
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrasound-assisted transition-metal-free catalysis: a sustainable route towards the synthesis of bioactive heterocycles
Biplob Borah
 and
L. Raju Chowhan
*
and
L. Raju Chowhan
*
School of Applied Material Sciences, Centre for Applied Chemistry, Central University of Gujarat, Gandhinagar-382030, India. E-mail: rchowhan@cug.ac.in
First published on 11th May 2022
Abstract
Heterocycles of synthetic and natural origin are a well-established class of compounds representing a broad range of organic molecules that constitute over 60% of drugs and agrochemicals in the market or research pipeline. Considering the vast abundance of these structural motifs, the development of chemical processes providing easy access to novel complex target molecules by introducing environmentally benign conditions with the main focus on improving the cost-effectiveness of the chemical transformation is highly demanding and challenging. Accordingly, sonochemistry appears to be an excellent alternative and a highly feasible environmentally benign energy input that has recently received considerable and steadily increasing interest in organic synthesis. However, the involvement of transition-metal-catalyst(s) in a chemical process often triggers an unintended impact on the greenness or sustainability of the transformation. Consequently, enormous efforts have been devoted to developing metal-free routes for assembling various heterocycles of medicinal interest, particularly under ultrasound irradiation. The present review article aims to demonstrate a brief overview of the current progress accomplished in the ultrasound-assisted synthesis of pharmaceutically relevant diverse heterocycles using transition-metal-free catalysis.
 Biplob Borah | Biplob Borah was born in 1995 in Garukhunda, a small village in the Nagaon district of Assam, India. He had graduated with a BSc in Chemistry from Nowgong College (Gauhati University), Assam, in 2017 and received his Master's degree in Industrial Chemistry from the Central University of Gujarat, India, in 2019. Then, he joined as a PhD scholar in the School of Applied Material Sciences at the Central University of Gujarat under the guidance of Dr L. Raju Chowhan. His research interest includes organocatalysis, asymmetric synthesis, mechanochemistry, multicomponent reactions (MCRs), green chemistry, synthesis of medicinally privileged heterocycles, and hybrid molecules in the aqueous medium. |
 L. Raju Chowhan | Dr L. Raju Chowhan obtained his BSc degree from Osmania University, Hyderabad, in 2005, and a Master's degree in Chemical Science from Hyderabad Central University, Hyderabad, in 2007. He received his PhD from CSIR-Indian Institute of Chemical Technology, in association with Hyderabad Central University. He joined as an Assistant Professor in the Centre for Applied Chemistry, Central University of Gujarat, Gandhinagar, on 5th September, 2012. His research interests include stereoselective synthesis of natural products, development of novel methodologies for asymmetric synthesis, multicomponent reactions, and 1,3-dipolar cycloaddition reactions. |
1. Introduction
Over 60% of the organic compounds produced by various chemical industries in the form of fine chemicals, drugs, pharmaceutical targets, and agrochemicals consist of heterocycles as key active ingredients.1 These structural motifs are not only encountered in the architecture of numerous drug candidates2 but are also well distributed as precursors of numerous natural products3,4 and optoelectronic materials.5 Indeed, heterocyclic compounds hold a pivotal position in the area of medicinal chemistry on account of their remarkable therapeutic potential, including anticancer, antimicrobial, antidiabetic, antioxidant, antituberculosis, antimalarial, anti-HIV, anti-hyperglycemic, and anti-dyslipidemic activity.6–13 Some representative examples of naturally occurring molecules and synthetic heterocycles with potential pharmacological applications are presented in Fig. 1.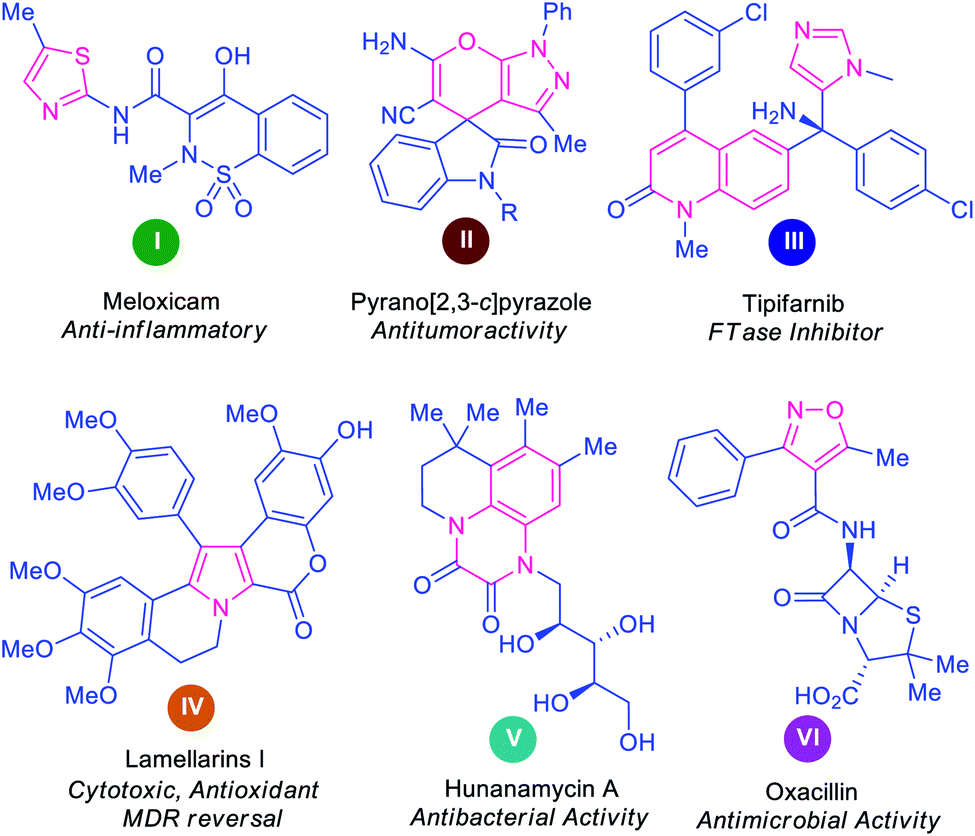 | ||
| Fig. 1 Some examples of bioactive natural and synthetic heterocyclic compounds. | ||
Notwithstanding the tremendous pharmaceutical application, a diversified structural scaffold comprising a heterocyclic core has been demonstrated to be a crucial element in multiple materials science sectors. For instance, organic light-emitting diodes (OLEDs),14 organic optoelectronic devices,15 organic solar cells,16 organic semiconductors,17 potential fluorescent materials,18 electroluminescent components,19 and dye-sensitized solar cells are some of the promising applications of heterocyclic compounds in materials science, among others.20
Considering the wide abundance of heterocyclic compounds and their well-established potentiality in synthetic, organic and medicinal chemistry and materials science, the development of efficient synthetic routes providing easy access to these structural features has remained a continuous challenge for chemists in academia and industry. However, with the ever-increasing public awareness about protecting the living environment from chemical pollution associated with the chemical process, the fields of synthetic, organic and medicinal chemistry have witnessed tremendous growth in the last two decades towards making the chemical process environmentally benign and more sustainable. In this pursuit, the use of ultrasound irradiation as a highly feasible and alternative eco-friendly activation method in organic synthesis has increased substantially owing to its capability to reduce reaction times and enhance product yields by avoiding higher energy requirements.21,22 The ability to accomplish or promote chemical reactions using sound waves triggered by ultrasound can make the sonochemical-assisted synthetic method superior to the conventional method in terms of green and sustainable chemistry.23–30 Ultrasound irradiation produced substantial cavitation bubbles upon exposure to a reaction mixture after the pressure reached a certain threshold. These cavitation bubbles underwent violent collapse after growing up rapidly to facilitate the formation of a fine emulsion between the starting materials and elevate the local temperature of the reaction mixture to cross the reaction's activation energy.31–34
On the other hand, catalysts hold a significant position in organic chemistry for accelerating a diverse range of chemical transformations. Mainly, transition-metal-catalysts have found tremendous application in organic synthesis over the past decades for their successful utilization in the assembly of natural product analogs and therapeutically promising compounds featuring heterocycles as the critical template. However, their exploitation in organic synthesis sometimes somewhat affects the eco- and environmentally benign nature of the chemical transformation. Not all but sometimes transition-metal-catalyzed processes are found to be very sensitive to air and moisture, and a high cost is required for the preparation of the catalyst(s). In addition, non-commercial supporting ligands, co-catalysts, and sometimes several additives are necessary to achieve transition-metal-catalyzed chemical transformation. Furthermore, the elimination of trace amounts of metal catalyst from the desired product upon completion of the reaction, which is crucial in the pharmaceutical industry, is costly and frequently becomes a daunting issue. These factors associated with transition-metal-catalyzed transformations indicate the failure of an environmentally benign and sustainable synthesis.35–37 As a result, the development of efficient chemical procedures for the synthesis of the molecular structure providing high atom- and step-economy with the main focus to minimize or avoid the use of harmful metal catalysts, co-catalysts, and any additives is highly needed to ensure the sustainability of our environment.
In line with this, transition-metal-free catalysis has recently proven to be an extremely efficient and ecologically friendly technique for synthesizing diverse structural complexity of significant therapeutic interest. It has emerged as a promising field in synthetic organic chemistry. Organic conversions employing transition-metal-free catalyst(s) have numerous benefits: eco-friendly, simple handling in reaction, no tedious work-up procedure, toxic-free, ligand-free, metal-free, additive-free, waste-free, etc.38–40
Recognizing the significance of ultrasound irradiation as an alternative eco-friendly method and the synthetic efficiency associated with transition-metal-free catalysis, the last decades have witnessed outstanding growth in the application of sonochemical activation in the synthesis of a library of heterocyclic compounds, especially under transition-metal-free catalysis. Several review articles demonstrated the application of ultrasound irradiation in diverse synthetic organic transformations.41–52 Banerjee summarized the ultrasound-assisted organic reactions under catalyst-free conditions in 2017.53 To our delight, no review articles existed for the transition-metal-free synthesis of diverse heterocyclic compounds based on the sonochemical activation approach. Herein, we have demonstrated a current overview of the transition-metal-free synthesis of diverse five-membered and six-membered heterocycles and complex-fused and spiro-heterocycles of potential therapeutic interest under ultrasound irradiation covering the literature from 2015 to date. Besides demonstrating the remarkable progress made in this fascinating area, we have also emphasized the limitations and challenges connected with reaction discovery to encourage further research.
2. Ultrasound irradiation-promoted transition-metal-free synthesis of five-membered heterocycles
2.1 Synthesis of five-membered heterocycles containing one-heteroatom
From the perspective of green and more sustainable chemistry, along with the possibilities of obtaining eco-and pot economic nature of the transformation via the well-known multicomponent reaction approach, Gui et al. developed an ultrasound-assisted tandem one-pot protocol for the preparation of polysubstituted pyrroles 4 from the three-component reaction of alkenes 1, N,N-disubstituted formamides 2 and TMSCN 3 under solvent-free conditions (Scheme 1).55 Using iodine as the catalyst and the oxidant, the corresponding products 4 were obtained in 77–92% yield within 40 minutes. The use of ultrasound reduced the reaction time from hours to minutes, making this approach energy-efficient and environmentally friendly. The overall process can proceed through the ultrasound-assisted iodine catalyzed formation of azomethine ylide Int-1 and resonating structure Int-2 from 2 and 3 that can then undergo regioselective [3 + 2] cycloaddition with 1 followed by in situ dehydration to produce intermediate Int-3. The subsequent oxidative dehydrogenation and aromatization of Int-3 yield the final products 4.
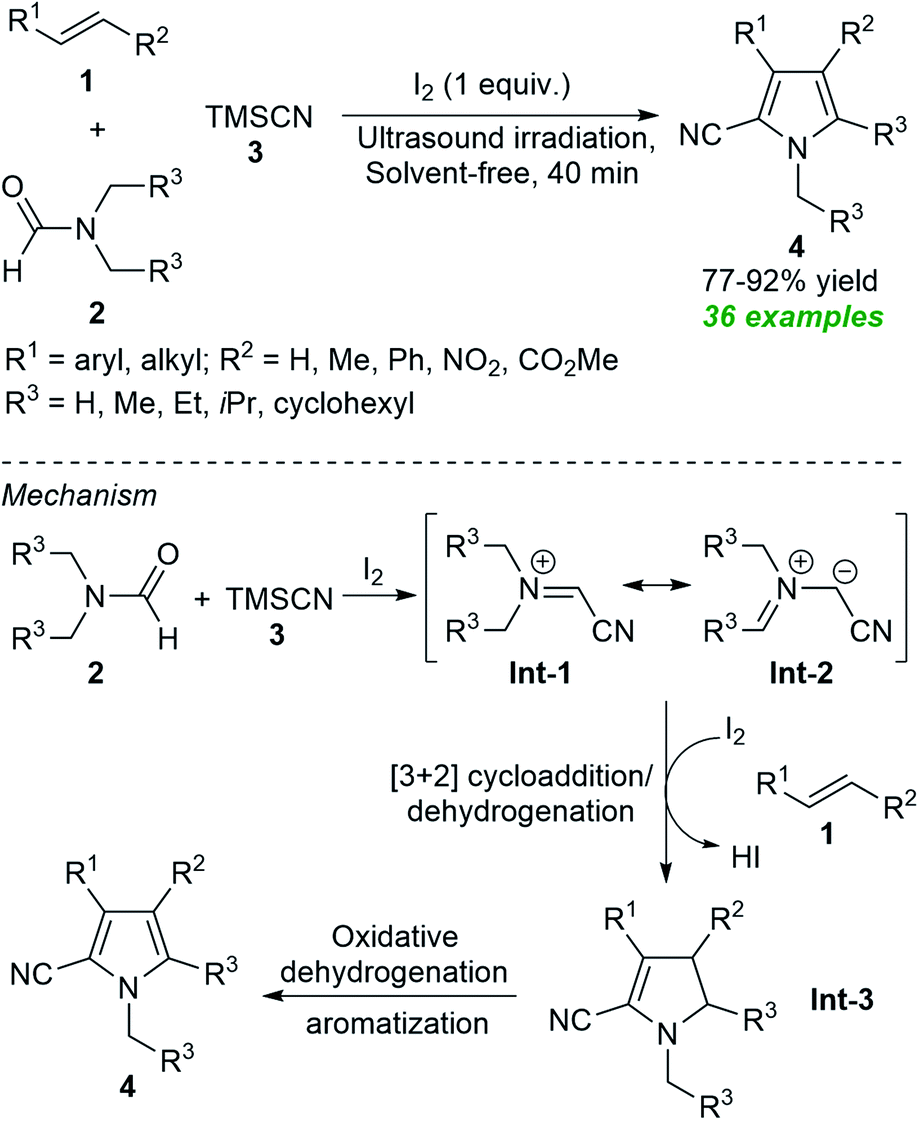 | ||
| Scheme 1 Ultrasound-assisted iodine-catalyzed one-pot three-component routes to access pyrroles 4. | ||
In 2021, Nazeri et al.56 disclosed a highly chemoselective multicomponent strategy towards synthesizing polysubstituted pyrroles 9 by introducing ultrasound irradiation as the green energy source (Scheme 2). With the help of 5 mol% of PTSA·H2O as the organocatalyst, the desired products 9 derived from various aldehydes 5, dialkyl acetylene dicarboxylate 6, isocyanides 7, and 5-amino-pyrazoles 8, have been obtained in moderate to excellent yield. Among them, most of the synthesized compounds have been confirmed to have fluorescence activities. The broad substrate scope, excellent yield, operational simplicity, and short reaction time are some of the critical features of this methodology.
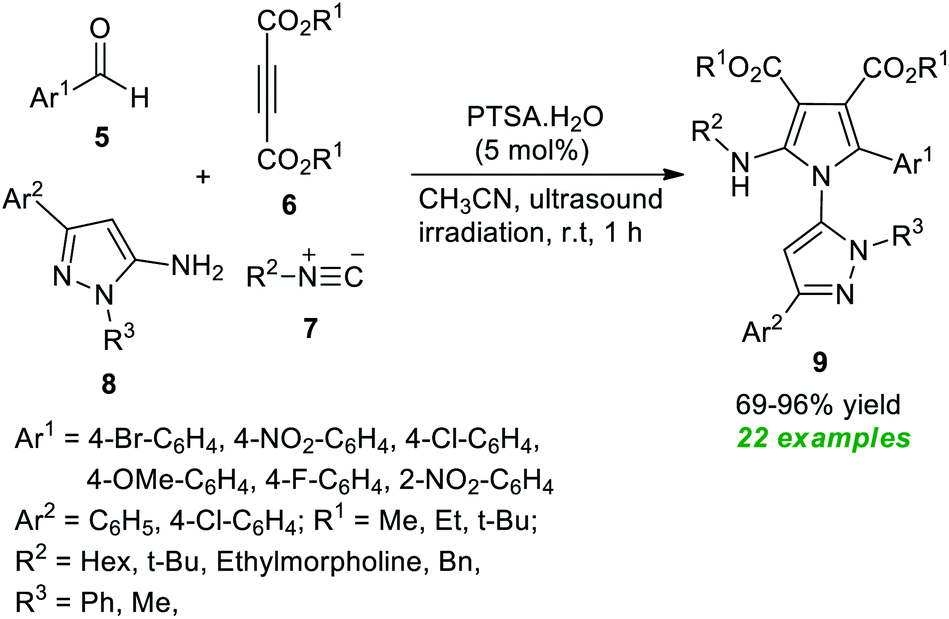 | ||
| Scheme 2 Acid-catalyzed chemoselective synthesis of pyrroles 9 under ultrasound irradiation. | ||
In 2020, Mangaonkar and co-workers demonstrated a highly convenient ultrasound-assisted strategy for the construction of functionalized benzofurans 11 via the cyclization of 2-hydroxystilbenes 10 (Scheme 3).58 Under the influence of 10 mol% of PhI as the pre-catalyst, m-CPBA as the oxidant, and TFA as additives, the products 11 were achieved in 63–87% yield at room temperature. In this reaction, the required iodine(III) species were produced in situ from the oxidation of PhI by m-CPBA and TFA. This iodine(III) species activates the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of 10 to yield intermediate Int-4, which then experiences intramolecular cyclization and subsequent reductive elimination to deliver the products 11 and precatalyst PhI via intermediate Int-5.
C bond of 10 to yield intermediate Int-4, which then experiences intramolecular cyclization and subsequent reductive elimination to deliver the products 11 and precatalyst PhI via intermediate Int-5.
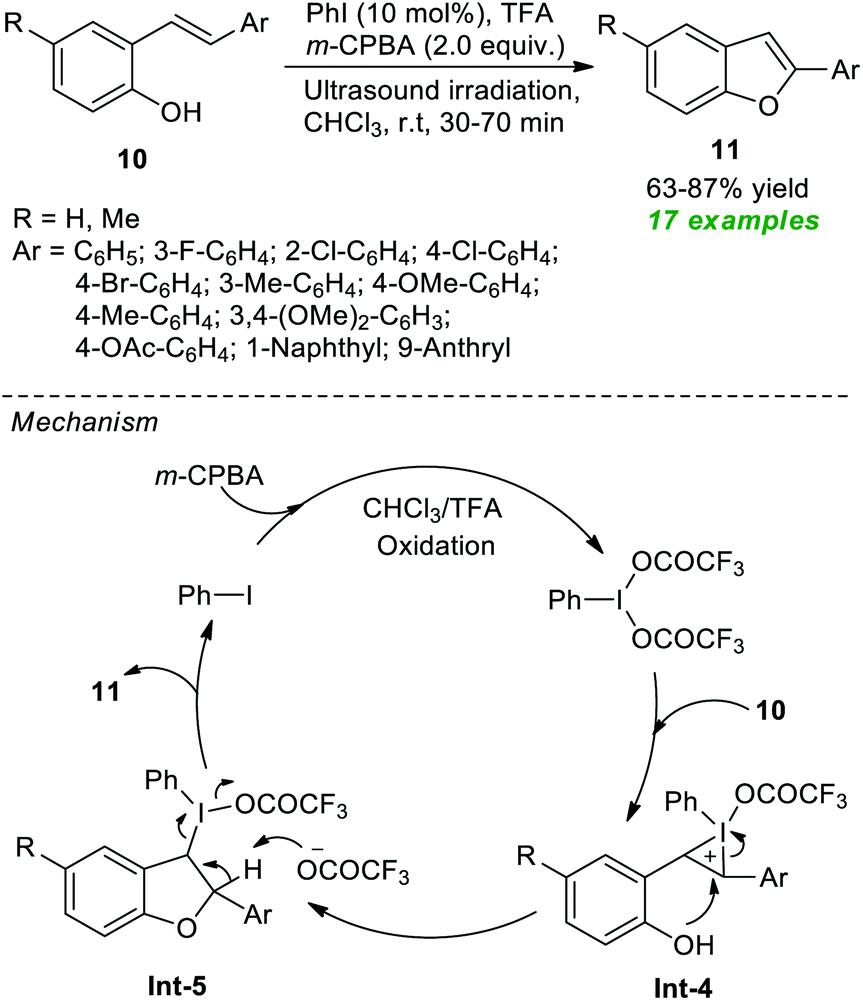 | ||
| Scheme 3 Ultrasound-assisted in situ generated hypervalent iodine-catalyzed synthesis of benzofurans. | ||
Because of their importance, a rapid and eco-compatible strategy for the assembly of diverse polysubstituted thiophenes 15 via a three-component Gewald reaction of carbonyl compounds 12, active methylene compounds 13, and elemental sulfur 14 in the presence of polyethylene glycol-600 (PEG-600) under ultrasound irradiation at room temperature was developed by Akbarzadeh and Dekamin in 2017 (Scheme 4).60 Using this catalyst-free protocol, 16 target compounds were accomplished in poor to excellent yield in comparatively short reaction durations.
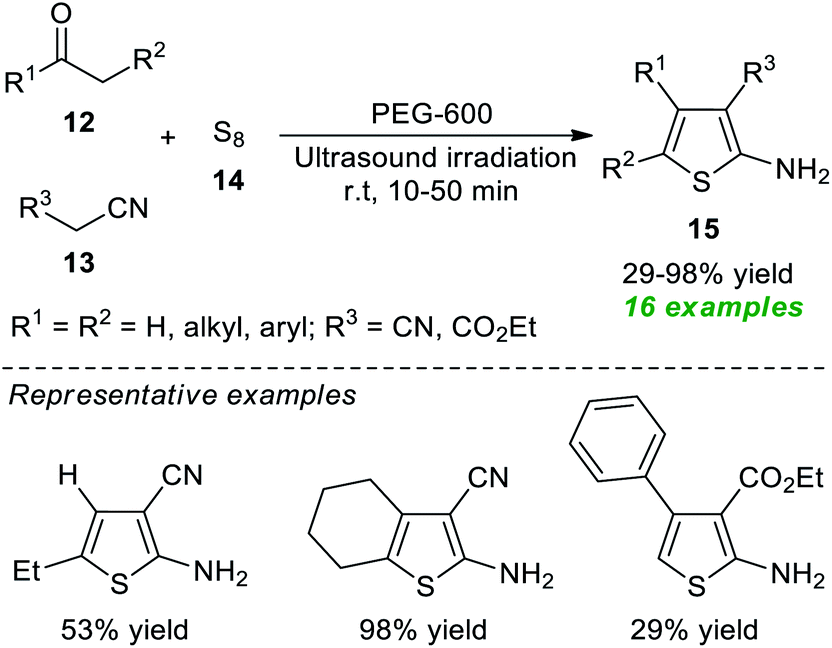 | ||
| Scheme 4 Ultrasound-assisted three-component synthesis of thiophenes 15. | ||
In 2021, Suárez et al.61 disclosed a one-step push–pull synthetic route to access various thiophene derivatives 18 from readily available α-brominated acetamides 16 and amino mercaptoacrylates 17 by introducing ultrasound irradiation as a powerful green technique (Scheme 5). A comparison of both conventional and ultrasound irradiation techniques for the synthesis of 18 suggests the use of ultrasound condition as the method of choice, which not only increased the product yield (66–85%) but also reduced reaction times, while the conventional technique required a longer reaction time and also decreased the product yield (35–72%).
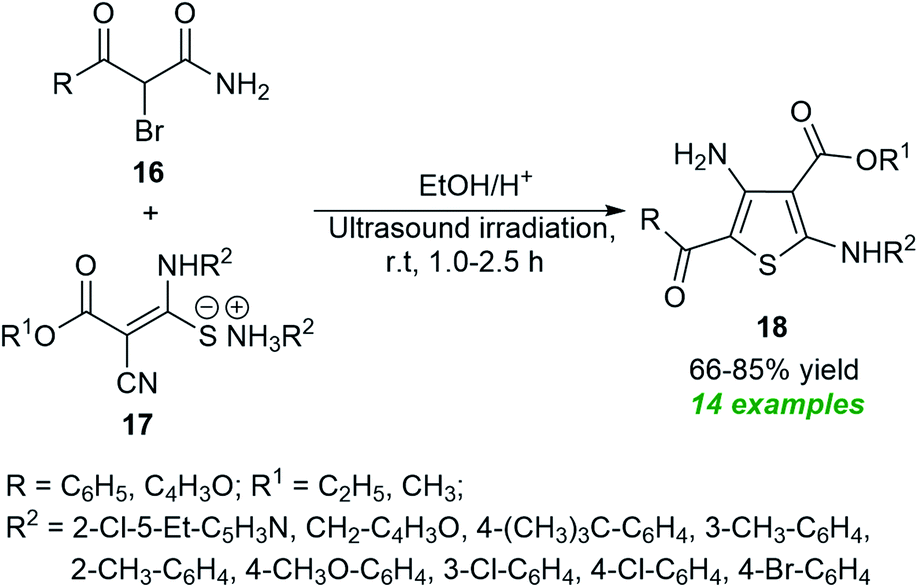 | ||
| Scheme 5 Ultrasound-assisted one-step synthesis of tetrasubstituted thiophenes 18. | ||
2.2 Synthesis of five-membered heterocycles containing two-heteroatoms
Recognizing their importance, various attempts to synthesize this moiety have been made. In 2020, Piltan and co-workers disclosed an organocatalytic tandem three-component approach for the facile access to trisubstituted imidazoles 22 from the treatment of benzil 19, aldehydes 20, and urea 21 in a one-pot fashion under ultrasonication (Scheme 6).67 A broad spectrum of substituted aromatic, heteroaromatic, and aliphatic aldehydes was well tolerated for this reaction under the influence of 1 mol% of triphenylphosphine (PPh3) as the organocatalyst and delivered the respective imidazole product 22 in good to high yields at room temperature. Compared to conventional heating conditions, the use of ultrasound technology enhances overall yields and reduces reaction times.
