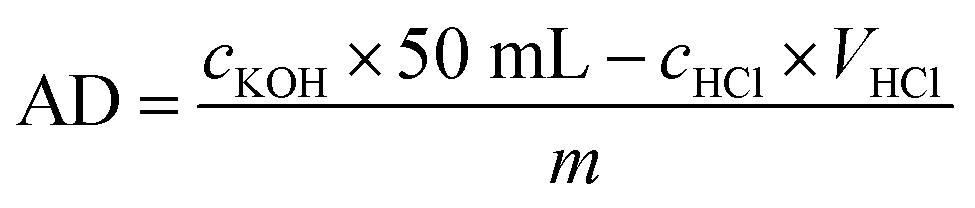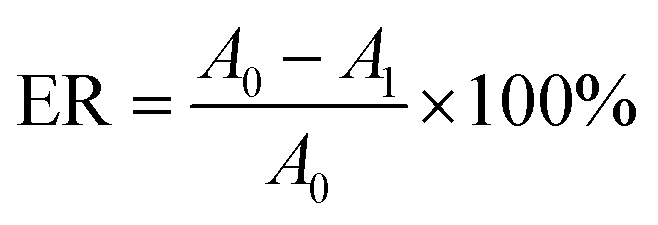DOI:
10.1039/D2RA01610A
(Paper)
RSC Adv., 2022,
12, 12363-12370
Preparation of a reusable and pore size controllable porous polymer monolith and its catalysis of biodiesel synthesis†
Received
11th March 2022
, Accepted 15th April 2022
First published on 25th April 2022
Abstract
A sulfonated porous polymer monolith (PPM-SO3H) has been prepared via the polymerisation of styrene (St) and divinyl benzene (DVB) with organic microspheres as pore-forming agents, followed by sulfonation with concentrated sulfuric acid. It was characterized by acid–base titration in order to determine its acid density, scanning electron microscopy (SEM), transmission electron microscopy (TEM), Fourier transform infrared (FT-IR) spectroscopy, mercury intrusion porosimetry (MIP) and thermogravimetric analysis (TG). The PPM-SO3H showed an acid density of 1.89 mmol g−1 and pore cavities with an average diameter of 870 nm. The catalytic activity of PPM-SO3H in practical biodiesel synthesis from waste fatty acids was investigated and the main reaction parameters were optimized through orthogonal experiment. The best reaction conditions obtained for the optimization of methanol to oil ratio, catalyst concentration, reaction temperature and reaction time were 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 20%, 80 °C and 8 h, respectively. PPM-SO3H showed excellent catalytic activity. In biodiesel synthesis, the esterification rate of PPM-SO3H is 96.9%, which is much higher than that of commercial poly(sodium-p-styrenesulfonate) (esterification rate 29.0%). The PPM-SO3H can be reused several times without significant loss of catalytic activity; the esterification rate was still 90.8% after 6 cycles. The pore size of this porous polymer monolith can be controlled. The dimension and shape of this porous polymer monolith were also adjustable by choosing a suitable polymerisation container.
:1, 20%, 80 °C and 8 h, respectively. PPM-SO3H showed excellent catalytic activity. In biodiesel synthesis, the esterification rate of PPM-SO3H is 96.9%, which is much higher than that of commercial poly(sodium-p-styrenesulfonate) (esterification rate 29.0%). The PPM-SO3H can be reused several times without significant loss of catalytic activity; the esterification rate was still 90.8% after 6 cycles. The pore size of this porous polymer monolith can be controlled. The dimension and shape of this porous polymer monolith were also adjustable by choosing a suitable polymerisation container.
1. Introduction
Porous materials usually refer to materials with interconnected pores including micropores, mesopores and macropores. During the past decades, porous materials including organic, inorganic and organic–inorganic materials have been extensively studied1–10 due to their special physicochemical properties. Porous materials have been used in various fields, such as energy storage,11–14 energy conversion,15–18 adsorption,19–25 catalysis,26–28 sensing,29–31 CO2 capture,32–36 etc. In recent years, the investigation of porous organic polymer (a class of highly crosslinked amorphous polymers possessing nano-pores) based catalysts has attracted significant attention owing to their confined pore space and high surface area which is beneficial for improving catalytic performance and stability. For example, Roy et al. synthesised a pristine conjugated microporous polymer with semi-conduction and redox-activity and used it in a metal-free oxygen reduction reaction.37 Singh et al. synthesised Co2+ and Zn2+ phthalocyanine based redox active metal–organic conjugated microporous polymers for oxygen evolution reaction catalysis through a Schiff base condensation reaction.38 Alkordi et al. reported the synthesis of novel porous organic polymers using functionalized Cr and Co–salen complexes as molecular building blocks and the porous polymers were used as heterogeneous catalysts for CO2-epoxide insertion.39 Alavinia et al. synthesised a kind of porous alginate-g-poly(p-styrene sulfonamide-co-acrylamide) catalyst, which showed high catalytic activity for the one-step synthesis of 1,3,4-oxadiazoles from the reaction of hydrazides and aryl iodides through isocyanide insertion/cyclization.40 Therefore, we considered that functionalized porous organic polymers may exhibit desirable catalytic activity and stability in biodiesel preparation.
Biodiesel (fatty acid alkyl esters), as a kind of renewable energy, has received considerable attention during the past decades because of its excellent features, such as nontoxic, low sulphur content, free of polycyclic aromatic hydrocarbons, biodegradable and environment-friendly.41,42 In industrial production process, biodiesel is typically prepared by esterification and transesterification of waste animal and vegetable oils with methanol in the presence of catalyst. Catalyst is one of the key roles to influence the biodiesel yield and sustainability of the industrial process.43 Industrial biodiesel production using homogeneous catalysts has its intrinsic drawbacks, such as equipment corrosion, separation difficulty and low reusability of catalyst. Furthermore, base-catalysed transesterification process are not suitable for feedstocks with high free fatty acids (FFAs), because FFAs cannot be converted into biodiesel through this process which will result in low yield of biodiesel.44 Immobilized enzyme as well as free enzyme also has problems on a large industrial scale, such as cost, reusability and deactivation of enzymes.45,46 As an alternative, heterogeneous acid catalyst may overcome some of these problems since it can be isolated through simple procedure and convert triacylglycerols by transesterification and free fatty acids by esterification simultaneously.47,48 Developing high efficient heterogeneous acid catalyst is essential for practical process.49 Among many kinds of heterogeneous acid catalysts, sulfonated-solid catalysts exhibit desirable catalytic activity owing to the presence of active –SO3H. Such as sulfonated carbon-based catalyst made from biomass resource,50–52 sulfonated graphene and graphene oxide53,54 and sulfonated resin55 and so on. However, many of these heterogeneous acid catalysts are mainly powdery small particles which is also inconvenient to operate in the industrial production of biodiesel. Some conjugated microporous polymer materials derived from π-conjugated aromatic building block have been reported as catalysts due to their unique properties.3,37,38 It is presumed that functionalized porous organic polymers may exhibit desirable catalytic activity in biodiesel preparation. Hence, we designed a sulfonated porous polymer monolith catalyst synthesised from aromatic monomers.
