DOI:
10.1039/D2RA02833F
(Paper)
RSC Adv., 2022,
12, 24003-24013
Development of a fully automatic separation system coupled with online ICP-MS for measuring rare earth elements in seawater†
Received
4th May 2022
, Accepted 16th August 2022
First published on 24th August 2022
Abstract
Rare earth elements (REEs) are useful geological indicators of marine geochemistry. However, extremely low concentrations (sub-ng L−1) and high-salt matrices result in inefficient measurements. A fully automatic separation system (ELSPE-2 Precon) is used in the online determination of ultra-trace REEs in seawater using inductively coupled plasma mass spectrometry. This system mainly comprises three sections: (i) an auto-sampler (eas-2A) with 120 positions; (ii) a poly(styrene-divinylbenzene) resin column (Prin-Cen Col007) with iminodiacetic and ethylenediaminetriacetic acid functional groups to eliminate the high-salt matrix (e.g., Na, Ca, K, Mg, Al, Ba, Fe, Sr, P, and S) and preserve the target REEs; and (iii) a Trp002 cleanup column for the reduction of the reagent and procedural blank values. The detection limits (3σ) were in the range 0.002 (Dy)–0.097 ng L−1 (La), and the long-term reproducibility (8 h) was between 80% and 120% for all REEs in a 3.5% NaCl matrix solution. The accuracy of this method was verified using a seawater reference material (NASS-6), and the measured REE concentrations were consistent with those previously reported. The proposed online system was used to investigate coastal water samples with varying salinities from the Pearl River Estuary (Guangdong, China). Variations in the REE distribution patterns of different layers of seawater were observed, which could be due to the mixing of potentially light rare earth element-enriched bottom seawater. Moreover, a positive Gd anomaly in river water and seawater might be attributed to anthropogenic pollution from hospitals and the pharmaceutical industry.
1. Introduction
Rare earth elements (REEs) exhibit highly similar geochemical characteristics, but their ionic radii decrease with increasing atomic number, resulting in systematically varying properties. These properties, in addition to the anomalies of Ce and Eu, are useful geological indicators in the field of marine geochemistry. They are widely used to study geochemical processes along the water column, solid–liquid interfacial interactions, water mixing, and natural or anthropogenic additives in estuaries.1–4 Meanwhile, REEs are widely used in numerous high-tech devices and modern industries (e.g., magnets, electronics, optics, lasers, rechargeable batteries, medicine, ceramics, and glassmaking).5–8 Therefore, the REE characteristics in the environment may be used to trace the pollution of urban, agricultural, and industrial wastewater in coastal waters.9–12 However, accurately measuring REE concentrations in coastal water or seawater remains a challenge due to their extremely low concentrations (sub-ng L−1) and the presence of high-salt matrices.
Owing to its high sensitivity, inductively coupled plasma mass spectrometry (ICP-MS) is widely used to determine trace elements in various matrices.13–28 However, this technique is not easily applicable to a high-salt matrix (i.e., 3.5% NaCl) because of polyatomic mass spectral interferences, instrumental drift, signal suppression, and clogging of the sample introduction system.29–32 Therefore, various offline separation and/or preconcentration procedures (e.g., chelating resins,32–35 solvent extraction,36–39 or co-precipitation40–43) are applied before measuring the REE concentrations using ICP-MS. However, developing a standard operating procedure for laboratory usage is often challenging due to the concomitant challenges with offline procedures, such as the required complicated, time-consuming techniques and large sample volumes, and an increased possibility of sample contamination.
An alternative online procedure based on flow injection44–46 that uses ion exchange47,48 or chelating resins45,49 was coupled with ICP-MS to measure the REE concentrations in natural water samples. This technique is commonly used owing to its automatization potential, low required sample volume, and enhanced accuracy compared with those of the traditional method.50–52 Recently, two commercially available automated devices, the CETAC DSX-100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 53 and seaFAST™ systems,50 were developed for use in the online analyses of trace elements in seawater samples. The CETAC DSX-100 incorporates a closed chemical cycle that preconcentrates analytes and removes matrix components using a suspended particulate reagent (SPR) comprising polymeric beads within a cartridge that exhibits chelating iminodiacetic functional groups.53 Unfortunately, the lifetime of this SPR cartridge is limited because the resin is destroyed during analysis. In contrast, the seaFAST™ system separates REEs and matrices (e.g., alkali and alkaline earth cations and anions) using a cartridge of polymeric beads with iminodiacetic and ethylenediaminetriacetic acid functional group resins (particle size: 75 μm). The seaFAST™ coupled with ICP-MS was used to determine the elemental concentrations (including REEs) in undiluted seawater samples.54–58 However, its practicality is limited by its speed (∼4 samples per h), bulkiness (1.5 × 1 × 0.6 m), and high cost (>US$100 000).
53 and seaFAST™ systems,50 were developed for use in the online analyses of trace elements in seawater samples. The CETAC DSX-100 incorporates a closed chemical cycle that preconcentrates analytes and removes matrix components using a suspended particulate reagent (SPR) comprising polymeric beads within a cartridge that exhibits chelating iminodiacetic functional groups.53 Unfortunately, the lifetime of this SPR cartridge is limited because the resin is destroyed during analysis. In contrast, the seaFAST™ system separates REEs and matrices (e.g., alkali and alkaline earth cations and anions) using a cartridge of polymeric beads with iminodiacetic and ethylenediaminetriacetic acid functional group resins (particle size: 75 μm). The seaFAST™ coupled with ICP-MS was used to determine the elemental concentrations (including REEs) in undiluted seawater samples.54–58 However, its practicality is limited by its speed (∼4 samples per h), bulkiness (1.5 × 1 × 0.6 m), and high cost (>US$100 000).
Herein, we developed a novel commercially available automated system (ELSPE-2 Precon) (detailed information could be found from https://www.princensci.com/en) coupled with ICP-MS for use in measuring REE concentrations in coastal water and seawater. The system was designed with a higher throughput (>14 samples per h) and lower sample consumption (<1 mL) at a lower cost than those of the currently used systems, CETAC DSX-100 and seaFAST™. The excellent performance of the ELSPE-2 Precon was obtained by efficiently removing the high-salt matrices and reducing the procedural blank value. The long-term reproducibility and accuracy were verified by respectively analyzing a 10 ng L−1 mixed REE standard solution in a 3.5% NaCl matrix and a seawater reference material, NASS-6. Furthermore, the proposed system was used to analyze river water and seawater samples collected from the Pearl River Estuary (Guangdong, China).
