
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactions of [60]fullerene with alkynes promoted by OH−†
Wei-Wei Chang ab,
Fa-Hui He*bc,
Alberto García-Peñasd,
Mehdihasan I. Shekhe and
Zong-Jun Lib
ab,
Fa-Hui He*bc,
Alberto García-Peñasd,
Mehdihasan I. Shekhe and
Zong-Jun Lib
aAnalysis & Testing Center, Shandong University of Technology, Zibo 255049, China
bState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Changchun 130022, China
cState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: hefg@dicp.ac.cn
dDepartment of Materials Science and Engineering and Chemical Engineering, IAAB, University Carlos III of Madrid, Madrid 28911, Spain
eNew Energy Materials Laboratory, College of Materials Science, Shenzhen University, Shenzhen 518055, China
First published on 10th May 2022
Abstract
In the current work, the reactions of [60]fullerene with alkynes promoted by OH− (base) are addressed. The treatment of C60 with alkynes in the presence of TBAOH produces alkynylation products (R-C60–H) with high selectivity in o-DCB at 100 °C. Plausible reaction mechanisms were proposed. This work provides a convenient and environmental friendly method for the functionalization of fullerenes.
Fullerenes functionalized with alkynyl groups are particularly valuable in materials science and organic chemistry due to their excellent electrochemical properties, among others. The combination of fullerene and acetylene units is not only a potential precursor for developing novel fullerene-based dimers or polymers,1,2 but also can promote the conjunction pathway with a supramolecular framework providing new properties. For example, it has been reported that fullerene covalently linked to acetylenic polythiophene produces unique photoelectronic properties.3,4 On the other hand, alkynes are considered as one of the most outstanding building blocks for unsaturated molecular scaffolds in organic synthesis, owing to their transformation potential into various functional groups.5–8
Up to now, the easiest method to achieve acetylene deriva-tives of C60 is based on reactions of fullerene with alkynyllithium. Komatsu et al. reported the synthesis, characterization and properties of the first acetylene derivative of C60 containing a trimethylsilyl ethynyl group or a phenylethynyl group.9,10 Diedrish et al. prepared a series of alkynylfullerenes using the same method.1,11 However, the use of organolithium reagents led to complex experimental conditions, due to them being extremely sensitive to air and moisture.12 Hence, new methods for the preparation of alkynylfullerene compounds are necessary, reducing the steps involved in the synthesis, and responding to new regulations associated with sustainability. In this sense, these new procedures would not only be efficient, selective, high-yielding, but also easy to operate and environmentally friendly.
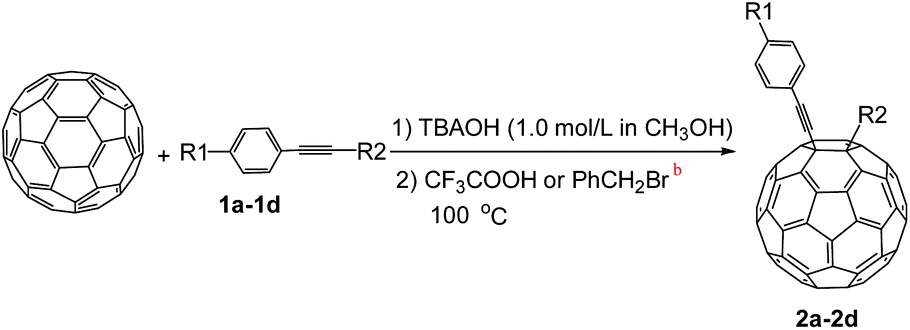
In this work, our group deeply worked on the new functionalized chemistry of fullerenes with alkynes and found that the nucleophilic carbon functionalization of C60 with alkynes could be mediated by OH−. In this sense, TBAOH (tetra-n-butyl-ammoniumhydroxide) was used as a source of OH−, because it is cheap, environment friendly, and allows getting a high efficiency in the transformations. This new process allows getting highly functionalized alkynylfullerene derivatives through a one-step reaction. Furthermore, the protocol is simple, and works with a wide range of alkynes. Interestingly, different types of alkynes undergo diverse reaction pathways.