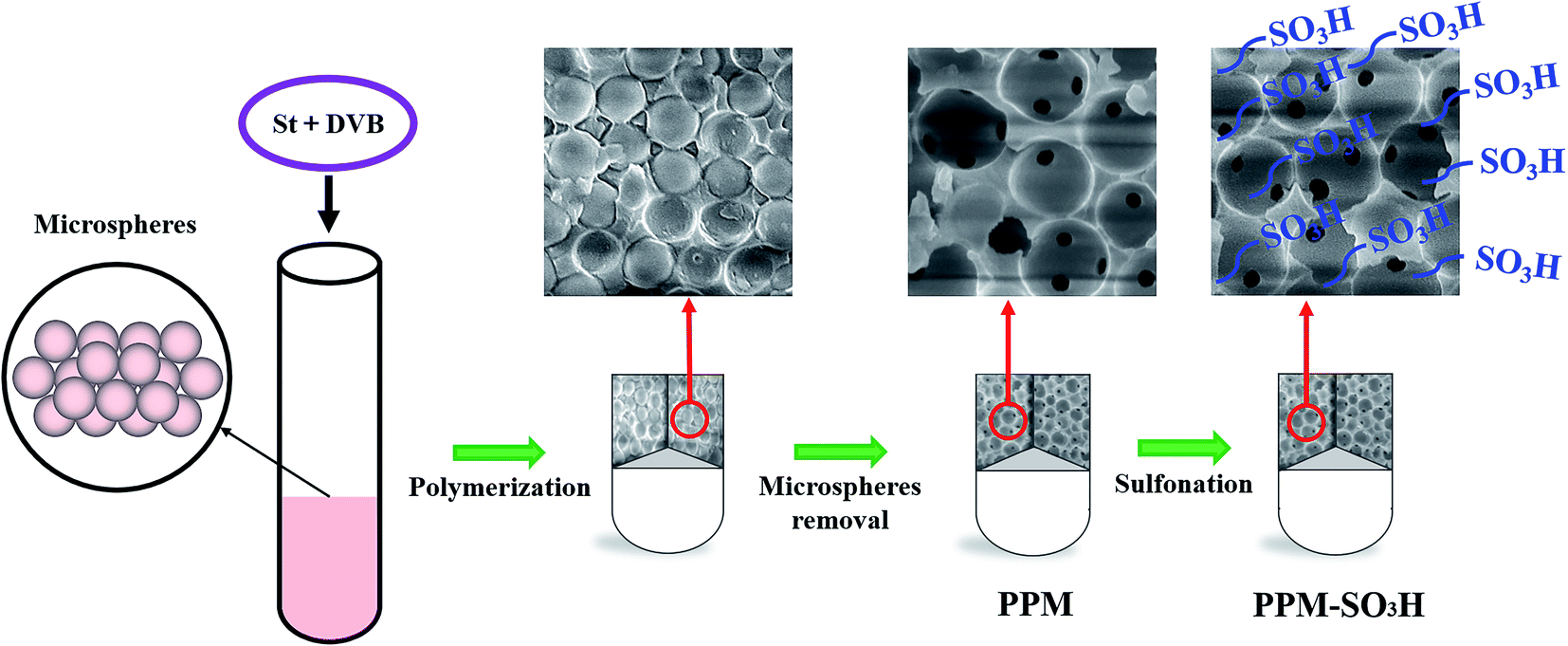
In this study, a porous polymer monolith catalyst was synthesised through the polymerisation of styrene (St) and divinyl benzene (DVB) with microspheres as pore-forming agent and followed by sulfonation (Scheme 1). As shown in Scheme 1, the mixture of St and DVB was added into the glass tube filled with microspheres (polymerization of maleic anhydride and vinyl acetate) for polymerisation. After polymerisation, the tube was broken and left a composite cylinder of crosslinked polystyrene/microspheres. Then, the porous polymer monolith (PPM) was available after removal of microspheres. At last, the PPM-SO3H was obtained through the sulfonation of PPM. The PPM-SO3H showed excellent catalytic activity in biodiesel synthesis, which is much higher than that of commercial poly(sodium-p-styrenesulfonate). The PPM-SO3H can be reused for several times without significant loss of catalytic activity. In addition, the pore size of this porous polymer monolith can be controlled by adjusting the diameter of microspheres. The dimension and shape of this porous polymer monolith were also adjustable by choosing suitable polymerisation container. The PPM-SO3H has the potential to be used as both catalyst and reactor in industrial biodiesel production.
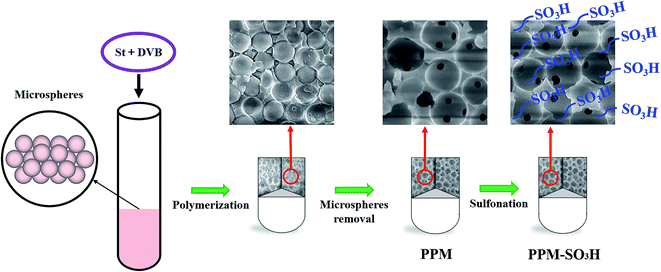 |
| | Scheme 1 Preparation of PPM-SO3H via polymerisation of St and DVB followed by sulfonation. | |
2. Materials and methods
2.1. Materials
The following chemicals were purchased from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China): benzoyl peroxide (BPO, 99.0% purity), vinyl acetate (VAc, 99.0% purity), styrene (St, 99.0% purity, contains 10–15 ppm of 4-tert-butylcatechol) and divinyl benzene (DVB, 80% mixture of isomers, contains 1000 ppm of 4-tert-butylcatechol). Maleic anhydride (MAn, >99.0% purity), n-butyl acetate (99.0% purity), N,N-dimethylformamide (DMF, ≥99.9% purity), methanol (99.5% purity) and poly(sodium-p-styrenesulfonate)(PSS, average molecular weight 1000000 Da, powder) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd (Shanghai, China). Sodium hydroxide (NaOH), potassium hydroxide (KOH), hydrochloric acid (HCl), chloroform, acetone and sulfuric acid (H2SO4, 98% purity) were purchased from Institute of Tianjin Chemical Reagent (Tianjin, China). Waste fatty acids was prepared from waste cooking oils. All other reagents (NaOH, KOH, HCl, chloroform, acetone, etc.) were of analytical grade. All reagents were used as received except VAc, St, DVB and BPO. VAc was refined by distillation. St and DVB were purified by sequential thoroudslfghly washing with NaOH aqueous solution and distilled water and reduced pressure distillation. BPO was purified through a typical procedure: 5 g benzoyl peroxide was dissolved in 20 mL chloroform and filtered, then the filtrate was poured into ca. 50 mL methanol for recrystallisation, the needle-like crystal was obtained by filtration.