2. Experimental
2.1. ELSPE-2 Precon system and measurement procedures
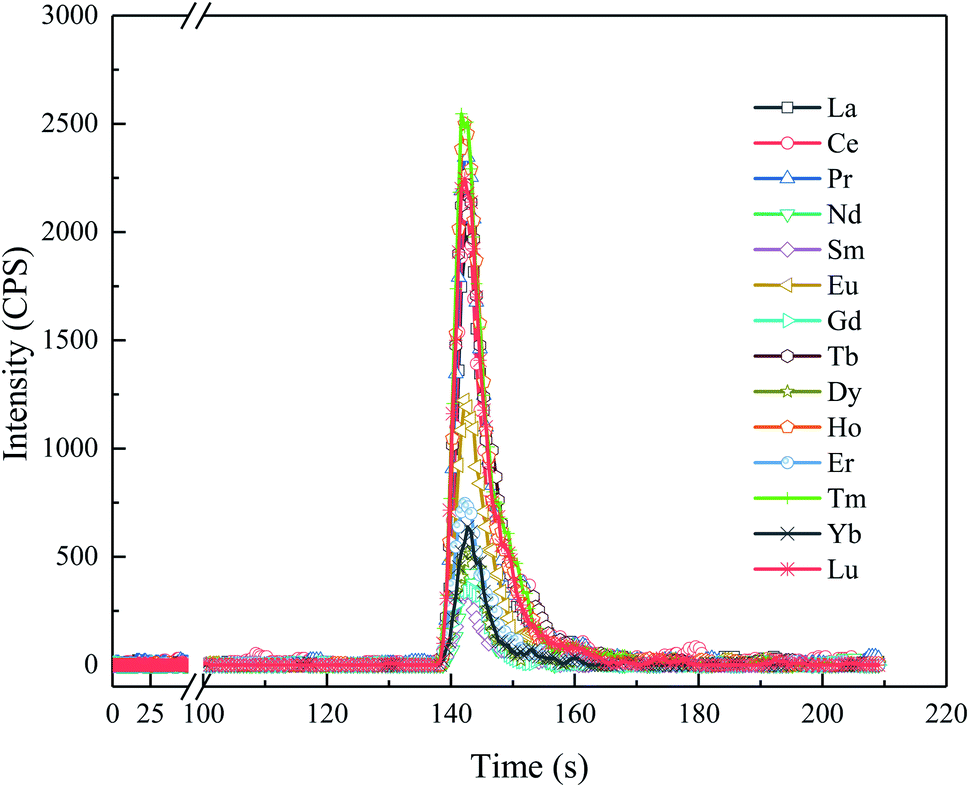
As shown in Fig. 1, the ELSPE-2 Precon system comprises an eas-2A autosampler (Prin-Cen, Guangzhou, China) with 120 positions, a Col007 separation column (Prin-Cen), Trp002 trace metal cleanup column (Prin-Cen, Guangzhou, China), one syringe pump (2.5 mL), three inert 6-port valves (Prin-Cen, Guangzhou, China), and three ion chromatography pumps (Prin-Cen, Guangzhou, China). All tubes and valves are fabricated using corrosion-resistant polyetheretherketone (PEEK) materials, and the pressure resistance of the system is up to 35 MPa. The dimensions of the entire system are only 0.5 × 0.5 × 0.4 m. The Col007 separation column uses poly(styrene-divinylbenzene) (PS-DVB) resin (5 μm, 200 mg) (Prin-Cen, Guangzhou, China) with iminodiacetic (IDA) and ethylenediaminetriacetic (ED3A) acid functional groups. High specific surface areas of the resins and adequate efficiency of the separation column are developed to ensure a sharp chromatographic peak (Fig. 2). The eluent is injected online into the introduction system (a glass concentric nebulizer and cyclonic spray chamber) of a NexION™ 350D ICP-M spectrometer (PerkinElmer, Waltham, MA, USA). The separation (ELSPE-2 Precon) and detection (ICP-MS) systems are simultaneously controlled using customized software (EasySpec, Guangzhou, China). The operating parameters of ELSPE-2 Precon and ICP-MS are shown in Table 1.
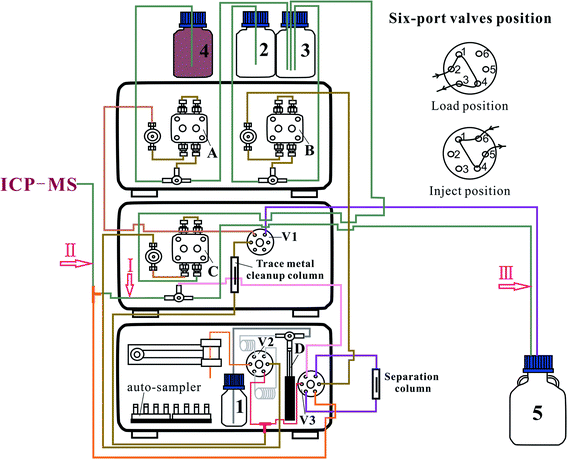 |
| | Fig. 1 Schematic configuration of the ELSPE-2 Precon system. 1 and 3: deionized water; 2: 5% HNO3; 4: ammonium acetate buffer; 5: waste; I and II: PTEE pipes with inner diameters of ≥0.25 mm; III: polytetrafluoroethylene pipe with a 0.5 mm inner diameter; A: ammonium acetate pump; B: HNO3 pump; C: water pump; D: syringe pump; V1, V2, and V3: six-port valves. | |
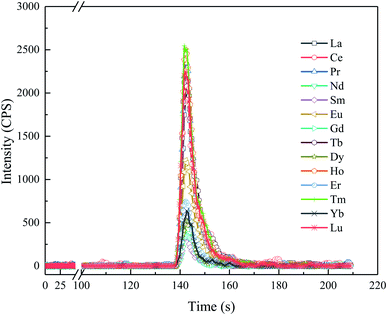 |
| | Fig. 2 Typical chromatograms of the REEs in a mixed REE standard solution (5 ng L−1), as obtained using the ELSPE-2 Precon automated separation system and online ICP-MS. | |
Table 1 Operating conditions of the ICP-MS automated separation system
| Automated separation ELSPE-2 Precon system |
Parameters |
| Mode of analysis |
Online |
| Column resin |
PS-DVB resin |
| Buffer |
2 moL L−1 ammonium acetate, pH 5.8–6 |
| Eluent |
5% HNO3 |
| Sample acidity |
1% HNO3 |
| Initial volume of the sample |
900 μL |
| Sample throughput |
4 min 10 s per sample |
| NexION™ 350D ICP-MS |
Parameters |
| ICP RF power |
1300 W |
| Nebulizer gas flow |
0.64 L min−1 |
| Auxiliary gas flow |
1.4 L min−1 |
| Plasma gas flow |
15 L min−1 |
| Sensitivity of 1 μg L−1 115In |
>45000 cps |
| Background on mass 220 |
0.67 cps |
| Oxide formation CeO+/Ce |
1.6% |
| Sweep |
20 |
| Reading |
480 |
| Replicates |
1 |
| Dwell time |
26 ms |
| Internal standard |
103Rh |
| Isotopes monitored |
139La, 140Ce, 141Pr, 143Nd, 147Sm, 153Eu, 157Gd, 159Tb, 163Dy, 165Ho, 166Er, 169Tm, 172Yb, and 175Lu |
| Control software |
EasySpec |
The detailed analytical procedures of the ELSPE-2 Precon system are shown in Table 2. Only 4 min 10 s is required to analyze each sample, the target REE chromatogram peaks last for 15–20 s (Fig. 2), and the sample injection volume is optimized (Fig. S1†). Based on the signal intensity (cps), signal-to-background ratio (SBR), and relative standard deviation (RSD), a sample injection volume of 900 μL is necessary in this study.