A series of phenylacetylene compounds were chosen as model substrates to optimize the reaction. The experimental procedure for the new nucleophilic reaction is very simple: a mixture composed by C60 (36 mg) and a series of alkynes (20 equiv.) was stirred in o-DCB (o-dichlorobenzene) with a solution 1.0 mol L−1 of TBAOH/CH3OH (150 μL, 3 equiv.), as the OH− source, under argon gas for 1 h at 100 °C. The colour of the reaction gradually changed from purple to dark green after the addition of TBAOH/CH3OH. It was observed that the use of an excess TBAOH or longer reaction time did not improve the yield because of the formation of polar side products, like multiple addition compounds.13 Subsequently, a trifluoroacetic acid treatment, produced the different alkynylfullerenes 2a–2d with 1,2-addition and the yield is 35.2%, 29%, 21.1%, 31.5%, respectively (listed in Table 1) after separation by Buckyprep column. The trifluoroacetic acid is working as the donor of protons. The 1,4-substituted phenylacetylene with both electron-donating and electron-withdrawing groups on the phenyl could be used in the reaction as it can be observed in Table 1.
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | R1 | R2 | 2a–2db | Yield (%)c |
a All reactions were performed in o-DCB using an oil bath at 100 °C. Molar ratio of compound 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C60 = 20:1.b CF3COOH was used in the reaction of C60 with 1a–1c. While PhCH2Br was used in the case of C60 with 1d.c Isolated yield. :C60 = 20:1.b CF3COOH was used in the reaction of C60 with 1a–1c. While PhCH2Br was used in the case of C60 with 1d.c Isolated yield. |
|||||
| 1 | 1a | H | H | 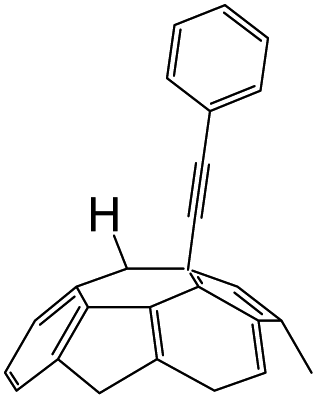 |
35.2 |
| 2 | 1b | OMe | H | 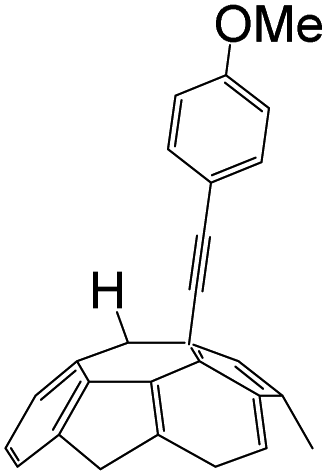 |
29.0 |
| 3 | 1c | CCH | H | 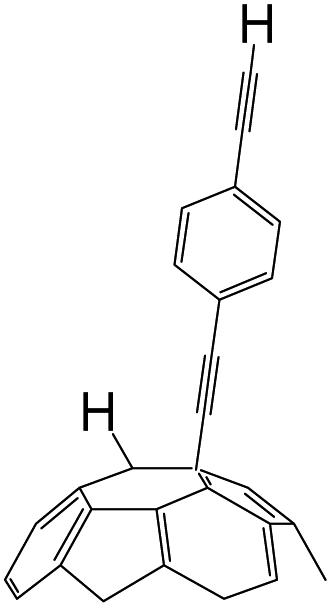 |
21.1 |
| 4 | 1d | H | CO2Et | 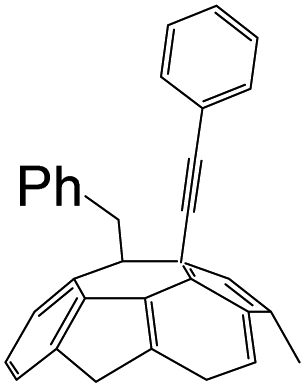 |
31.5 |
The structures of 1,2-addition C60 compounds 2a–2d were fully characterized and elucidated by their HRMS, 1H NMR, 13C NMR, and UV-vis spectra. The products 2a–2c exhibited proper molecular weights in their mass spectra. The 13C NMR spectra for 2a–2c clearly exhibited no more than 29 peaks in the range of 134–152 ppm, associated with the sp2-carbons of the fullerene cage, which is consistent with the Cs symmetry of their molecule structure, and the peak placed at 54–55 ppm for the sp3-carbon of fullerene and alkyne. The UV-vis spectra for all products showed a peak at 434 nm, which is the characteristic peak of the 1,2-adducts of C60.