2.2. Methods
2.2.1. Preparation of polymer microspheres. MAn (9.8 g, 0.10 mol), VAc (8.3 g, 0.10 mol), BPO (96.9 mg, 0.40 mmol), 41 mL n-butyl acetate and 6 drops of DMF were added into a 250 mL three-necked flask equipped with a condenser tube and two rubber plugs. After bubbling with N2 for 30 min through a long needle, the reaction flask was kept in a water bath at 80 °C for 4 h without agitation. The reaction system was naturally cooled to room temperature at the end of the reaction. Then, the resultant mixture was poured into several glass tubes, and the synthesised polymer microspheres were separated by centrifugation (3000 rpm × 8 min) and washed with n-butyl acetate for several times to eliminate unreacted monomers. Finally, the polymer microspheres in glass tubes were obtained after pouring out the supernatant and drying naturally. Furthermore, the diameter of the microspheres is controllable by adjusting monomer concentration and the molar ratio of MAn to VAc.56
2.2.2. Preparation of porous polymer monolith (PPM). The mixture comprised of St, DVB and BPO (the molar ratio of St to DVB and BPO was 1000:800:13) was added into the glass tube filled with polymer microspheres. The liquid level of the mixture added should properly higher than the height of the microspheres in the glass tube. Then, the tube was sealed with rubber plug and placed for several hours to make the solution penetrated into the gaps between the polymer microspheres. The state of the mixture in the glass tube would become translucent after full penetration. Next, the polymerisation reaction was performed by keeping the glass tube at 70 °C for 12 h. After polymerisation, the tube was broken carefully and left a composite cylinder of crosslinked polystyrene/microspheres. The cylinder was extracted by acetone for 72 h to obtain a porous polystyrene monolith, which was subsequently vacuum-dried at 40 °C for 24 h.
2.2.3. Preparation of sulfonated porous polymer monolith (PPM-SO3H). The porous polystyrene monolith was sulfonated by concentrated sulfuric acid (H2SO4) to prepare a catalyst for biodiesel production. The detailed process was as follows: 1 g PPM and 30 mL H2SO4 were added to a flask, then the sulfonation reaction procedure was carried out at 40 °C for 8 h with magnetic stirring. At the end of the reaction, the sulfonated porous polymer monolith was separated and cooled to room temperature. Then, it was washed and socked with deionized water for many times until SO42− couldn't be detected in the washing water. Finally, the sulfonated porous polymer monolith (PPM-SO3H) was vacuum-dried at 40 °C for 24 h.
2.2.4. Determination of acid density. The acid density of the sulfonated porous polymer monolith was determined by acid–base retro titration according to some relevant literature.57–59 In this study, 0.1 g porous polymer monolith, which has been cut into pieces, was immersed in 50 mL aqueous solution of KOH (0.01 mol −1). The mixture was then placed under ultrasonication for 2 h to make the KOH solution penetrate into the pores of monolith catalyst faster and better. After soaking for 24 h, the mixture was titrated with aqueous solution of HCl (0.01 mol −1). The acid density (AD) of PPM-SO3H was calculated by eqn (1):| |
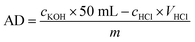 | (1) |
where cKOH is the concentration of KOH standard solution; cHCl is the concentration of HCl standard solution.
2.2.5. Characterization. The surface morphology was observed by a field emission scanning electron microscope (Hitachi SU8010, Hitachi, Japan) with an acceleration voltage of 5 kV. The microstructure of the porous monolith was characterized by a transmission electron microscope (Hitachi H-7650, Hitachi, Japan) operating at 80.0 kV. The FTIR spectra were recorded on an infrared spectrometer (Tensor 27, Bruker, Germany) using KBr tablet sample in the range of 400–4000 cm−1 with a resolution of 2 cm−1. Thermogravimetric analysis was performed on a thermogravimetric/differential thermal analyzer (STA 409 PC Luxx, Netzsch, Germany), the measurement was carried out at a heating rate of 10°C min−1 with nitrogen flow rate of 50 mL min−1. The pore size and volume of the sulfonated porous monolith was analysed by a mercury intrusion porosimeter (Autopore IV 9500, Micromeritics, USA) from pressure 0.10 to 33000.00 psia.
2.2.6. General procedure for biodiesel preparation from waste fatty acids. In a typical experiment, 5 g waste fatty acids, a prefixed amount of methanol and catalyst (PPM-SO3H or PSS) were placed in a glass flask equipped with a condenser, a magnetic stirrer and a thermometer. The esterification reaction was carried out under a pre-set reaction temperature for a certain time. At the end of the reaction, the system was naturally cooled to room temperature. Then the catalyst was isolated from the resultant crude biodiesel through simple filtration process and thoroughly washed with methanol for reusing. Finally, the crude biodiesel was purified through vacuum-rotary evaporation to remove the unreacted methanol. The esterification rate (ER) was calculated by eqn (2):| |
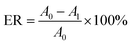 | (2) |
where A0 is the acid value of waste fatty acids used for biodiesel preparation; A1 is the acid value of biodiesel.
3. Results and discussion
3.1. Characterization
3.1.1. Acid density. Acid density is another critical factor affecting the catalytic performance of porous polymer monolith in addition to pore size, specific surface area, etc. The acid density of the sulfonated porous polymer monolith measured by the method in Section 2.2.4. was 1.89 mmol −1. This result not only demonstrates the successful sulfonation of porous polymer monolith, but also predicts its good catalytic performance, which will be confirmed in subsequent experiments.
3.1.2. SEM. Fig. 1 shows the SEM image for the morphology of polymer microspheres which were used as pore-forming agent in the preparation of PPM-SO3H. The SEM image was analysed through Image J software in order to obtaining the average diameter of the polymer microspheres. It can be seen that the morphology of the polymer was spherical with uniform and the average diameter of the polymer microsphere was about 870 nm. Furthermore, the diameter of the polymer microspheres could be controlled by changing polymerisation conditions such as the monomer concentration, feed ratio and solvent (reaction medium) and so on.56 Therefore, the pore size of PPM-SO3H is controllable.
 |
| | Fig. 1 SEM image of polymer microspheres. | |
PPM and PPM-SO3H were successfully prepared. Fig. 2 shows scanning electron microscopy images for the morphology of the PPM and its sulfonated counterpart (PPM-SO3H). As observed from Fig. 2, the morphology of PPM and PPM-SO3H was similar, the pore cavities previously occupied by polymer microspheres in PPM and PPM-SO3H were spherical. The pore structure has no obvious deformation after sulfonation. Furthermore, there were some other small pores between the pore cavities connecting the pore cavities. The SEM images were analysed using Image J software for obtaining the size of pore cavities and small pores. The average diameter of pore cavities was about 870 nm, which was basically consistent with the polymer microspheres in size. In addition, the average diameter of small pores was about 145 nm.