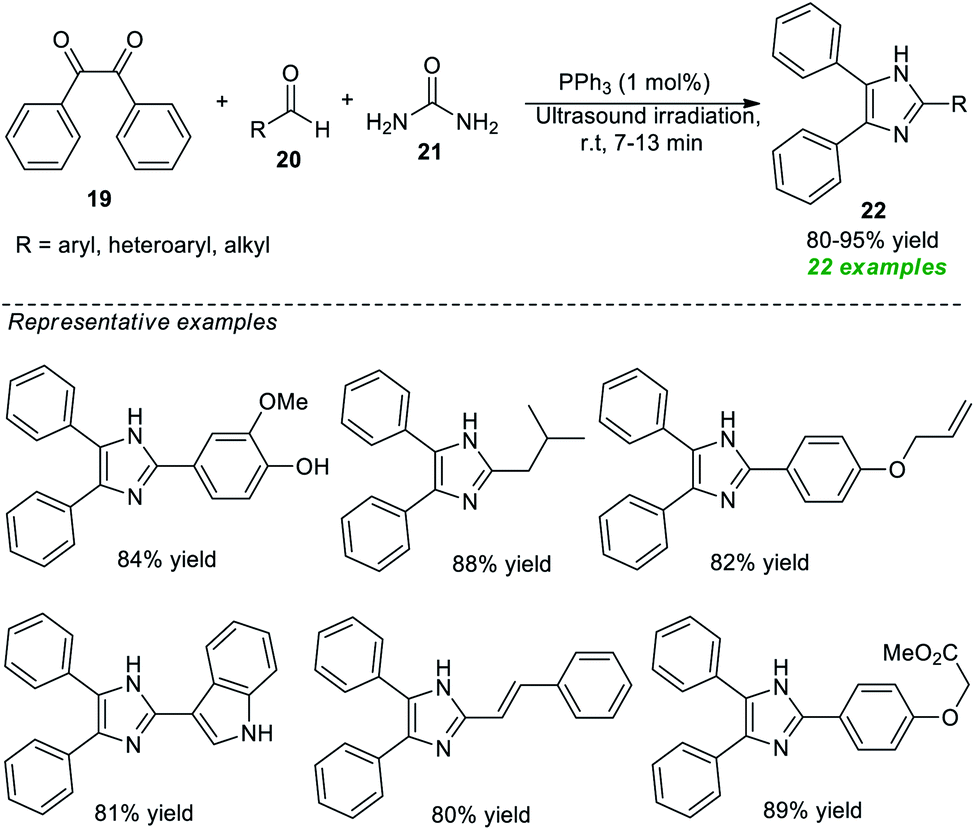 | ||
| Scheme 6 Ultrasound-assisted organocatalytic syntheses of tri-substituted imidazoles as reported by Piltan et al. | ||
Almost at the same time, another milestone in the synthesis of substituted imidazoles was gained by Agrawal et al.68 They disclosed a one-pot ultrasound-assisted reaction of benzil 19, aryl aldehydes 23, and ammonium acetate 24 in an aqueous ethanolic solution with 10 mol% of C-1 as the catalyst (Scheme 7). Pleasingly, this three-component reaction afforded the respective imidazoles 25 in good to high yield within 15–30 minutes. The same group further extends their three-component reaction strategy to synthesize some tetrasubstituted imidazoles. Using 10 mol% of C-1, the corresponding tetrasubstituted imidazoles 27 derived from benzil 19, aldehydes 23, ammonium acetate 24, and substituted amine 26 were formed in 88–94% yield. The exploitation of energy-saving ultrasound techniques features a clean pathway for the reaction, reduces the reaction time, and increases the product yield. Other significant aspects of this approach include the wide functional group tolerance, green reaction conditions, recyclable catalysts with low loading, etc. The unusually required prolonged reaction time and the low yield of the products by the thermal process make ultrasound a very attractive and powerful green strategy for this reaction in contrast to the thermal method.
 | ||
| Scheme 7 3-N-Morpholinopropanesulfonic acid (C-1)-catalyzed ultrasound-assisted synthesis of imidazoles. | ||
After noticing this, many synthetic protocols have been discovered for the efficient construction of various types of isoxazoles, particularly α, β unsaturated isoxazole-5(4H)-one's derivatives. However, most of the existing methods deal with several significant issues due to which Joshi et al.73 in 2019 developed an ultrasound-assisted rapid, efficient one-pot three-component method for the practical synthesis of diverse isoxazole derivatives (Scheme 8). Using pyridine as the organocatalyst, the three-component reaction of several pyrazole aldehydes 28, methyl 4-methyl-3-oxovalerate 29, and hydroxylamine hydrochloride 30 in aqueous ethanolic solution at room temperature yielded a total of 9 new novel isoxazole derivatives 31 with 82–96% yields. A comparison study between the conventional and ultrasound conditions concluded the ultrasound method as the best efficient method in terms of product yield and reaction times. The mechanism begins with the formation of intermediate Int-6 from the reaction of 29 and 30, which undergo nucleophilic addition with 28 under the influence of pyridine to deliver the intermediate Int-8. The final products 31 have been achieved via cyclization of Int-8 followed by elimination of water and methanol.
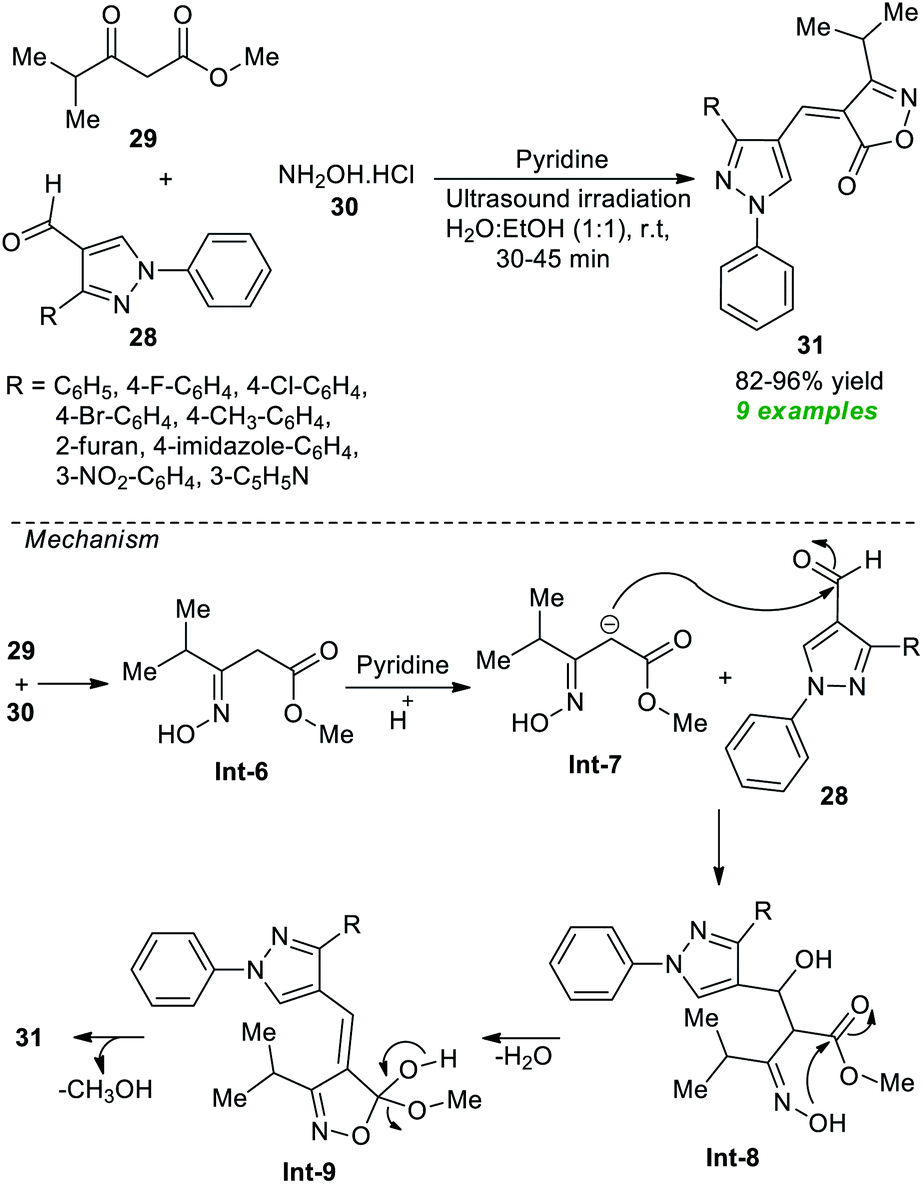 | ||
| Scheme 8 Secondary amine-catalyzed synthesis of substituted isoxazole under ultrasound irradiation. | ||
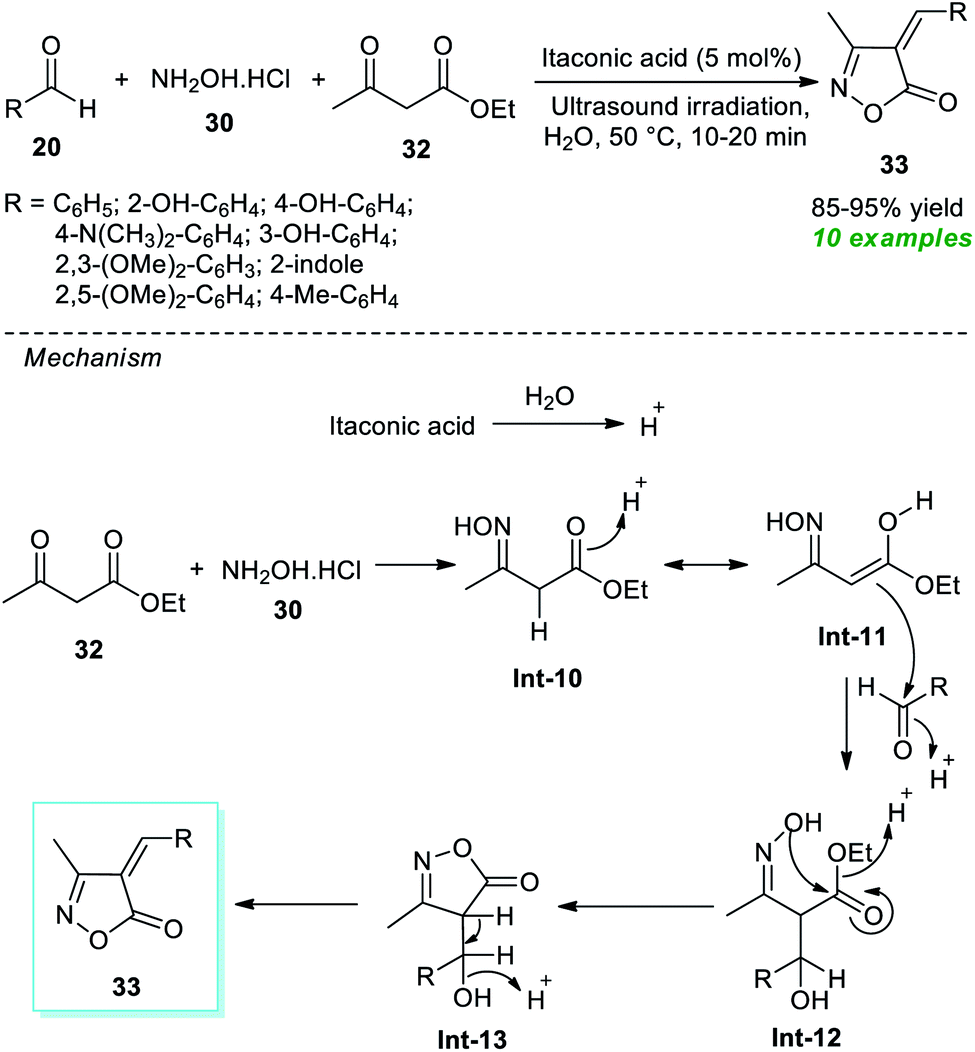
Around the same time, Thopate and Kasar also demonstrated an expedient organocatalytic one-pot technique to produce various isoxazole-5(4H)-one derivatives (Scheme 9).74 The three-component reaction of a variety of aldehydes 20, hydroxylamine hydrochloride 30, and ethyl acetoacetate 32 in water as the solvent at 50 °C using 5 mol% of itaconic acid as the organocatalyst was found to proceed smoothly to form the corresponding isoxazole derivatives 33 in 85–95% yields in a concise duration of time under ultrasonication. Broad functional group tolerance and a reusable catalytic system with low loading and metal- and waste-free nature are several key highlights of this approach. The author postulated a mechanism to realize this transformation, which entails the generation of the key intermediate Int-11 via acid-mediated reactions of 30 and 32. This intermediate Int-11 reacts with 20 to produce Int-12, which further experiences intramolecular cyclization under the influence of itaconic acid to yield the final products 33.
 | ||
| Scheme 9 Itaconic acid-catalyzed three-component synthesis of isoxazoles under ultrasound irradiation. | ||
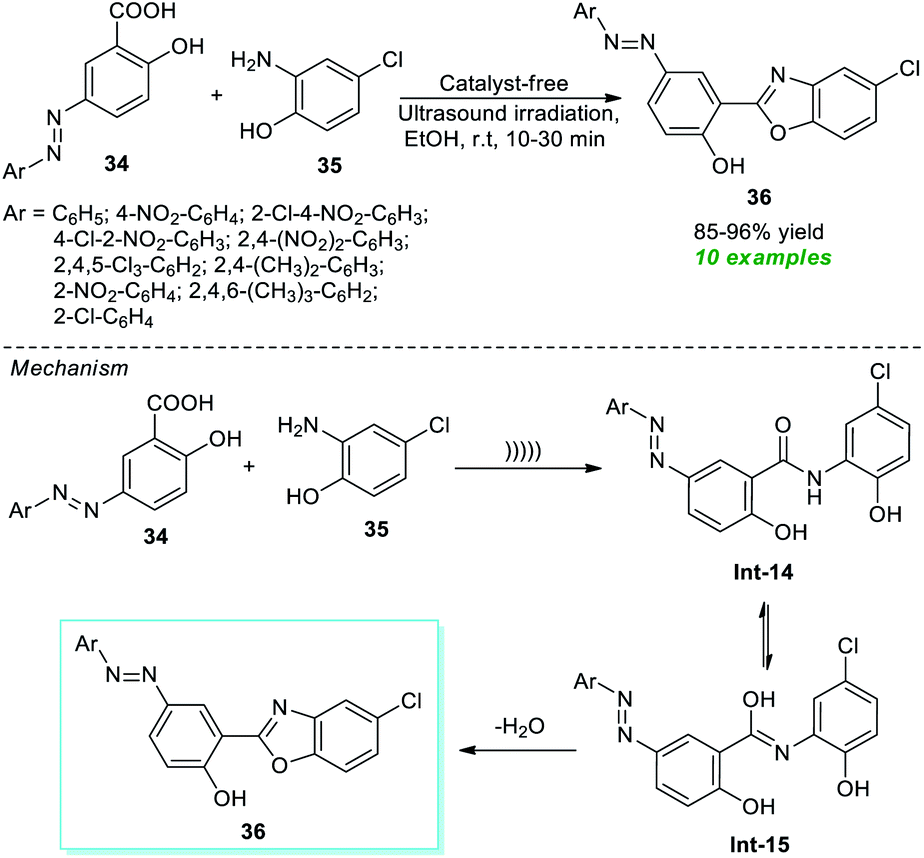
In this regard, Nikpassand et al.76 developed a facile one-pot procedure for the construction of a series of novel benzoxazole derivatives with the help of ultrasound irradiation (Scheme 10). Using the catalyst-free conditions, the reaction of various azo-linked salicylic acid derivatives 34 and 2-amino-4-chlorophenol 35 in ethanol was found to proceed under ambient conditions under ultrasound irradiation to deliver highly functionalized benzoxazole derivatives 36 in 85–96% yields in 10–30 minutes. The primary advantages of this approach include cost-effectiveness, operational simplicity, sustainability, etc. A mechanism is depicted in Scheme 10 for this reaction. Initially, an intermediate Int-14 is formed from the nucleophilic addition of 35 to 34 under ultrasonication. Consequently, the tautomerization of Int-14 to Int-15 and its dehydration lead to the desired products 36.
 | ||
| Scheme 10 Ultrasound-assisted azo-linked benzoxazoles as reported by Nikpassand et al. | ||
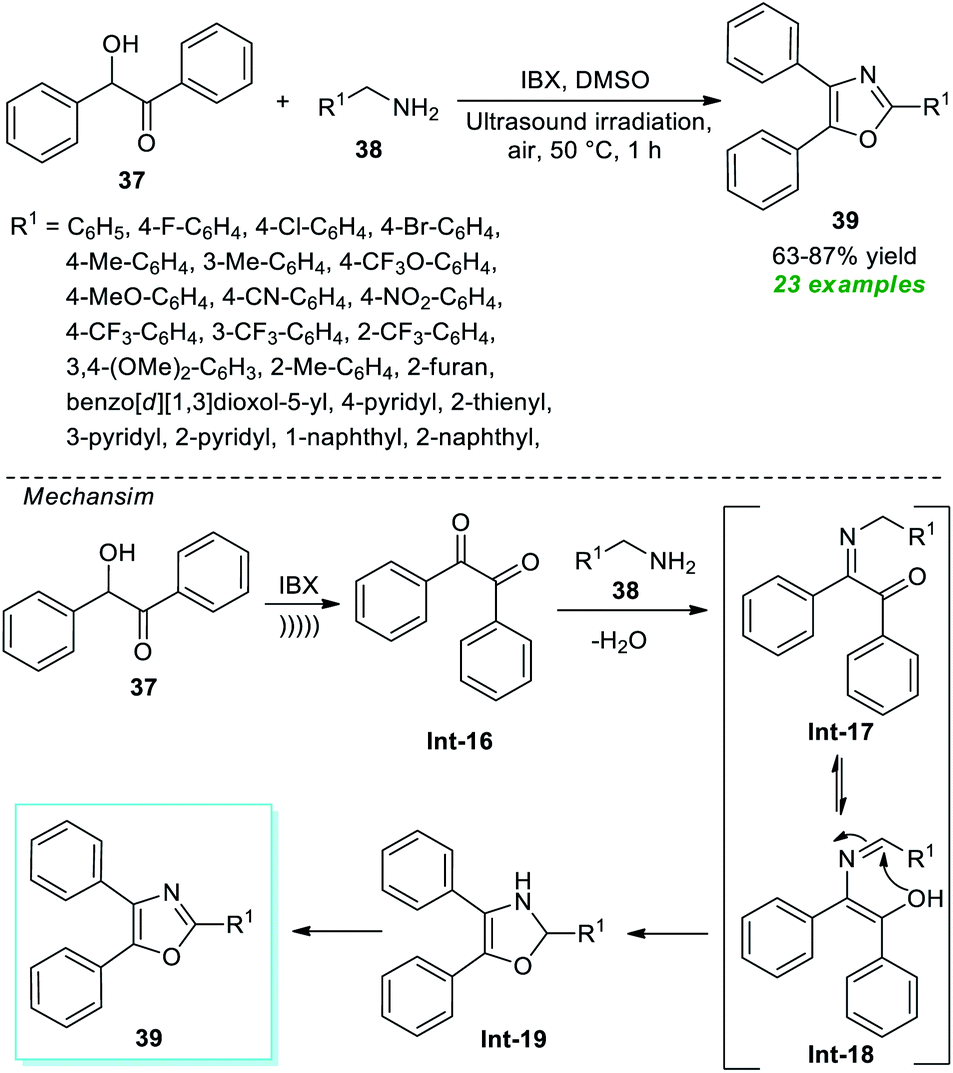
Another achievement in the synthesis of oxazoles derivatives was accomplished by Dandela, Pal, and their group in 2021 by employing the ultrasound technique as an efficient green strategy. Treatment of commercially available benzoin 37 with various amines 38 in the presence of IBX was found to occur in DMSO under air at 50 °C to deliver the respective oxazole derivatives 39 in 63–87% yields (Scheme 11).77 This method was successfully proceeded not just with aryl amines but also with alkyl and heteroaryl amines. The overall reaction could be initiated via the IBX mediated transformation of benzoin 37 to benzil Int-16 and then reaction with amines 38 to afford the adduct Int-17 which after tautomerization and intramolecular cyclization delivers Int-19. Its final aromatization in the presence of air yields the desired products 39.
 | ||
| Scheme 11 IBX-mediated synthesis of 2-aryl substituted oxazoles under ultrasound irradiation. | ||
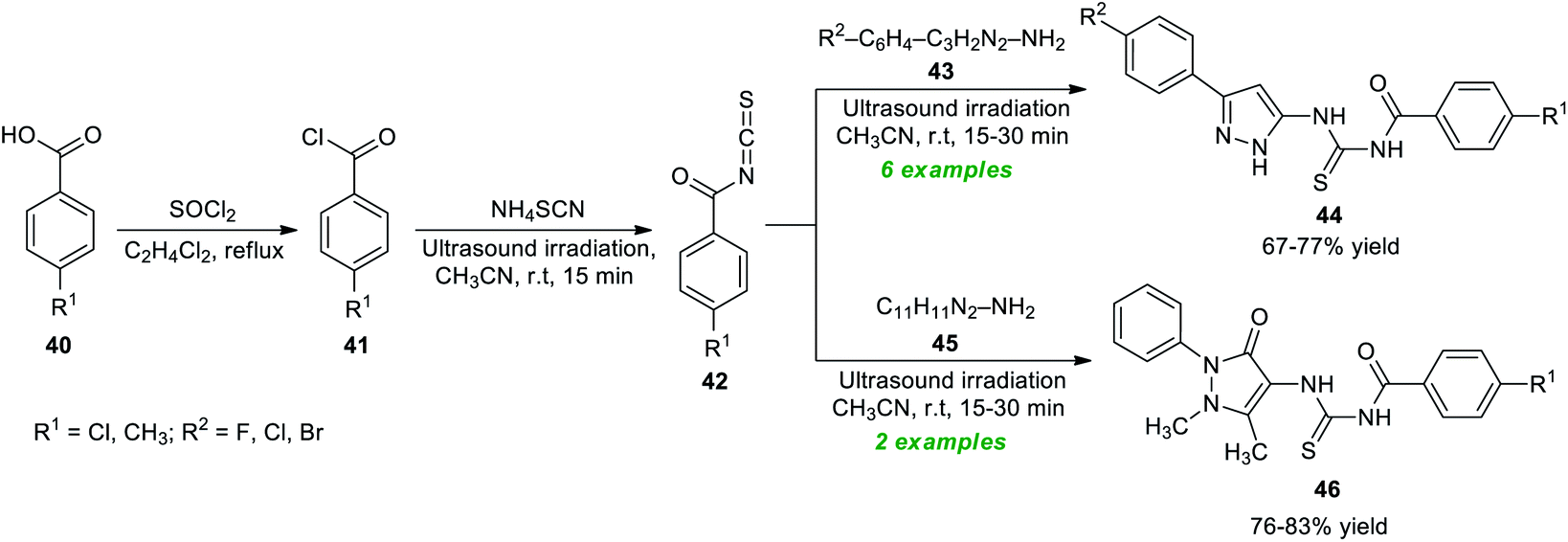
Considering these importances, Bleotu et al.80 demonstrated an eco- and environmentally benign strategy for the construction of diverse pyrazoles and pyrazolone derivatives under ultrasound irradiation conditions (Scheme 12). The overall synthetic process involves the initial reaction of substituted benzoic acid 40 with thionyl chloride, which delivers the desired benzoyl chloride 41. The ultrasound-assisted reaction of 41 with ammonium isothiocyanate in CH3CN at room temperature provided benzoyl isothiocyanates 42. The definitive treatment of 42 with 43 or 45 yields the corresponding pyrazole products 44 and pyrazolone products 46 in 67–77% and 76–83% yield, respectively. Many synthesized compounds have been recognized as the inhibitors of cell cycle kinases that mark the key features of the present protocol.
 | ||
| Scheme 12 Ultrasound-assisted step-wise synthesis of thiourea-linked pyrazole derivatives. | ||
Recently, Sarkate and co-workers have devised the synthesis of a library of tetrazole-based pyrazoline derivatives by employing ultrasound irradiation as an eco-friendly activation method with the aid of metal-free conditions, and the synthesized compounds were found to exhibit anticancer properties (Scheme 13).81 The desired pyrazoline products were achieved via a two-step procedure starting from the initial Claisen–Schmidt condensation of tetrazole linked carbonyl compound 47 and various aldehydes 23 in ethanol using NaOH as the base catalyst at room temperature to afford the adduct 48 in 76–90% yields in a short duration of time, which on further treatment with hydrazine hydrate 49 under ultrasound irradiation offers the respective products 50 in 93–98% yields.
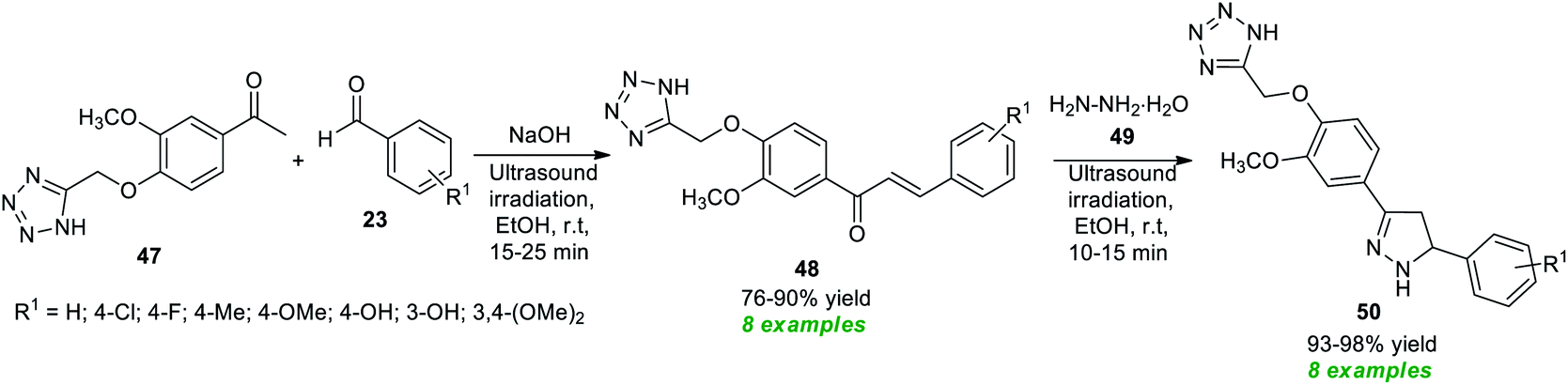 | ||
| Scheme 13 One-pot synthesis of tetrazole-linked pyrazole derivatives under ultrasound irradiation. | ||
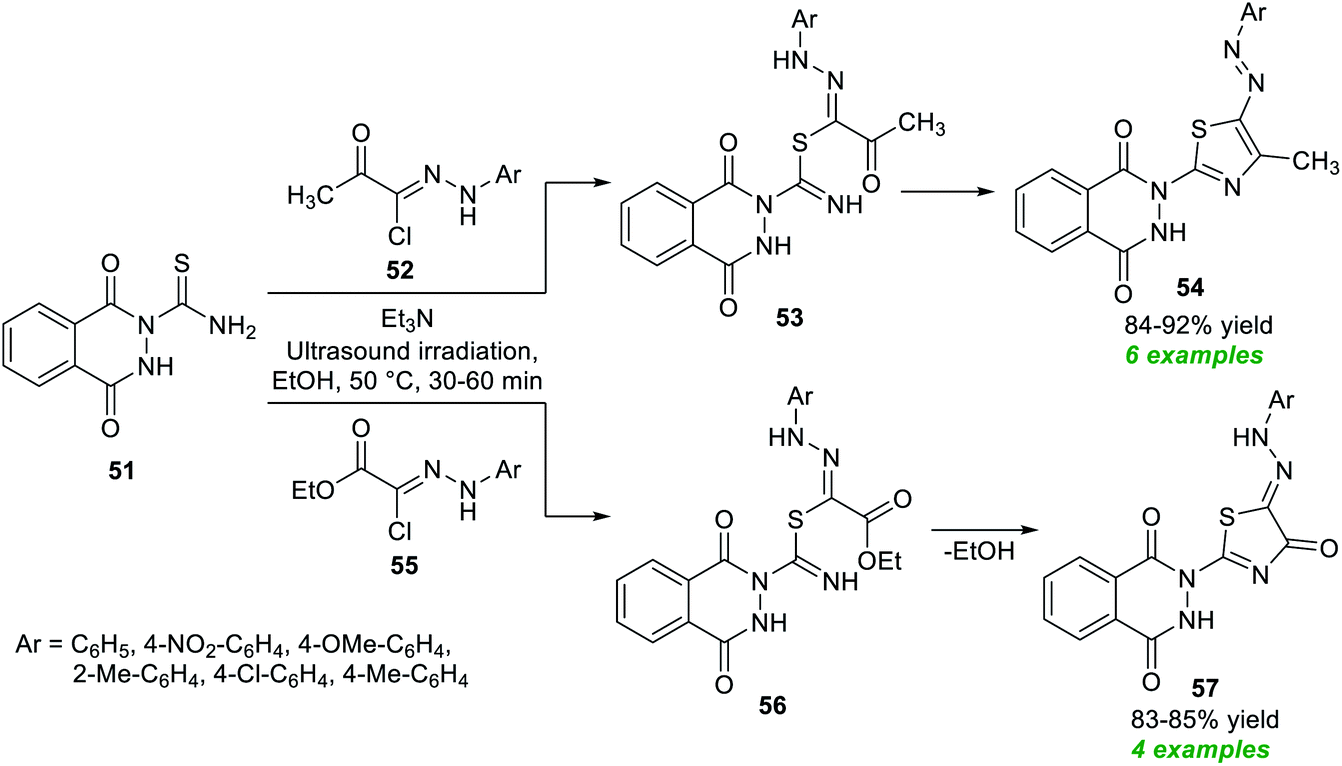 | ||
| Scheme 14 Ultrasound-assisted triethylamine-catalyzed assembly of functionalized thiazoles as reported by Farghaly et al. | ||
A simple, facile, and highly convenient method to access a variety of Hantzsch thiazole derivatives has been reported by the Rachedi group.84 With the help of their prepared silica-supported tungstosilisic acid (SiW·SiO2) catalyst, a one-pot three-component reaction between bromoacetyl substituted-pyran-2-one 58, thiourea 59, and aryl aldehydes 60 was performed with ultrasound irradiation in aqueous ethanolic solution, which delivers the desired thiazole derivatives 61 in 79–90% yield at room temperature within 1.5–2 hours (Scheme 15). Substitutions in different positions of the benzaldehyde rings revealed no significant influence on the reaction rate or product yield.