The previous analysis allows proposing a reaction mechanism for the formation of 2a–2d as is shown in Scheme l. First, the reaction of terminal alkynes 1 with TBAOH/CH3OH generates an intermediate compound 3, which suffers a nucleophilic addition to produce 4 (RC60−). The protonation of the intermediate species 4 leads to the formation of the product 2.
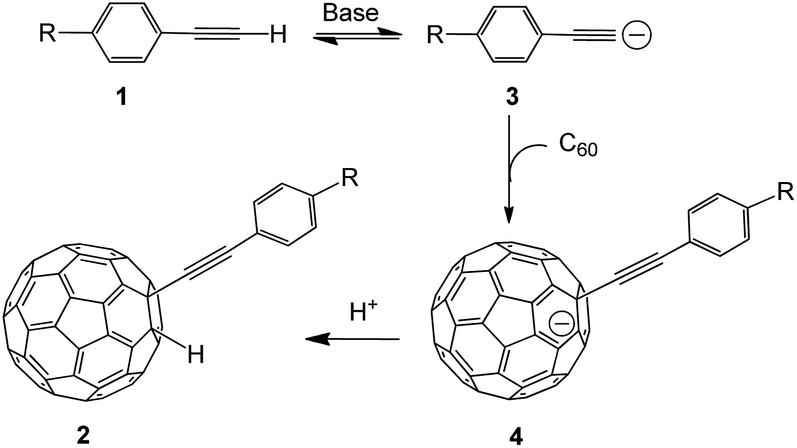 | ||
| Scheme 1 Proposed mechanism for the formation of 2a–2c. | ||
The reactivity of other types of alkynes in the same conditions was also studied. The derivatives 2a (in general conditions), and 2d (when CF3COOH changed to PhCH2Br) were curiously obtained when the terminal alkynes were transformed into ethylphenylpropiolate. The structures composed by two different C60-with alkynes were studied and determined through their HRMS, 1H NMR, 13C NMR, and UV-vis spectra. The results indicated that the cleavage of ethylphenylpropiolate occurred at the C–C bond, specifically, between phenylpropynyl and ethoxycarbonyl. This fact is confirmed by the crystal structure of 2d (Fig. 1).
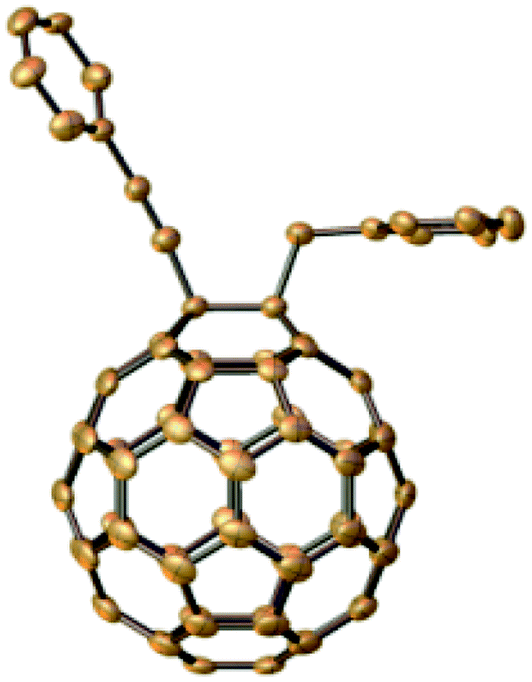 | ||
| Fig. 1 Single crystal structure of 2d with 30% ellipsoid probability (hydrogens are omitted). The CCDC number of the crystal structure is: 2082000. | ||
A plausible mechanism for the formation of 2a and 2d compounds is deduced in the Scheme 2. The substrate 5 undergoes hydrolysis (under base conditions) to produce a carboxylic anion, which decomposes to CO2. Because the conjugated phenylethynyl group can disperse the negative charge, it makes it possible to form the intermediate 6. The α,β-unsaturated carboxylic acid 6 undergo decarboxylation14 and generates a carbanion 3 in base condition to attack C60. The protonation or nucleophilic addition of benzyl bromide produces 2a and 2d, respectively.