 |
| | Fig. 2 SEM images of porous polymer monolith (a and b) and sulfonated porous polymer monolith (c and d). | |
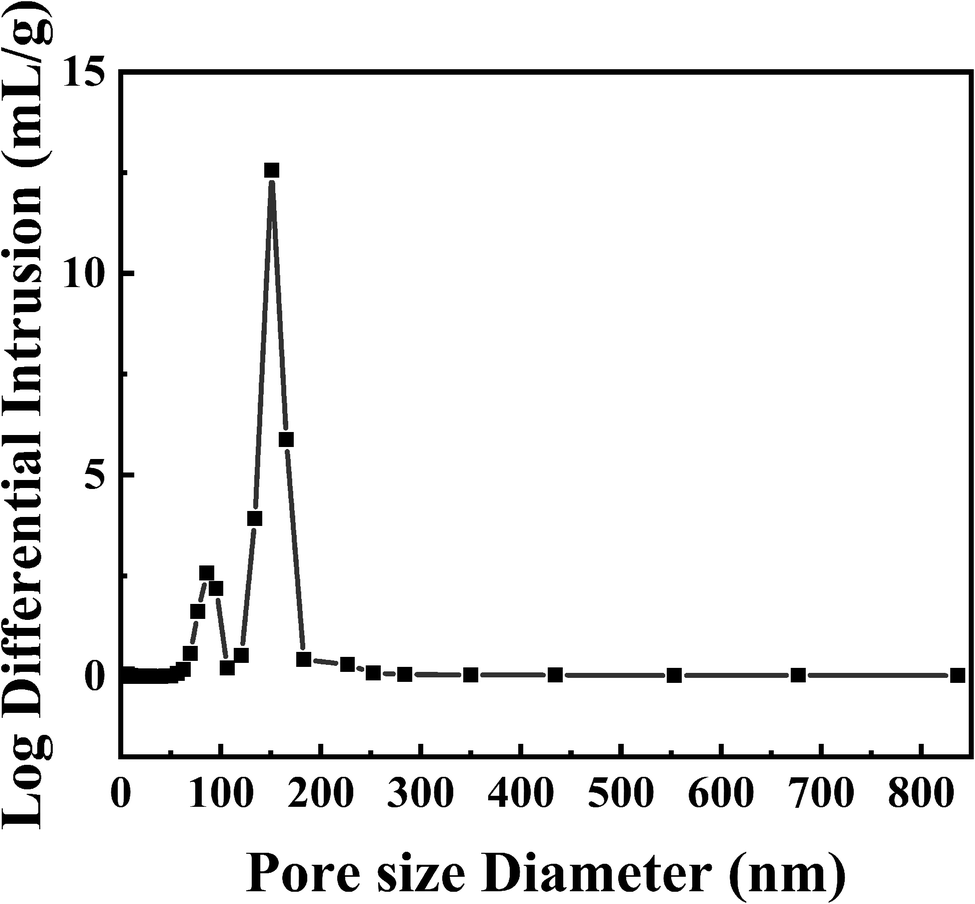
3.1.3. Mercury intrusion porosimetry. Pore diameters and volumes of PPM-SO3H were measured by mercury intrusion porosimetry using MICROMERITICS AutoPore IV 9500 instrument. Since the MIP measures only open pores and solely the entryway between the surface of the sample and the pore cavities and not the radius of the pore cavities itself,60,61 the pore size distribution (Fig. 3) should be assigned to the small pores connecting pore cavities. As presented in Fig. 3, the pore size of PPM-SO3H was 85–151 nm, which is consistent with the result from the SEM image (Fig. 2d).
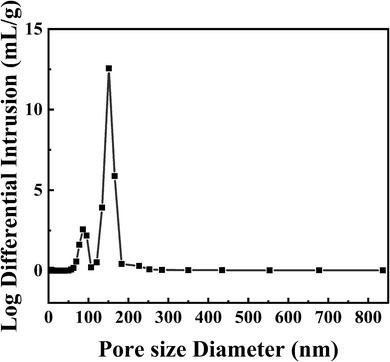 |
| | Fig. 3 Pore size distribution of PPM-SO3H measured by mercury intrusion porosimetry. | |
3.1.4. TEM. Fig. 4 displays the TEM images for the morphology and microstructure of PPM and PPM-SO3H. Through observing the close-up view of the structure of the porous monolith prepared via using polymer microspheres as pore-forming agent, there should be no difficulty to find that the monolith is porous and the pores are interconnected. These micrographs (Fig. 2 and 4) illustrated that the sulfonation did not cause significant collapse to the surface and the pores of the PPM.
 |
| | Fig. 4 TEM images of PPM (a) and PPM-SO3H (b). | |
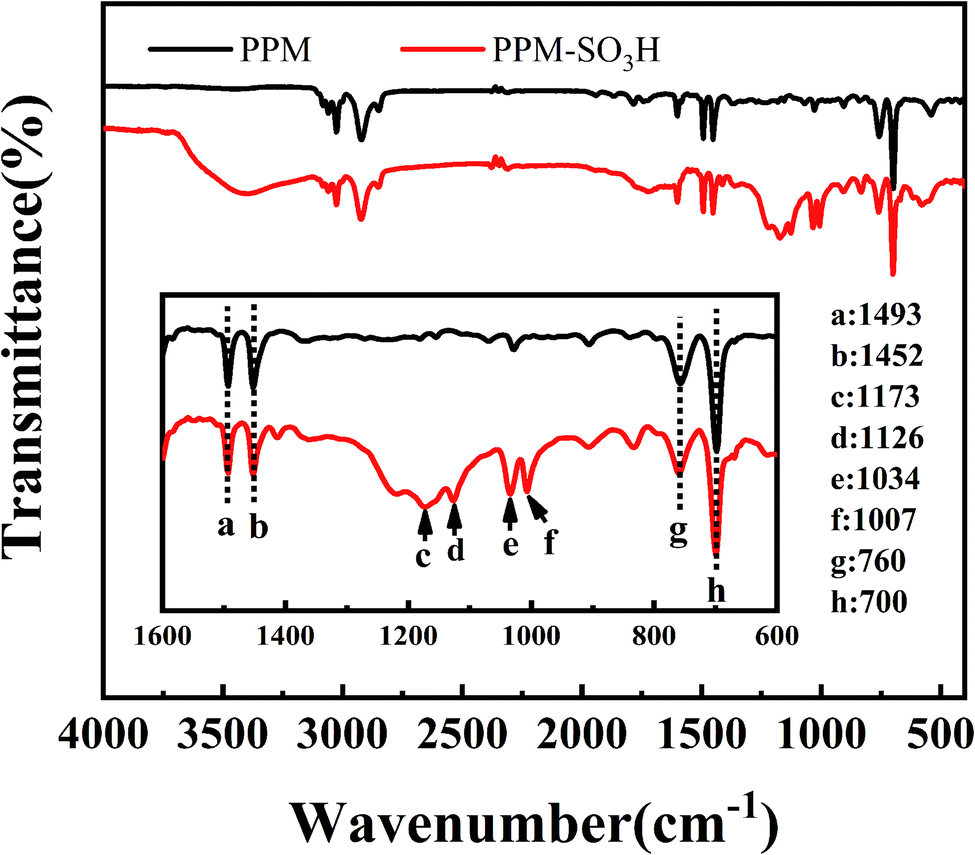