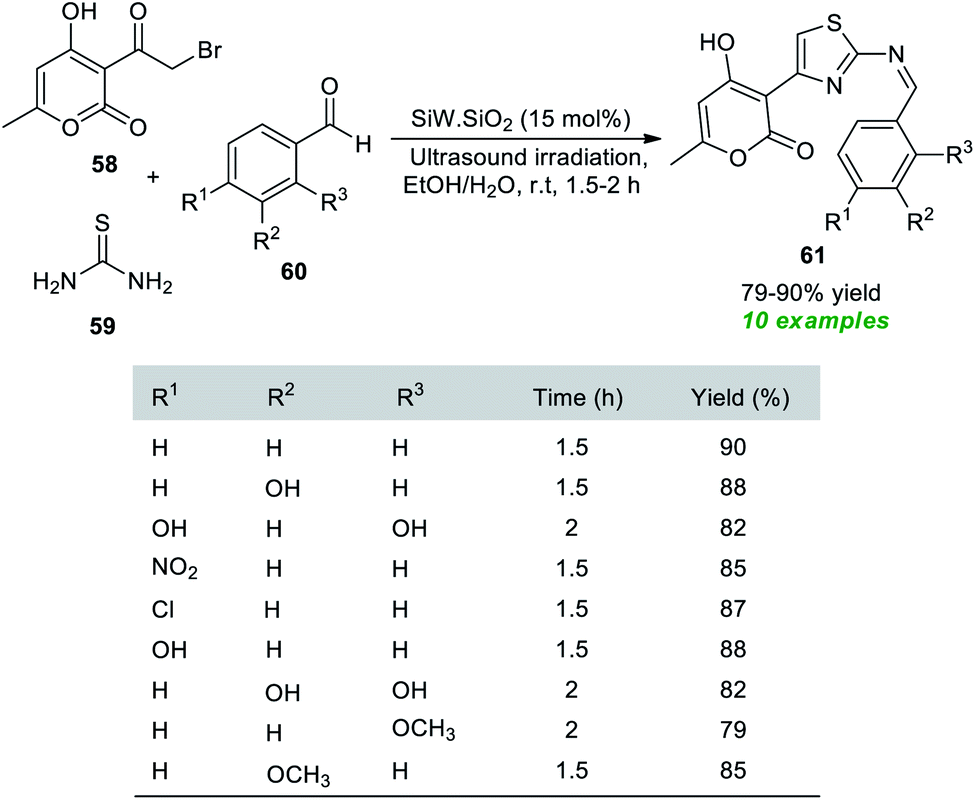 | ||
| Scheme 15 SiW·SiO2-catalyzed ultrasound-irradiated assembly of thiazole derivatives. | ||
2.3 Synthesis of five-membered heterocycles containing three-heteroatoms
 | ||
| Scheme 16 Step-wise synthesis of diverse 1,3,4-oxadiazoles under ultrasound irradiation. | ||
A very straightforward method to access a variety of 1,3,4-oxadiazole derivatives under ultrasound irradiation was reported by Santos, Machado, and their group (Scheme 17).87 Using NBS-NaOAc as the oxidizing system, the corresponding 1,3,4-oxadiazole products 70 derived from the cyclization of various semicarbazones 69 in the presence of acetic acid at 25–110 °C were obtained in 53–92% yield within only 15 minutes. This protocol starts with the initial NBS promoted reaction of semicarbazones 69 to form an intermediate Int-20 which, after 1,3-dipolar elimination on treatment with NaOAc, delivers nitrilimines Int-21. Consequently, the 1,5-electrocyclization of the adduct Int-21 furnishes the desired products 70. Broad functional group tolerance, simple work-up procedure, eco-friendly nature, scalable synthesis, and being environmentally benign are a few glimpses of the protocol's most essential features.
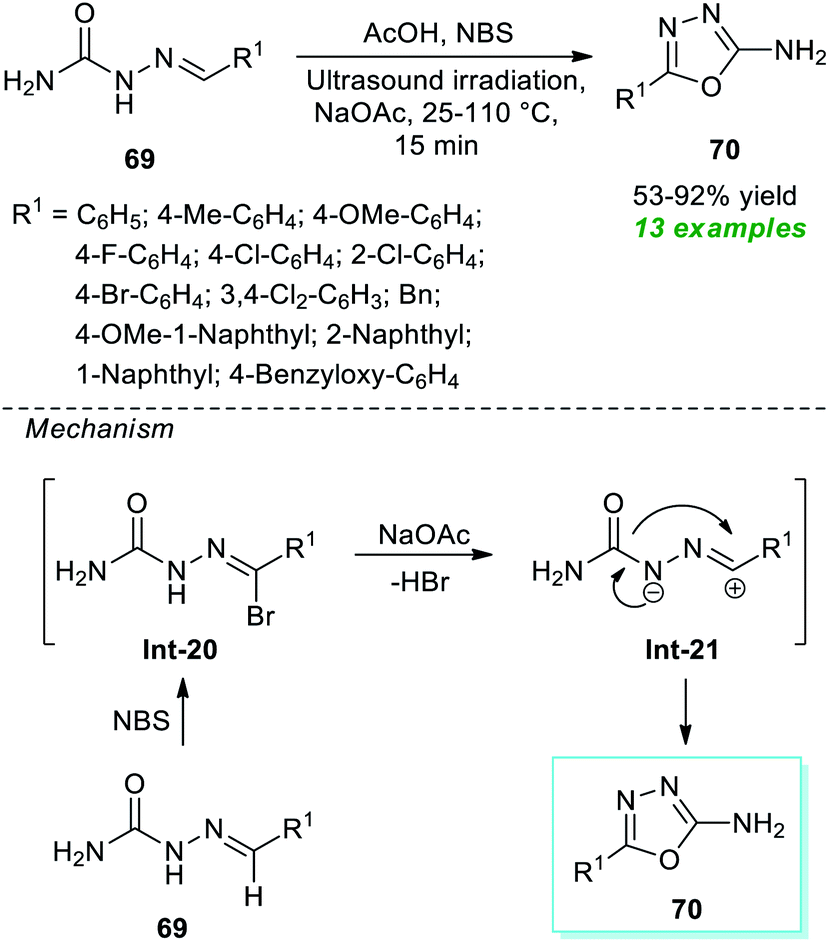 | ||
| Scheme 17 Ultrasound-assisted oxidative cyclization approach to access amino-substituted oxadiazoles. | ||
Treatment of various hydrazides 71 and cyanogen bromide 72 in the presence of potassium bicarbonate as an efficient catalyst in ethanol under ultrasonication at 50 °C was found to lead to a variety of 1,3,4-oxadiazole derivatives 73 in 81–93% yields after 2.5–7 hours (Scheme 18).88 Alkyl and heteroaryl substituted aldehydes situated on the hydrazide ring have been well sustained by this approach, like aromatic aldehydes with varied electron-withdrawing and electron-donating groups. The significant outcome of the present method was established by authenticating the antioxidant properties of most of the synthesized compounds.
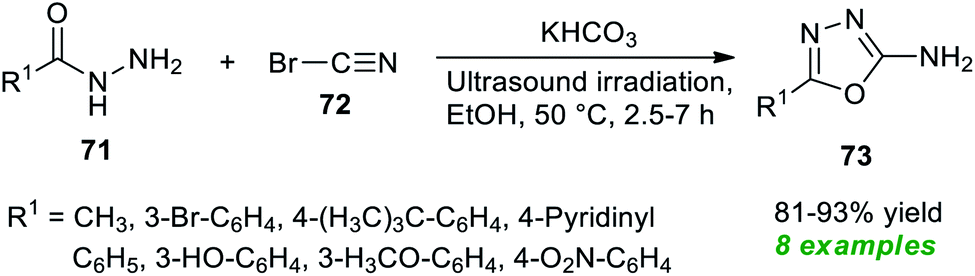 | ||
| Scheme 18 Potassium bicarbonate-catalyzed ultrasound-assisted synthesis of 1,3,4-oxadiazole derivatives. | ||
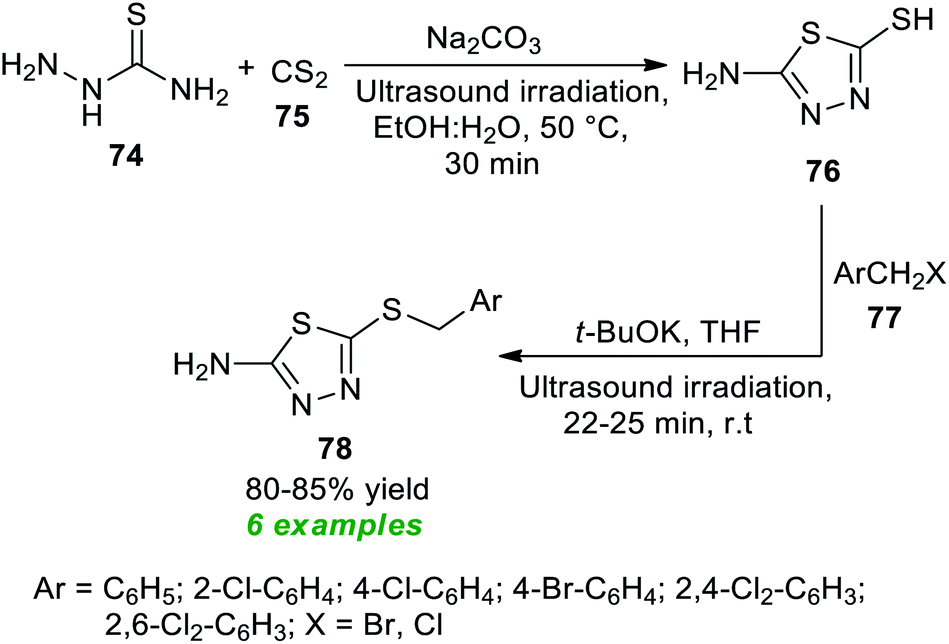 | ||
| Scheme 19 Ultrasound-assisted synthesis of amino-substituted thiadiazole scaffolds. | ||
Recently, a simple and expeditious method for the green synthesis of several 1,2,4-thiadiazole derivatives has been accomplished by Srivastava et al.91 by employing metal-as well as catalyst-free and ultrasound irradiation as the eco-friendly and environmentally benign reaction condition (Scheme 20). With the help of their optimized reaction condition, the corresponding thiadiazole derivatives 81 derived from thioamides 79 and chloranil 80 have been obtained in good to excellent yield in a water medium. By employing ultrasound techniques, a total of ten products were isolated in a short interval of time. The proposed mechanism for this reaction involves the oxidative addition of thioamide 79a to chloranil 80 to form an intermediate Int-22 which dimerizes to generate the intermediate Int-23. Consequently, the cyclization of intermediate Int-23 yields the final products 81.
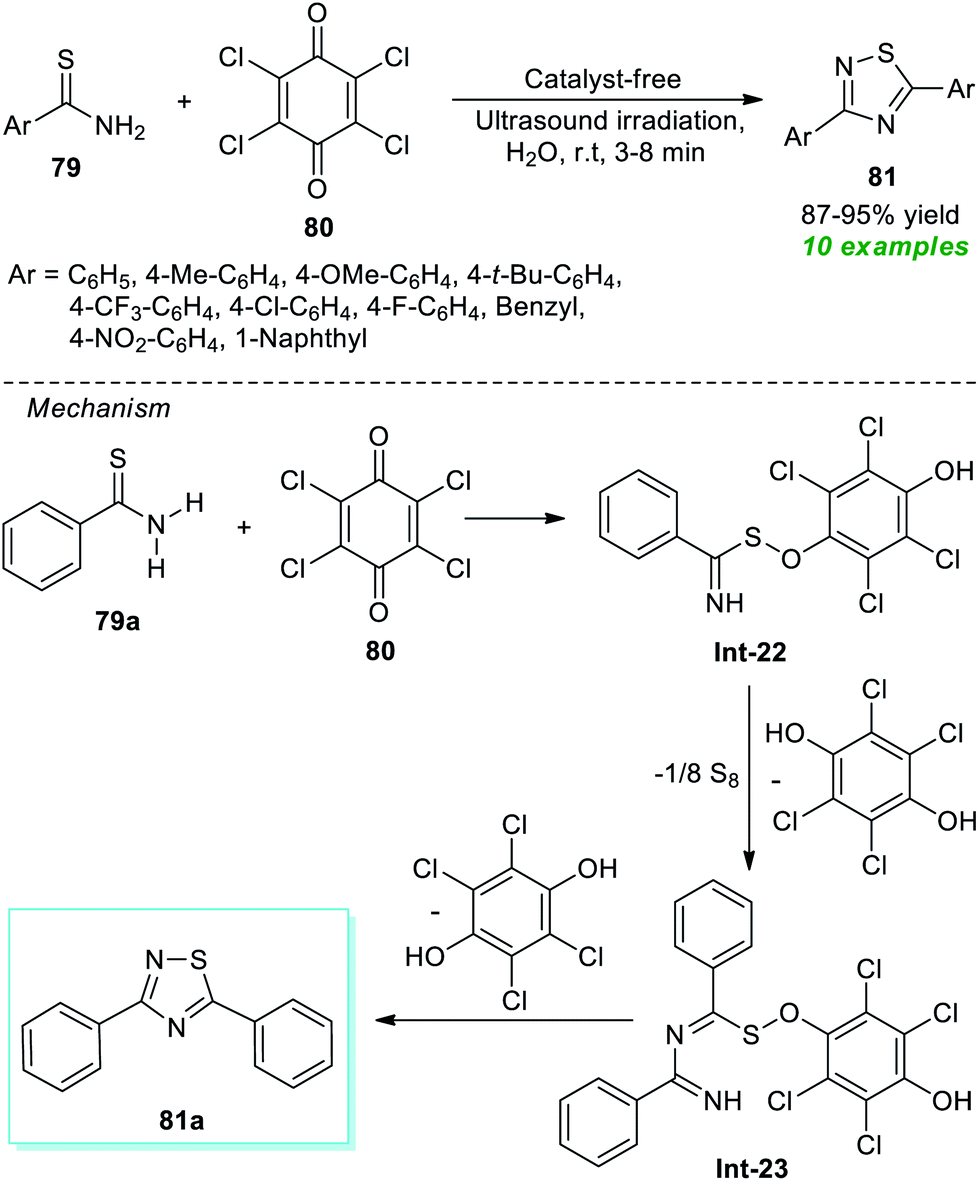 | ||
| Scheme 20 Ultrasound-assisted conversion of thioamides to thiadiazoles in an aqueous medium. | ||
In 2017, Alves et al.94 disclosed an organocatalytic [3 + 2] cycloaddition reaction for the rapid access to a variety of 1,2,3-triazole derivatives under ultrasound irradiation (Scheme 21). The corresponding triazole products 84 produced from a variety of β-oxo-amides 82 and substituted aryl azides 83 were achieved in reasonable to outstanding yields by employing 5 mol% of diethylamine as an organocatalyst. The appearance of an electron-withdrawing group on the aryl ring of β-oxo-amides reduces the product yield, while an electron-releasing substituent enhances it. On the other hand, aryl azides bearing different substituents seemed to have no negative impact on the rate of the reaction except methyl- and fluoro-substituted aryl azide (R3 = 4-Me, 2-F), which offers the product with a lower yield. The mechanistic route for realizing this transformation begins with the generation of enamine intermediate Int-24 from the condensation of Et2NH and 82a, which then undergoes 1,3-dipolar cycloaddition with 83a to yield intermediate Int-25. Following the removal of Et2NH, Int-25 experiences a 1,3-hydride shift to generate the intermediate Int-26, which rapidly undergoes its zwitterionic form Int-27 and produces the desired product 84a.
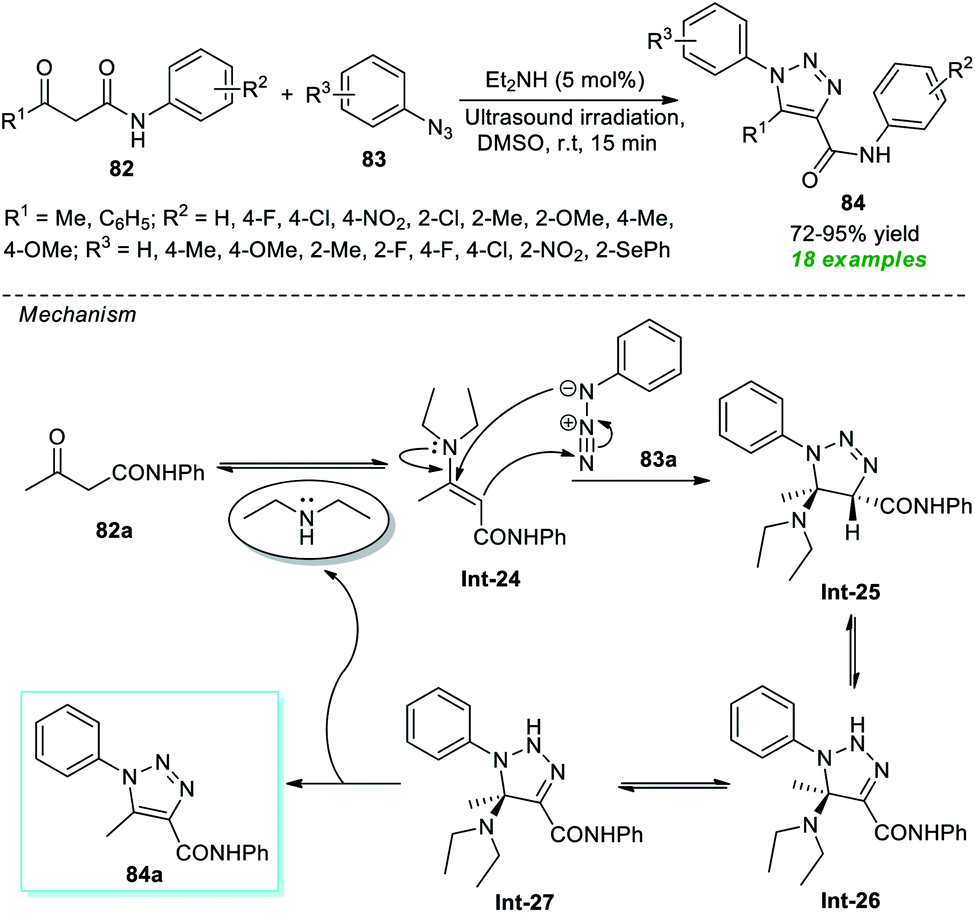 | ||
| Scheme 21 Organocatalytic ultrasound-assisted synthesis of substituted 1,2,3-triazole derivatives. | ||
Recently, Karthikeyan et al.95 demonstrated a convenient and straightforward strategy to access a vast array of 1,5-substituted triazoles 87 and 1,4-substituted triazoles 89 (Scheme 22). By employing metal- and catalyst-free conditions, the ultrasound-assisted treatment of various azides 85 and nitroolefins 86 or β-enaminones 88 with water as the green solvent at room temperature efficiently produced the respective products 87 and 89 in 72–92% and 72–90% yield respectively at 30 minutes. A range of alkyl, aryl, and heteroaryl substituted azides were discovered to operate very well with this approach. The formation of 87 was proposed via the key intermediate Int-28 generated by a regioselective [3 + 2] cycloaddition of azide 85 with nitroolefins 86 and the subsequent aromatization of Int-28. In the case of products 89, an inverse-electron-demand [3 + 2] cycloaddition between azide 85 and β-enaminones 88 was proposed to occur to form the stable key intermediate Int-29, which leads to the production of the preferred product 89 once HNMe2 was removed.
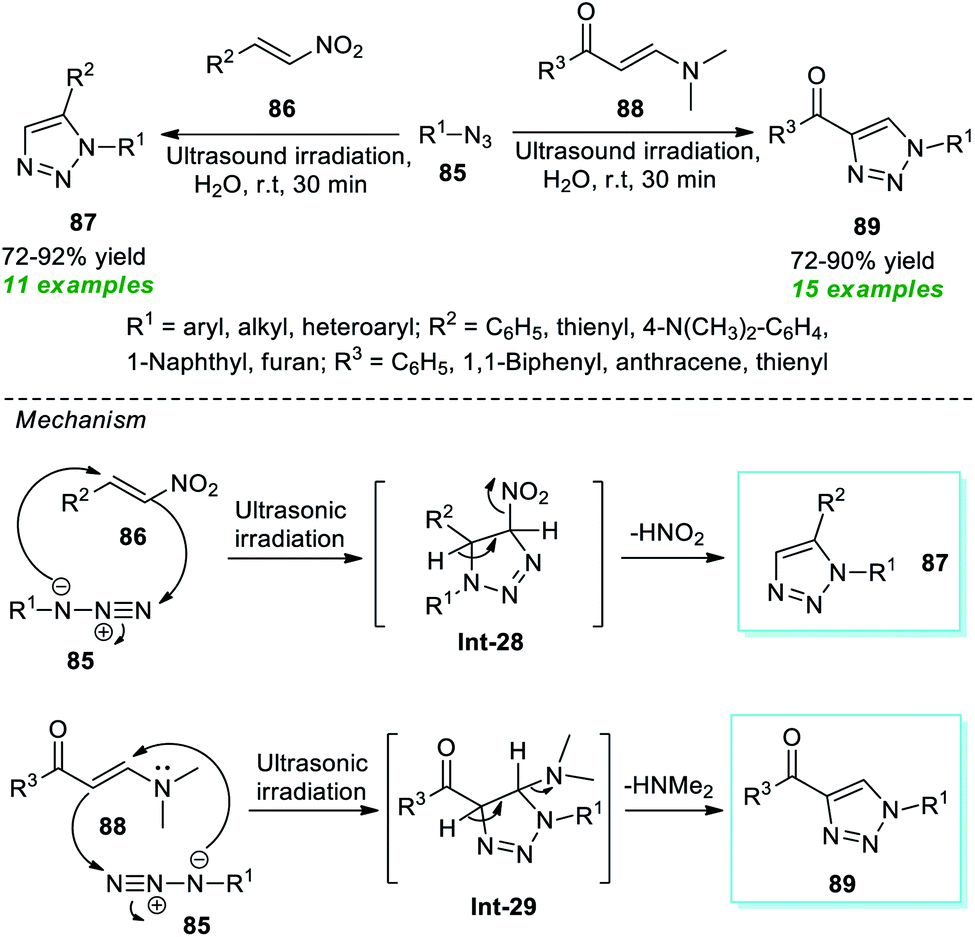 | ||
| Scheme 22 Water-mediated sonochemical-assisted synthesis of 1,5- and 1,4-substituted 1,2,3-triazoles. | ||
2.4 Synthesis of five-membered heterocycles containing four-heteroatoms
In 2017, Arafa et al.99 demonstrated a one-pot procedure for synthesizing bis-tetrazoles 91 from the ultrasound irradiated sequential reaction of dialdehydes 90, hydroxylamine hydrochloride, phosphorous pentoxide, and sodium azide exploring transition-metal-free conditions in dry DMF at 70 °C for 55–75 minutes (Scheme 23). By employing this mild reaction condition, six compounds have been accomplished in 88–95% yields without using column chromatography techniques. The entire reaction can begin with the generation of oximes Int-30 from dialdehydes 90 and hydroxylamine, which can react in situ with phosphorous pentoxide to form bis-nitriles Int-31. The final reaction of sodium azide with bis-nitriles Int-31 afforded the corresponding bis-tetrazoles 91.
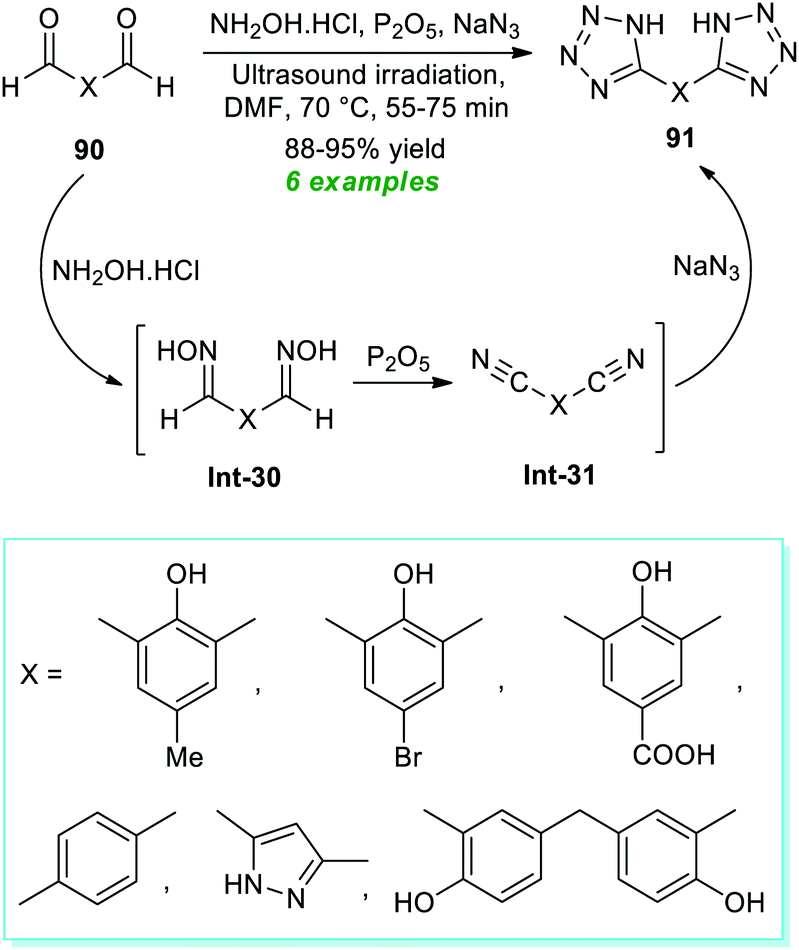 | ||
| Scheme 23 Ultrasound-irradiated one-pot synthesis of bis-tetrazole derivatives. | ||
Later, an isocyanide-based multicomponent click reaction towards constructing diverse substituted tetrazole derivatives by employing ultrasound techniques under catalyst-and solvent-free conditions was devised by Gámez-Montaño and co-authors (Scheme 24).100 With the help of their optimized reaction conditions, the corresponding tetrazole products 95 were derived from various aromatic and aliphatic substituted isocyanides 92, TMSN3 93, and water 94; the products were unaffected by the presence of different substituents on various positions of the isocyanide ring. Broad functional group tolerance, reduced reaction time, simple handling, mild set-up, and column-free are some salient features of this strategy.
 | ||
| Scheme 24 Tetrazoles obtained by the ultrasound-assisted isocyanide-based multicomponent click reaction. | ||
3. Ultrasound irradiation-promoted transition-metal-free synthesis of six-membered heterocycles
3.1 Synthesis of six-membered heterocycles containing one-heteroatom
In a continuous effort to establish an eco-friendly and environmentally benign protocol, Pagadala et al.115 in 2020 disclosed a one-pot ultrasound-assisted multicomponent strategy for the construction of various diversely substituted pyridine derivatives (Scheme 25). The authors initially performed a four-component reaction between benzaldehyde 23, malononitrile 96, ammonium hydroxide 97, and ethyl methyl ketone 98 or cycloheptanone 99 in the presence of different catalyst systems such as acetic acid, gold, MgO, and iodine, and other solvent systems such as ethanol and acetonitrile under sonication at room temperature. The observation discovered that the reaction comprising iodine as a catalyst and ethanol or acetonitrile as a solvent efficiently offered high yields of the requisite compounds. Although both solvent systems worked well in the present protocol, ethanol has been recognized as the ideal medium to realize this reaction based on a green chemistry point of view, as acetonitrile was not recommended as a green solvent. With these conditions in hand, various substituted aryl aldehydes were examined to establish the efficacy of their protocol. Accordingly, all the tested aldehydes smoothly provided the products 100 and 101 in 91–97% and 91–96% yield, respectively, at 1.5–2 hours. The authors proposed a mechanism to recognize this reaction, which started with the initial Knoevenagel condensation between aldehydes 23 and malononitrile 96 under the influence of iodine to give the adduct Int-32 that can then experience nucleophilic addition from 98 via Michael addition to form the intermediate Int-33. Treatment of Int-33 with NH4OH leads to the formation of intermediate Int-34. The intermediate Int-34 after cyclization forms 1,4-dihydropyridine Int-35 and yields the final product 100.