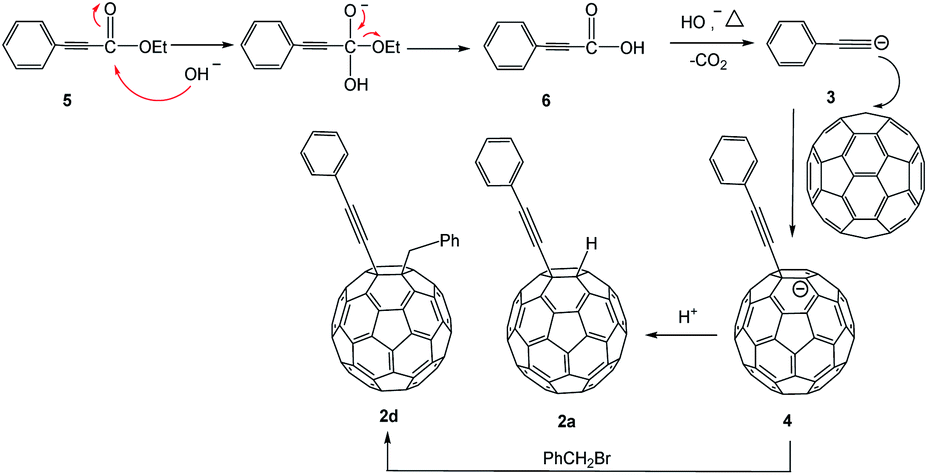 | ||
| Scheme 2 Proposed mechanism for the formation of 2d. | ||
The reactivity of the ester group was deeply studied using ethyl propiolate as substrate, which has a weak acid proton and an electron-withdrawing ester group in its structure. It is interesting to observe that the reactions of C60 with 7 using TBAOH/CH3OH (1.0 mol L−1) and TBAOH/isopropanol (1.0 mol L−1) led to different products as the unprecedented product 7a and 7b with an isolated yield of 35.2% and 11.8% respectively. In addition, three CH3O− groups were incorporated in the adduct 7a, apparently, methanol participates in the reaction and two bonds of alkyne are broken. Nevertheless, the alkynylfullerene derivative 7c with an isolated yield of 27.5% was obtained at room temperature (Fig. 2). The reaction between C60 with 7 and TBAOH/CH3OH (1.0 mol L−1), suggests that methanol participates in the reaction. Obviously, the reactions follow different routes in the presence of methanol and isopropanol.