3.1.5. FT-IR. Fig. 5 shows the FT-IR spectra of PPM and PPM-SO3H. The main peaks at 700, 760, 1452 and 1493 cm−1 are due to the polystyrene component of PPM.62 After sulfonation, the characteristic peaks at 1173 cm−1 and 1126 cm−1 attributed to the vibrational absorption of sulfuric acid group (–SO3H) appeared.63,64 The successful sulfonation of the PPM can also be demonstrated by the bands at 1034 cm−1 and 1007 cm−1, which can be assigned to the –SO3H group.65,66
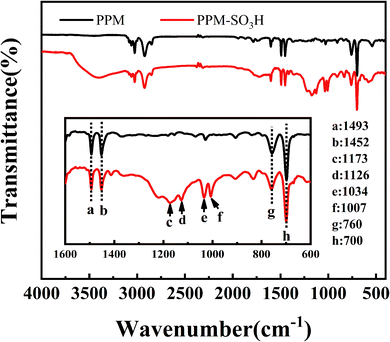 |
| | Fig. 5 FT-IR spectra of PPM and PPM-SO3H (top: original spectra; bottom: partial amplification of wave numbers 1600 to 600). | |
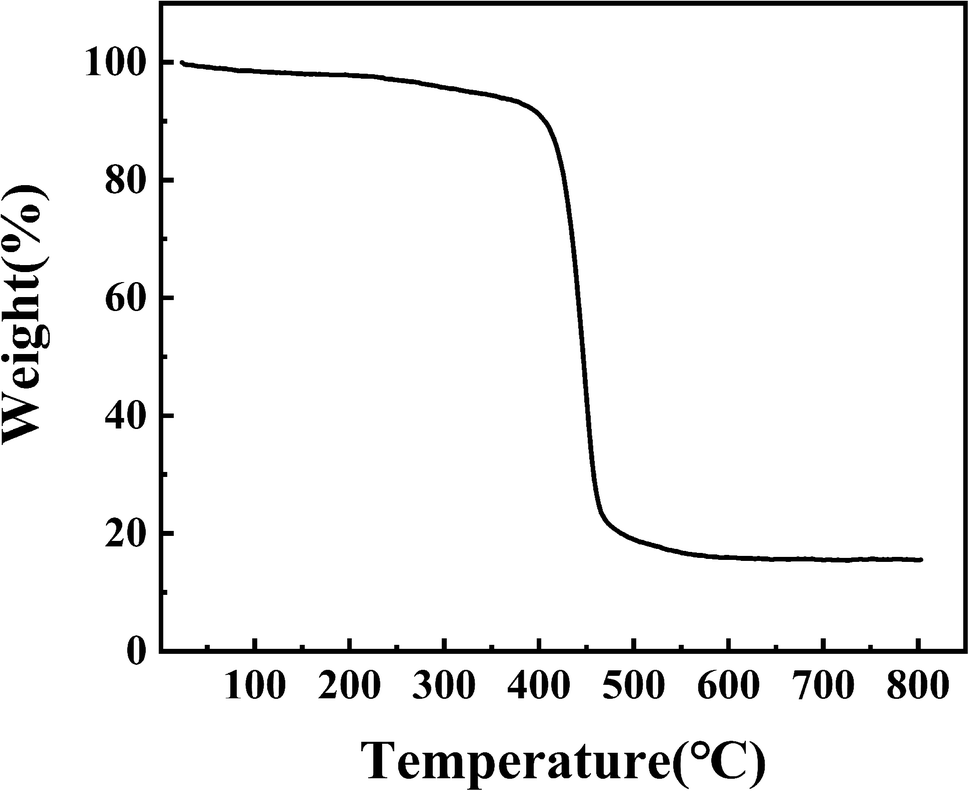
3.1.6. TG. The thermal stability of PPM-SO3H was measured by TG analyses. As shown in Fig. 6, no obvious weight loss (less than 5% of total weight) was observed before 325 °C, which suggesting that PPM-SO3H was chemically stable. The peak temperature of thermal decomposition for PPM-SO3H was 445 °C. In addition, PPM-SO3H lost weight rapidly from 350 °C to 480 °C due to the decomposition of the polystyrene chain, and the final residual masses was 15.55%. The results suggest that PPM-SO3H can be stable below 350 °C, so it can be used to catalyse reactions at temperatures below 350 °C. The reaction temperatures for biodiesel preparation were usually much lower than 350 °C. Hence, PPM-SO3H is fully qualified for catalysis of biodiesel synthesis.