Table 2 Steps of the separation procedures
| Step |
Valve (V) positions |
Syringe (S) action, (flow rate – run time) |
Action |
| (1) Load sample on coil and clean columns |
V1: load, V2: load, V3: load |
A: 1 mL min−1 – 30 s, B: 1 mL min−1 – 30 s, C: 1 mL min−1 – 30 s, D: Adjusted for each sample set – 30 s |
Sampling probe penetrated sample solution, vacuum pump turned on and aspirated sample into 1 mL sample coil. Meanwhile, DI water was passed through the tandem columns (trace metal cleanup column Trp002 and separation column Col007) |
| (2) Condition columns |
V1: load, V2: load, V3: load |
A: 1 mL min−1 – 20 s, B: 1 mL min−1 – 20 s, C: 1 mL min−1 – 20 s |
Ammonium acetate buffer (2 mol L−1) was passed through the tandem columns. The REE background of this buffer was reduced by the first column (Trp002), and the purified ammonium acetate was passed through the Col007 (separation) column to maintain a pH of 6.0 |
| (3) Load sample on resin and rinse matrix |
V1: load, V2: inject, V3: load |
A: 1 mL min−1 – 70 s, B: 1 mL min−1 – 70 s, C: 1 mL min−1 – 70 s, D: 1 mL min−1 – 70 s |
Sample (0.9 mL) and buffer were mixed before injection into the Col007 separation column. After injecting the sample, the matrix elements (i.e., Na, K, and alkaline earth elements) were eluted using ammonium acetate (2 mL min−1), while the target REEs were chelated onto the Col007 column |
| (4) rinse out buffer |
V1: inject, V2: inject, V3: load |
A: 0.6 mL min−1 – 20 s, B: 1 mL min−1 – 20 s, C: 2 mL min−1 – 20 s |
DI water was pumped through the Col007 column to rinse out the ammonium acetate |
| (5) Elution |
V1: inject, V2: inject, V3: inject |
A: 0.6 mL min−1 – 110 s, B: 1 mL min−1 – 110 s, C: 0.6 mL min−1 – 110 s |
The Col007 column was reverse flushed with 5% HNO3. The eluent was introduced into the ICP-MS detector, and the target REEs were measured |
2.2. Reagents, standards, and samples
All experiments were conducted in a class-1000 ultra-clean laboratory (State Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences, Wuhan, China). High-purity HNO3 and 2 mol L−1 ammonium acetate (pH = 5.8–6) was obtained from Sinopharm Chemical Reagent (Shanghai, China) and Prin-Cen, respectively. HNO3 was further distilled twice using a custom-built PFA sub-boiling distillation system, and deionized (DI) water (18.2 MΩ cm) was obtained using a Milli-Q purification system (MilliporeSigma, Burlington, MA, USA). The REE standard solutions were diluted from a 10 μg mL−1 multi-REE mixed standard solution (SPEX CertiPrep, Metuchen, NJ, USA) using 5% HNO3. All reagents, standard solutions, and samples were stored in Teflon-fluorinated ethylene propylene (FEP) bottles. All bottles and tubes were soaked for one day in 10% HNO3 and rinsed three times with DI water. The seawater reference material NASS-6 was purchased from the National Research Council of Canada (NRC, Ottawa, Canada) for the evaluation of the method. Eighteen coastal samples (river water and seawater) were collected from the Pearl River Estuary from the 9th to the 11th of July 2021, with ∼50 mL of each sample collected and the sampling sites shown in Fig. S2.† After filtration using 0.45 μm filters, the pH values and conductivities of the samples were measured (Table S1†), and each sample was acidified in situ to form a 1% distilled HNO3 solution and stored in an FEP bottle.
3. Results and discussion
3.1. Removal of the high-salt matrix
The most challenging issue in accurately measuring sub-ng L−1 REEs in seawater is the presence of the high-salt matrix, which leads to significant signal suppression, polyatomic mass spectral interferences (i.e., the Eu signal is interfered with by that of BaO), instrumental drift, and clogging of the sample introduction system.29–32 Thus, the main objective of the proposed automated separation system, ELSPE-2 Precon, is to eliminate the high-salt matrix (i.e., alkali and alkaline earth metals and anions) within the seawater. In this study, three different layers of seawater (i.e., S5 surface, S1 intermediate, and S1 bottom seawater) were used to evaluate the removal rates of several major matrix elements and ions (i.e., Ba, Na, Mg, Ca, K, Sr, Fe, PO43−, and SO42−). At pH = 5–6, the poly(styrene-divinylbenzene) (PS-DVB) resins with iminodiacetate (IDA) and ethylenediaminetriacetate (ED3A) functional groups can absorb ∼100% REE elements rather than alkali and alkaline earth elements and anion. Therefore, the resin would use to remove the major matrix elements and ions of seawater.50,51 As shown in Table 3, the removal rates of all ions are >99.1%, and even 99.9% of Na (the most abundant element in seawater) is effectively removed. Notably, the interference of BaO with Eu is ignored because of the low Ba concentrations in seawater samples (<200 μg L−1), high removal efficiency of the system (>99.3%), and low oxidation yield during ICP-MS (1.6%). Therefore, the removal efficiencies of the high-salt matrix and Ba within seawater using the proposed automated separation system enables accurate determination of sub-ng L−1 REE concentrations in seawater.
Table 3 Removal rates of the high-salt matrices in three seawater samples using the ELSPE-2 Precon system, %
| Matrix |
S5 Surface seawater |
S1 Intermediate seawater |
S1 Bottom seawater |
| Before separation mg L−1 |
After separation mg L−1 |
Removal rate % |
Before separation mg L−1 |
After separation mg L−1 |
Removal rate % |
Before separation mg L−1 |
After separation mg L−1 |
Removal rate, % |
| Concentration units are μg L−1. |
| Baa |
140 |
0.90 |
99.4 |
70.0 |
0.38 |
99.5 |
80.0 |
0.45 |
99.4 |
| Ca |
164 |
1.43 |
99.1 |
438 |
1.62 |
99.6 |
426 |
2.00 |
99.5 |
| Fe |
1.57 |
0.01 |
99.5 |
2.28 |
0.02 |
99.1 |
2.40 |
0.01 |
99.5 |
| K |
66 |
0.03 |
100 |
25.7 |
0.16 |
99.4 |
85.2 |
0.06 |
99.9 |
| Mg |
516 |
1.50 |
99.7 |
1416 |
2.41 |
99.8 |
1420 |
3.12 |
99.8 |
| Na |
3285 |
0.33 |
100 |
9510 |
0.95 |
100 |
9626 |
1.93 |
100 |
| Sr |
2.84 |
0.01 |
99.7 |
7.75 |
0.02 |
99.8 |
7.81 |
0.02 |
99.7 |
| PO43– |
23.5 |
0.06 |
99 |
12.9 |
0.10 |
99.3 |
14.2 |
0.13 |
99.1 |
| SO42– |
329 |
0.03 |
100 |
888 |
0.00 |
100 |
909 |
0.09 |
100 |
3.2. Reduction of the procedural blank
Another challenge in accurately analyzing sub-ng L−1 REEs in seawater is to reduce the blank value of the entire process (or procedural blank value) as much as possible, which may result in a high SBR and low detection limit (DL). In this study, the procedural blank values are the signal intensities of REEs in a 5% HNO3 blank sample and mainly include reagent and manifold blanks. Because the signal of the distilled HNO3 blank is very low, the source of the main reagent blank is ammonium acetate (as a buffer). In the ELSPE-2 Precon system, a special purification module equipped with a trace metal cleanup Trp002 column (Fig. 1) is designed to minimize the REE background of ammonium acetate. The performance before and after the purification were shown in Table 4, the ratio of the blank value for each target after purification to those of without purification (100 × reagent blank with purification/reagent blank without purification, %) ranged from 22% to 92%. The calculated SBR of La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb and Lu increased 3.7-, 1.9-, 1.6-, 1.1-, 1.2-, 3.2-, 1.3-, 4.8-, 3.7-, 1.2-, 1.8-, 6.6-, 8.9- and 3.2-fold (SBRwith purification/SBRwithout purification, in Table 4), respectively, indicating the improved DL of this method. The manifold blank was reduced as follows: (i) a syringe pump was used instead of the conventional peristaltic pump to minimize the possibility of contamination. (ii) Clean tubes and valves fabricated using corrosion-resistant PEEK materials were used. (iii) All preparation steps for reagents and samples were performed in the clean laboratory (class 1000). (iv) The Trp002 and Col007 columns were cleaned for >0.5 h with 5% HNO3 before sample analysis to ensure that the signal intensities of the most abundant elements in the procedural blank (e.g., La and Ce) were 1200 cps.
