of titanium dispersion state on distribution and reactivity of active centers in propylene polymerization with MgCl2-supported Ziegler-Natta catalysts: a kinetic study based on active center counting, ChemCatChem, 2020, 12(20), 5140–5148 CrossRef CAS.
- S. I. Chowdhury, R. Tanaka, Y. Nakayama and T. Shiono, Copolymerization of norbornene and conjugated dienes using anilinonaphthoquinone-ligated nickel complexes, Polymer, 2020, 187, 122094 CrossRef CAS.
- S.-S. Choi and Y.-K. Kim, Analysis of 5-ethylidene-2-norbornene in ethylene-propylene-diene terpolymer using pyrolysis-GC/MS, Polym. Test., 2011, 30(5), 509–514 CrossRef CAS.
- S. Ahmadjo, H. Arabi, M. Nekoomanesh, G. H. Zohuri, M. M. Mortazavi and G. Naderi, Terpolymerization of Ethylene/Propylene/Diene Monomers Using (2-PhInd)2ZrCl2 Metallocene Catalysts, Macromol. React. Eng., 2010, 4(11–12), 707–714 CrossRef CAS.
- M. I. Jamil, Q. Wang, A. Ali, M. Hussain, T. Aziz, X. Zhan and Q. Zhang, Slippery Photothermal Trap for Outstanding Deicing Surfaces, Journal of Bionic Engineering, 2021, 18(3), 548–558 CrossRef.
- G. Rasool, A. Shafiq, Y.-M. Chu, M. S. Bhutta and A. Ali, Optimal Homotopic Exploration of Features of Cattaneo-Christov Model in Second Grade Nanofluid Flow via Darcy-Forchheimer Medium Subject to Viscous Dissipation and Thermal Radiation, Comb. Chem. High Throughput Screening, 2021, 34477515 Search PubMed.
- B. Jongsomjit, S. Phoowakeereewiwat, S. Thongyai, T. Shiono and P. Praserthdam, Impact of diene addition on properties for ethylene–propylene copolymerization with rac-Et[Ind]2ZrCl2/MAO catalyst, Mater. Lett., 2005, 59(28), 3771–3774 CrossRef CAS.
- A. Ali, A. Uddin, M. I. Jamil, X. Shen, M. Abbas, T. Aziz, M. Hussain, S. Hussain, R. Fang and Z. Fan, Kinetics and mechanistic investigations of ethylene-propylene copolymerizations catalyzed with symmetrical metallocene and activated by TIBA/borate, J. Organomet. Chem., 2021, 121929 CrossRef CAS.
- S. Marathe and S. Sivaram, Regioselective copolymerization of 5-vinyl-2-norbornene with ethylene using zirconocene-methylaluminoxane catalysts: a facile route to functional polyolefins, Macromolecules, 1994, 27(5), 1083–1086 CrossRef CAS.
- J. C. Randall, A review of high resolution liquid 13carbon nuclear magnetic resonance characterizations of ethylene-based polymers, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1989, 29(2–3), 201–317 CrossRef.
- A. Ali, M. Nadeem, J. Lu, J. M. Moradian, T. Rasheed, T. Aziz, C. Maouche, Y. Guo, M. Awais and F. Zhiqiang, Correction: rapid kinetic evaluation of homogeneous single-site metallocene catalysts and cyclic diene: how do the catalytic activity, molecular weight, and diene incorporation rate of olefins affect each other?, RSC Adv., 2021, 11(53), 33301 RSC.
- A. Ali, X. Liu, Y. Guo, M. A. Akram, H. Wu, W. Liu, A. Khan, B. Jiang, Z. Fu and Z. Fan, Kinetics and mechanism of ethylene and propylene polymerizations catalyzed with ansa-zirconocene activated by borate/TIBA, J. Organomet. Chem., 2020, 922, 121366 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01264b |
| This journal is © The Royal Society of Chemistry 2022 |