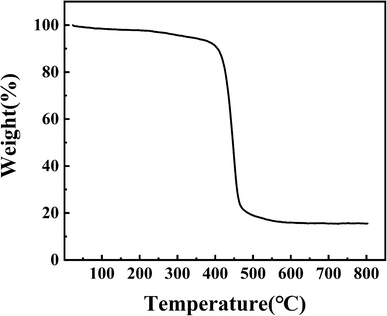 |
| | Fig. 6 TG curve of PPM-SO3H. | |
3.2. Orthogonal experiment of biodiesel preparation from waste fatty acids
The esterification of waste fatty acids with methanol catalysed by PPM-SO3H was planned using a four-level, five factor (L16(45)) orthogonal design. Four important parameters (MeOH to oil ratio, catalyst concentration, reaction temperature and reaction time), which have been identified to have larger effects on the yield of biodiesel produced from other feedstocks,67 was investigated. The results are shown in Table S1 (see ESI†), it can be observed that the range of the esterification rate varies from 21.3% to 92.4%; these data were taken as the original data and used in range analysis and univariate analysis. The range analysis results for the orthogonal experiment are listed in Table S2 (see ESI†). It can be concluded that reaction time has significant effect on the esterification rate and the optimal condition is A2B4C4D4. Therefore, the optimum conditions were obtained as follows: MeOH to oil ratio, 1:1; catalyst concentration, 20.0%; reaction temperature, 80 °C; reaction time, 8 h. It reached an esterification rate of 96.9%. Furthermore, the univariate analysis results for the orthogonal experiment are shown Table S3 (see ESI†). For each factor, a higher F value or R indicates that the level has a larger effect on esterification rate. According to the F value, the factors influencing esterification rate were listed in a decreasing order as follows: reaction time > reaction temperature > catalyst concentration > MeOH to oil ratio. This result was consistent with the judgment according to the R in Table S2.†
3.3. Comparison of the catalytic performance of PPM-SO3H with PSS
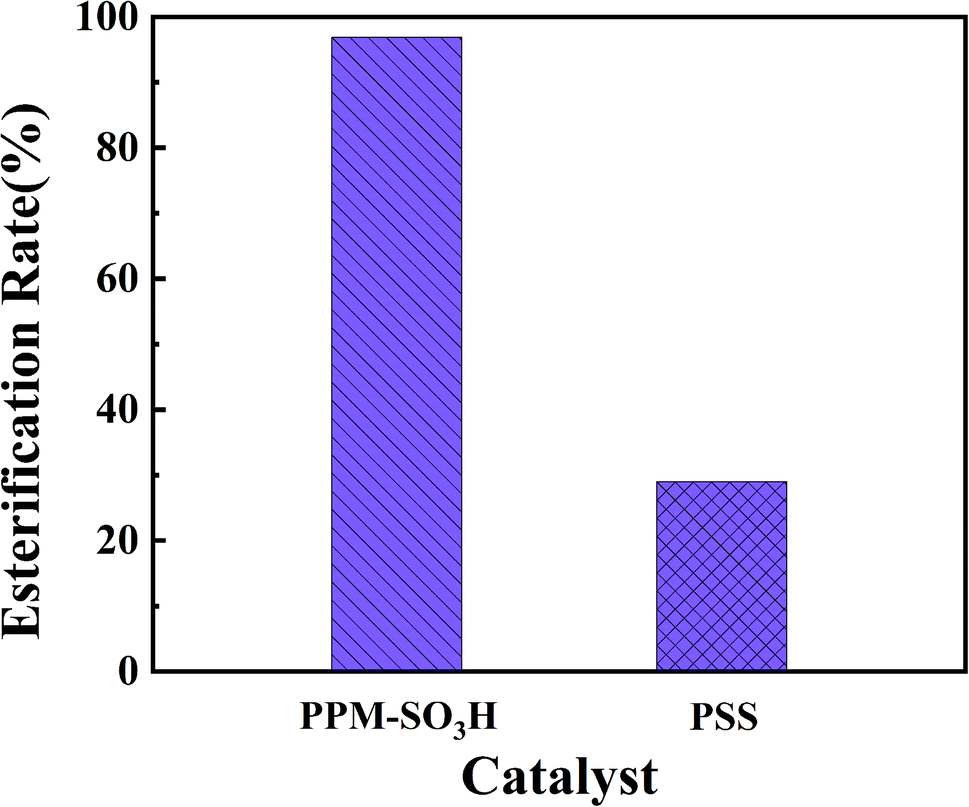
In order to further study the catalytic performance of PPM-SO3H, we compared the esterification rates in the synthesis of biodiesel from waste fatty acids using PPM-SO3H and PSS as catalysts respectively according to method Section 2.2.6. The results are presented in Table 1 and Fig. 7. Under the same reaction conditions, the esterification rate of PPM-SO3H is 96.9%, whereas commercial poly(sodium-p-styrenesulfonate) is 29.0%. It is quite clear that the PPM-SO3H showed much better catalytic performance than PSS.
Table 1 Catalytic performance of PPM-SO3H and PSS waste fatty acids 5 g, methanol 5 g, reaction temperature 80 °C, reaction time 8 h
| Trial number |
Catalyst |
Esterification rate (%) |
| 1 |
PPM-SO3H (1 g) |
96.9% |
| 2 |
PSS (1 g) |
29.0% |
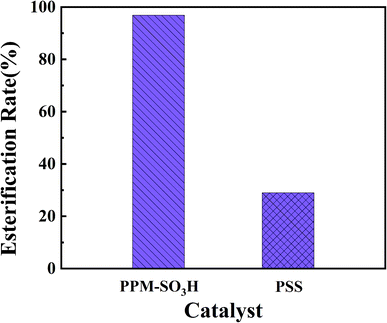 |
| | Fig. 7 Catalytic performance of PPM-SO3H and PSS. | |
3.4. Catalyst reusability
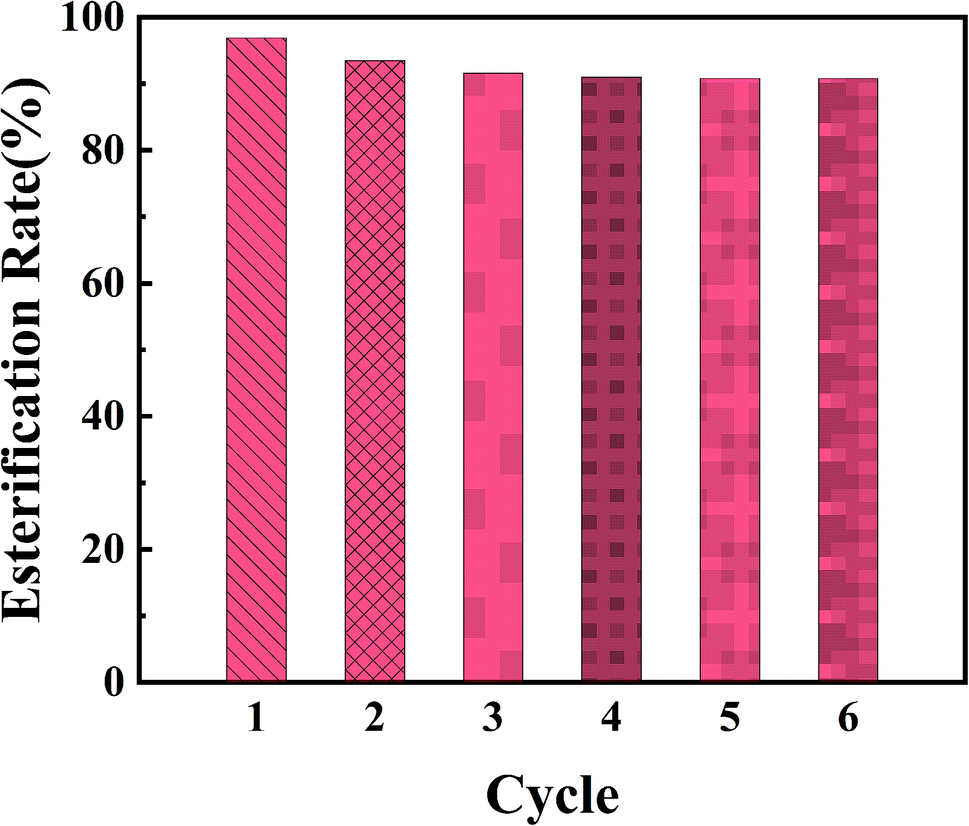
Reusability is one of the most important properties of heterogeneous catalyst. In order to investigate the reusability of PPM-SO3H, the catalyst was recovered by simple filtration, washing with methanol and drying in a vacuum oven at 40 °C. Then, PPM-SO3H was reused in a subsequent esterification reaction. The esterification reaction experiments were performed for six times under the optimal conditions. The results indicate that esterification rate was still 90.8% after 6 cycles (Fig. 8). Therefore, the PPM-SO3H we synthesised have great potential to be a stable and highly active solid acid catalyst used for biodiesel preparation.