Table 4 Reagent blank signal intensities and signal-to-background ratios (SBRs) of 2 mol L−1 ammonium acetate with and without purification using the Trp002 column
| REEs |
Without purification |
With purification |
Performance comparison |
| Reagent blank, cps |
Sensitivity of 1 ng L−1 standard, cps |
SBR a |
Reagent blank, cps |
Sensitivity of 1 ng L−1 standard, cps |
SBR |
100 × reagent blank with purification/reagent blank without purification, % |
SBRwith purification/SBRwithout purificati |
| SBR = (sensitivity of 1 ng L−1 standard reagent blank)/reagent blank signal intensity. |
| La |
2853 ± 560 |
6379 ± 661 |
1.2 |
628 ± 110 |
3405 ± 163 |
4.4 |
22 |
3.7 |
| Ce |
2722 ± 798 |
5577 ± 603 |
1 |
1165 ± 106 |
3428 ± 617 |
1.9 |
43 |
1.9 |
| Pr |
308 ± 97 |
2723 ± 355 |
7.8 |
220 ± 36 |
2913 ± 404 |
12.2 |
71 |
1.6 |
| Nd |
287 ± 133 |
933 ± 90 |
2.3 |
158 ± 17 |
561 ± 68 |
2.6 |
55 |
1.1 |
| Sm |
38 ± 32 |
370 ± 106 |
8.8 |
28 ± 10 |
330 ± 104 |
10.7 |
74 |
1.2 |
| Eu |
102 ± 49 |
1322 ± 313 |
12 |
37 ± 14 |
1446 ± 297 |
38 |
36 |
3.2 |
| Gd |
38 ± 20 |
505 ± 50 |
12.2 |
35 ± 19 |
613 ± 106 |
16.3 |
92 |
1.3 |
| Tb |
77 ± 69 |
2482 ± 529 |
31.4 |
18 ± 17 |
2736 ± 568 |
149.4 |
23 |
4.8 |
| Dy |
44 ± 40 |
467 ± 227 |
9.5 |
18 ± 1 |
660 ± 89 |
34.9 |
41 |
3.7 |
| Ho |
23 ± 6 |
2677 ± 297 |
117 |
20 ± 6 |
2897 ± 385 |
142.2 |
87 |
1.2 |
| Er |
121 ± 55 |
1025 ± 572 |
7.5 |
73 ± 30 |
1081 ± 212 |
13.8 |
60 |
1.8 |
| Tm |
38 ± 19 |
2807 ± 186 |
72.8 |
7 ± 9 |
3166 ± 244 |
478.4 |
18 |
6.6 |
| Yb |
57 ± 58 |
406 ± 328 |
6.1 |
13 ± 9 |
736 ± 309 |
54.3 |
23 |
8.9 |
| Lu |
45 ± 48 |
2806 ± 132 |
61.6 |
17 ± 18 |
3372 ± 295 |
200 |
38 |
3.2 |
3.3. Detection limit, accuracy, and long-term reproducibility
Calibration curves were constructed using mixed REE standard solutions (0.5–20 ng L−1), and linear correlation coefficients (R2 > 0.999) were calculated for all REEs (Table 5). Low DLs are obtained (0.002–0.097 ng L−1), with the DL defined as three times the standard deviation of the procedural blank value divided by the sensitivity (i.e., the slope of the calibration curve). The proposed procedural blank values, SBRs, and DLs are superior to those reported by Hathorne et al.50 and Zhu et al.,59 using the seaFAST S1 and S2 systems, respectively.
Table 5 Linearity coefficients, procedural blanks, detection limits, and relative standard deviations
| REEs |
Linearity coefficient (R2) |
Procedural blank (ng L−1) |
SBRa |
Detection limit (ng L−1) |
RSDb (%) |
| SBR is the signal-to-background ratio. RSD is the relative standard deviation. |
| La |
0.9999 |
0.185 |
4.4 |
0.097 |
0.17 |
| Ce |
0.9997 |
0.340 |
1.9 |
0.093 |
0.09 |
| Pr |
0.9999 |
0.076 |
12.2 |
0.037 |
0.16 |
| Nd |
0.9994 |
0.281 |
2.6 |
0.089 |
0.11 |
| Sm |
0.9997 |
0.085 |
10.7 |
0.093 |
0.36 |
| Eu |
0.9998 |
0.026 |
38.0 |
0.029 |
0.37 |
| Gd |
0.9996 |
0.058 |
16.3 |
0.091 |
0.52 |
| Tb |
0.9999 |
0.007 |
149 |
0.018 |
0.91 |
| Dy |
0.9998 |
0.028 |
34.9 |
0.002 |
0.03 |
| Ho |
0.9999 |
0.007 |
142 |
0.006 |
0.29 |
| Er |
0.9994 |
0.068 |
13.8 |
0.083 |
0.41 |
| Tm |
0.9997 |
0.002 |
478 |
0.009 |
1.39 |
| Yb |
0.9997 |
0.018 |
54.3 |
0.035 |
0.64 |
| Lu |
0.9997 |
0.005 |
200 |
0.016 |
1.06 |
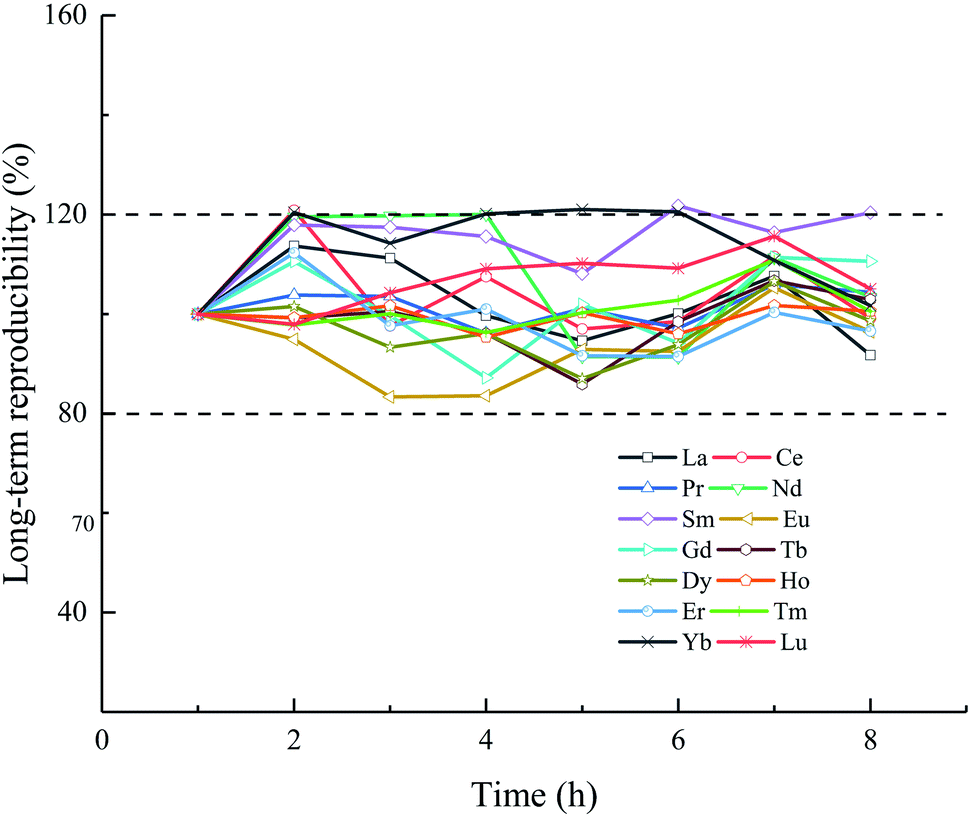
The accuracy of the proposed method was evaluated using the seawater reference material NASS-6. Because no certified values of the REEs were obtained from the producer (NRC), the accuracies of the NASS-6 values obtained during this study are compared with those previously reported.31,59–62 As shown in Table 6, the measured values are consistent with the reference values, which confirms the reliability of the proposed method based on the ELSPE-2 Precon automated separation system. The short-term precision (2SD, n = 3) of NASS-6 analysis is in the range 0.02 (Tm)–1.75 ng L−1 (La). The long-term reproducibility was also verified using a 10 ng L−1 mixed REE standard solution in a 3.5% NaCl matrix for 8 h. As shown in Fig. 3, the long-term reproducibility of this mixed solution is 80–120% (normalized results of measured values to added concentration) for all REEs.