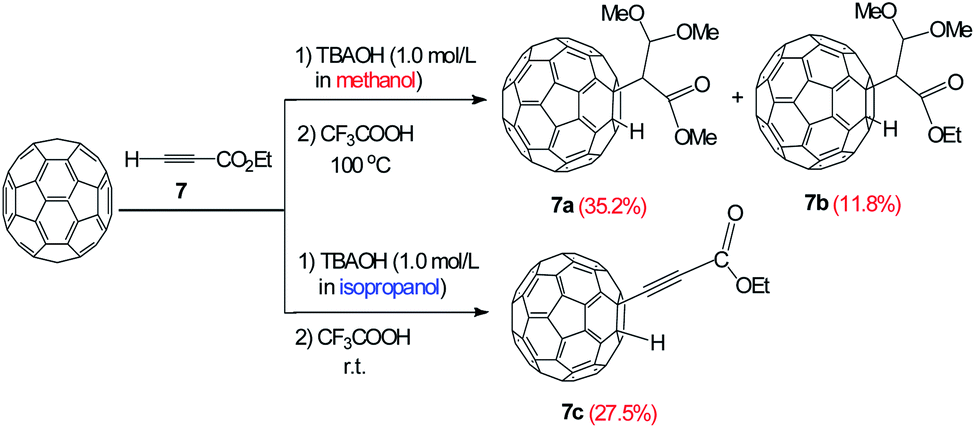 | ||
| Fig. 2 Reaction of C60 and 7 in the presence of OH−. | ||
The structure of derivatives 7a–7c were estimated through HRMS, 1H NMR, 13C NMR, HMBC, HSQC and UV-vis spectra. HRMS (+ESI) of 7a and HRMS (-ESI) of 7c show the protonated molecular ions ([M + H]+) and ([M–H]−), which are placed at 869.6473 and 817.0260, respectively (Fig. S23 and S29 in ESI†). These are in good agreement with the assigned structures (7a: C66H13O4+, calcd. 869.0814; 7c: C65H5O2−, calcd. 817.0290). The 1,2-addition pattern of 7a and 7c was further confirmed through UV-vis spectra which shown a characteristic sharp peak placed at 435 nm. Fig. 3 displays the 1H NMR spectrum of 7a, where the methoxy protons are characterized by the three singlets situated at 3.48, 3.51, and 3.65 ppm, respectively.13,15 The C60–H proton was observed at 6.97 ppm. The ratio defined by peak area of the methoxy protons and the C60–H proton is 3:1, consist with the addends added to C60 is methoxy groups, and indicated that the methanol from TBAOH solution took part in the reaction. Two methoxy protons (3.51 and 3.65 ppm) show 3JCH correlations with the carbon of the methenyl group respectively (Fig. S26 in ESI†), which indicates that these two methoxyl groups were linked to the same carbon of methenyl. HSQC NMR (Fig. S27 in ESI†) confirmed the structure of 7a. It should be noted that there is another peak with a retention time at 6.8 min associated with a HPLC trace of the reaction of C60 with TBAOH/CH3OH (1.0 mol L−1) (Fig. S5 in ESI†). Data extracted from HRMS, 1H NMR, 13C NMR, HMBC, HSQC and UV-vis spectra, lead us to inferred that the peak placed before 7a belongs to product 7b (Fig. S29–S33 in ESI†).
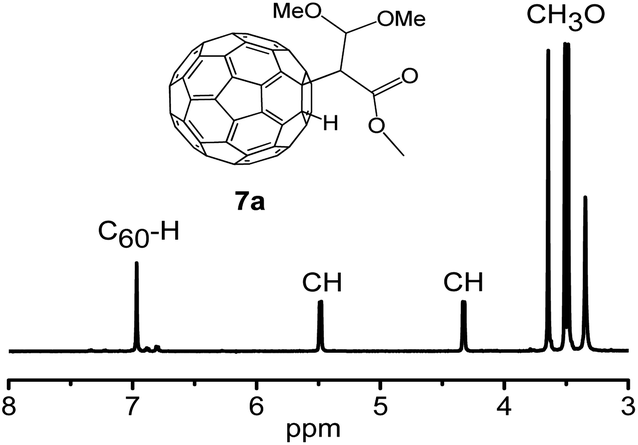 | ||
| Fig. 3 Expanded 1H NMR spectrum of 7a recorded using CS2/(DMSO-d6 as the external lock) measured in a 600 MHz instrument. | ||
A reaction mechanism is proposed and shown in the Scheme 3. The key is that the alcoholic solution (in the TBAOH) could affect the structure of the final products. Ethyl propiolate 7 is an α,β-unsaturated carbonyl compound in the reaction. Methanol attacks to β-carbon (basic conditions), and produces the intermediate 8. To verify the alcohol exchange reaction, a control experiment between 7 and TBAOH/CH3OH (1.0 mol L−1, 150 μL, 3 equiv.) in o-DCB was carried out for 1 h at 100 °C. As expected, 1H NMR spectra of the reaction shows peaks at 4.26 ppm (q, 2H) and 1.32 ppm (t, 3H), which are associated with methylene and methyl protons of compound 7, respectively. In addition, the peak