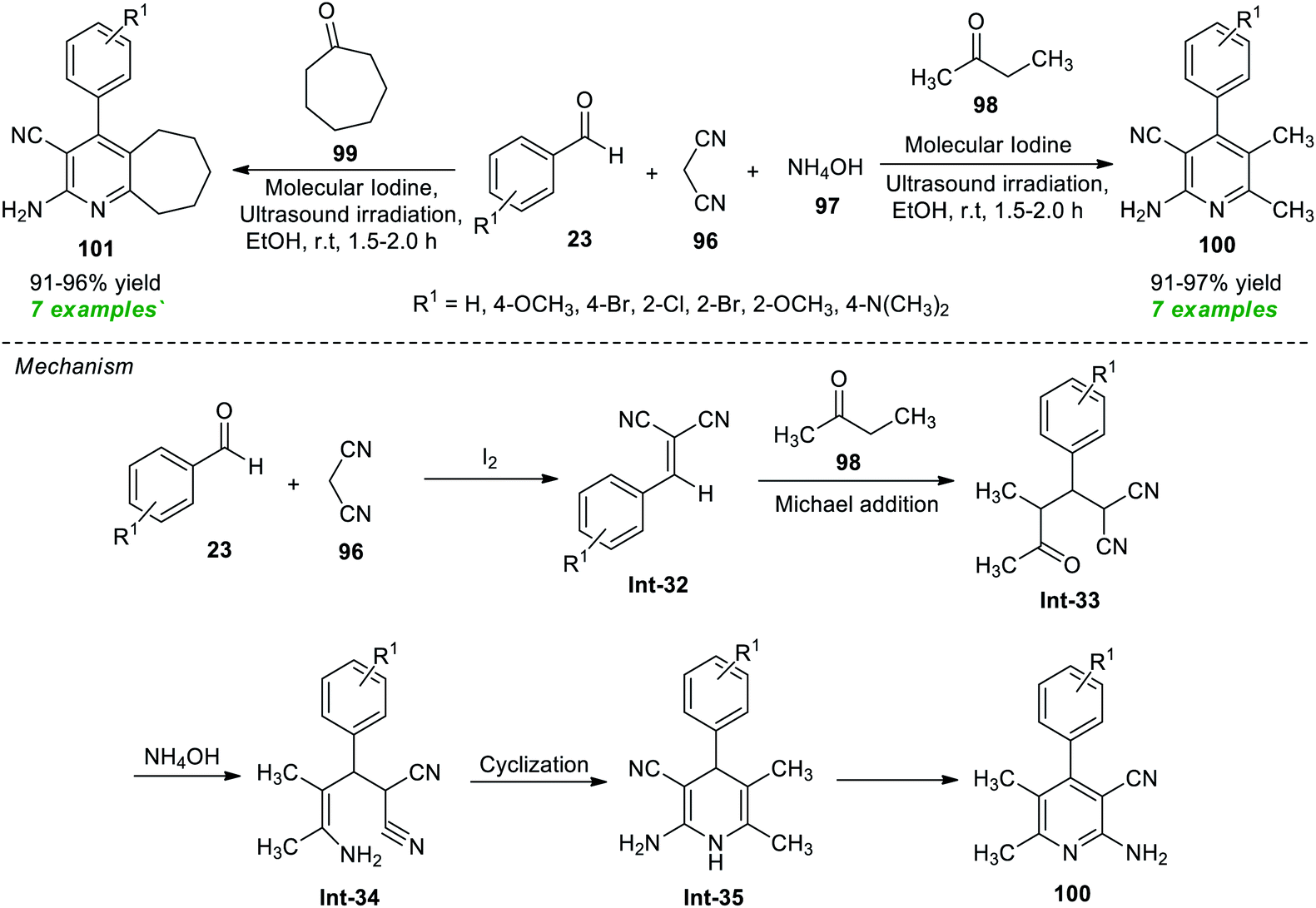 | ||
| Scheme 25 Iodine-catalyzed ultrasound-assisted multicomponent synthesis of substituted pyridines. | ||
At the same time, Dastjerdi and Ghanbari have prepared a novel nanohybrid catalyst via the functionalization of H5PW10V2O40 (HPA) with 2-APTS-4,6-bis(3,5-dimethyl-1H-pyrazol-1-yl)-1,3,5-triazine (ADMPT) linked SBA-15 mesoporous silica and the formation of the nanohybrid was entirely confirmed by FT-IR, TEM, SEM, BET, XRD, and EDX techniques (Scheme 26).116 The catalytic performance of the nanohybrid (SBA-15@ADMPT-HPA) was examined in the ultrasound irradiated one-pot reaction between aromatic/heteroaromatic aldehydes 23, with an amine source 24 or 26, malononitrile 96, and cyclic ketones 102 in ethanol as the solvent at room temperature. The catalyst was discovered to be very consistent in producing the respective pyridine products 103 and it could be separated easily and utilized for further consecutive cycles with a negligible loss in product yields. With the help of such an expeditious condition, a total of fifteen pyridine products 103 were synthesized in 79–95% yield in a relatively short time. The combination of the sonochemical activation strategy and the utilization of SBA-15@ADMPT/HPA as a heterogeneous catalyst efficiently makes this protocol eco-friendly and environmentally sustainable.
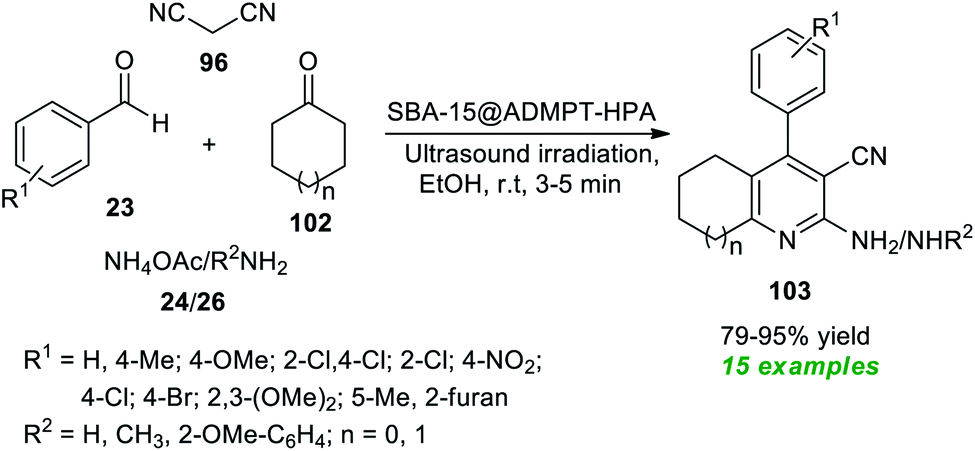 | ||
| Scheme 26 Sonochemical-assisted assembly of diverse pyridine derivatives. | ||
The Rajanna group has demonstrated the synergic combination of 2,4,6-trichloro-1,3,5-triazine (TCTA) with N,N-dimethylformamide (DMF) as an effective Vilsmeier–Haack (VH) reagent for the ultrasound-assisted cyclization of acetanilides to form the substituted quinolines in high yield within 35–95 minutes (Scheme 27).127 This reaction condition was proven to be extremely successful in delivering eleven different compounds. Although the reaction could be accomplished using the traditional approach, it would take a considerably longer reaction time. Therefore, ultrasound irradiation was regarded as the method of choice for this reaction with respect to product derivation and environmental friendliness. Some of the appealing features of this protocol include the use of TCTA as an environmentally benign, easily accessible, and cost-effective substance, a simple reaction set-up, and broad substrate scope. The overall reaction can proceed through the formation of TCTA–DMF adduct Int-36 that could be transformed to chloromethyleniminum cation Int-37 for further reaction with acetanilides 104 to form the final products 105 via intermediate Int-39.
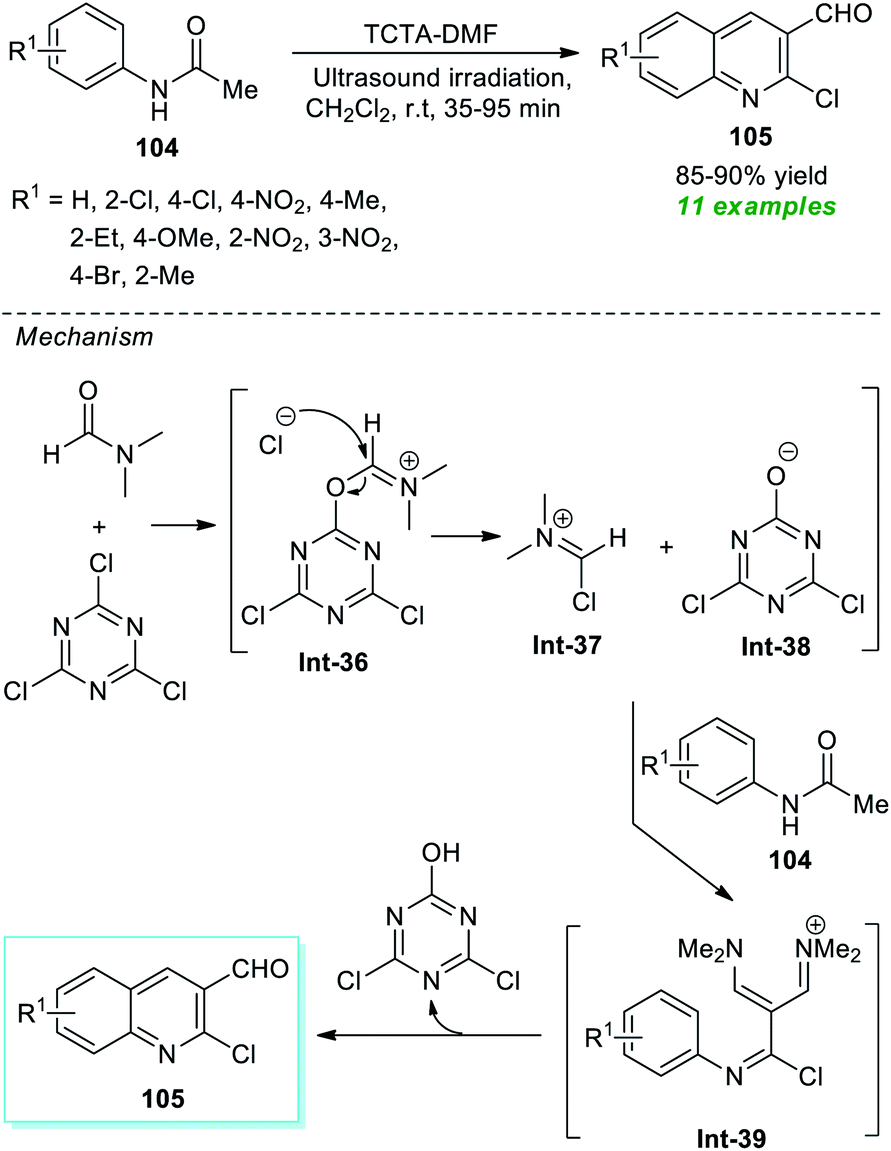 | ||
| Scheme 27 Expedient sonochemical synthesis of substituted quinolines. | ||
Bazine et al.128 in 2020 synthesized a plethora of novel quinoline scaffolds bearing the α-aminophosphonate moiety via Kabachnik–Fields reaction by employing ionic liquid triethylammonium acetate (TEAA) as the catalyst and the solvent system under sonication (Scheme 28). The entire procedure begins with the formation of chloro-substituted formyl quinolines 105 from the condensation of acetanilides 104 with Vilsmeier–Haack reagent (DMF–POCl3) through the Meth–Cohn reaction.129 The so formed quinoline derivatives 105 on treatment with substituted amines 26 and PO(OEt)3 in the presence of TEAA in ultrasonication afforded the desired quinoline derivatives 106 bearing an α-aminophosphonate core in moderate to good yield. While executing the reaction without using TEEA, the rate of the reaction was found to be extremely slow, and the yield of the products was comparatively low. This protocol has many advantages, including a simple workup procedure, a fast completion rate, mild set-up, energy efficiency, and so on. However, the limited substrate scope and reasonably low product yields point toward the drawback of this method that necessitates future developments.
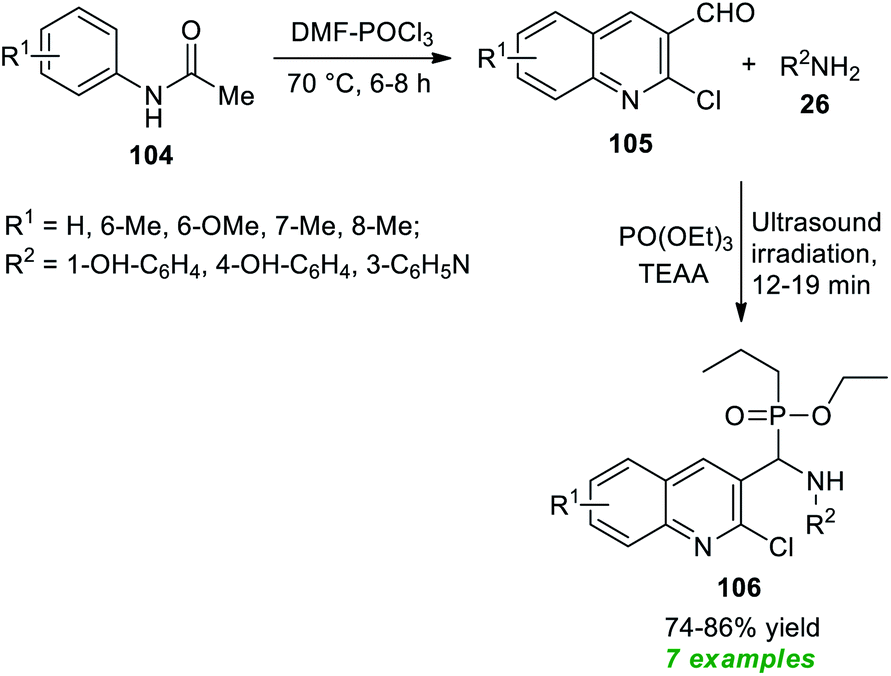 | ||
| Scheme 28 Ultrasound-assisted rapid access to quinolines bearing α-aminophosphonate. | ||
To realize the importance of 4H-pyrans, particularly 2-amino-3-cyano-4H-pyrans, with the main focus on developing a green pathway, Pasha and co-workers have reported the utilization of iodine as a highly reactive and efficient catalyst in the sonochemical-assisted cyclo-condensation reaction of commercially accessible aldehydes 20, malononitrile 96, and substituted 2,3-diketones 107 in an aqueous medium to synthesize the respective 2-amino-3-cyano-4H-pyran products 108 only in 10 minutes (Scheme 29).139 The methodology was demonstrated to be feasible for a variety of 1,3-diketones and aldehydes, and a total of twelve target compounds 108 were accomplished in 85–97% yields. The utilization of ultrasound irradiation makes the protocol energy-efficient and reduces reaction times. The mechanism behind this reaction begins with the iodine-mediated Knoevenagel condensation of 20 and 96, thereby delivering the intermediate Int-40. In this step, the presence of iodine activates the carbonyl compounds towards the nucleophilic attack of malononitrile. Michael's addition of the enolate form Int-41 of 107 with the intermediate Int-40 delivers Int-42, which can then be cyclized to provide the intermediate Int-43. The consequent tautomerization of Int-43 yields the final products 108.
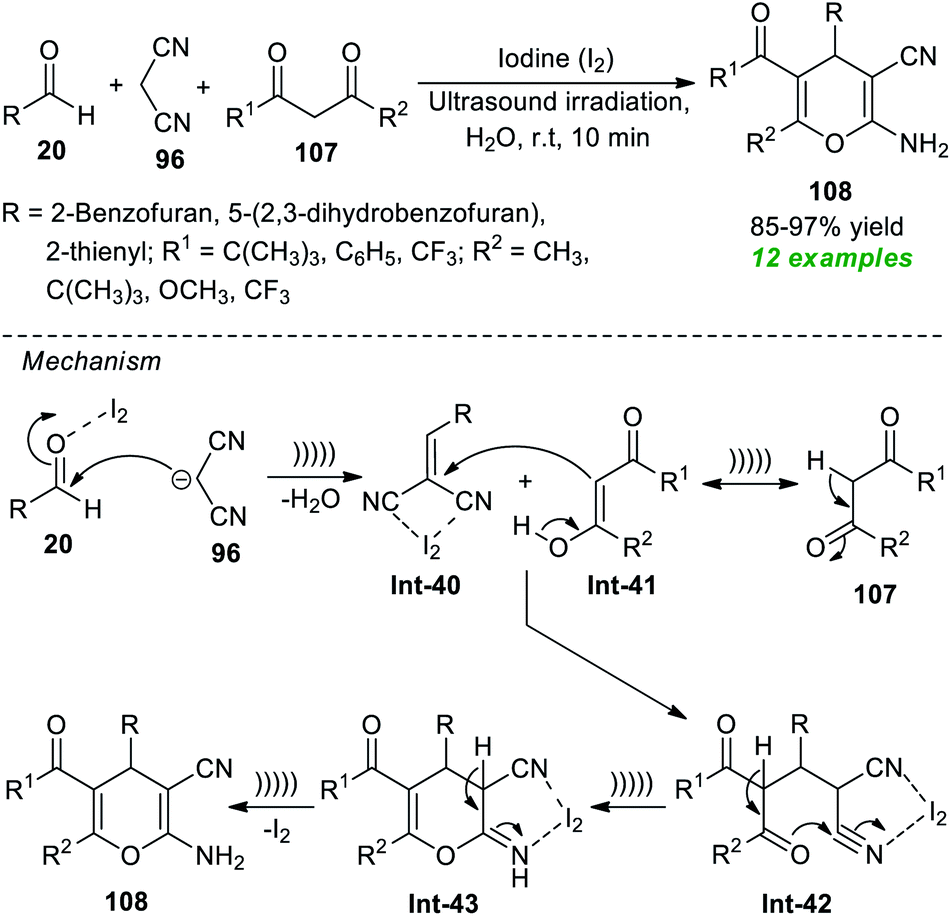 | ||
| Scheme 29 Iodine-catalyzed three-component sonochemical-assisted synthesis of 4H-pyrans. | ||
Another successful application of ultrasound as an eco-friendly activation method to access 2-amino-3-cyano-4H-pyrans was demonstrated by the group of Herrera (Scheme 30).140 Under the influence of 20 mol% of Et3N as the organic base catalyst, an ultrasound-assisted three-component reaction of readily available aldehydes 20, malononitrile 96, and 1,3-diketones 107 was executed in water at room temperature for 60 minutes, which efficiently afforded the desired 4H-pyran products 108 in 44–98% yields. This operationally easy reaction worked well not only with aryl aldehydes but also with heteroaryl substituted aldehydes, resulting in a total of 15 compounds.
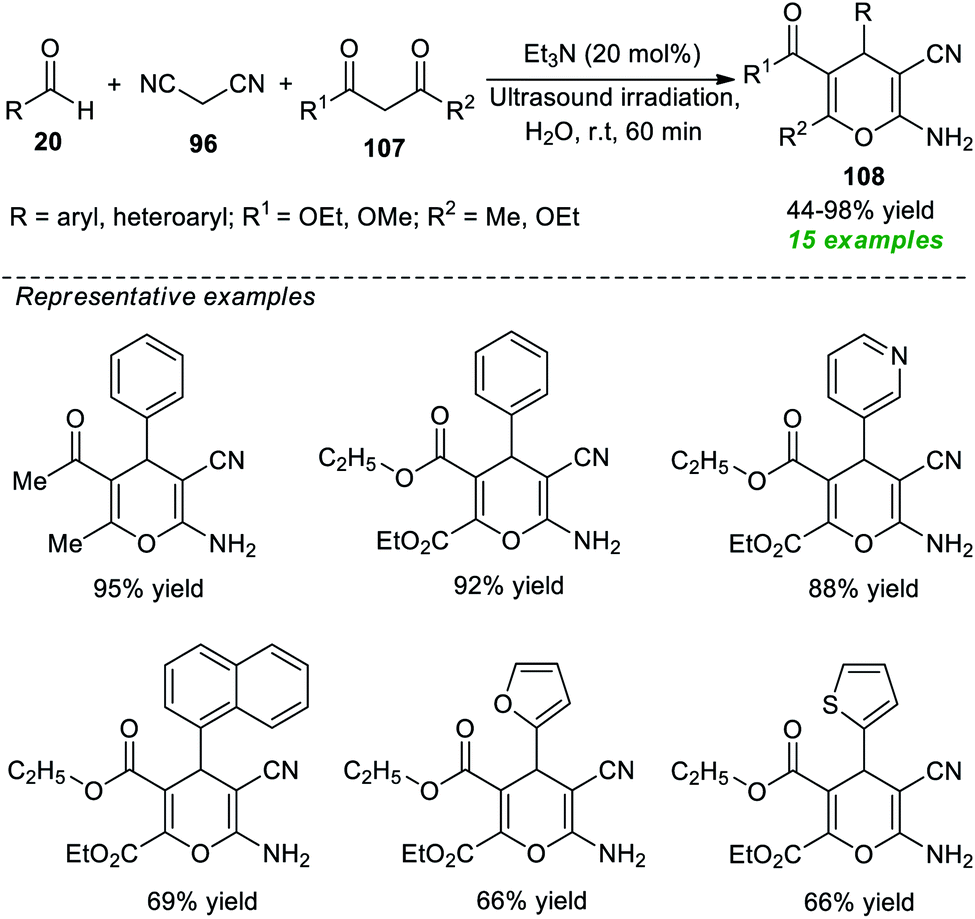 | ||
| Scheme 30 One-pot ultrasound-assisted construction of 4H-pyrans using Et3N as a catalyst. | ||
3.2 Synthesis of six-membered heterocycles containing two-heteroatoms
In 2018, Nikalje et al.150 disclosed an ultrasound irradiated step-wise strategy to realize the construction of 2-amino pyrimidine derivatives 114 coupled with indolin-2-ones (Scheme 31). Initially, the synthesis involves the ultrasound irradiation promoted condensation of 4-chloroacetophenone 109 with aryl/heteroaryl aldehydes 20 using 40 mol% of KOH as the base catalyst in EtOH at room temperature to form the enones 110 in 84–92% yield in 15–25 minutes. Although the reaction is achieved with conventional heating conditions, it takes a lot of time (4–6 hours) to deliver the products. In the next step, the ultrasound-assisted reaction of enones 110 and guanidine 111 was performed with the help of the same catalytic amount of KOH in ethanol at 50 °C for a time of 20–30 minutes that efficiently afforded the 2-amino substituted pyrimidine 112, which on further treatment with isatin 113 using glacial acetic acid as the catalyst in ethanol at 50 °C furnished the respective products 114 in 86–94% yield. Similarly, the formation of 112 and 114 from 110 can be done using thermal heating conditions; however, due to the requirements of a long reaction time in every step, sonochemical activation was recognized as the most effective approach in terms of reaction time as well as from the perspective of sustainable chemistry.
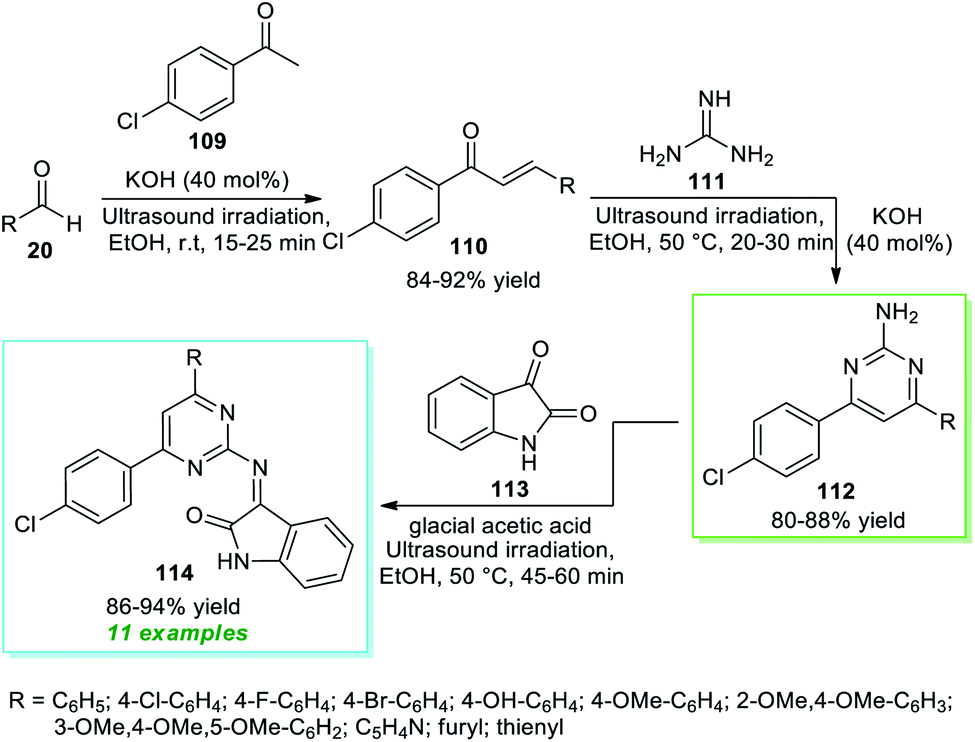 | ||
| Scheme 31 Ultrasound-assisted multistep synthesis of diversely substituted pyrimidines 114. | ||
A straightforward and energy-efficient green protocol for the construction of various 2-amino substituted pyrimidines was disclosed by Güllü and co-workers in 2019 (Scheme 32).151 Under ultrasound irradiation, the treatment of different β-diketone compounds 115 with guanidine hydrochloride 116 using various base catalysts including Na2CO3, NaOH, and NaOEt in an aqueous medium was found to smoothly proceed at 60–70 °C to deliver the corresponding 2-aminopyrimidine derivatives 117 in 72–80% yield. The presence of various substituents on the β-diketone ring plays a crucial role in this reaction as well as in the yield of the products. The β-diketone ring bearing ester moiety (R1 = OEt or R2 = OEt) underwent the reaction in the presence of NaOEt, and hydrolysis of the ester moiety has occurred, thereby delivering the 2-aminopyrimidine ring with an –OH group at 4- and 6-positions (R4 = R5 = OH), whereas methyl-substituted β-diketones (R1 or R2 = Me) were effectively worked under the influence of NaOH or Na2CO3, and methyl-substituted 2-aminopyrimidine derivatives were observed. A comparison study of both conventional and ultrasound techniques revealed that a high-temperature condition of around 100 °C for a time of 5–6 hours was required when the reaction was carried out conventionally rather than sonochemically that was completed at only 60–70 °C in 30 minutes.