Table 6 REE concentrations in the seawater reference material NASS-6 in this work and those previously reported (ng L−1)
| REEs |
This worka |
Zhu15 |
Zhu et al.43 |
Raso et al.44 |
Wang et al.45 |
Wysocka et al.46 |
| Mean ± 2SD (N = 3). |
| La |
11.94 ± 1.75 |
9.93 ± 0.14 |
11.82 ± 0.62 |
12.70 ± 0.8 |
10.70 ± 1.25 |
10.18 ± 1.06 |
| Ce |
5.10 ± 0.63 |
3.35 ± 0.19 |
6.35 ± 0.49 |
6.20 ± 0.60 |
4.48 ± 0.84 |
3.93 ± 0.42 |
| Pr |
1.72 ± 0.09 |
1.46 ± 0.03 |
1.83 ± 0.14 |
2.00 ± 4.00 |
1.69 ± 0.28 |
1.56 ± 0.17 |
| Nd |
6.52 ± 0.98 |
5.88 ± 0.26 |
7.84 ± 0.36 |
6.00 ± 6.00 |
6.78 ± 1.15 |
6.75 ± 0.72 |
| Sm |
1.05 ± 0.42 |
1.08 ± 0.04 |
1.42 ± 0.10 |
0.80 ± 0.14 |
1.35 ± 0.15 |
1.17 ± 0.13 |
| Eu |
0.26 ± 0.06 |
0.25 ± 0.05 |
0.27 ± 0.07 |
0.24 ± 0.08 |
0.27 ± 0.03 |
0.26 ± 0.030 |
| Gd |
1.20 ± 0.15 |
1.39 ± 0.03 |
1.85 ± 0.08 |
1.00 ± 4.00 |
1.34 ± 0.28 |
1.39 ± 0.14 |
| Tb |
0.23 ± 0.01 |
0.21 ± 0.02 |
0.26 ± 0.05 |
0.20 ± 0.40 |
0.24 ± 0.10 |
0.23 ± 0.025 |
| Dy |
1.71 ± 0.04 |
1.46 ± 0.03 |
1.80 ± 0.08 |
1.60 ± 0.40 |
1.63 ± 0.16 |
1.67 ± 0.18 |
| Ho |
0.46 ± 0.04 |
0.36 ± 0.02 |
0.45 ± 0.06 |
0.31 ± 0.04 |
0.40 ± 0.12 |
0.42 ± 0.044 |
| Er |
1.45 ± 0.31 |
1.25 ± 0.05 |
1.47 ± 0.16 |
1.70 ± 0.60 |
1.34 ± 0.17 |
1.39 ± 0.15 |
| Tm |
0.24 ± 0.02 |
0.19 ± 0.01 |
0.21 ± 0.05 |
0.24 ± 0.18 |
0.19 ± 0.05 |
0.20 ± 0.022 |
| Yb |
1.38 ± 0.24 |
1.21 ± 0.15 |
1.40 ± 0.14 |
1.30 ± 0.40 |
1.31 ± 0.28 |
1.37 ± 0.14 |
| Lu |
0.22 ± 0.03 |
0.19 ± 0.01 |
0.24 ± 0.07 |
0.26 ± 0.08 |
0.21 ± 0.07 |
0.22 ± 0.025 |
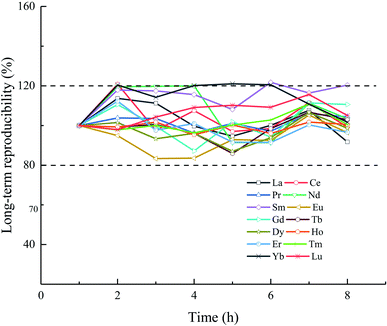 |
| | Fig. 3 Normalized results of measured concentration: added concentration (10 ng L−1 mixed REE standard solution with a 3.5% NaCl matrix) over 8 h. | |
3.4. Applications to seawater collected from the Pearl River Estuary
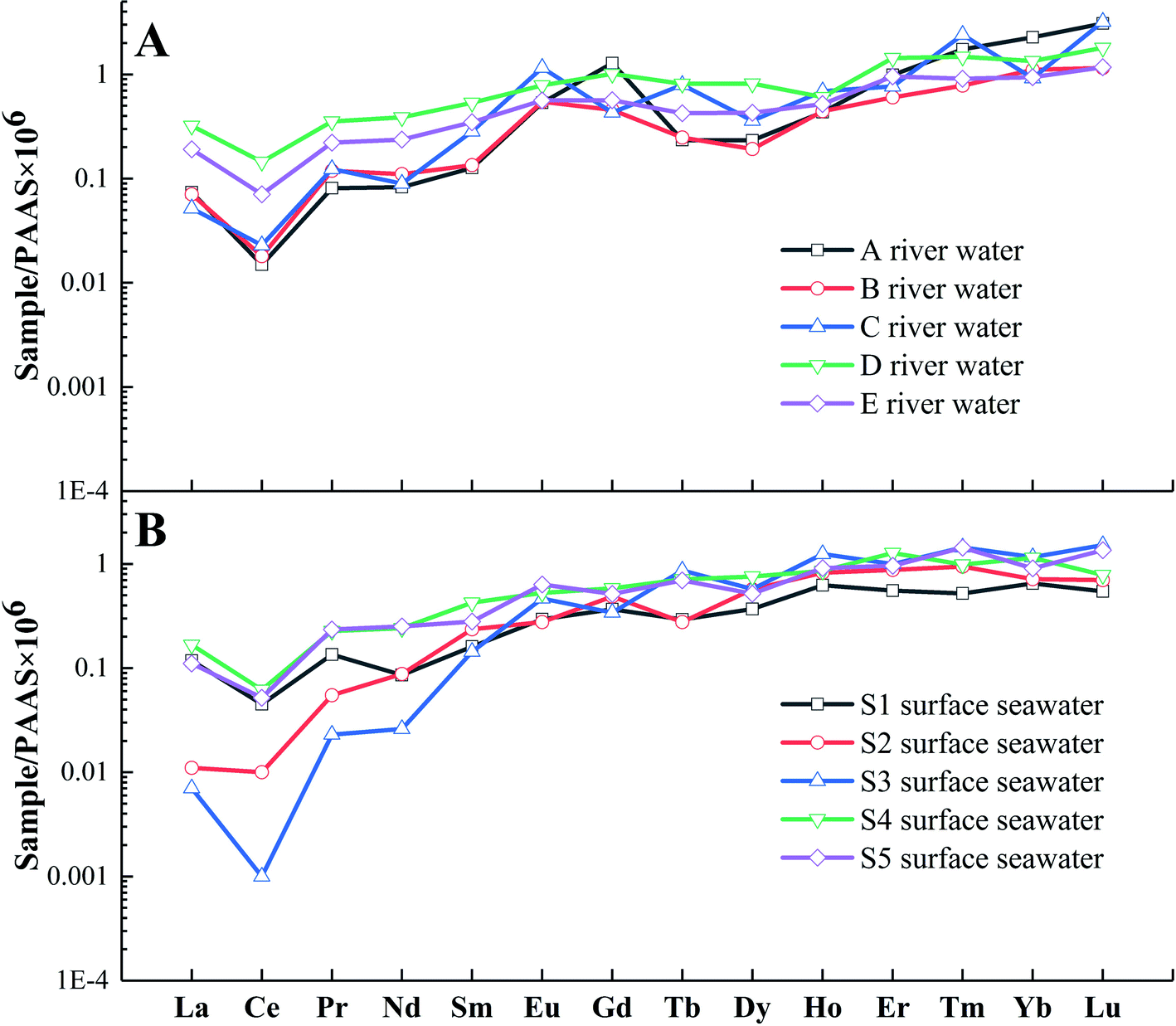
The proposed system was applied to measure the REE concentrations of 18 coastal water samples (5 river water and 13 seawater samples) to trace their sources to determine whether they originated from geochemical processes, anthropogenic pollution, or a combination. To interpret the geochemical behaviors of REEs in coastal waters, the results of REE assessment, based on the distribution patterns after normalization to the Post-Archean Australian Shale (PAAS) values63 and the GdN/GdN* values (the calculation method is previously reported7), are shown in Tables S2 and S3.†
As shown in Fig. 4, typical light rare earth element (LREE)-depleted (LREEN/MREEN = 0.03–0.54) and high rare earth element (HREE)-enriched (HREEN/MREEN = 1.29–11.9) distributions are observed within the river water (Fig. 4A) and seawater samples (Fig. 4B). The most negative bias of LREEs occurs at Point A of the river water, which indicates that this area is most affected by seawater mixing, and the higher conductivity of this sample compared to those of the other river water samples supports this explanation (Table S1†). During land-sea mixing, the notable change in water salinity leads to the migration of more LREEs from the water body to the soluble organic colloids64,65 and inorganic complex flocculation.66 This contributes to an increasing LREE loss in samples removed closer to the sea.