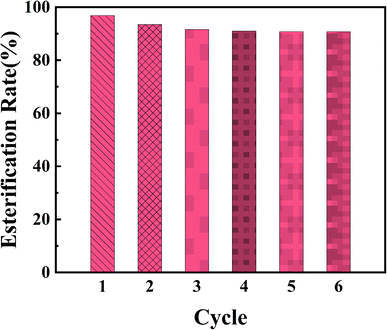 |
| | Fig. 8 Reusability of PPM-SO3H under optimal conditions. | |
4. Conclusions
A novel pore size controllable sulfonated porous polymer monolith (PPM-SO3H) has been prepared via the polymerisation of St and DVB with polymer microspheres as pore-forming agent, followed by sulfonation. The prepared PPM-SO3H was successfully applied to the synthesis of biodiesel from waste fatty acids. The reaction parameters were optimized through orthogonal experiment. The best reaction conditions obtained in the optimization of methanol to oil ratio, catalyst concentration, reaction temperature and reaction time were 1:1, 20%, 80 °C and 8 h, respectively. The sulfonated porous polymer monolith (esterification rate 96.9%) showed much better catalytic activity than that of commercial PSS (esterification rate 29.0%) in the synthesis of biodiesel. It can also be reused for several times without significant loss of activity, esterification rate still maintains 90.8% after 6 cycles. The PPM-SO3H has the potential to be used as both catalyst and reactor in industrial biodiesel production. In addition, the pore size, dimension and shape of this porous polymer monolith can be controlled. Consequently, according to process requirements and raw material characteristics, we can choose polymer monolith with different pore size, dimension and shape. This is of great significance from the perspective of industrial biodiesel production.
Author contributions
Weiqing Chen: investigation, data curation, writing-original draft. Zhaoji Wu: visualization. Zhengge Wang: formal analysis. Changjiu Chen: validation. Zhigang Zhang: funding acquisition, supervision, writing-review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by Department of Education of Hebei Province [No. ZD2020326].
References
- J. Castro-Gutiérrez, A. Sanchez-Sanchez, J. Ghanbaja, N. Díez, M. Sevilla, A. Celzard and V. Fierro, Green Chem., 2018, 20, 5123–5132 RSC.
- D. I. Fried, F. J. Brieler and M. Fröba, ChemCatChem, 2013, 5, 862–884 CrossRef CAS.
- A. Singh, D. Samanta, M. Boro and T. K. Maji, Chem. Commun., 2019, 55, 2837–2840 RSC.
- J. Wang, G. Wang, Z. Zhang, G. Ouyang and Z. Hao, RSC Adv., 2021, 11, 36577–36586 RSC.
- S. Wang, L. Tan, C. Zhang, I. Hussain and B. Tan, J. Mater. Chem. A, 2015, 3, 6542–6548 RSC.
- Y. Wang, B. Wang, J. Wang, Y. Ren, C. Xuan, C. Liu and C. Shen, J. Hazard. Mater., 2018, 344, 849–856 CrossRef CAS PubMed.
- J. Zhang, J. Chen, S. Peng, S. Peng, Z. Zhang, Y. Tong, P. W. Miller and X.-P. Yan, Chem. Soc. Rev., 2019, 48, 2566–2595 RSC.
- H. Zheng, F. Gao and V. Valtchev, J. Mater. Chem. A, 2016, 4, 16756–16770 RSC.
- W. Zhou, Y. Zhang, J. Wang, H. Li, W. Xu, B. Li, L. Chen and Q. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 46767–46778 CrossRef CAS PubMed.
- B. Zheng, X. Lin, X. Zhang, D. Wu and K. Matyjaszewski, Adv. Funct. Mater., 2020, 30, 1907006 CrossRef CAS.
- D. G. Atinafu, W. Dong, C. Hou, R. S. Andriamitantsoa, J. Wang, X. Huang, H. Gao and G. Wang, Mater. Today Energy, 2019, 12, 239–249 CrossRef.
- J. Min, K. Kierzek, X. Chen, P. K. Chu, X. Zhao, R. J. Kaleńczuk, T. Tang and E. Mijowska, New J. Chem., 2017, 41, 13553–13559 RSC.
- B. Xue, Z. Wang, Y. Zhu, X. Wang and R. Xiao, Ind. Crop. Prod., 2021, 170, 113724 CrossRef CAS.
- J. Zhou and W. Bo, Chem. Soc. Rev., 2017, 46, 6927–6945 RSC.
- L. Kong, M. Zhong, W. Shuang, Y. Xu and X.-H. Bu, Chem. Soc. Rev., 2020, 49, 2378–2407 RSC.
- Y.-F. Shen, C. Zhang, C.-G. Yan, H.-Q. Chen and Y.-J. Zhang, Chin. Chem. Lett., 2017, 28, 1312–1317 CrossRef CAS.
- D. Wei, F. Xu, J. Xu, J. Fang, S. W. Koh, K. Li and Z. Sun, Ceram. Int., 2020, 46, 1396–1402 CrossRef CAS.
- H. Zhong, Y. Su, X. Chen, X. Li and R. Wang, ChemSusChem, 2017, 10, 4855–4863 CrossRef CAS PubMed.
- Y. He, W. Bao, Y. Hua, Z. Guo, X. Fu, B. Na, D. Yuan, C. Peng and H. Liu, RSC Adv., 2022, 12, 5587–5594 RSC.
- G. Li, C. Yao, J. Wang and Y. Xu, Sci. Rep., 2017, 7, 13972 CrossRef.
- Y. Li, H. Wang, W. Zhao, X. Wang, Y. Shi, H. Fan, H. Sun and L. Tan, J. Appl. Polym. Sci., 2019, 136, 47987 CrossRef.
- S. Lu, Q. Liu, R. Han, M. Guo, J. Shi, C. Song, N. Ji, X. Lu and D. Ma, J. Environ. Sci., 2021, 105, 184–203 CrossRef CAS PubMed.
- R. Matsuda, Bull. Chem. Soc. Jpn., 2013, 86, 1117–1131 CrossRef CAS.
- W. Peng, S. Liu, H. Sun, Y. Yao, L. Zhi and S. Wang, J. Mater. Chem. A, 2013, 1, 5854–5859 RSC.
- K. V. Rao, S. Mohapatra, C. Kulkarni, T. K. Maji and S. J. George, J. Mater. Chem., 2011, 21, 12958–12963 RSC.
- A. Munyentwali, C. Li, H. Li and Q. Yang, Chem. Asian J., 2021, 16, 2041–2047 CrossRef CAS PubMed.
- S.-S. Qin, Z.-K. Wang, L. Hu, X.-H. Du, Z. Wu, M. Strømme, Q.-F. Zhang and C. Xu, Nanoscale, 2021, 13, 3967–3973 RSC.