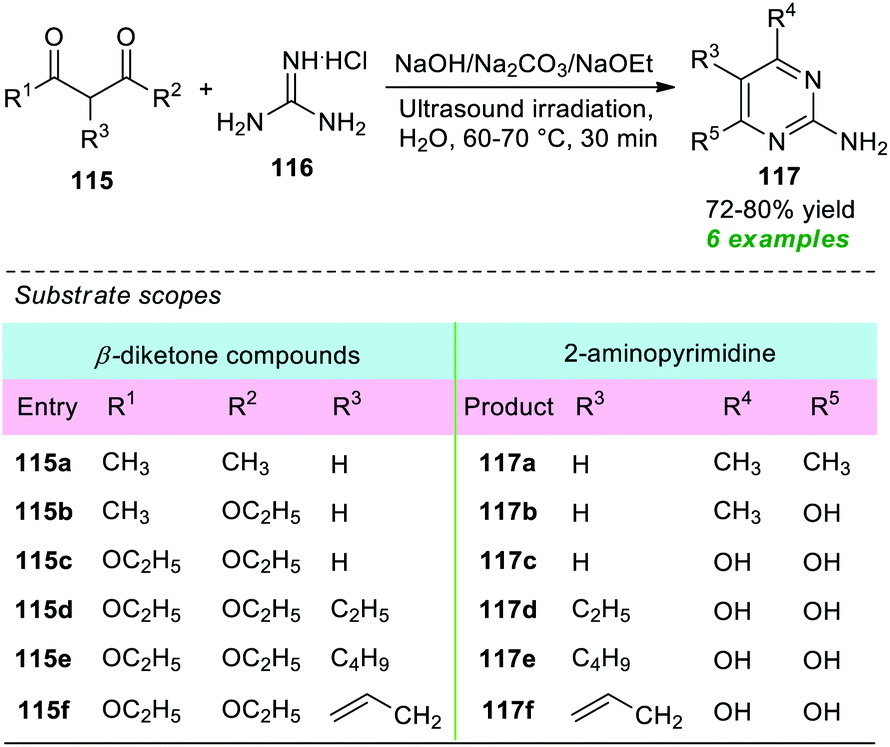 | ||
| Scheme 32 Base-catalyzed sonochemical synthesis of 2-aminopyrimidine in an aqueous medium. | ||
To realize the outstanding participation of quinoxalines in drug design and discovery, Rouhani and Ramazani in 2018 disclosed an isocyanide-based multicomponent approach for rapid access to a variety of highly functionalized quinoxalines by introducing ultrasound irradiation as a robust alternative activation approach (Scheme 33).163 In this regard, a one-pot three-component reaction between aldehydes 23, with 1,2-diamine 118, and isocyanide 119 in the presence of perlite-SO3H nanoparticles as the efficient catalyst was performed in ethanol under ultrasound irradiation for a time of 37–42 minutes at room temperature. The reaction condition tolerates a wide variety of aryl aldehydes with different electron-poor, and electron-rich substituents, and a total of nine quinoxaline products 120 were synthesized in 91–94% yield. Although the preferred quinoxaline products could be synthesized using the thermal method, the reaction rate was found to be exceedingly sluggish, and the yield was not satisfactory, which recommended the sonochemical approach as the method of consideration from the viewpoint of green and synthetic chemistry.
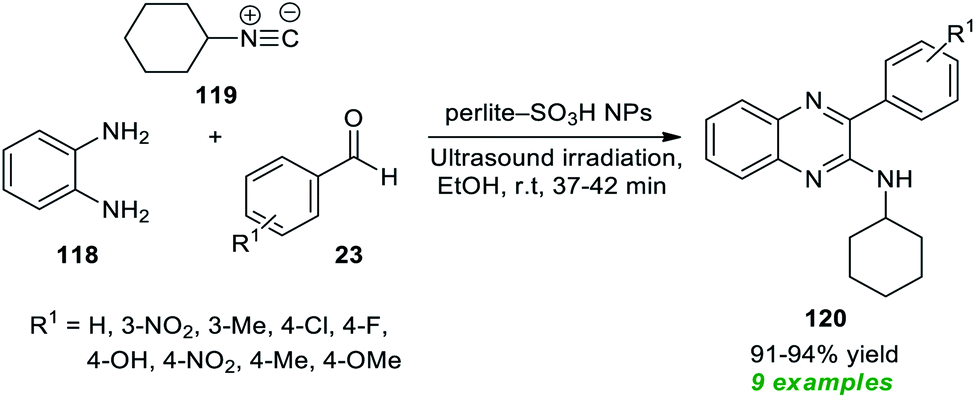 | ||
| Scheme 33 One-pot three-component sonochemical synthesis of quinoxalines. | ||
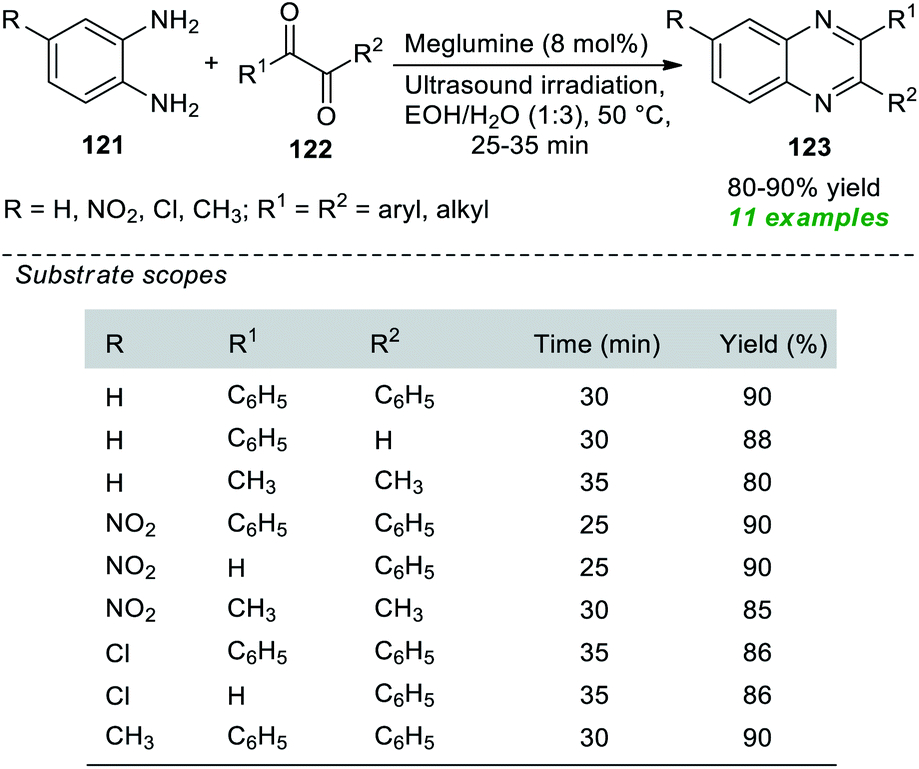
Nongkhlaw et al. in 2019 pioneered the use of meglumine as a biodegradable organocatalyst in association with the application of sonochemical activation for the production of various quinoxaline derivatives 123 (Scheme 34).164 Initial optimization of the appropriate amount of the catalytic system and reaction medium for the reaction of 1,2-diamine 121 with 1,2-diketone 122 revealed that only 8 mol% of the catalyst was sufficient for catalyzing this reaction and aqueous ethanol was found to be the best solvent under sonication as well as under conventional heating conditions. However, the reaction requires much more time under traditional heating conditions, and the desired quinoxaline products have been achieved in low yield in contrast to the reaction executed under ultrasound irradiation. A variety of o-phenylenediamines, as well as aryl and alkyl-substituted 1,2-diketone, were well tolerated by this mild approach, and a total of eleven quinoxalines were obtained in 80–90% yield. Broad functional group tolerance, operational simplicity, cost-effectiveness, being environmentally benign, and toxic-free are some of the advantages of this protocol.
 | ||
| Scheme 34 Organocatalytic ultrasound-assisted two-component synthesis of quinoxalines. | ||
Given the properties mentioned above, Latip, Seo, and co-workers devised an atom-economical and practical one-pot multicomponent strategy to access a plethora of highly functionalized quinazolinones under ultrasound irradiation conditions (Scheme 35).178 With the help of 20 mol% of p-TSA as the catalytic system, the desired quinazolinone products 127, derived from the reaction of isatoic anhydride 124, 2-furoic hydrazides 125, and various substituted salicylaldehydes 126 in aqueous ethanolic solution under ultrasound irradiation at ambient temperature for 55–70 minutes, were obtained in 71–96% yields. The salicylaldehydes with various electron-rich and electron-poor groups have seemed to be efficiently worked under the standard conditions and have no negative impact on the yield of the products. Some of the prepared compounds were also identified as inhibitors of the tyrosinase enzyme. The utilization of sound waves as an efficient green method, mixed green solvents, short reaction time, and easy isolation process are some of the key highlights of this approach.
 | ||
| Scheme 35 Acid-catalyzed multicomponent synthesis of quinazolinones under sonication. | ||
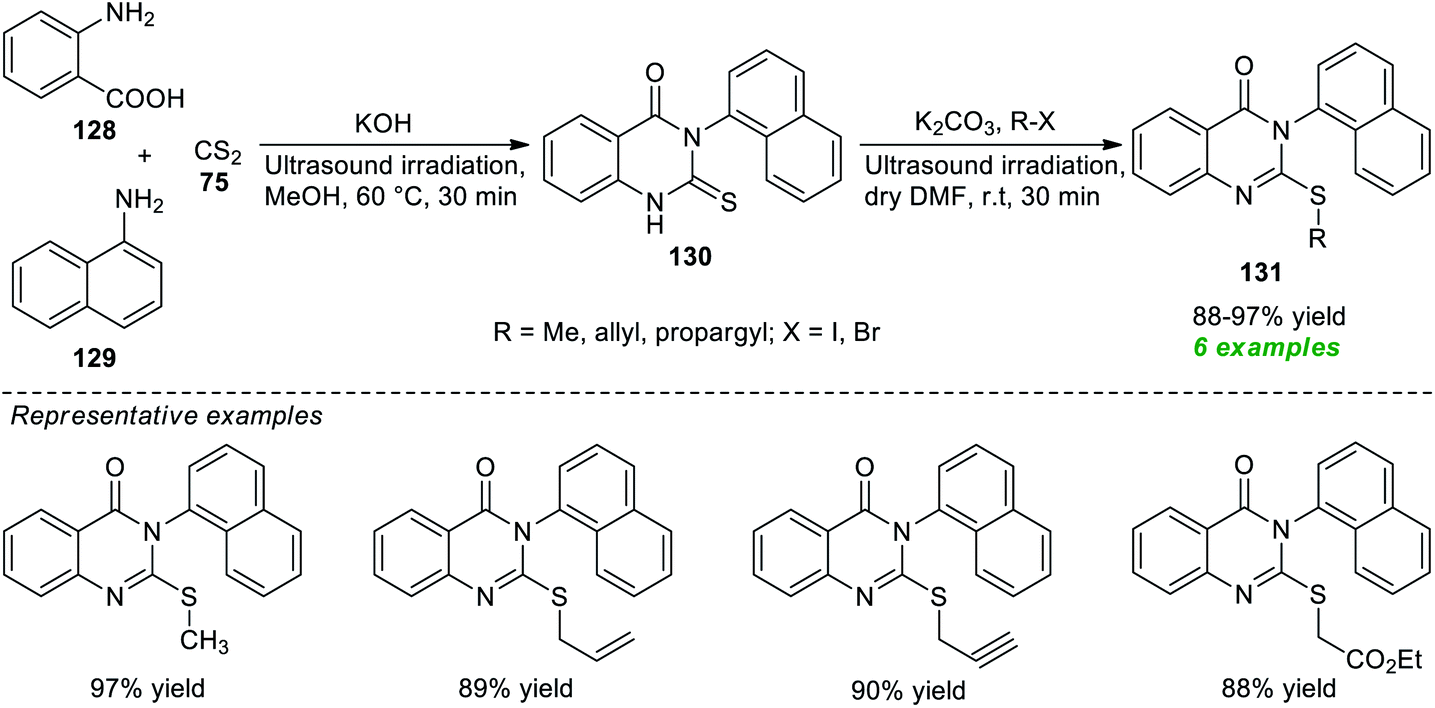
A facile and straightforward protocol for expedient access to a variety of alkyl-substituted quinazolinones has been disclosed by Hagar, Ashry, and their group in 2021 (Scheme 36).179 With the help of KOH as a base catalyst, an ultrasound-assisted treatment of anthranilic acid 128, 1-naphthylamine 129, and carbon disulfide 75 was found to occur smoothly at 60 °C to lead to the products 130, which on further reaction with different alkyl halides catalyzed by K2CO3 in dry DMF under ultrasound irradiation at room temperature furnished the corresponding products 131 in 88–97% yields. Although the present methodology was categorized as efficient, eco-compatible, and sustainable, and the products were easily obtained without using column chromatography, the limited number of substrates denotes a shortcoming of this procedure, demanding further advancements in broadening the substrate scope, otherwise outstanding developments.
 | ||
| Scheme 36 Ultrasound-assisted multicomponent reaction mediated two-step synthesis of quinazolinones. | ||
4. Ultrasound irradiation-promoted transition-metal-free synthesis of complex fused poly-heterocycles
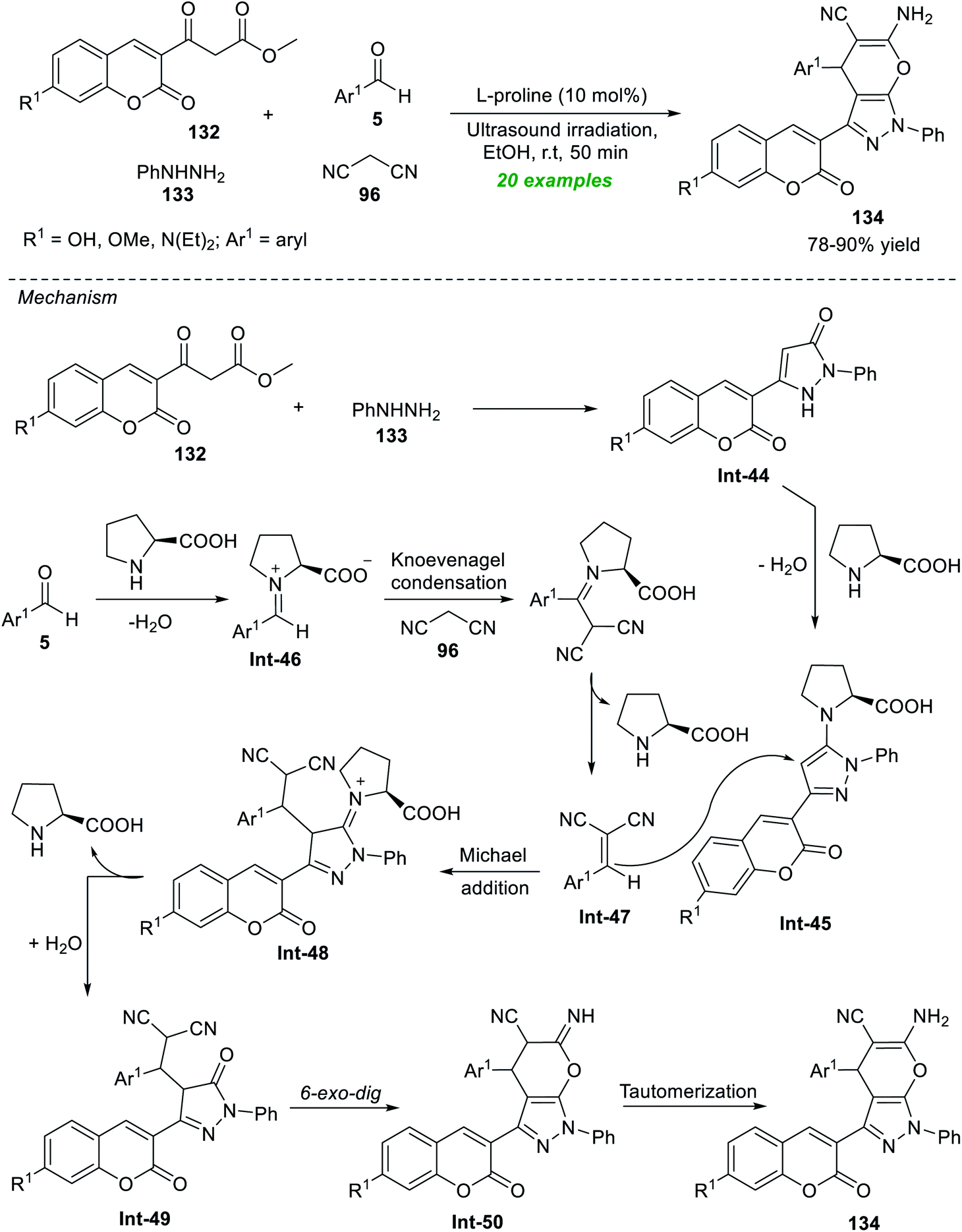
A broad spectrum of complex fused heterocyclic compounds in transition-metal-free catalysis under ultrasound irradiation have been achieved in the last few years. The synthesis of these compounds where several heterocycles are combined in such a way that leads to the formation of new single molecules from easily available simple starting materials has recently received considerable attention from chemists and pharmacologists for the outstanding properties of the targeted compounds.180–182The development in this regard realizes the outstanding contribution of Seydimemet et al. to the expedient synthesis of a plethora of coumarin-containing pyrano[2,3-c]pyrazoles by introducing the sonochemical activation strategy under organocatalytic conditions (Scheme 37).183 Under the influence of 10 mol% of L-proline, the one-pot domino reaction of β-ketoesters 132 with phenylhydrazine 133, aromatic aldehydes 5, and active methylene 96 in ethanol was carried out in sonication, which provided the respective products 134 in 78–90% yield after 50 minutes. Broad functional group tolerance, efficiency, fast reaction rate, cost-effectiveness, and toxic-free nature are some highlights of this methodology. The formation of 134 can be initiated via the initial reaction of 132 with 133 to produce the coumarin–pyrazolone intermediate Int-44, which afforded the intermediate Int-45 under the influence of L-proline. In the meantime, L-proline catalyzed condensation of 5 with 96 yields the intermediate Int-47. The subsequent conjugate addition of Int-45 to Int-47 afforded the intermediate Int-49. The final 6-exo-dig cyclization of intermediate Int-49 and then tautomerization via intermediate Int-50 yields the actual product 134.
 | ||
| Scheme 37 Organocatalytic sonochemical synthesis of pyrano[2,3-c]pyrazoles linked with coumarin. | ||
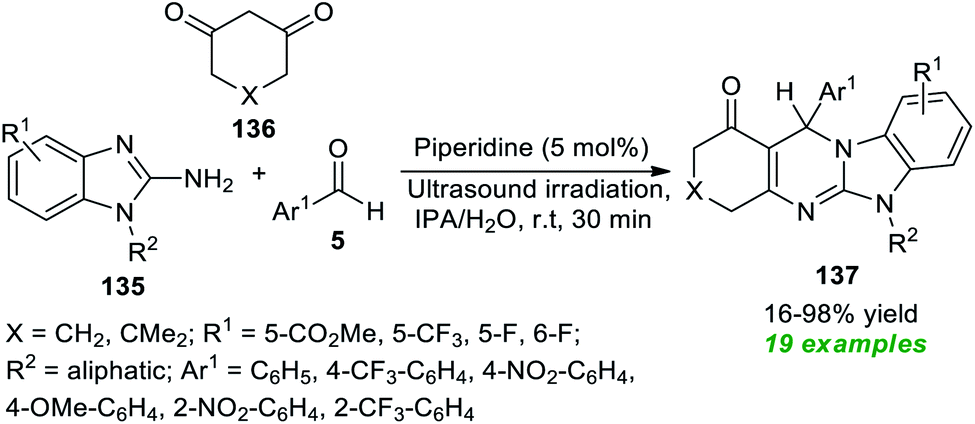
Sun et al.184 disclosed an ultrasound-irradiated three-component reaction of 2-amino-benzimidazoles 135, aryl aldehydes 5, and 1,3-dione 136 by employing 5 mol% of organic base piperidine as the catalyst for the assembly of diversely substituted imidazo[2,1-b]quinazolinones 137 (Scheme 38). During the optimization of the reaction parameters, a variety of solvents were tested, including CH2Cl2, MeOH, THF, toluene, CH3CN, DMF, IPA, H2O, and IPA/H2O; among them, IPA/H2O turned out to be an efficient solvent system for this reaction, delivering the products in excellent yield in a short duration of time. A diverse set of aromatic aldehydes with varied electron-poor and electron-rich substituents were examined to broaden the substrate scope, and all of them have been discovered to proceed efficiently under optimal conditions. Similarly, substitution on the 2-amino-benzimidazole ring and 1,3-diketone was found to have no substantial effect on the yield of the products, and all have smoothly participated in the reaction. A total of nineteen benzimidazo[2,1-b]quinazolin-1(1H)-one products 137 were synthesized in poor to excellent yield. This procedure has a broad spectrum of substrate scope, is less time-consuming, and is cost-effective. The comparatively low yield of the product 137 (R1 = 5-CO2Me, R2 = cyclohexane, X = CH2, Ar1 = 4-OMe-C6H4) somewhat denotes a limitation of this approach. This is most likely because of the presence of the 4-OMe group on the aldehyde ring, which leads to the formation of 4-methoxybenzyl alcohol and 4-methoxy benzoic acid as by-products.
 | ||
| Scheme 38 Three-component organocatalyzed synthesis of benzimidazo[2,1-b]quinazolin-1(1H)-ones under sonication. | ||
Construction of pyrimidine fused bioactive compounds, namely pyrido[2,3-d]pyrimidine 140 through a domino Knoevenagel–Michael addition initiated multicomponent reaction under ultrasound conditions has been achieved by Bhat and co-authors (Scheme 39).185 By employing 20 mol% of DMAP (4-dimethylaminopyridine) as an organocatalyst, the corresponding products 140, accomplished from the three-component reaction of readily available aldehydes 23, active methylene compounds 138, and 6-aminouracil 139 in DMF at room temperature, were obtained in 82–93% yields. The reaction was initially performed under various catalysts such as DMAP, DBU, DABCO, piperidine, and morpholine in different solvent systems like H2O, MeOH, EtOH, CH2Cl2 and DMF. Among the catalysts and solvents tested, the utilization of DMAP as the catalytic system and DMF as the solvent system was found to be highly efficient for the fast reaction rate and for providing a good yield of the products. The optimal condition was extremely compatible for a variety of aromatic aldehydes containing electron-rich and electron-poor substituents on the different positions of the aromatic ring of the aldehydes. When compared to the traditional thermal method, which took nearly 2–6 hours, ultrasound irradiation reduces the reaction time from 32 minutes to 18 minutes. Similarly, when applying ultrasound techniques, product yields were boosted by up to 93%, compared to the conventional method, which only provides 67–89% yields.
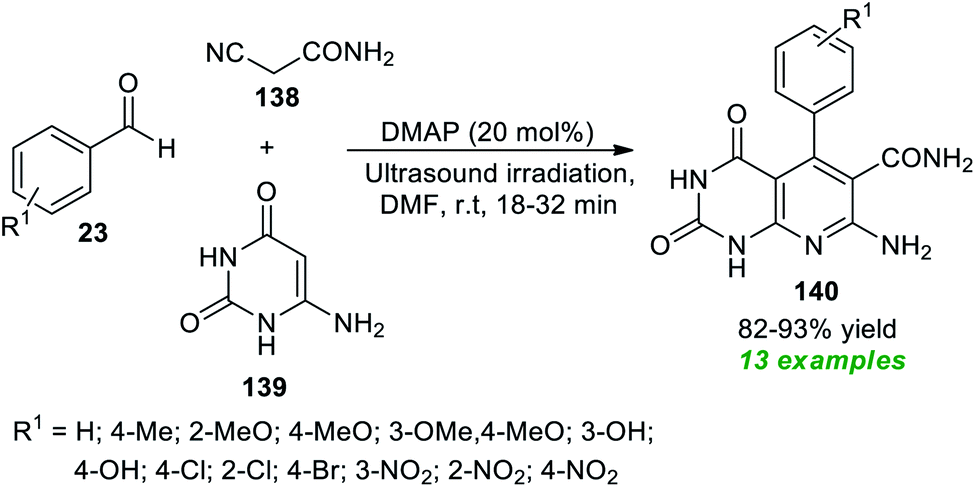 | ||
| Scheme 39 DMAP-catalyzed ultrasound-assisted synthesis of pyrido[2,3-d]pyrimidines 140. | ||
Considering their wide-ranging chemical landscape and biological properties, pyrazolone moieties were recognized as a versatile building block for the creation of diverse heterocyclic compounds. Among them, dihydropyrano[2,3-c]pyrazole is a well established fused heterocycle synthesized from pyrazolone, which displays various pharmacological properties like anti-bacterial, anti-HIV, insecticidal, anti-infective, anti-platelet, anti-fungal, anti-cancer, anti-microbial, antioxidant, analgesic activity, etc.79,181,186,187
Due to the above mentioned pivotal significance of pyrano[2,3-c]pyrazoles, Kotha and his co-workers demonstrated an atom-economic approach for the rapid access to a library of pyrano[2,3-c]pyrazoles 142 from a three-component ultrasound-assisted reaction of aldehydes 23, malononitrile 96, and pyrazolone 141 in aqueous ethanolic solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at room temperature using sodium fluoride (NaF) as the base catalyst (Scheme 40).188 A diverse range of aryl aldehydes bearing different substituents were efficiently worked by this method, and overall, 12 target compounds have been accomplished in 88–98% yield within only 5–10 minutes. The broad functional group tolerance, reduced reaction time, cost-effectiveness, clean reaction profile, low catalyst loading, and green solvents mark the highlights of this protocol.
:1, v/v) at room temperature using sodium fluoride (NaF) as the base catalyst (Scheme 40).188 A diverse range of aryl aldehydes bearing different substituents were efficiently worked by this method, and overall, 12 target compounds have been accomplished in 88–98% yield within only 5–10 minutes. The broad functional group tolerance, reduced reaction time, cost-effectiveness, clean reaction profile, low catalyst loading, and green solvents mark the highlights of this protocol.
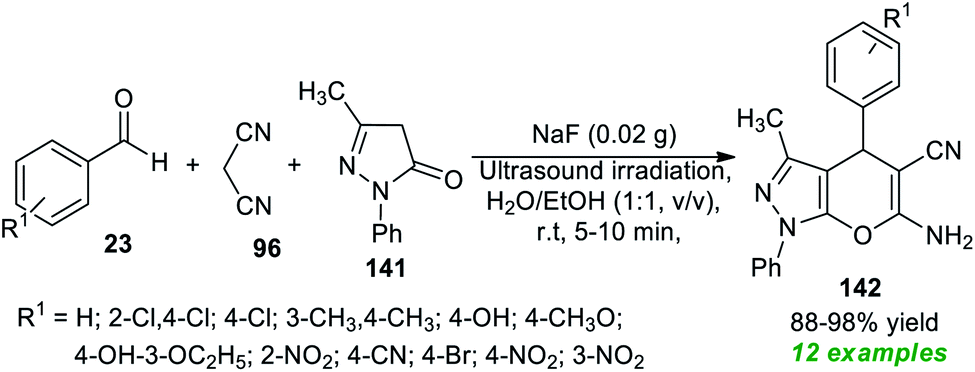 | ||
| Scheme 40 NaF-catalyzed MCRs to access pyrano[2,3-c]pyrazoles under sonication. | ||
Subsequent to this report, a three-component tandem reaction of triazolyl aldehydes 143, malononitrile 96, and pyrazolone 144 was carried out in ultrasound irradiation by employing 20 mol% of NaHCO3 as the base catalyst in water at 30 °C for 5–7 minutes (Scheme 41).189 This reaction afforded the 1,2,3-triazolyl based pyrano[2,3-c]pyrazoles 145 in 92–98% yields. Although different mild bases such as Na2CO3 and K2CO3 could tolerate the reaction, it took a lot of time, and excellent yield was achieved only when NaHCO3 was used rather than Na2CO3 or K2CO3. The authors evaluated the potential therapeutic properties of the synthesized compounds, and most of the compounds were found to be efficient anti-fungal and antioxidant agents.