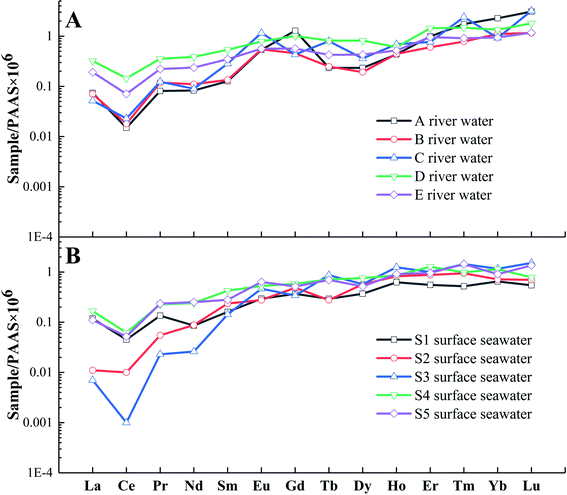 |
| | Fig. 4 REE patterns (PAAS normalized values) in river water (A) and surface seawater (B) samples. | |
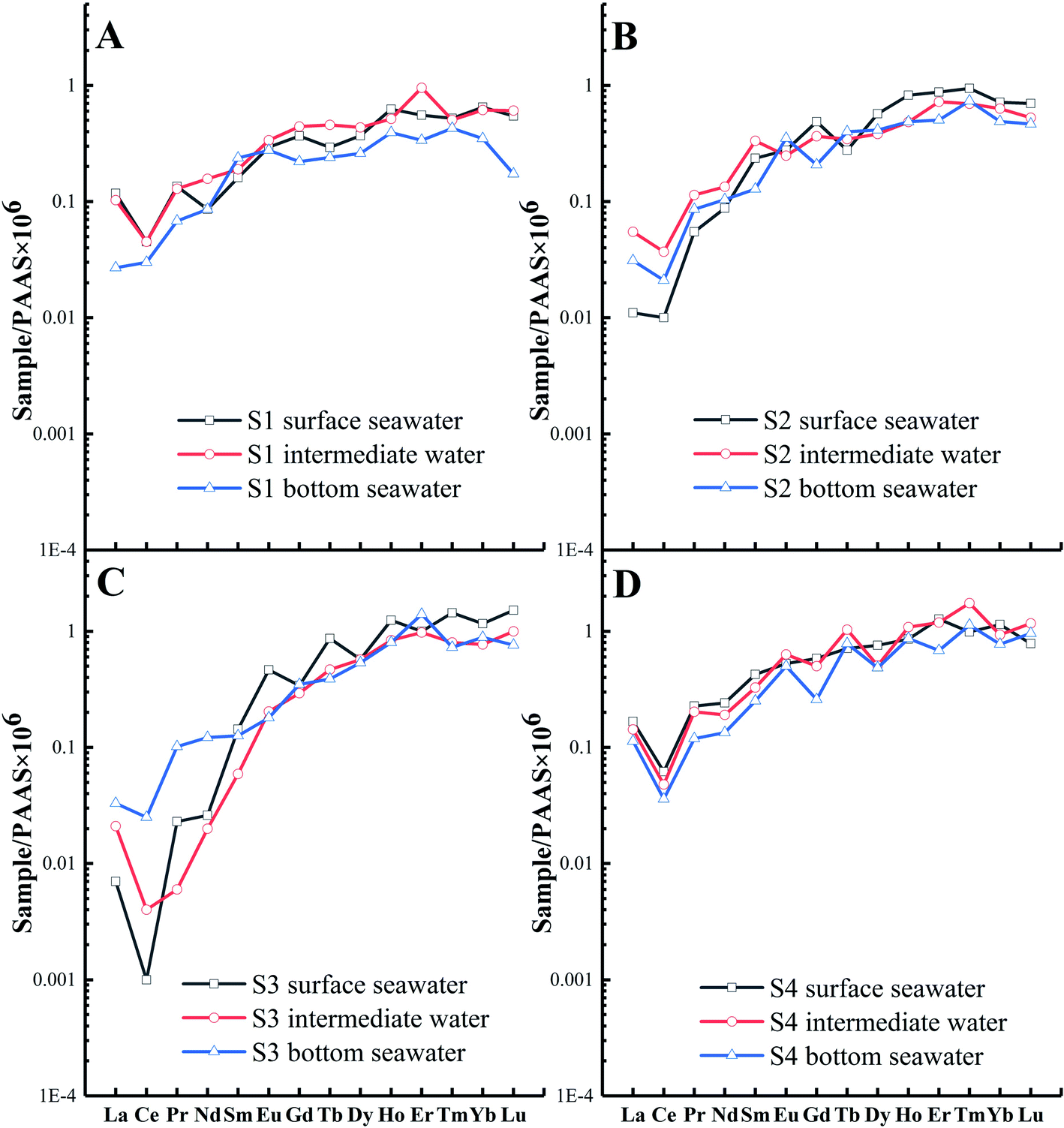
Based on the results, the correlations between the ∑REE and pH values and salinities of the seawater samples are not apparent. This is inconsistent with previous studies67–69 that report that the physical and chemical indicators of water (such as salinity and pH) may affect the REE concentration in coastal seawater, which may be due to the general consistency in the salinities and pH values of most seawater bodies (2.91–3.30% salinity and pH values of 7.88–8.26, Table S1 and Fig. S3†). Moreover, several studies reveal that the precipitation of suspended solids leads to REE fractionation between the different water layers.68,70 However, the REE distribution patterns of seawater in different layers (i.e., S1, S2, and S4) display no significant variations (Fig. 5A, B, and D) in this study, which may be related to the shallow seawater depth or strong hydrodynamic force of the coastal area. However, the LREEs of the S3 bottom seawater are exceptions owing to a richer composition compared to those of the surface and intermediate seawater (Fig. 5C). Considering the significant salinity variations in the vertical direction of the water column (Table S1†), the observed pattern may be affected by the mixing of potentially LREE-enriched and low-salinity water with bottom seawater.