around 3.80 ppm is associated with methyl proton of methyl propiolate (Fig. S42 in ESI†), and it reveals that the crude contains both ethyl propiolate 7 and methyl propiolate, confirming the alcohol exchange reaction. Therefore, 7a and 7b products are both detected. However, similar reactions do not take place in the case of phenylacetylene and ethylphenylpropiolate are used as substrates, probably due to the steric hindrance of the phenyl ring in the molecule. The produced intermediate 8 generates 9 via Michael addition.15,16 A part of 9 produces 11 through alcohol exchange reaction. The Michael addition, and subsequent protonation, would afford the generation of 7a and 7b. However, the Michael addition reaction was restricted when methanol was changed to isopropanol. This fact was ascribed to two reasons: (1) the steric effect of isopropanol; (2) the lower reactivity of isopropanol.17–19 Thus, a deprotonation reaction of 7 happens when isopropanol is present. The generated intermediate 13 undergoes through nucleophilic addition and protonation to produce 7c.
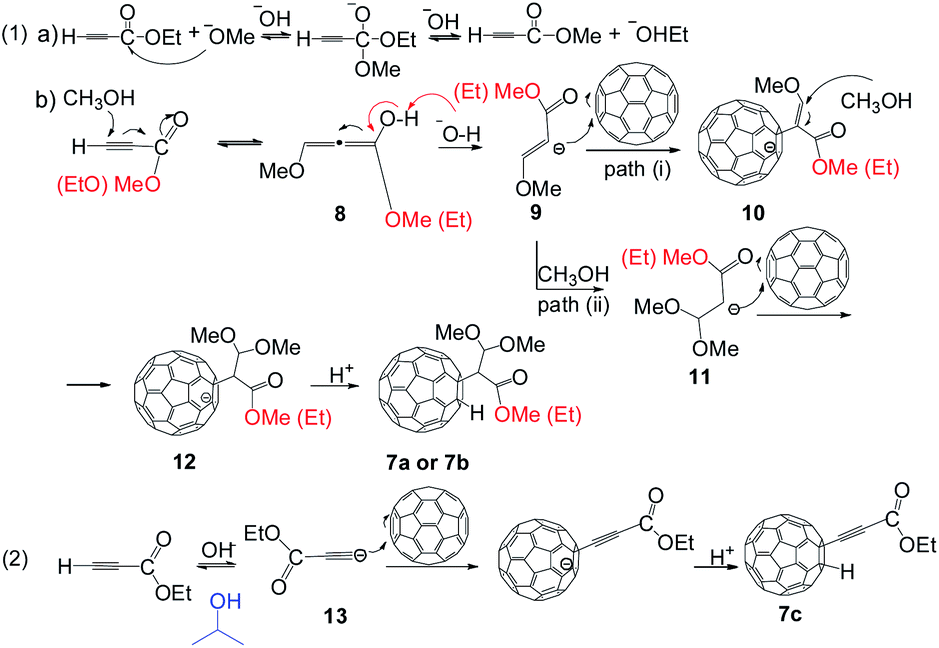 | ||
| Scheme 3 Proposed mechanism for the formation of 7a–7c. | ||
Conclusions
In summary, we have found that the addition of alkynes to C60 can now be achieved without the use of heavy metals and alkynyllithium, under simple reactiond which were mediated by OH−. In the reaction of C60 with ethyl propiolate, the use of alcoholic solution in the TBAOH was found to be crucial. The reaction performed under methanol afforded a Michael-addition product, but Michael-addition was restrained while the reaction under isopropanol. Through the nucleophilic addition, we have introduced alkynes, a versatile functional group to C60 under mild conditions, and the monoadducts may also prove to be useful for other applications.Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province in China (BS2015CL011 for WWC) and the National Natural Science Foundation of China (21202157 for ZJL). The authors gratefully acknowledge the financial support of the China Scholarship Council (CSC), PRC, and German Academic Exchange Service (DAAD), Germany. We thank Dr Wang Shentang (School of Chemistry and Chemical Engineering, and Chongqing Key Laboratory of Soft-Matter Material Chemistry and Function Manufacturing, Southwest University, Chongqing 400715, PR China) for help with the refinement of crystal structure.Notes and references
- P. Timmerman, H. L. Anderson, R. Faust, J. Nierengarten, T. Habicher, P. Seiler and F. Diederich, Tetrahedron, 1996, 52, 4925–4947 CrossRef CAS.