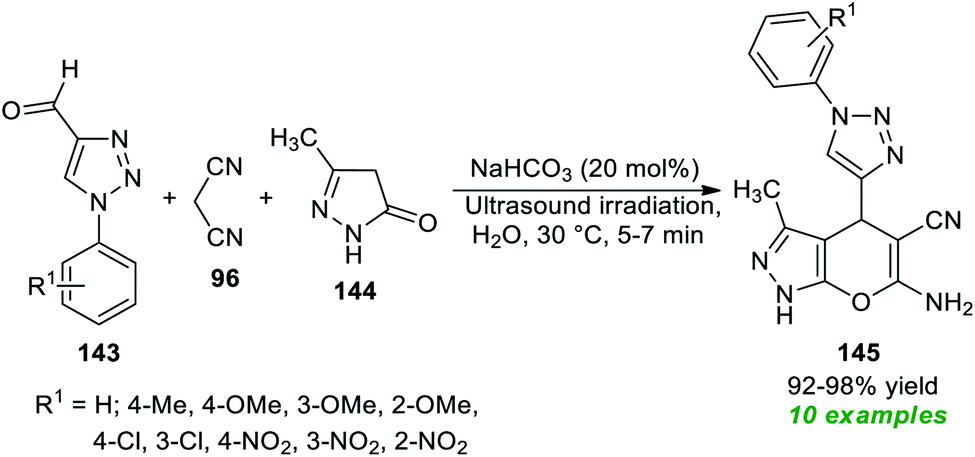 | ||
| Scheme 41 NaHCO3-catalyzed sonochemical synthesis of triazole-based pyrano[2,3-c]pyrazoles. | ||
Brahmachari et al.190 established a simple, straightforward, and environmentally benign multicomponent approach for the practical construction of structurally divergent chromeno[4,3-d]pyrido[1,2-a]pyrimidin-6(7H)-ones (Scheme 42). By introducing 20 mol% of sulphamic acid (NH2SO3H) as an organocatalyst, they executed an atom- and pot-economic reaction between readily accessible 4-hydroxycoumarin 146, substituted aldehydes 20, and 2-aminopyridine 147 with an equimolar mixture of aqueous ethanolic solution (1:1, v/v) under ultrasound irradiation under ambient conditions, which provided the respective products 148 in 70–97% yields. Both aromatic aldehydes featuring diverse substitution on C-3 and C-4 positions and substituted heteroaromatic aldehydes reveal this reaction condition to be entirely compatible. The use of sonochemistry in this reaction shortens the reaction time and results in a clean, high-yield synthesis of the products. It's worth noting that no co-catalyst, additives, or reagents are required for the reaction. The broad substrate scope, clean and green solvents, high reaction mass efficiency, and high atom efficiency mark the highlights of this protocol. The overall transformation starts with the initial NH2SO3H catalyzed Claisen–Schmidt condensation reaction between 20 and 146, producing 52. The subsequent nucleophilic addition of Int-52 with 147 delivers the adduct Int-53 that can then undergo tautomerization and cyclization to afford the intermediate Int-55. Finally, the elimination of water from the intermediate Int-55 yields the corresponding product 148.
 | ||
| Scheme 42 Sulfamic acid-catalyzed multicomponent synthesis of chromeno[4,3-d]pyrido[1,2-a]pyrimidin-6(7H)-ones under sonication. | ||
A simple and highly convenient strategy to synthesize a diverse set of fused quinoxaline scaffolds under ultrasound irradiation from readily available o-phenylenediamine was devised by Srivastava et al. (Scheme 43).191 The synthesis involves the treatment of o-phenylenediamine 149 with isatin 13 under the catalyst-free conditions in the presence of water as the green reaction medium in sonication at room temperature, which afforded the desired complex-fused quinoxaline products, namely indolo[2,3-b]quinoxalines 151 in good to excellent yields within 3.2–4.1 minutes. Similarly, under the standard experimental parameter, the treatment of 149 with ninhydrin 152 yielded a variety of indeno[1,2-b]quinoxalines 153 in outstanding yields in a short interval of time. This reaction effectively proceeded for a range of diverse substitutions on the isatin ring and the 1,2-diamine ring. Both unsubstituted isatins (R1 = H) and substituted isatins (R1 = Et, Bn, and Pr) reacted efficiently under the standard reaction conditions.
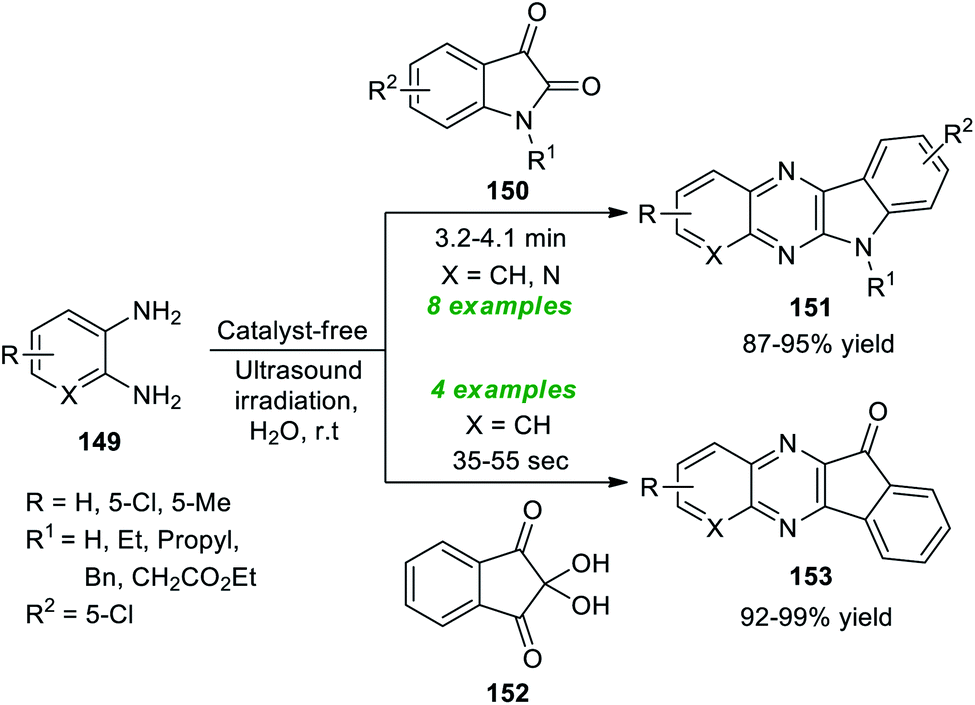 | ||
| Scheme 43 Ultrasound-assisted synthesis of diverse fused quinoxalines. | ||
Safaei-Ghomi et al.192 demonstrated the ultrasound irradiation technique as an eco-friendly activation method for the highly diastereoselective synthesis of coumarin-fused furans, namely furo[3,2-c]coumarins 157 (Scheme 44). Under the influence of 3 mol% of magnesium oxide nanoparticles (MgO NPs) as a highly reactive catalyst, the respective products 157 were accomplished from readily available substituted 4-hydroxycoumarin 154, aromatic aldehydes 23, bromo-substituted acetophenone 155, and pyridine 156 in ethanol at room temperature. Overall ten compounds have been synthesized by this reaction as a single diastereomer in an 85–92% yield. The reaction was very fast and completed within only 10–15 minutes under sonication compared to the conventional heating condition, which marks the advantage of this approach. The synthetic potentiality of the present method was established from the successful gram-scale synthesis of the model compounds with reduced catalyst loading. Other notable features of this technique include broad substrate scope, simple work-up procedures, easy product isolation with no tedious column chromatography techniques, easy separation of the catalyst, and re-utilization for further reactions with negligible loss in catalytic activity. On the other hand, the catalyst necessitates multistep synthesis at significantly higher temperatures, which indicates the drawback of this method. The postulated mechanism for realizing this reaction involves the MgO promoted Knoevenagel condensation of 154 and 23, thereby producing the intermediate Int-56 on the surface of the catalyst. The pyridinium ylide Int-57 is produced from the reaction of 155 and 156, and then undergoes Michael addition with Int-56. The final product 157 was obtained via intramolecular cyclization of the resultant intermediate Int-58, along with the elimination of pyridines.
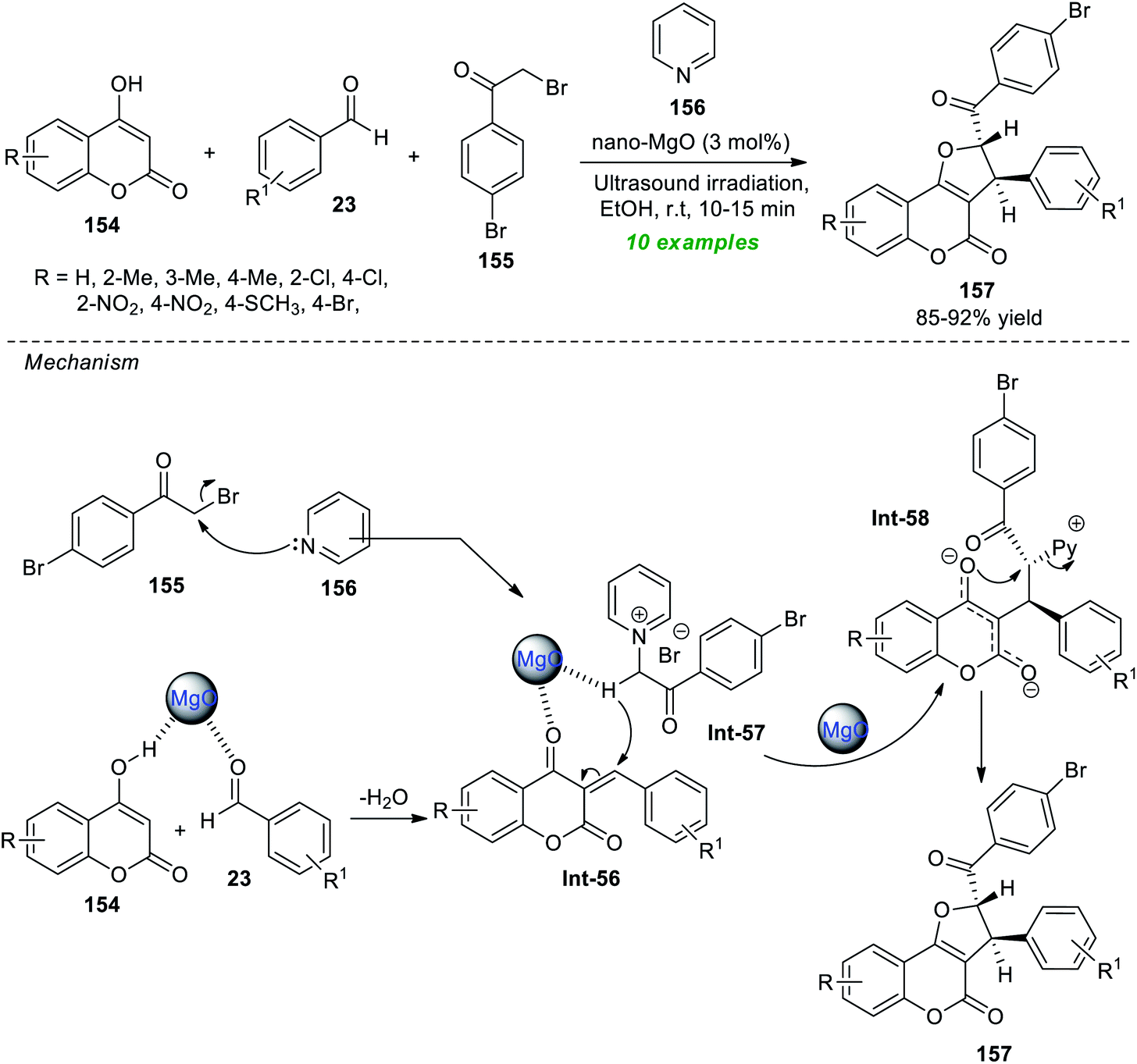 | ||
| Scheme 44 MgO-catalyzed ultrasound-assisted synthesis of furo[3,2-c]coumarin. | ||
Kumari and co-workers illustrated an eco-compatible and sustainable protocol by demonstrating the prolific activity of the ionic liquid as a solvent as well as catalytic system towards the synthesis of a library of dihydro-6H-chromeno[3,4-e]isoxazolo[5,4-b]pyridin-6-ones 159 (Scheme 45).193 Under the influence of [C4mim][HSO4] as the ionic liquid, both traditional thermal methods and ultrasound techniques were used to execute a three-component reaction between 4-hydroxycoumarin 154, aryl/heteroaryl aldehydes 5, and amino-substituted isoxazoles 158. From the experimental outcome, it was revealed that the reaction conducted with conventional heating at 80 °C took almost 20–45 minutes to yield the corresponding products 159 in 90–95% yield, whereas the same reaction when irradiated with ultrasound at room temperature took only 5–15 minutes to produce the expected product 159 in nearly the same quantity. Even though both of these parameters were proven to be effective, the ultrasound technique surpassed the conventional one with respect to reaction time and energy consumption. A total of 15 compounds bearing different substituents on the aryl- and heteroaryl rings of the aldehydes were synthesized by this method. In this regard, the authors proposed a mechanism that starts with the initial condensation of aldehydes 5 with amino-isoxazole 158 to afford the intermediate Int-59 that experiences nucleophilic attack from 4-hydroxycoumarin 154, thereby offering intermediate Int-61. The subsequent 1,2-addition of Int-61 with 158 followed by cyclization leads to the formation of intermediate Int-63. Consequently, a [1,3]-H shift of Int-62 provides the corresponding product 159.
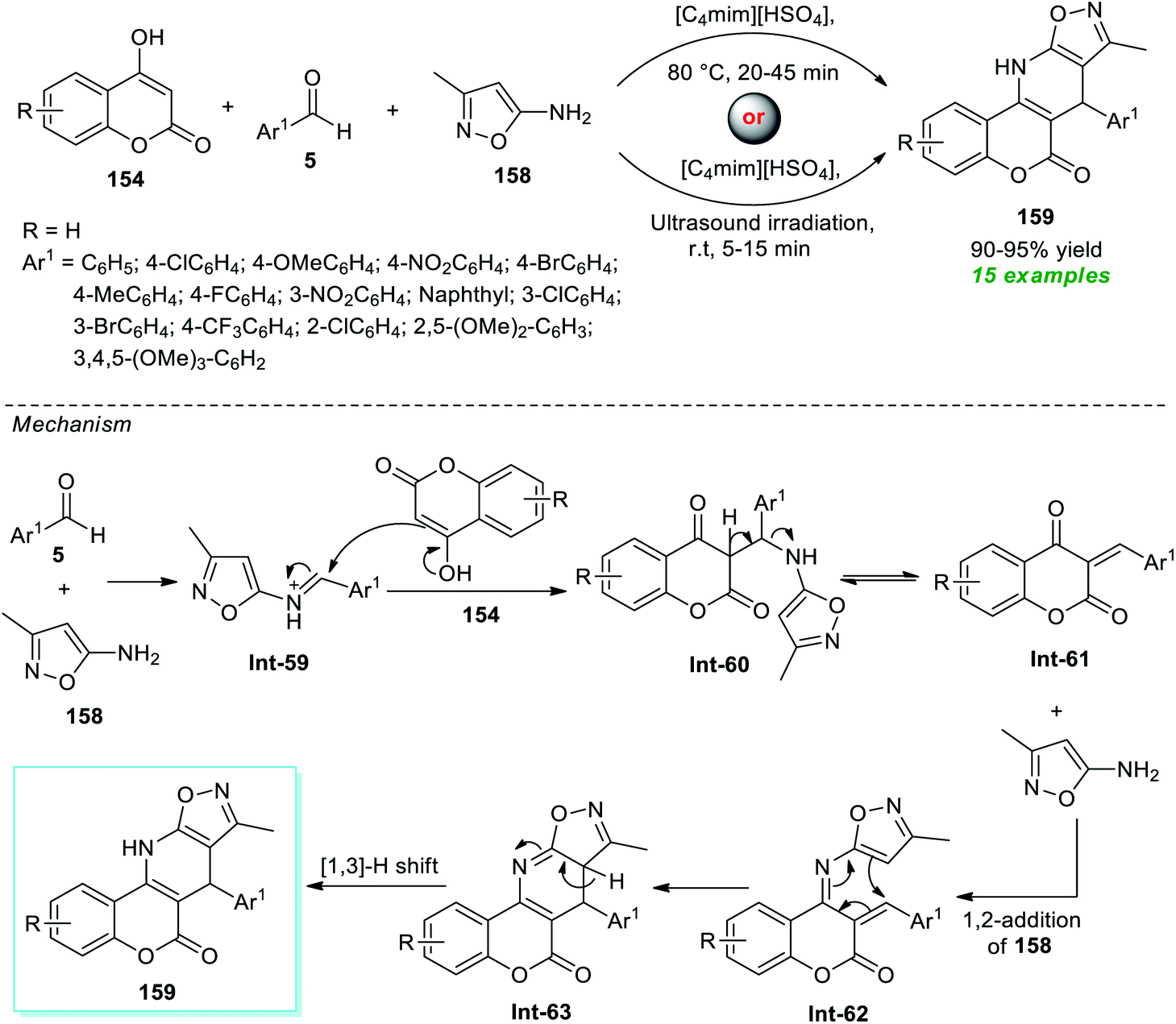 | ||
| Scheme 45 Sonochemical access to chromeno[3,4-e]isoxazolo[5,4-b]pyridin-6-one using ionic liquid catalysis. | ||
Construction of a diverse set of pyrano[3,2-c]coumarin derivatives 161 has been accomplished by Al-bogami et al.194 from the piperidine catalyzed one-pot atom-economical reaction of substituted benzaldehyde 23, methylene compounds 160, and highly reactive 4-hydroxycoumarin 154 under ultrasound irradiation (Scheme 46). The reaction was performed at room temperature with ethanol as the best optimum solvent. Noticeably, all reactants were found to efficiently participate in the reaction in 30–45 minutes, and the target compounds 161 were achieved in 86–94% yields. To broaden the substrate scope, a variety of aromatic aldehydes with varied electron-donating and electron-withdrawing groups and various substitutions on methylene compounds were employed. All are suitably worked under the optimized reaction condition. Although it is possible to execute the reaction using the conventional method to synthesize the desired products, due to the required long reaction time and high energy input, it has limitations from the perspective of synthetic potentiality and sustainability. The sonochemical approach sped up the reaction and increased the product yield, and the entire process took only a few minutes. To explain this transformation, the authors proposed a mechanism that begins with piperidine catalyzed Knoevenagel condensation of active methylene compounds 160a with aldehydes 23a. The resulting intermediate Int-64 undergoes Michael addition with 4-hydroxycoumarin 154a to generate the intermediate Int-65. The final product 161a is obtained via intramolecular cyclization of Int-65 and concurrent tautomerization of resultant intermediate Int-66.
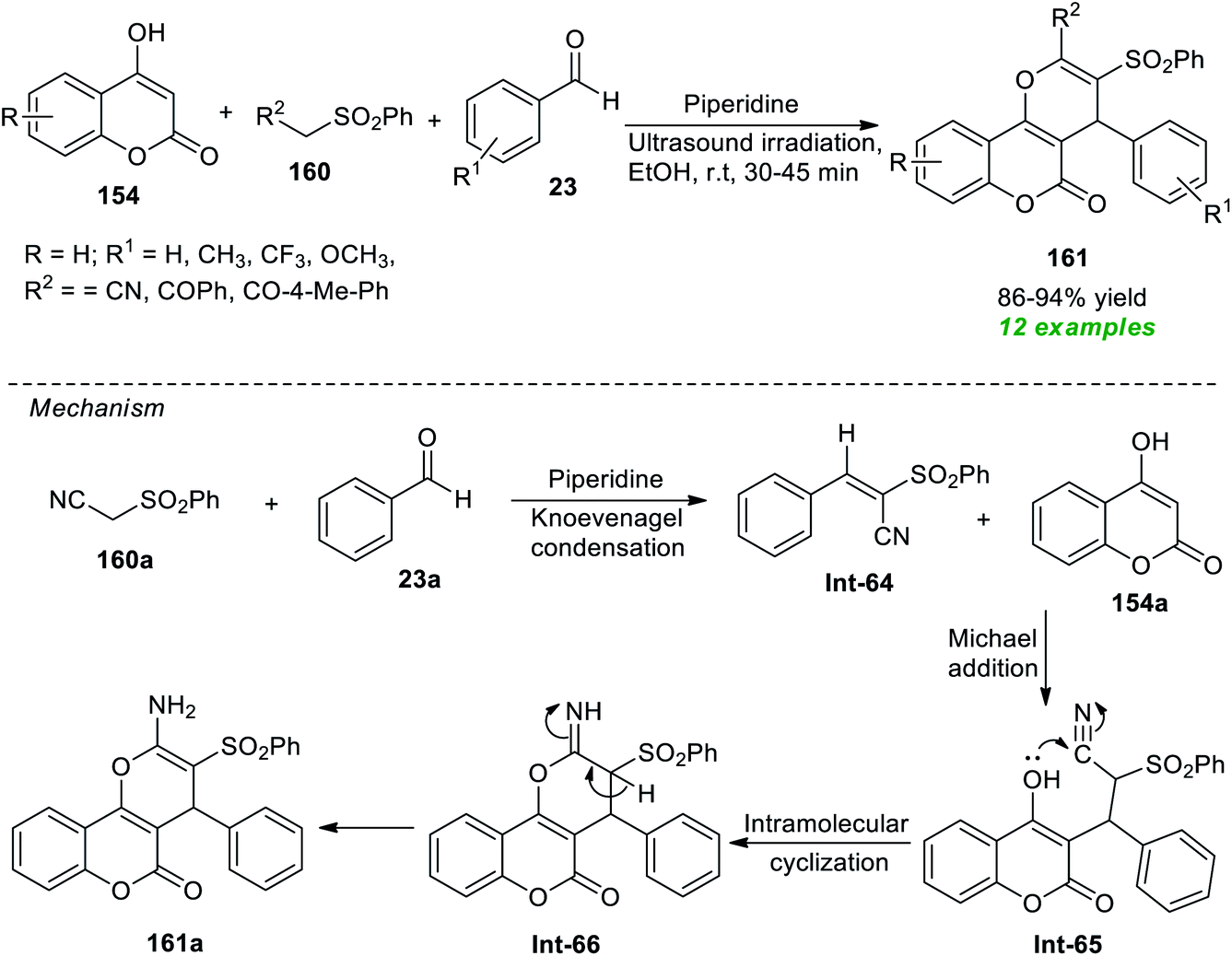 | ||
| Scheme 46 Piperidine-catalyzed ultrasound-assisted multicomponent assembly of pyrano[3,2-c]coumarin. | ||
Application of supramolecular catalysis in the ultrasound-assisted expedient construction of pyrazolo-pyrano[2,3-d]pyrimidine framework 163 has been demonstrated by Shingate and co-workers in 2020 (Scheme 47).195 In this regard, a one-pot reaction of aldehydes 23, barbituric acid 162, ethyl acetoacetate 32, and hydrazine 65 in an aqueous medium was performed with the help of 20 mol% of β-cyclodextrin as a supramolecular catalyst under sonication at 50 °C for 25–70 minutes. This four-component reaction furnished the corresponding products 163 in 84–93% yields. This approach was successful in synthesizing a total of 21 molecules possessing various electron-poor and electron-rich groups on the aromatic as well as the heteroaromatic ring of aldehydes. The feasibility of the current approach was validated by proving the catalyst's recyclability and reusability for the next successive reaction sequences without altering the significant outcome of the protocol.
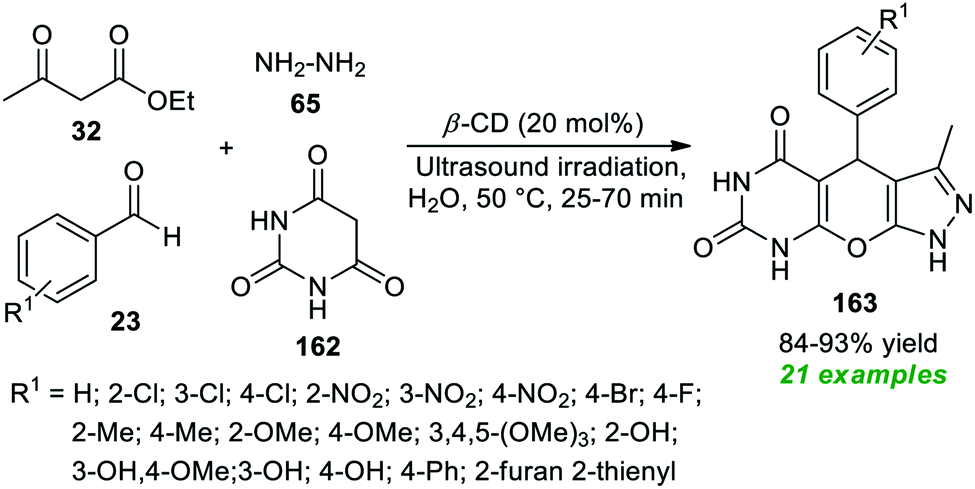 | ||
| Scheme 47 Supramolecular catalysis in the sonochemical synthesis of pyrazolo-pyrano[2,3-d]pyrimidine. | ||
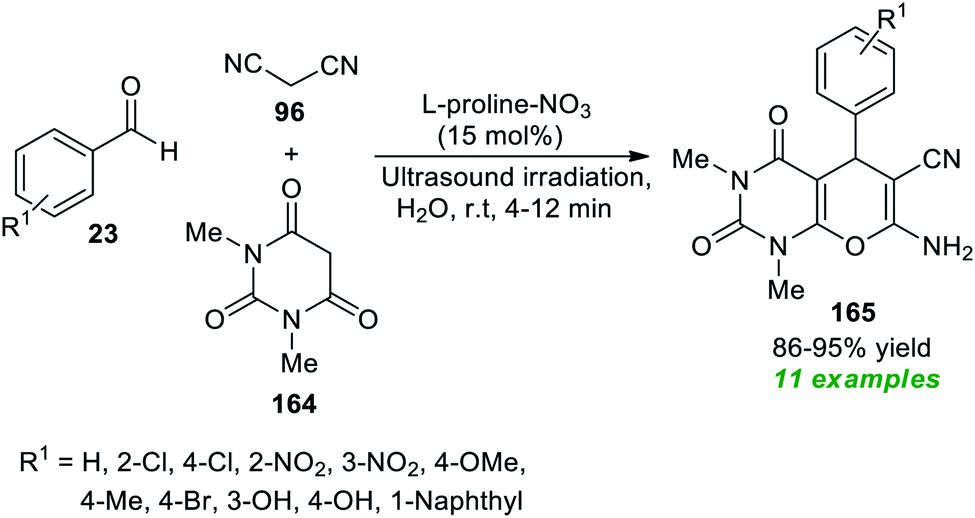
Another great achievement for the utilization of ultrasound irradiation in the construction of complex-fused heterocycles by introducing L-proline-based ionic liquids as the efficient homogeneous catalyst was disclosed by More et al. at the same time (Scheme 48).196 Initially, the ionic liquid L-proline-NO3 was prepared by the authors from the reaction of L-proline with HNO3 in water at 60 °C for 24 hours. After the successful formation of the catalyst, a three-component reaction between aryl/heteroaryl aldehydes 23, malononitrile 96, and 1,3-dimethyl barbituric acid 164 in water was executed using both traditional heating and ultrasonication to examine the catalytic activity of the prepared catalyst. Only 15 mol% of the catalyst was found to be sufficient for catalyzing the reaction, as per the observations. This reaction yielded the corresponding pyrano[2,3-d]pyrimidine products 165 in 86–95% yields in 4–12 minutes with ultrasound irradiation at room temperature. In contrast, the reaction under conventional heating conditions (80 °C) produced the respective products in a lower yield than the ultrasound method.