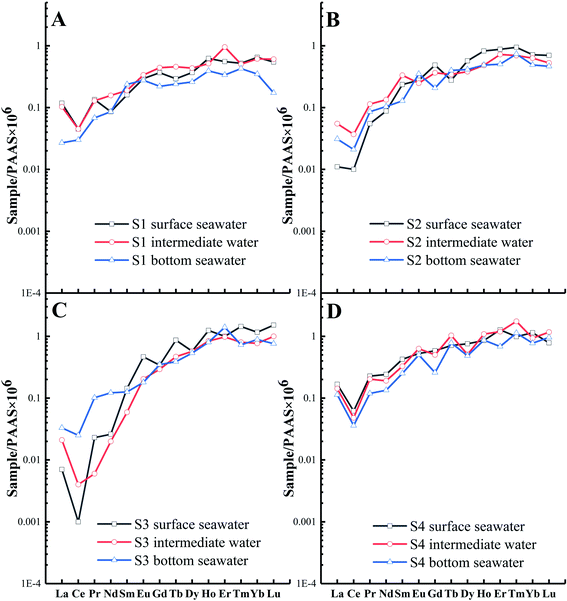 |
| | Fig. 5 REE patterns (PAAS normalized values) in different layers (depths) at four sampling points: (A) S1, (B) S2, (C) S3, and (D) S4. | |
A positive Gd anomaly (GdN/GdN* = 1.08–6.44) is observed in surface water (Table S3†), which is consistent with an earlier study using samples collected from the same area between December 2016 and August 2017.71 Several researchers state that the extensive use of gadopentetic acid contrast agents in magnetic resonance imaging is the main cause of Gd emission into the environment.6–8,10 In the Pearl River Delta region, effluents from hospitals and the pharmaceutical industry may be one of the main pollution sources, leading to serious Gd pollution of river water and causing the Gd anomaly. Several seawater samples also exhibit positive Gd anomalies, which may be caused by mixing with river water. Therefore, the observed Gd anomaly is useful in tracking anthropogenic pollution in seawater, and stricter pollution control measures should be implemented in the Pearl River Estuary.
4. Conclusions
Compared with those of the CETAC DSX-10053 and SeaFAST™ systems,50 the proposed ELSPE-2 Precon system (Table S4†) exhibited a higher throughput (>14 samples per h), lower sample consumption (∼1 mL) and cost (<US$50 000), and smaller dimensions (0.5 × 0.5 × 0.4 m). The comparatively superior analytical performance demonstrated by the system (based on procedural blanks, DLs, accuracy, and long-term reproducibility) confirmed its suitability for use in routine laboratory analyses. The ELSPE-2 Precon system was successfully applied to investigate the REE concentrations in coastal water samples collected from the Pearl River Estuary. The results showed that the mixing of coastal waters (river water or seawater) is a natural phenomenon—i.e., the mixing of bottom seawater and anthropogenic pollution from hospitals or pharmaceutical industries.
Author contributions
H. T. Li. and R. Tong wrote the manuscript and did most of the works. W. Guo designed the framework of the work and polish the manuscript. Q. H. Xu and L. L. Jin assisted with the experiments. W. Guo, Q. H. Xu, and S. H. Hu conceived the idea. D. Y. Tao and Yang L. gleaned the material and helped with literature and data processing. All authors contributed to the article and approved the submitted version.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (41873072, 41521001, and 4202010400), the Guangdong Prin-Cen Scientific Project (20220101), the Natural Science Foundation of Hubei Province (2020CFA045).
References
- D. S. Alibo and Y. Nozaki, Geochim. Cosmochim. Acta, 1999, 63, 363–372 CrossRef CAS.
- T. M. Conway, T. J. Horner, Y. Plancherel and A. G. González, Chem. Geol., 2021, 580, 120381 CrossRef CAS.
- S. Piarulli, B. H. Hansen, T. Ciesielski, A.-L. Zocher and A. Malzahn, Environ. Pollut., 2021, 291, 118230 CrossRef CAS PubMed.
- Y. Nozaki and D. S. Alibo, Earth Planet. Sci. Lett., 2003, 205, 155–172 CrossRef CAS.
- R. Kaegi, A. Gogos, A. Voegelin, S. J. Hug, L. H. E. Winkel, A. M. Buser and M. Berg, Water Res.: X, 2021, 11, 100092 CAS.
- R. L. B. Andrade, V. Hatje, R. M. A. Pedreira, P. Böning and K. Pahnke, Chem. Geol., 2020, 532, 119303 CrossRef CAS.
- H. Song, W. J. Shin, J. S. Ryu, H. S. Shin, H. Chung and K. S. Lee, Chemosphere, 2017, 172, 155–165 CrossRef CAS PubMed.
- N. Tepe, M. Romero and M. Bau, Appl. Geochem., 2014, 45, 191–197 CrossRef CAS.
- M. Desrosiers, G. Pelletier, D. Dieme, J. Cote, M. Jomaa, A. Nong and M. Bouchard, Environ. Int., 2020, 155, 106685 CrossRef PubMed.
- G. Klaver, M. Verheul, I. Bakker, E. Petelet-Giraud and P. Négrel, Appl. Geochem., 2014, 47, 186–197 CrossRef CAS.
- K. Schmidt, M. Bau, G. Merschel and N. Tepe, Sci. Total Environ., 2019, 687, 1401–1408 CrossRef CAS PubMed.
- G. Merschel and M. Bau, Sci. Total Environ., 2015, 533, 91–101 CrossRef CAS PubMed.
- Y. Wu, D. M. Huang, T. Feng, L. L. Jin, D. Xiong, J. Yu, Q. Xu, R. M. Huang and S. H. Hu, At. Spectrosc., 2021, 42, 374–382 CAS.
- A. Kierulf, C. Pytyck and D. Beauchemin, At. Spectrosc., 2021, 42, 365–373 CAS.
- R. A. Althobiti and D. Beauchemin, At. Spectrosc., 2021, 42, 271–277 CrossRef CAS.
- Y. Hua, S. Zhang, H. Min, J.-Y. Li, X.-H. Wu, D. Sheng, X.-B. Cui, Y.-J. Chen, C. Li, H.-Z. Lian and S. Liu, At. Spectrosc., 2021, 42, 217–226 CAS.
- F. F. Jia, Q. Luo, Y. Y. Zhang, Y. Zhao, K. Wu and F. Y. Wang, At. Spectrosc., 2021, 42, 166–174 CAS.
- O. V. Kuznetsova and A. R. Timerbaev, At. Spectrosc., 2021, 42, 85–90 CrossRef CAS.
- Y. T. Xiao, J. Yang, J. Deng, W. Wang, Y. Q. Ke and Y. J. Sun, At. Spectrosc., 2021, 42, 36–42 CrossRef CAS.
- N. Lv, K. Y. Chen, Z. A. Bao, K. Wu, D. B. Lei and H. L. Yuan, At. Spectrosc., 2021, 42, 51–61 CrossRef CAS.
- J. H. Liu, L. N. Zheng, J. W. Shi, X. Wei, X. Li, M. L. Chen, M. Wang, J. H. Wang and W. Y. Feng, At. Spectrosc., 2021, 42, 114–119 CAS.
- J. Z. Zhou, X. Ni, J. B. Fu, Y. T. Li, W. Guo, L. L. Jin, Y. E. Peng and S. H. Hu, At. Spectrosc., 2021, 42, 210–216 CrossRef CAS.
- J. T. Li, H. Qin and T. Yang, At. Spectrosc., 2021, 42, 359–364 CAS.