- F. Giacalone and N. Martín, ChemInform, 2006, 106, 5136–5910 CAS.
- T. Yamazaki, Y. Murata, K. Komatsu, K. Furukawa, M. Morita, N. Maruyama, T. Yamao and S. Fujita, Org. Lett., 2004, 6, 4865–4868 CrossRef CAS PubMed.
- N. Bucci and T. J. J. Müller, Tetrahedron Lett., 2006, 47, 8329–8332 CrossRef CAS.
- L. Huang, Q. Wang, X. Liu and H. Jiang, Angew. Chem., Int. Ed., 2012, 51, 5696–5700 CrossRef CAS PubMed.
- N. Okamoto, T. Sueda, H. Minami, Y. Miwa and R. Yanada, Org. Lett., 2015, 17, 1336–1339 CrossRef CAS PubMed.
- B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937–2980 CrossRef CAS PubMed.
- R. Chinchilla and C. Nájera, Chem. Rev., 2013, 114, 1783–1826 CrossRef PubMed.
- Y. Murata, K. Motoyama, K. Komatsu and T. S. M. Wan, Tetrahedron, 1996, 52, 5077–5090 CrossRef CAS.
- K. Komatsu, N. Takimoto, Y. Murata, T. S. M. Wan and T. Wong, Tetrahedron Lett., 1996, 37, 6153–6156 CrossRef CAS.
- Y.-Z. An, Y. Rubin, C. Schaller and S. W. McElvany, J. Org. Chem., 1994, 59, 2927–2929 CrossRef CAS.
- H. J. Reich, Chem. Rev., 2013, 113, 7130–7178 CrossRef CAS PubMed.
- W.-W. Chang, Z.-J. Li, W.-W. Yang and X. Gao, Org. Lett., 2012, 14, 2386–2389 CrossRef CAS PubMed.
- R. A. Fursule, P. O. Patil, B. D. Shewale, S. B. Kosalge, P. K. Deshmukh and D. A. Patil, Chem. Pharm. Bull., 2009, 11, 1243–1245 CrossRef PubMed.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- V. L. Heasley, D. F. Shellhamer, A. E. Chappell, J. M. Cox, D. J. Hill, S. L. McGovern, C. C. Eden and C. L. Kissel, J. Org. Chem., 1998, 63, 4433–4437 CrossRef CAS.
- K. Chandrasekaran and J. K. Thomas, Chem. Phys. Lett., 1983, 97, 357–360 CrossRef CAS.
- T. Yamanaka, M. Kawasaki, M. D. Hurley, T. J. Wallington, W. F. Schneider and J. Bruce, Phys. Chem. Chem. Phys., 2007, 9, 4211–4217 RSC.
- B. Rajakumar, D. C. McCabe, R. K. Talukdar and A. R. Ravishankara, Int. J. Chem. Kinet., 2010, 42, 10–24 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2082000. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra01300b |
| This journal is © The Royal Society of Chemistry 2022 |