 | ||
| Scheme 48 L-Proline-NO3-catalyzed three-component synthesis of pyrano[2,3-d]pyrimidine under sonication. | ||
5. Ultrasound-assisted synthesis of complex spiro-heterocycles under transition-metal-free conditions
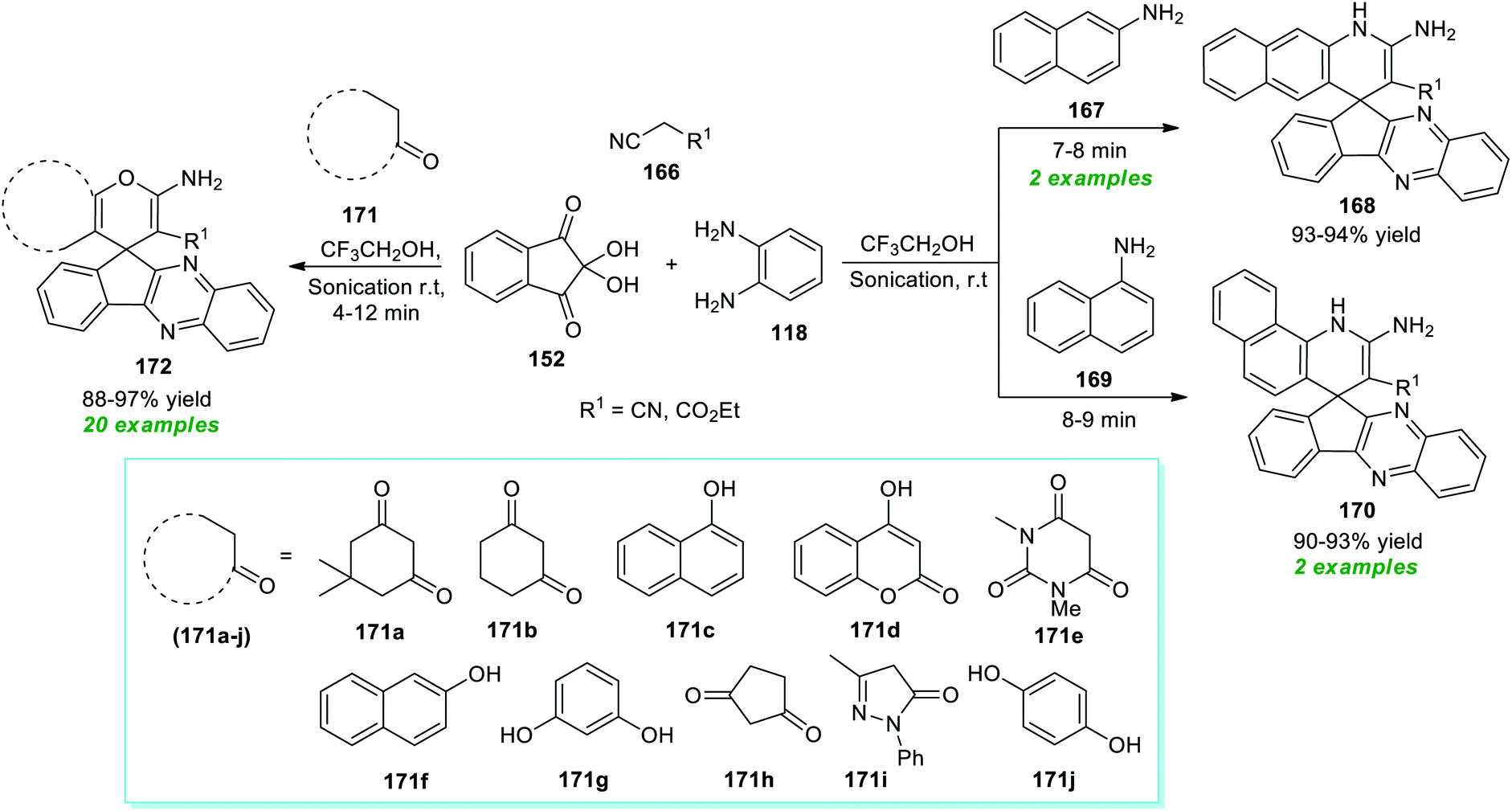
Spiro heterocycles are considered the privileged molecular framework commonly encountered in the basic skeleton of a diverse range of naturally occurring molecules and synthetic pharmaceutical compounds.197–200 A vast array of compounds possessing a spirocyclic core in their structure are known for their antitumor, antimicrobial, antibiotic, antitubercular, and anticancer activities.In this perspective, Heravi and Norouzy successfully demonstrated the use of ultrasound irradiation in the construction of a library of spiro-heterocycles via a multi-component approach by employing catalyst-free conditions (Scheme 49).201 Using trifluoroethanol (TFE) as the reaction medium, the four-component reaction between readily accessible 2,2-dihydroxy-1H-indene-1,3(2H)-dione 152, benzene-1,2-diamine 118, active methylene compound 166, and naphthalen-2-amine 167 or naphthalen-1-amine 169 yielded the corresponding spiro-benzoquinoline-indenoquinoxaline 168 and spiro-benzoquinoline-indenoquinoxaline 170 in 93–94% and 90–93% yield at 7–9 minutes respectively. Interestingly, replacing the amines with various CH-activated acidic compounds 171 was found to efficiently react with 2,2-dihydroxy-1H-indene-1,3(2H)-dione 152, benzene-1,2-diamine 118, and active methylene compound 166 under the same reaction condition, and a series of diverse highly functionalized fused pyran substituted spiro-indeno[2,3-b]quinoxaline products 172 were accomplished in 88–97% yield within 4–12 minutes. The extraordinary demands of relatively high energy conditions along with the prolonged timeframe associated with the conventional method for the completion of the reaction as compared to ultrasound irradiation methods that take place only in a short duration of time make the ultrasound method best for the synthesis of these compounds from the synthetic as well as green chemistry point of view.
 | ||
| Scheme 49 Construction of a library of spiro-heterocycles under ultrasound irradiation. | ||
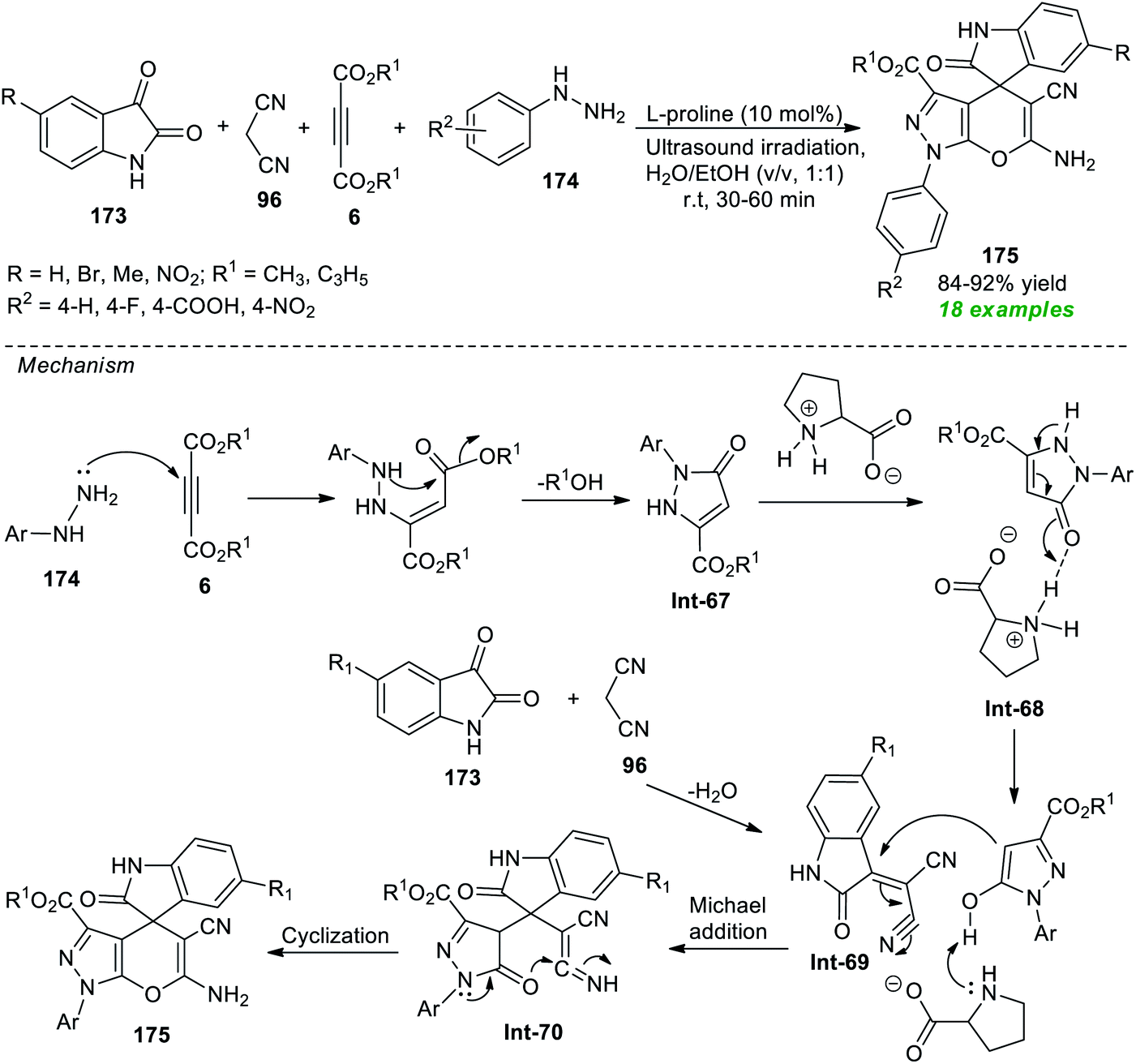
A rapid, and straightforward one-pot ultrasound-assisted secondary amine L-proline catalyzed approach for the synthesis of spiro[indoline-3,4-pyrano[2,3-c]pyrazoles] was devised by Liju and co-workers from the four-component reaction of various isatins 173, malononitrile 96, dialkyl acetylene dicarboxylates 6, and phenyl substituted hydrazines 174 in aqueous ethanolic solution at room temperature (Scheme 50).202 This reaction required a very low loading of the catalyst and following this condition, a diverse range of target compounds 175 have been synthesized in 84–92% yield within 30–60 minutes. The methodology was found to be compatible only with N-unsubstituted isatin. Therefore, there is a need for extending the protocol to N-substituted isatin. To explain the possible formation routes to 175, the authors proposed a mechanism that is depicted in Scheme 50. Initially, an exothermic reaction of 174 with 6 can take place to form pyrazolone derivatives Int-67 which then produce the L-proline activates enolate intermediate Int-68. The Michael addition of the resulting –OH form of pyrazolone with Int-69 (produced from the reaction between 173 and 96) yields the intermediate Int-70. The subsequent cyclization of intermediate Int-70, as well as tautomerization, furnished the corresponding products 175.
 | ||
| Scheme 50 Secondary amine-catalyzed one-pot sonochemical synthesis of amino-substituted spiro-pyrano[2,3-c]pyrazoles. | ||
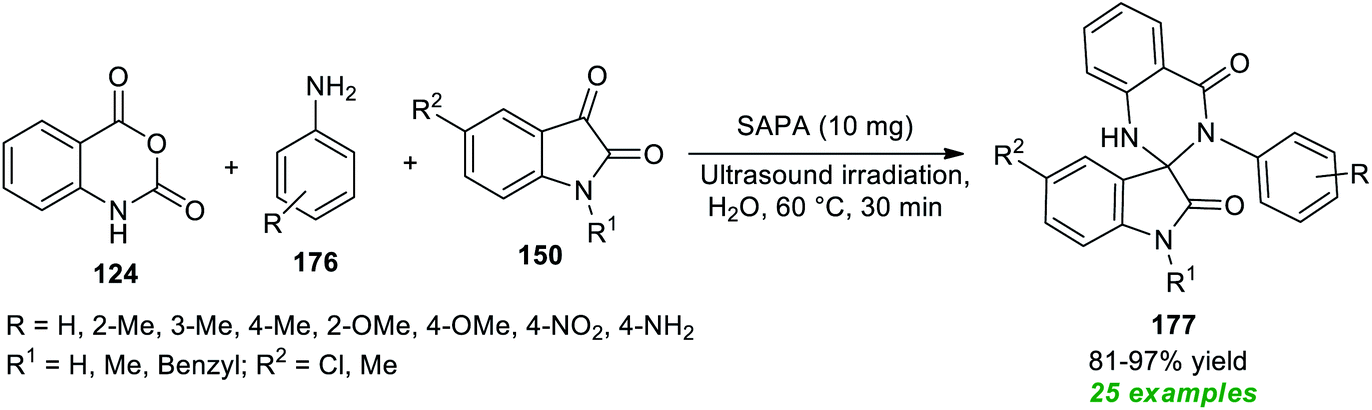
As illustrated in Scheme 51, treatment of isatoic anhydride 124, various arylamines 176, and isatins 150 in water under the influence of a phytic acid-based solid catalyst (SAPA) in sonication at 60 °C afforded a plethora of spiro-oxindole embedded dihydro-quinazolinones 177.203 The catalyst was found to be very efficient for this reaction and could be recycled and reused for further successive reaction sequences without altering the synthetic potentiality of the protocol. This reaction offers a total of 25 compounds in 81–97% yield within only 30 minutes. Variations in the substitution of different groups on the aryl ring of amines had no substantial effect on the reaction rate or product yield. Both N-substituted and N-unsubstituted isatins were found to be quite compatible with this approach.
 | ||
| Scheme 51 Ultrasound-assisted acid-catalyzed construction of spiro-oxindole-based quinazolines. | ||
An eco-compatible and facile constructive approach for expedient access to a variety of spiropyrazoline derivatives 180 via reverse 1,3-dipole mediated [3 + 2]cycloaddition of 2,2-dimethyl-5-[(4-oxo-4H-chromen-3-yl)methylene]-1,3-dioxane-4,6-dione 178 with nitrile imines produced in situ from hydrazonoyl chlorides 179 in the presence of Et3N as the catalyst under ultrasound irradiation was developed by Yavari and Fadakar (Scheme 52).204 The authors screened different types of catalytic systems including DIPEA, K2CO3, Et3N, Cs2CO3, DBU, and DABCO, and solvent systems such as EtOH, CH2Cl2, DMF, EtOAc, and H2O at room temperature for this reaction and identified that Et3N in EtOH under sonication at room temperature was the best combination for this reaction.
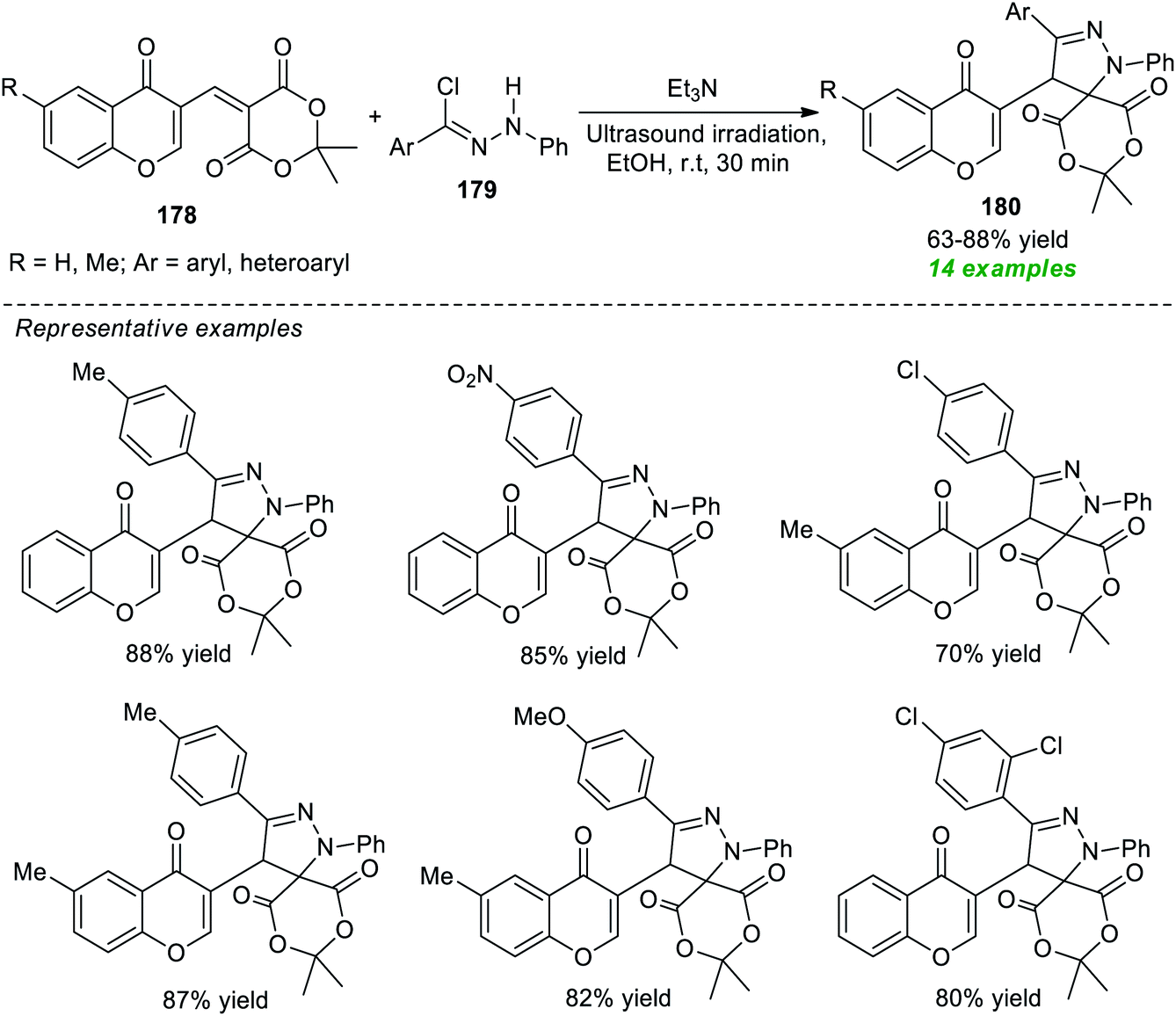 | ||
| Scheme 52 Reverse 1,3-dipole-mediated [3 + 2]cycloaddition for the synthesis of spiropyrazolines under sonication. | ||
6. Conclusion and future perspective
Recognizing the frequent occurrences of heterocyclic compounds in the basic skeleton of diverse ranges of natural products, potential bioactive compounds, pharmaceutical agents, and optoelectronic materials, the development of efficient methodologies to realize their synthesis as well as for converting easily accessible raw materials into significant structural scaffolds that find huge application in many branches of chemistry has remained a formidable challenge for chemists and pharmacologists. Nevertheless, the people of this millennium are fully conscious of the importance of protecting their living environment from pollution caused by various hazardous reagents and solvents in the form of chemical waste during a chemical process both on industrial and laboratory scales.In this pursuit, the field of synthetic organic chemistry has witnessed outstanding growth in the last decades for modifying synthetic chemical processes with the main focus to reduce the cost-effectiveness of the transformation by introducing environmentally benign conditions to offer a hazard-free and sustainable environment. Accordingly, the emergence of ultrasound irradiation as an alternative efficient activation method has opened up new frontiers for carrying out diverse organic transformations. It has a lot of advantages over traditional heating conditions, being eco-friendly and consuming less amount of energy. Consequently, the development of potential synthetic routes that can be operated in sonication is becoming extremely significant both scientifically and technically. In particular, sonochemistry in transition-metal-free catalysis offers a lot of opportunities in the recent efforts to establish green and more sustainable chemistry. The successful combination of the features of ultrasound irradiation as an environmentally acceptable activation method and the efficiency of transition-metal-free catalysis in promoting chemical reactions is predominantly relevant in achieving the ambition of green and sustainable chemistry.
Concerning this prominent significance, the present review article aims to highlight the recent progress accomplished in the application of ultrasound irradiation as a non-conventional energy source for the synthesis of a wide variety of oxygen, nitrogen, and sulfur-containing five-membered and six-membered heterocycles as well as complex-fused heterocycles and spiro-heterocycles by employing transition-metal free catalysis. We endeavored to highlight the drawbacks and limitations of the existing protocols in addition to demonstrating the successful improvements made in the creation of diverse heterocycles using these ecologically friendly approaches. The possible scope of future developments is also highlighted.
From the aforementioned observation made in this review, it is clear to conclude that the appearance of sonochemistry in transition-metal-free catalysis allows for all the reactions to be carried out in the absence of high energy conditions, toxic and expensive metal catalysts, or co-catalysts, ligands, volatile organic solvents or hazardous reagents. The use of water, ionic liquid, deep eutectic solvents, and other solvents such as ethanol, PEG, and aqueous ethanol makes the reported work environmentally and eco-friendly benign. In addition, solvent-free methods are being developed in metal-free environments using ultrasound irradiation. Furthermore, the majority of the synthesized compounds were discovered to have significant therapeutic activities. Notwithstanding these developments, some of the previously existing techniques have drawbacks such as narrow substrate scope, low product yields, and high catalyst loading. Consequently, considerable attention still needs to be paid to improving and expanding the scope of the reactions, with an outstanding product selectivity, by introducing readily accessible raw materials that are easy to handle and cost-effective and developing a scalable protocol with ultralow catalyst loading that will find a wider application to the industrial area.
We hope the information presented here will help researchers for identifying the current evolution accomplished in the synthesis of diverse-heterocyclic scaffolds under sonication under metal-free conditions as well as in developing many more precise and concise efficient synthetic routes using various metal-free catalysts especially organocatalysts, and stimulating further inventive developments that could find immense application to many branches of chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
BB thanks UGC for the fellowship and Central University of Gujarat, Gandhinagar for the support.Notes and references
- (a) A. R. Katritzky, Chem. Heterocycl. Compd., 1992, 28, 241–259 CrossRef; (b) N. Sahiba and S. Agarwal, Top. Curr. Chem., 2020, 378, 44 CrossRef CAS PubMed; (c) A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Handbook of Heterocyclic Chemistry, Elsevier, 2010 Search PubMed; (d) K. Hemming, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2009, 105, 129–149 RSC.
- (a) A. Gomtsyan, Chem. Heterocycl. Compd., 2012, 48, 7–10 CrossRef CAS; (b) A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
- (a) K. C. Majumdar and S. K. Chattopadhyay, Heterocycles in Natural Product Synthesis, John Wiley & Sons, 2011 CrossRef; (b) J. A. Joule, Adv. Heterocycl. Chem., 2016, 119, 81–106 CrossRef CAS.
- J. Cossy and A. Guerinot, Adv. Heterocycl. Chem., 2016, 119, 107–142 CrossRef CAS.
- G. Cooke, I. R. Evans and P. J. Skabara, J. Mater. Chem. C, 2019, 7, 6492 RSC.
- I. Ali, M. Nadeem Lone, Z. AAl-Othman, A. Al-Warthan and M. Marsin Sanagi, Curr. Drug Targets, 2015, 16, 711–734 CrossRef CAS PubMed.
- M. Fesatidou, A. Petrou and G. Athina, Curr. Pharm. Des., 2020, 26, 867–904 CrossRef CAS PubMed.
- R. H. Bahekar, M. R. Jain, A. A. Gupta, A. Goel, P. A. Jadav, D. N. Patel and P. R. Patel, Arch. Pharm., 2007, 340, 359–366 CrossRef CAS PubMed.
- K. Haider, M. R. Haider, K. Neha and M. S. Yar, Eur. J. Med. Chem., 2020, 204, 112607 CrossRef CAS PubMed.
- R. S. Keri, B. S. Sasidhar, B. M. Nagaraja and M. A. Santos, Eur. J. Med. Chem., 2015, 100, 257–269 CrossRef CAS PubMed.
- A. Chugh, A. Kumar, A. Verma, S. Kumar and P. Kumar, Med. Chem. Res., 2020, 29, 1723–1750 CrossRef CAS.
- R. Wang, K. Xu and W. Shi, Arch. Pharm., 2019, 352, 1900045 CrossRef PubMed.
- A. Kumar, R. A. Maurya, S. Sharma, P. Ahmad, A. B. Singh, G. Bhatia and A. K. Srivastava, Bioorg. Med. Chem. Lett., 2009, 19, 6447–6451 CrossRef CAS PubMed.
- D. Chen, S. J. Su and Y. Cao, J. Mater. Chem. C, 2014, 2, 9565–9578 RSC.
- C. Y. Chan, Y. C. Wong, M. Y. Chan, S. H. Cheung, S. K. So and V. W. W. Yam, ACS Appl. Mater. Interfaces, 2016, 8, 24782–24792 CrossRef CAS PubMed.
- X. Ke, L. Meng, X. Wan, Y. Sun, Z. Guo, S. Wu, H. Zhang, C. Li and Y. Chen, Mater. Chem. Front., 2020, 4, 3594–3601 RSC.
- X. Zhao, S. T. Chaudhry and J. Mei, Adv. Heterocycl. Chem., 2017, 121, 133–171 CrossRef CAS.
- S. Schramm and D. Weiss, Adv. Heterocycl. Chem., 2019, 128, 103–179 CrossRef CAS.
- K. Tanaka, H. Osuga, N. Tsujiuchi, M. Hisamoto and Y. Sakaki, Bull. Chem. Soc. Jpn., 2002, 75, 551–557 CrossRef CAS.
- H. Hayashi, A. S. Touchy, Y. Kinjo, K. Kurotobi, Y. Toude, S. Ito, H. Saarenpää, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, ChemSusChem, 2013, 6, 508–517 CrossRef CAS PubMed.
- G. Chatel, Ultrason. Sonochem., 2018, 40, 117–122 CrossRef CAS PubMed.
- P. Cintas and J. L. Luche, Green Chem., 1999, 1, 115–125 RSC.
- D. H. Bremner, Ultrason. Sonochem., 1994, 1, S119–S124 CrossRef CAS.
- T. J. Mason, Chem. Soc. Rev., 1997, 26, 443–451 RSC.
- R. Patil, P. Bhoir, P. Deshpande, T. Wattamwar, M. Shirude and P. Chaskar, Ultrason. Sonochem., 2013, 20, 1327–1336 CrossRef CAS PubMed.
- P. K. Jaiswal, V. Sharma, J. Prikhodko, I. V. Mashevskaya and S. Chaudhary, Tetrahedron Lett., 2017, 58, 2077–2083 CrossRef CAS.
- E. M. Hussein and K. S. Khairou, Rev. J. Chem., 2014, 4, 221–251 CrossRef.
- F. Penteado, B. Monti, L. Sancineto, G. Perin, R. G. Jacob, C. Santi and E. J. Lenardão, Asian J. Org. Chem., 2018, 7, 2368–2385 CrossRef CAS.
- A. Talha, H. Tachallait, R. Benhida and K. Bougrin, Tetrahedron Lett., 2021, 81, 153366 CrossRef CAS.
- R. Cella and H. A. Stefani, Tetrahedron, 2009, 65, 2619–2641 CrossRef CAS.
- C. Einhorn, J. Einhorn and J. L. Luche, Synthesis, 1989, 1989, 787–813 CrossRef.
- S. Asgharzadehahmadi, A. A. A. Raman, R. Parthasarathy and B. Sajjadi, Renewable Sustainable Energy Rev., 2016, 63, 302–314 CrossRef CAS.
- M. Vinatoru and T. J. Mason, Ultrason. Sonochem., 2019, 52, 2–5 CrossRef CAS PubMed.
- W. Phakhodee, C. Duangkamol, N. Wiriya and M. Pattarawarapan, Tetrahedron Lett., 2016, 57, 5290–5293 CrossRef CAS.
- C. H. Jun, Chem. Soc. Rev., 2004, 33, 610–618 RSC.
- L. Ping, D. S. Chung, J. Bouffard and S. G. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC.
- P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452–1461 RSC.
- C. L. Sun and Z. J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed.
- A. Ibrar, I. Khan, N. Abbas, U. Farooq and A. Khan, RSC Adv., 2016, 6, 93016–93047 RSC.