- X. Lin, W. Guo, L. L. Jin and S. H. Hu, At. Spectrosc., 2020, 41, 1–10 CrossRef CAS.
- W. X. Wang, X. Dai, W. Guo, L. L. Jin and S. H. Hu, At. Spectrosc., 2020, 41, 74–80 CrossRef CAS.
- S. Z. Chen, Y. X. Liu, J. T. Yan, C. L. Wang and D. B. Lu, At. Spectrosc., 2020, 41, 169–174 CrossRef CAS.
- L. W. Liu, Y. Cheng, J. H. Yuan and Y. Su, At. Spectrosc., 2020, 41, 242–248 CAS.
- J. Lu, W. Chen, S.-Y. Jiang, K.-D. Zhao, A. Simonetti and D.-H. Pi, At. Spectrosc., 2020, 41, 223–233 CrossRef CAS.
- C.-H. Chung, I. Brenner and C.-F. You, Spectrochim. Acta, Part B, 2009, 64, 849–856 CrossRef.
- N. M. Raut, L.-S. Huang, S. K. Aggarwal and K.-C. Lin, Spectrochim. Acta, Part B, 2003, 58, 809–822 CrossRef.
- Y. Zhu, Talanta, 2020, 209, 120536 CrossRef CAS PubMed.
- Y. Zhu, K. Nakano, Y. Shikamori and A. Itoh, Spectrochim. Acta, Part B, 2021, 179, 106100 CrossRef CAS.
- K. Schmidt, M. Bau, G. Merschel and N. Tepe, Sci. Total Environ., 2019, 687, 1401–1408 CrossRef CAS PubMed.
- G. Merschel and M. Bau, Sci. Total Environ., 2015, 533, 91–101 CrossRef CAS PubMed.
- I. Wysocka, Talanta, 2021, 221, 121636 CrossRef CAS PubMed.
- M. B. Shabani, T. Akagi, H. Shimizu and A. Masuda, Anal. Chem., 1990, 62, 2709–2714 CrossRef CAS.
- H. Amakawa, D. S. Alibo and Y. Nozaki, Geochim. Cosmochim. Acta, 2000, 64, 1715–1727 CrossRef CAS.
- H. P. Neves, G. M. D. Ferreira, G. M. D. Ferreira, L. R. D. Lemos, G. D. Rodrigues, V. A. Leão and A. B. Mageste, Sep. Purif. Technol., 2021, 282, 120064 CrossRef.
- J. Yin, B. Hu, M. He and Z. Jiang, Anal. Chim. Acta, 2007, 594, 61–68 CrossRef CAS PubMed.
- T. C. C. Rousseau, J. E. Sonke, J. Chmeleff, F. Candaudap, F. Lacan, G. Boaventura, P. Seyler and C. Jeandel, J. Anal. At. Spectrom., 2013, 28, 573–584 RSC.
- N. Freslon, G. Bayon, D. Birot, C. Bollinger and J. A. Barrat, Talanta, 2011, 85, 582–587 CrossRef CAS PubMed.
- D. Rahmi, Y. B. Zhu, E. Fujimori, T. Umemura and H. Haraguchi, Talanta, 2007, 72, 600–606 CrossRef CAS PubMed.
- G. Bayon, D. Birot, C. Bollinger and J. A. Barrat, Geostand. Geoanal. Res., 2011, 35, 145–153 CrossRef CAS.
- X.-P. Yan, R. Kerrich and M. J. Hendry, J. Anal. At. Spectrom., 1999, 14, 215–221 RSC.
- L. Halicz, I. Gavrieli and E. Dorfman, J. Anal. At. Spectrom., 1996, 9, 811–814 RSC.
- J. Ruzicka and E. H. Hansen, Trends Anal. Chem., 2008, 27, 390–393 CrossRef CAS.
- E. Douville, J. L. Charlou, E. H. Oelkers and P. Bienvenu, Chem. Geol., 2002, 184, 37–48 CrossRef CAS.
- B. A. Haley and G. P. Klinkhammer, Mar. Chem., 2003, 82, 197–220 CrossRef CAS.
- T. Kajiya, M. Aihara and S. Hirata, Spectrochim. Acta, Part B, 2004, 59, 543–550 CrossRef.
- E. C. Hathorne, B. Haley, T. Stichel, P. Grasse, M. Zieringer and M. Frank, Geochem., Geophys., Geosyst., 2012, 13, Q01020 CrossRef.
- Y. Zhu, T. Umemura, H. Haraguchi, K. Inagaki and K. Chiba, Talanta, 2009, 78, 891–895 CrossRef CAS PubMed.
- K. Pyrzynska, A. Kubiak and I. Wysocka, Talanta, 2016, 154, 15–22 CrossRef CAS PubMed.
- M. K. M. Kriews, Fresenius. J. Anal. Chem., 2000, 367, 440–444 CrossRef.
- T. V. D. Flierdt, K. Pahnke, H. Amakawa, P. Andersson and C. Basak, Limnol. Oceanogr.: Methods, 2012, 10, 234–251 CrossRef.
- M. Molina-Kescher, M. Frank and E. Hathorne, Geochim. Cosmochim. Acta, 2014, 127, 171–189 CrossRef CAS.
- B. A. Haley, M. Frank, E. Hathorne and N. Pisias, Geochim. Cosmochim. Acta, 2014, 126, 455–474 CrossRef CAS.
- E. C. Hathorne, M. Frank and P. M. Mohan, Front. Mar. Sci., 2020, 6, 767 CrossRef.
- I. Rapp, C. Schlosser, D. Rusiecka, M. Gledhill and E. P. Achterberg, Anal. Chim. Acta, 2017, 976, 1–13 CrossRef CAS PubMed.
- X. Zhu, J. Lin, A. Gao, D. H. Wang and M. Zhu, At. Spectrosc., 2017, 38, 77–85 CrossRef CAS.
- M. Raso, P. Censi and F. Saiano, Talanta, 2013, 116, 1085–1090 CrossRef CAS PubMed.
- B.-S. Wang, C.-P. Lee and T.-Y. Ho, Talanta, 2014, 128, 337–344 CrossRef CAS PubMed.
- I. Wysocka and E. Vassileva, Spectrochim. Acta, Part B, 2017, 128, 1–10 CrossRef CAS.
- A. Pourmand, N. Dauphas and T. J. Ireland, Chem. Geol., 2012, 291, 38–54 CrossRef CAS.
- T. Duncan and T. J. Shaw, Aquat. Geochem., 2003, 9, 233–255 CrossRef CAS.
- J. Hoyle, H. Elderfield, A. Gledhill and M. Greaves, Geochim. Cosmochim. Acta, 1984, 48, 143–149 CrossRef CAS.
- M. Davranche, O. Pourret, G. Gruau and A. Dia, J. Colloid Interface Sci., 2004, 277, 271–279 CrossRef CAS PubMed.
- Z. Xu and G. Han, Appl. Geochem., 2009, 24, 1803–1816 CrossRef CAS.
- H. Elderfield, R. Upstill-Goddard and E. R. Sholkovitz, Geochim. Cosmochim. Acta, 1990, 54, 971–991 CrossRef CAS.
- H. Elderfield and M. J. Greaves, Nature, 1982, 296, 214–219 CrossRef CAS.
- G. Han and C.-Q. Liu, Aquat.
Geochem., 2006, 13, 95–107 CrossRef.
- L. Ma, D. H. Dang, W. Wang, R. D. Evans and W.-X. Wang, Environ. Pollut., 2019, 244, 190–201 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Quanhui Xub,
Danyang Taoc,
Yang Laia,
Lanlan Jina and
Shenghong Hu
*a,
Quanhui Xub,
Danyang Taoc,
Yang Laia,
Lanlan Jina and
Shenghong Hu