- W. Tong, W.-H. Li, Y. He, Z.-Y. Mo, H.-T. Tang, H.-S. Wang and Y.-M. Pan, Org. Lett., 2018, 20, 2494–2498 CrossRef CAS PubMed.
- C. Dai, H.-L. Qian and X.-P. Yan, J. Hazard. Mater., 2021, 416, 125860 CrossRef CAS PubMed.
- L. Guo and D. Cao, J. Mater. Chem. C, 2015, 3, 8490–8494 RSC.
- Z. Song, Y. Ma, A. Morrin, C. Ding and X. Luo, TrAC, Trends Anal. Chem., 2021, 135, 116155 CrossRef CAS.
- S. Shingdilwar, S. Dolui, D. Kumar and S. Banerjee, Mater. Adv., 2022, 3, 665–671 RSC.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- M. Niu, H. Yang, X. Zhang, Y. Wang and A. Tang, ACS Appl. Mater. Interfaces, 2016, 8, 17312–17320 CrossRef CAS PubMed.
- H. He, W. Li, M. Zhong, D. Konkolewicz, D. Wu, K. Yaccato, T. Rappold, G. Sugar, N. E. David and K. Matyjaszewski, Energy Environ. Sci., 2013, 6, 488–493 RSC.
- C. Rosu, S. H. Pang, A. R. Sujan, M. A. Sakwa-Novak, E. W. Ping and C. W. Jones, ACS Appl. Mater. Interfaces, 2020, 12, 38085–38097 CrossRef CAS PubMed.
- S. Roy, A. Bandyopadhyay, M. Das, P. P. Ray, S. K. Pati and T. K. Maji, J. Mater. Chem. A, 2018, 6, 5587–5591 RSC.
- A. Singh, S. Roy, C. Das, D. Samanta and T. K. Maji, Chem. Commun., 2018, 54, 4465–4468 RSC.
- M. H. Alkordi, Ł. J. Weseliński, V. D'Elia, S. Barman, A. Cadiau, M. N. Hedhili, A. J. Cairns, R. G. AbdulHalim, J.-M. Basset and M. Eddaoudi, J. Mater. Chem. A, 2016, 4, 7453–7460 RSC.
- S. Alavinia and R. Ghorbani-Vaghei, RSC Adv., 2021, 11, 29728–29740 RSC.
- A. Demirbas, Energy Pol., 2007, 35, 4661–4670 CrossRef.
- X. Tang, S. Niu, S. Zhao, X. Zhang, X. Li, H. Yu, C. Lu and K. Han, J. Ind. Eng. Chem., 2019, 77, 432–440 CrossRef CAS.
- H. H. Mardhiah, H. C. Ong, H. H. Masjuki, S. Lim and H. V. Lee, Renewable Sustainable Energy Rev., 2017, 67, 1225–1236 CrossRef CAS.
- A. Rajalingam, S. P. Jani, A. S. Kumar and M. A. Khan, J. Chem. Pharm. Res., 2016, 8, 170–173 CAS.
- A. Bajaj, P. Lohan, P. N. Jha and R. Mehrotra, J. Mol. Catal. B: Enzym., 2010, 62, 9–14 CrossRef CAS.
- A. Guldhe, B. Singh, T. Mutanda, K. Permaul and F. Bux, Renewable Sustainable Energy Rev., 2015, 41, 1447–1464 CrossRef CAS.
- B.-X. Peng, Q. Shu, J.-F. Wang, G.-R. Wang, D.-Z. Wang and M.-H. Han, Process Saf. Environ. Prot., 2008, 86, 441–447 CrossRef CAS.
- W. Xie and H. Wang, Renew. Energy, 2020, 145, 1709–1719 CrossRef CAS.
- Q. Guan, Y. Li, Y. Chen, Y. Shi, J. Gu, B. Li, R. Miao, Q. Chen and P. Ning, RSC Adv., 2017, 7, 7250–7258 RSC.
- W. Y. Lou, Q. Guo, W. J. Chen, M. H. Zong, H. Wu and T. J. Smith, ChemSusChem, 2012, 5, 1533–1541 CrossRef CAS PubMed.
- H. H. Mardhiah, H. C. Ong, H. H. Masjuki, S. Lim and Y. L. Pang, Energy Convers. Manage., 2017, 144, 10–17 CrossRef CAS.
- Q. Shu, J. Gao, Z. Nawaz, Y. Liao, D. Wang and J. Wang, Appl. Energy, 2010, 87, 2589–2596 CrossRef CAS.
- J. Cheng, Y. Qiu, J. Zhang, R. Huang, W. Yang and Z. Fan, Bioresour. Technol., 2017, 244, 569–574 CrossRef CAS PubMed.
- M. C. Nongbe, T. Ekou, L. Ekou, K. B. Yao, E. Le Grognec and F.-X. Felpin, Renew. Energy, 2017, 106, 135–141 CrossRef CAS.
- V. Trombettoni, D. Lanari, P. Prinsen, R. Luque, A. Marrocchi and L. Vaccaro, Prog. Energy Combust. Sci., 2018, 65, 136–162 CrossRef.
- C. M. Xing and W. T. Yang, J. Polym. Sci., Polym. Chem. Ed., 2005, 43, 3760–3770 CrossRef CAS.
- R. R. C. Bastos, A. P. da Luz Corrêa, P. T. S. da Luz, G. N. da Rocha Filho, J. R. Zamian and L. R. V. da Conceição, Energy Convers. Manage., 2020, 205, 112457 CrossRef CAS.
- K. Dong, Q. Sun, X. Meng and F.-S. Xiao, Catal. Sci. Technol., 2017, 7, 1028–1039 RSC.
- Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC.
- H. Giesche, Part. Part. Syst. Charact., 2006, 23, 9–19 CrossRef.
- C. Voigt, J. Hubálková, H. Giesche and C. G. Aneziris, Microporous Mesoporous Mater., 2020, 299, 110125 CrossRef CAS.
- Y. Yang, Y. Chu, F. Yang and Y. Zhang, Mater. Chem. Phys., 2005, 92, 164–171 CrossRef CAS.
- D. Li, W. D. Yan, Z. Z. Yang and J. M. Zhang, Acta Polym. Sin., 2001, 561–564 CAS.
- J. C. Yang, M. J. Jablonsky and J. W. Mays, Polymer, 2002, 43, 5125–5132 CrossRef CAS.
- S. B. Brijmohan, S. Swier, R. A. Weiss and M. T. Shaw, Ind. Eng. Chem. Res., 2005, 44, 8039–8045 CrossRef CAS.
- S. Hosseini, J. Janaun and T. S. Y. Choong, Process Saf. Environ. Prot., 2015, 98, 285–295 CrossRef CAS.
- D. Y. C. Leung and Y. Guo, Fuel Process. Technol., 2006, 87, 883–890 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Zhaoji Wu,
Zhengge Wang,
Changjiu Chen and
Zhigang Zhang*
,
Zhaoji Wu,
Zhengge Wang,
Changjiu Chen and
Zhigang Zhang*