- A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140–3152 RSC.
- (a) B. Török, K. Balázsik, K. Felföldi and M. Bartók, Ultrason. Sonochem., 2001, 8, 191–200 CrossRef; (b) B. Borah, J. Bora, P. Ramesh and L. R. Chowhan, RSC Adv., 2022, 12, 12843–12857 RSC.
- B. Banerjee, Ultrason. Sonochem., 2017, 35, 15–35 CrossRef CAS PubMed.
- B. Banerjee, J. Serb. Chem. Soc., 2017, 82, 755–790 CrossRef CAS.
- N. Kaur, Synth. Commun., 2018, 48, 1715–1738 CrossRef CAS.
- G. M. Ziarani and P. Gholamzadeh, Mol. Diversity, 2020, 24, 771–820 CrossRef PubMed.
- N. Kaur, Synth. Commun., 2018, 48, 1235–1258 CrossRef CAS.
- M. Mittersteiner, F. F. Farias, H. G. Bonacorso, M. A. Martins and N. Zanatta, Ultrason. Sonochem., 2021, 79, 105683 CrossRef CAS PubMed.
- N. Kaur, Mini-Rev. Org. Chem., 2018, 15, 520–536 CrossRef CAS.
- S. Saranya, S. Radhika, C. M. Afsina Abdulla and G. Anilkumar, J. Heterocycl. Chem., 2021, 58, 1570–1580 CrossRef CAS.
- G. Kaur, A. Sharma and B. Banerjee, ChemistrySelect, 2018, 3, 5283–5295 CrossRef CAS.
- M. A. Schiel, G. F. Silbestri, M. B. Alvarez and C. E. Domini, Ultrasound-promoted metal-catalyzed synthesis of heterocyclic compounds of medicinal interest, in Green Synthetic Approaches for Biologically Relevant Heterocycles, Elsevier, 2021, vol. 1, pp. 461–496 Search PubMed.
- B. Borah, K. D. Dwivedi, B. Kumar and L. R. Chowhan, Arabian J. Chem., 2022, 15, 103654 CrossRef CAS.
- B. Banerjee, Ultrason. Sonochem., 2017, 35, 1–14 CrossRef CAS PubMed.
- B. Borah, K. D. Dwivedi and L. R. Chowhan, RSC Adv., 2021, 11, 13585–13601 RSC.
- Q. W. Gui, F. Teng, S. N. Ying, Y. Liu, T. Guo, J. X. Tang, J. Y. Chen, Z. Cao and W. M. He, Chin. Chem. Lett., 2020, 31, 3241–3244 CrossRef CAS.
- M. T. Nazeri, A. Shaabani and B. Notash, Org. Biomol. Chem., 2021, 19, 3722–3734 RSC.
- H. Khanam, Eur. J. Med. Chem., 2015, 97, 483–504 CrossRef CAS PubMed.
- S. R. Mangaonkar, S. E. Shetgaonkar, A. A. Vernekar and F. V. Singh, ChemistrySelect, 2020, 5, 10754–10758 CrossRef CAS.
- A. Singh, G. Singh and P. M. S. Bedi, J. Heterocycl. Chem., 2020, 57, 2658–2703 CrossRef CAS.
- A. Akbarzadeh and M. G. Dekamin, Green Chem. Lett. Rev., 2017, 10, 315–323 CrossRef CAS.
- J. A. Q. Suárez, A. P. Ricardo, J. M. Costa, C. P. Gonçalves, M. A. Tremante and M. S. A. Saavedra, Curr. Org. Chem., 2021, 25, 748–756 CrossRef.
- S. Kantevari, S. V. Vuppalapati, D. O. Biradar and L. Nagarapu, J. Mol. Catal. A: Chem., 2007, 266, 109–113 CrossRef CAS.
- K. Roy and J. T. Leonard, Bioorg. Med. Chem., 2005, 13, 2967–2973 CrossRef CAS PubMed.
- I. Ali, M. N. Lone and H. Y. Aboul-Enein, Med. Chem. Commun., 2017, 8, 1742–1773 RSC.
- D. Zampieri, M. G. Mamolo, E. Laurini, G. Scialino, E. Banfi and L. Vio, Bioorg. Med. Chem., 2008, 16, 4516–4522 CrossRef CAS PubMed.
- F. Bellina, S. Cauteruccio and R. Rossi, Tetrahedron, 2007, 63, 4571–4624 CrossRef CAS.
- S. Behrouz, M. Navid Soltani Rad, M. Abdollahzadeh and M. Amin Piltan, ChemistrySelect, 2020, 5, 7467–7473 CrossRef CAS.
- A. U. Khandebharad, S. R. Sarda, C. Gill and B. R. Agrawal, Org. Prep. Proced. Int., 2020, 52, 524–529 CrossRef CAS.
- G. Daidone, D. Raffa, B. Maggio, F. Plescia, V. M. C. Cutuli, N. G. Mangano and A. Caruso, Arch. Pharm., 1999, 332, 50–54 CrossRef CAS PubMed.
- R. M. Kumbhare, U. B. Kosurkar, M. J. Ramaiah, T. L. Dadmal, S. N. C. V. L. Pushpavalli and M. Pal-Bhadra, Bioorg. Med. Chem. Lett., 2012, 22, 5424–5427 CrossRef CAS PubMed.
- M. Ahmed, M. A. Qadir, A. Hameed, M. Imran and M. Muddassar, Chem. Biol. Drug Des., 2018, 91, 338–343 CrossRef CAS PubMed.
- B. L. Deng, M. D. Cullen, Z. Zhou, T. L. Hartman, R. W. Buckheit Jr, C. Pannecouque, E. De Clercq, P. E. Fanwick and M. Cushman, Bioorg. Med. Chem., 2006, 14, 2366–2374 CrossRef CAS PubMed.
- T. D. Bhatt, D. G. Gojiya, P. L. Kalavadiya and H. S. Joshi, ChemistrySelect, 2019, 4, 11125–11129 CrossRef CAS.
- S. B. Kasar and S. R. Thopate, Curr. Organocatal., 2019, 6, 231–237 CrossRef CAS.
- H. Z. Zhang, Z. L. Zhao and C. H. Zhou, Eur. J. Med. Chem., 2018, 144, 444–492 CrossRef CAS PubMed.
- M. Nikpassand, L. Z. Fekri and P. Farokhian, Ultrason. Sonochem., 2016, 28, 341–345 CrossRef CAS PubMed.
- V. R. Kandula, M. Pothireddy, K. S. Babu, R. Kapavarapu, R. Dandela and M. Pal, Tetrahedron Lett., 2021, 70, 153011 CrossRef CAS.
- Ş. G. Küçükgüzel and S. Şenkardeş, Eur. J. Med. Chem., 2015, 97, 786–815 CrossRef PubMed.
- B. Borah, K. D. Dwivedi and L. R. Chowhan, Arkivoc, 2021, part i, 273–328 Search PubMed.
- G. M. Nitulescu, L. Matei, I. M. Aldea, C. Draghici, O. T. Olaru and C. Bleotu, Arabian J. Chem., 2019, 12, 816–824 Search PubMed.
- V. S. Dofe, A. P. Sarkate, S. V. Tiwari, D. K. Lokwani, K. S. Karnik, I. A. Kale, S. Dodamani, S. S. Jalalpure and P. V. Burra, Bioorg. Med. Chem. Lett., 2020, 30, 127592 CrossRef CAS PubMed.
- P. C. Sharma, K. K. Bansal, A. Sharma, D. Sharma and A. Deep, Eur. J. Med. Chem., 2020, 188, 112016 CrossRef CAS PubMed.
- F. S. Elsharabasy, S. M. Gomha, T. A. Farghaly and H. S. Elzahabi, Molecules, 2017, 22, 319 CrossRef PubMed.
- H. Bouherrou, A. Saidoun, A. Abderrahmani, L. Abdellaziz, Y. Rachedi, F. Dumas and A. Demenceau, Molecules, 2017, 22, 757 CrossRef PubMed.
- A. Vaidya, D. Pathak and K. Shah, Chem. Biol. Drug Des., 2021, 97, 572–591 CrossRef CAS PubMed.
- U. D. Nimbalkar, S. G. Tupe, J. A. Seijas Vazquez, F. A. Khan, J. N. Sangshetti and A. P. G. Nikalje, Molecules, 2016, 21, 484 CrossRef PubMed.
- A. F. Borsoi, M. E. Coldeira, K. Pissinate, F. S. Macchi, L. A. Basso, D. S. Santos and P. Machado, Synth. Commun., 2017, 47, 1319–1325 CrossRef CAS.
- H. Beyzaei, S. Sargazi, G. Bagherzade, A. Moradi and E. Yarmohammadi, Acta Chim. Slov., 2021, 68, 109–117 CrossRef CAS PubMed.
- Y. Hu, C. Y. Li, X. M. Wang, Y. H. Yang and H. L. Zhu, Chem. Rev., 2014, 114, 5572–5610 CrossRef CAS PubMed.
- T. Erdogan, J. Iran. Chem. Soc., 2019, 16, 899–912 CrossRef CAS.
- S. Chauhan, P. Verma, A. Mishra and V. Srivastava, Chem. Heterocycl. Compd., 2020, 56, 123–126 CrossRef CAS.
- D. Dixit, P. K. Verma and R. K. Marwaha, J. Iran. Chem. Soc., 2021, 18, 2535–2565 CrossRef CAS.
- A. De Nino, L. Maiuolo, P. Costanzo, V. Algieri, A. Jiritano, F. Olivito and M. A. Tallarida, Catalysts, 2021, 11, 1120 CrossRef CAS.
- D. M. Xavier, B. S. Goldani, N. Seus, R. G. Jacob, T. Barcellos, M. W. Paixao, R. Luque and D. Alves, Ultrason. Sonochem., 2017, 34, 107–114 CrossRef CAS PubMed.
- T. Kiranmye, M. Vadivelu, S. Sampath, K. Muthu and K. Karthikeyan, Sustainable Chem. Pharm., 2021, 19, 100358 CrossRef.
- V. A. Ostrovskii, R. E. Trifonov and E. A. Popova, Russ. Chem. Bull., 2012, 61, 768–780 CrossRef CAS.
- Q. H. Lin, Y. C. Li, C. Qi, W. Liu, Y. Wang and S. P. Pang, J. Mater. Chem. A, 2013, 1, 6776–6785 RSC.
- S. Manzoor, X. Yin and J. G. Zhang, Def. Technol., 2021, 17, 1995–2010 CrossRef.
- W. A. A. Arafa and A. F. Abdel-Magied, Arkivoc, 2017, 5, 327–340 Search PubMed.
- S. G. Pharande, M. A. Rentería-Gómez and R. Gámez-Montaño, New J. Chem., 2018, 42, 11294–11298 RSC.
- A. R. Pinder, Nat. Prod. Rep., 1992, 9, 491–504 RSC.
- A. O. Plunkett, Nat. Prod. Rep., 1994, 11, 581–590 RSC.
- Y. W. Kim, J. C. Hackett and R. W. Brueggemeier, J. Med. Chem., 2004, 47, 4032–4040 CrossRef CAS PubMed.
- S. X. Lin, M. A. Curtis and J. Sperry, Bioorg. Med. Chem., 2020, 28, 115820 CrossRef CAS PubMed.
- E. A. Mohamed, N. S. Ismail, M. Hagras and H. Refaat, Future Journal of Pharmaceutical Sciences, 2021, 7, 1–17 CrossRef.
- T. Tahir, M. Ashfaq, M. Saleem, M. Rafiq, M. I. Shahzad, K. Kotwica-Mojzych and M. Mojzych, Molecules, 2021, 26, 4872 CrossRef CAS PubMed.
- P. Patil, S. P. Sethy, T. Sameena and K. Shailaja, Asian J. Res. Chem., 2013, 6, 888–899 Search PubMed.
- E. S. Ibrahim, G. E. H. Elgemeie, M. M. Abbasi, Y. A. Abbas, M. A. Elbadawi and A. M. E. Attia, Nucleosides, Nucleotides Nucleic Acids, 1995, 14, 1415–1423 CrossRef CAS.
- N. B. Patel and S. N. Agravat, Chem. Heterocycl. Compd., 2009, 45, 1343–1353 CrossRef CAS.
- M. Liu, C. Quan, M. Dang, Y. Ren, J. Ren, J. Xiang, X. Liu, L. He, W. Liu and A. Liu, J. Heterocycl. Chem., 2018, 55, 2061–2068 CrossRef CAS.
- S. E. Metobo, H. Jin, M. Tsiang and C. U. Kim, Bioorg. Med. Chem. Lett., 2006, 16, 3985–3988 CrossRef CAS PubMed.
- S. Narramore, C. E. Stevenson, A. Maxwell, D. M. Lawson and C. W. Fishwick, Bioorg. Med. Chem., 2019, 27, 3546–3550 CrossRef CAS PubMed.
- M. Mantri, O. de Graaf, J. van Veldhoven, A. Göblyös, J. K. von Frijtag Drabbe Künzel, T. Mulder-Krieger, R. Link, H. de Vries, M. W. Beukers, J. Brussee and A. P. IJzerman, J. Med. Chem., 2008, 51, 4449–4455 CrossRef CAS PubMed.
- C. Allais, J. M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114, 10829–10868 CrossRef CAS PubMed.
- R. Pagadala, V. Kasi and S. Perugu, J. Chin. Chem. Soc., 2020, 67, 1296–1302 CrossRef CAS.
- N. Mollakarimi Dastjerdi and M. Ghanbari, Green Chem. Lett. Rev., 2020, 13, 192–205 CrossRef.
- P. Y. Chung, Z. X. Bian, H. Y. Pun, D. Chan, A. S. Chan, C. H. Chui, J. C. Tang and K. H. Lam, Future Med. Chem., 2015, 7, 947–967 CrossRef CAS PubMed.
- G. Yernale, Bioorg. Med. Chem., 2021, 32, 115973 CrossRef PubMed.
- S. M. A. Hussaini, Expert Opin. Ther. Pat., 2016, 26, 1201–1221 CrossRef CAS PubMed.
- G. Lewinska, J. Sanetra and K. W. Marszalek, J. Mater. Sci.: Mater. Electron., 2021, 32, 18451–18465 CrossRef CAS.
- S. K. Kang, J. Woo, S. E. Lee, Y. K. Kim and S. S. Yoon, Mol. Cryst. Liq. Cryst., 2019, 685, 114–123 CrossRef CAS.
- X. Q. Ma, Y. Wang, T. B. Wei, L. H. Qi, X. M. Jiang, J. D. Ding, W. B. Zhu, H. Yao, Y. M. Zhang and Q. Lin, Dyes Pigm., 2019, 164, 279–286 CrossRef CAS.
- T. Chanda, R. K. Verma and M. S. Singh, Chem.–Asian J., 2012, 7, 778–787 CrossRef CAS PubMed.
- N. A. Harry, S. M. Ujwaldev and G. Anilkumar, Org. Biomol. Chem., 2020, 18, 9775–9790 RSC.
- V. F. Batista, D. C. Pinto and A. M. Silva, ACS Sustainable Chem. Eng., 2016, 4, 4064–4078 CrossRef CAS.
- D. Wang, L. Désaubry, G. Li, M. Huang and S. Zheng, Adv. Synth. Catal., 2021, 363, 2–39 CrossRef CAS.
- P. Venkanna, K. C. Rajanna, M. S. Kumar, M. B. Ansari and M. M. Ali, Tetrahedron Lett., 2015, 56, 5164–5167 CrossRef CAS.
- I. Bazine, Z. Cheraiet, R. Bensegueni, C. Bensouici and A. Boukhari, J. Heterocycl. Chem., 2020, 57, 2139–2149 CrossRef CAS.
- O. Meth-Cohn, B. Narine and B. Tarnowski, J. Chem. Soc., Perkin Trans. 1, 1981, 1, 1520–1530 RSC.
- O. V. Drygina and A. D. Garnovskii, Chem. Heterocycl. Compd., 1983, 19, 807–821 CrossRef.
- R. Pratap and V. J. Ram, Chem. Rev., 2014, 114, 10476–10526 CrossRef CAS PubMed.
- P. Nazari, A. Bazi, S. A. Ayatollahi, H. Dolati, S. M. Mahdavi, L. Rafighdoost and M. Amirmostofian, Iran. J. Pharm. Res., 2017, 16, 943 CAS.
- D. Kumar, P. Sharma, H. Singh, K. Nepali, G. K. Gupta, S. K. Jain and F. Ntie-Kang, RSC Adv., 2017, 7, 36977–36999 RSC.
- T. Zahra, M. K. Mohammad, H. Haleh, L. Bagher, A. Samira and M. Mohammad, Mol. Diversity, 2020, 24, 1385–1431 CrossRef PubMed.
- Y. M. Litvinov and A. M. Shestopalov, Adv. Heterocycl. Chem., 2011, 103, 175–260 CrossRef CAS.
- A. R. Saundane, K. Vijaykumar and A. V. Vaijinath, Bioorg. Med. Chem. Lett., 2013, 23, 1978–1984 CrossRef CAS PubMed.
- B. Borah, K. D. Dwivedi and L. R. Chowhan, Polycyclic Aromat. Compd., 2021, 1–45 Search PubMed.
- Z. J. Yang, Q. T. Gong, Y. Wang, Y. Yu, Y. H. Liu, N. Wang and X. Q. Yu, Mol. Catal., 2020, 491, 110983 CrossRef CAS.
- S. Tabassum, S. Govindaraju and M. A. Pasha, Ultrason. Sonochem., 2015, 24, 1–7 CrossRef CAS PubMed.
- F. Auria-Luna, V. Fernández-Moreira, E. Marqués-López, M. C. Gimeno and R. P. Herrera, Sci. Rep., 2020, 10, 1–17 CrossRef PubMed.
- J. Rani, S. Kumar, M. Saini, J. Mundlia and P. K. Verma, Res. Chem. Intermed., 2016, 42, 6777–6804 CrossRef CAS.
- S. B. Patil, Int. J. Pharma Sci. Res., 2018, 9, 44–52 CrossRef CAS.
- E. Zarenezhad, M. Farjam and A. Iraji, J. Mol. Struct., 2021, 1230, 129833 CrossRef CAS.
- W. B. Parker, Chem. Rev., 2009, 109, 2880–2893 CrossRef CAS PubMed.
- R. S. Dongre, A. R. Bhat and J. S. Meshram, Am. J. PharmTech Res., 2014, 4, 138–155 Search PubMed.
- M. Amir, S. A. Javed and H. Kumar, Indian J. Pharm. Sci., 2007, 69, 337 CrossRef CAS.
- M. Mahfoudh, R. Abderrahim, E. Leclerc and J. M. Campagne, Eur. J. Org. Chem., 2017, 2017, 2856–2865 CrossRef CAS.
- S. Mohana Roopan and R. Sompalle, Synth. Commun., 2016, 46, 645–672 CrossRef.
- H. ur Rashid, M. A. Martines, A. P. Duarte, J. Jorge, S. Rasool, R. Muhammad, N. Ahmad and M. N. Umar, RSC Adv., 2021, 11, 6060–6098 RSC.
- A. P. G. Nikalje, S. V. Tiwari, J. N. Sangshetti and M. D. Damale, Res. Chem. Intermed., 2018, 44, 3031–3059 CrossRef CAS.
- D. Bayramoğlu, G. Kurtay and M. Güllü, Synth. Commun., 2020, 50, 649–658 CrossRef.
- D. G. Martin, S. A. Mizsak, C. Biles, J. C. Stewart, L. Baczynskyj and P. A. Meulman, J. Antibiot., 1975, 28, 332–336 CrossRef CAS PubMed.
- S. Matsuura, J. Antibiot., Ser. A, 1965, 18, 43–46 CAS.
- M. Shin, K. Inouye and H. Otsuka, Bull. Chem. Soc. Jpn., 1984, 57, 2203–2210 CrossRef CAS.
- M. S. Abdelfattah, T. Kazufumi and M. Ishibashi, J. Nat. Prod., 2010, 73, 1999–2002 CrossRef CAS PubMed.
- S. Tariq, K. Somakala and M. Amir, Eur. J. Med. Chem., 2018, 143, 542–557 CrossRef CAS PubMed.
- M. Montana, F. Mathias, T. Terme and P. Vanelle, Eur. J. Med. Chem., 2019, 163, 136–147 CrossRef CAS PubMed.
- C. O. Knowles, Environ. Health Perspect., 1976, 14, 93–102 CrossRef CAS PubMed.
- A. Husain and D. Madhesia, J. Pharm. Res., 2011, 4, 924–929 CAS.
- T. Kaushal, G. Srivastava, A. Sharma and A. S. Negi, Bioorg. Med. Chem., 2019, 27, 16–35 CrossRef CAS PubMed.
- E. Vicente, L. M. Lima, E. Bongard, S. Charnaud, R. Villar, B. Solano, A. Burguete, S. Perez-Silanes, I. Aldana, L. Vivas and A. Monge, Eur. J. Med. Chem., 2008, 43, 1903–1910 CrossRef CAS PubMed.
- B. Borah and L. R. Chowhan, RSC Adv., 2021, 11, 37325–37353 RSC.
- M. Rouhani and A. Ramazani, J. Iran. Chem. Soc., 2018, 15, 2375–2382 CrossRef CAS.
- R. Nongrum, G. K. Kharmawlong, J. W. S. Rani, N. Rahman, A. Dutta and R. Nongkhlaw, J. Heterocycl. Chem., 2019, 56, 2873–2883 CrossRef.
- S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787–9826 CrossRef CAS.
- U. A. Kshirsagar, Org. Biomol. Chem., 2015, 13, 9336–9352 RSC.
- M. Sharma, K. Chauhan, R. Shivahare, P. Vishwakarma, M. K. Suthar, A. Sharma, S. Gupta, J. K. Saxena, J. Lal, P. Chandra and B. Kumar, J. Med. Chem., 2013, 56, 4374–4392 CrossRef CAS PubMed.
- A. Hameed, M. Al-Rashida, M. Uroos, S. A. Ali, M. Arshia Ishtiaq and K. M. Khan, Expert Opin. Ther. Pat., 2018, 28, 281–297 CrossRef CAS PubMed.
- M. Bhat, S. L. Belagali, S. V. Mamatha, B. K. Sagar and E. V. Sekhar, Stud. Nat. Prod. Chem., 2021, 71, 185–219 CAS.
- T. M. M. Maiden and J. P. A. Harrity, Org. Biomol. Chem., 2016, 14, 8014–8025 RSC.
- L. Mohammadkhani and M. M. Heravi, Front. Chem., 2020, 8, 921 Search PubMed.
- P. S. Auti, G. George and A. T. Paul, RSC Adv., 2020, 10, 41353–41392 RSC.
- L. He, H. Li, J. Chen and X. F. Wu, RSC Adv., 2014, 4, 12065–12077 RSC.
- K. P. Rakesh, N. Darshini, T. Shubhavathi and N. Mallesha, Organic & Medicinal Chemistry International Journal, 2017, 2, 41–45 Search PubMed.
- C. Fischer, S. Shah, B. L. Hughes, G. N. Nikov, J. L. Crispino, R. E. Middleton, A. A. Szewczak, B. Munoz and M. S. Shearman, Bioorg. Med. Chem. Lett., 2011, 21, 773–776 CrossRef CAS PubMed.
- M. M. Reddy and A. Sivaramakrishna, J. Heterocycl. Chem., 2020, 57, 942–954 CrossRef CAS.
- A. A. Al-Amiery, A. A. H. Kadhum, M. Shamel, M. Satar, Y. Khalid and A. B. Mohamad, Med. Chem. Res., 2014, 23, 236–242 CrossRef CAS.
- N. C. Dige, P. G. Mahajan, H. Raza, M. Hassan, B. D. Vanjare, H. Hong, K. H. Lee, J. Latip and S. Y. Seo, Bioorg. Chem., 2019, 92, 103201 CrossRef CAS PubMed.
- F. F. Mohammed, M. Hagar, S. Parveen, R. B. Alnoman, H. A. Ahmed, E. S. H. E. Ashry and H. A. Rasheed, Polycyclic Aromat. Compd., 2021 DOI:10.1080/10406638.2021.1878245.
- B. Borah, K. D. Dwivedi and L. R. Chowhan, Asian J. Org. Chem., 2021, 10, 3101–3126 CrossRef CAS.
- K. D. Dwivedi, B. Borah and L. R. Chowhan, Front. Chem., 2020, 7, 944 CrossRef PubMed.
- S. Zhi, X. Ma and W. Zhang, Org. Biomol. Chem., 2019, 17, 7632–7650 RSC.
- M. Seydimemet, K. Ablajan, M. Hamdulla, W. Li, A. Omar and M. Obul, Tetrahedron, 2016, 72, 7599–7605 CrossRef CAS.
- L. H. Chen, T. W. Chung, B. D. Narhe and C. M. Sun, ACS Comb. Sci., 2016, 18, 162–169 CrossRef CAS PubMed.
- A. R. Bhat, G. A. Naikoo, I. U. Hassan, R. S. Dongra and T. Ara, Beni-Suef University Journal of Basic and Applied Sciences, 2017, 6, 238–246 CrossRef.
- S. Sikandar and A. F. Zahoor, J. Heterocycl. Chem., 2021, 58, 685–705 CrossRef CAS.
- M. Mamaghani and R. Hossein Nia, Polycyclic Aromat. Compd., 2021, 41, 223–291 CrossRef CAS.
- R. Konakanchi, R. Gondru, V. B. Nishtala and L. R. Kotha, Synth. Commun., 2018, 48, 1994–2001 CrossRef CAS.
- S. P. Khare, T. R. Deshmukh, J. N. Sangshetti, V. M. Khedkar and B. B. Shingate, Synth. Commun., 2019, 49, 2521–2537 CrossRef CAS.
- G. Brahmachari, I. Karmakar and K. Nurjamal, ACS Sustainable Chem. Eng., 2018, 6, 11018–11028 CrossRef CAS.
- A. Mishra, S. Singh, M. A. Quraishi and V. Srivastava, Org. Prep. Proced. Int., 2019, 51, 345–356 CrossRef CAS.
- J. Safaei-Ghomi, P. Babaei, H. Shahbazi-Alavi and S. Zahedi, J. Saudi Chem. Soc., 2017, 21, 929–937 CrossRef CAS.
- S. Kumari, M. Rajeswari and J. M. Khurana, Aust. J. Chem., 2016, 69, 1049–1053 CrossRef CAS.
- A. S. Al-bogami, Res. Chem. Intermed., 2015, 41, 93–104 CrossRef CAS.
- S. V. Akolkar, N. D. Kharat, A. A. Nagargoje, D. D. Subhedar and B. B. Shingate, Catal. Lett., 2020, 150, 450–460 CrossRef CAS.
- P. G. Patil, Y. Satkar and D. H. More, Synth. Commun., 2020, 50, 3804–3819 CrossRef CAS.
- P. Saraswat, G. Jeyabalan, M. Z. Hassan, M. U. Rahman and N. K. Nyola, Synth. Commun., 2016, 46, 1643–1664 CrossRef CAS.
- G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed.
- T. L. Pavlovska, R. G. Redkin, V. V. Lipson and D. V. Atamanuk, Mol. Diversity, 2016, 20, 299–344 CrossRef CAS PubMed.
- M. S. Reddy, L. R. Chowhan, N. S. Kumar, P. Ramesh and S. B. Mukkamala, Tetrahedron Lett., 2018, 59, 1366–1371 CrossRef CAS.
- M. R. P. Heravi and F. Norouzy, Res. Chem. Intermed., 2017, 43, 4265–4282 CrossRef.
- W. Liju, K. Ablajan and F. Jun, Ultrason. Sonochem., 2015, 22, 113–118 CrossRef PubMed.
- X. Yang, X. Wang, T. Wang, W. Wang, J. Zhang and Y. Ma, Chem. Res. Chin. Univ., 2019, 35, 33–40 CrossRef CAS.
- I. Yavari and Y. Fadakar, Mol. Diversity, 2021, 1–10, DOI:10.1007/s11030-021-10240-4.
| This journal is © The Royal Society of Chemistry 2022 |