
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePost-synthetically modified metal–porphyrin framework GaTCPP for carbon dioxide adsorption and energy storage in Li–S batteries†
Nikolas Királya,
Dominika Capková b,
Miroslav Almášia,
Tomáš Kazdac,
Ondej Čechc,
Pavel Čudekc,
Andrea Straková Fedorkováb,
Maxim Lisnichukd,
Vera Meynene and
Vladimír Zeleňák*a
b,
Miroslav Almášia,
Tomáš Kazdac,
Ondej Čechc,
Pavel Čudekc,
Andrea Straková Fedorkováb,
Maxim Lisnichukd,
Vera Meynene and
Vladimír Zeleňák*a
aDepartment of Inorganic Chemistry, Faculty of Sciences, Pavol Jozef Šafárik University in Košice, Moyzesova 11, 04154, Košice, Slovak Republic. E-mail: vladimir.zelenak@upjs.sk
bDepartment of Physical Chemistry, Faculty of Sciences, Pavol Jozef Šafárik University in Košice, Moyzesova 11, 04154, Košice, Slovak Republic
cDepartment of Electrical and Electronic Technology, Faculty of Electrical Engineering and Communication, Brno University of Technology, Technická 10, 616 00, Brno, Czech Republic
dInstitute of Physics, Faculty of Sciences, Pavol Jozef Šafárik University in Košice, Park Angelinum 9, 04001 Košice, Slovak Republic
eLaboratory of Adsorption and Catalysis (LADCA), Department of Chemistry, University of Antwerp, Universiteitsplein 1, 2610 Wilrijk, Belgium
First published on 24th August 2022
Abstract
Lithium–sulphur batteries attract increasing interest due to their high theoretical specific capacity, advantageous economy, and “eco-friendliness”. In this study, a metal–organic framework (MOF) GaTCPP containing a porphyrinic base ligand was used as a conductive additive for sulphur. GaTCPP was synthesized, characterized, and post-synthetically modified by the transition metal ions (Co2+/Ni2+). The doping of GaTCPP ensured an increase in the carbon dioxide adsorption capacities, which were measured under different conditions. Post-synthetic modification of GaTCPP with Co2+/Ni2+ ions has been shown to increase carbon dioxide storage capacity from 22.8 wt% for unmodified material to 23.1 wt% and 26.5 wt% at 0 °C and 1 bar for Co2+ and Ni2+-doped analogues, respectively. As a conductive part of cathode material, MOFs displayed successful sulphur capture and encapsulation proven by stable charge/discharge cycle performances, high-capacity retention, and coulombic efficiency. The electrodes with pristine GaTCPP showed a discharge capacity of 699 mA h g−1 at 0.2C in the fiftieth cycle. However, the doping of GaTCPP by Ni2+ has a positive impact on the electrochemical properties, the capacity increased to 778 mA h g−1 in the fiftieth cycle at 0.2C.
Introduction
In the field of crystalline porous materials, metal−organic frameworks (MOFs) exhibit tuneable pore sizes and surface chemistry, which in turn lead to a wide range of chemical and physical properties. They are formed by the coordination of organic ligands to the metal sites. The metal sites can be ions of transition metals, p-block elements, alkaline earth metals, lanthanides, and actinide, while the organic ligands are usually divalent or polyvalent organic carboxylates. The high surface area, crystallinity, controllable pore size, flexibility, and functionalization of the porous surface are some of the main characteristics which determine the versatility of MOFs.1 Hence, MOFs are investigated in applications such as gas storage and separation,2–6 catalysis,7–9 drug delivery,10–12 proton conductivity,13–15 magnetism16–19 etc.20 In our research for discovering novel original porous frameworks with multifunctional linkers, we mainly focused on porphyrinic building blocks. Metallo/porphyrins represent a fascinating class of molecules as they play a crucial role in nature e.g., respiration process, enzymatic, metabolic, and redox reactions.21 When metallo/porphyrins are used as building blocks for MOFs, a subclass, namely metal/porphyrin frameworks (MPFs), is created. Facile molecular modification of porphyrins expands the possibilities of structural design.22 Compared with the most studied carbonaceous materials, the pores of MOFs can be decorated with chemically active sites, such as Lewis acidic sites and functional organic groups. They can be inserted into the porphyrin via post-synthetic modification and/or introduced via the various metal sites into the pore surface of MPFs without alteration of the framework topology.22 In addition, the physical and chemical properties of metallo/porphyrins can be controlled by appropriate functionalization of the porphyrin core through post-synthetic modifications.23,24 The development of MOFs for use in applications increasingly relies on the synthesis of sophisticated types of compounds. Given the free centre porphyrin structures, this site is the most suitable for post-synthetic modification to achieve targeted functionality. Wang et al. reported post-synthetic metal-ion exchange Cd2+ by Co2+ in porphyrin core for application in trans-stilbene reaction,25 another review focused on ion-exchange in secondary building units (SBUs) unit was published by Brozek.26 MOFs have been demonstrated as useful device material for energy storage after doping of Co2+ cations in Co-doped MOF-5 i.e. Co-MOF-5 as an electrode for supercapacitors. Doping has been applied previously to achieve higher adsorption capacities for H2, CH4, and CO2 (from 56 wt% to 67 wt% at 10 bar and 0 °C for CO2), at high pressure, in comparison to Co-free analogue.27 Accordingly, Co-doped MOF-5, after carbonization, also showed as a high-performance anode material for Li-ion batteries, with a reversible capacity of 725 mA h g−1 up to the 50th cycle, at a current density of 100 mA h g−1.28 Another example is a pair of isostructural compounds of the MOF-74 family (Ni2(p-dobdc) and Co2(p-dobdc), Ni2(m-dobdc) and Co2(m-dobdc), p-dobdc – 2,5-dioxido-1,4-benzenedicarboxylate, m-dobdc – 4,6-dioxido-isophthalate) having honeycomb frameworks with etb-topology containing hexagonal channels. Hydrogen storage properties for these materials were investigated under practical conditions at −75 to 100 °C and at different pressures up to 100 bar. Of all the materials tested, it has been shown that the highest H2 storage capacities were observed for Ni2+-containing compounds at all measured temperatures and pressures.29Carbon dioxide, as one of a group of greenhouse gases generated by the burning of fossil fuels, is being produced at an alarming rate by human society. To stabilize the level of CO2 in the environment, it is important to create a cost-effective group of carbon dioxide adsorbents. In this process, liquid organic amines are used, which have a high adsorption capacity for carbon dioxide, as it is chemisorption. However, their disadvantage is regeneration, which requires a high temperature (∼50 °C) and high pressure (20–60 bar). There is currently a growing interest in the adsorption of carbon dioxide by means of highly porous solids, and MOFs compounds are at the forefront of this interest due to their large specific surfaces and the ability to functionalize the pore walls.30 The first key criterion for choosing suitable MOFs for CO2 adsorption is that the MOFs' pores have to be compatible with the kinetic diameter of the CO2 molecules. The second criterion is the adsorption capacity. MOFs with polar (hydroxy, azo, amino, imino, etc. groups) pores have a higher CO2 adsorption capacity, with compare to MOFs without these groups, due to the quadrupole moments of CO2 molecules.
Electrochemical energy storage technologies, with their numerous advantages such as “eco-friendly”, high efficiency, and wide applicability, have attracted tremendous attention as important possible solutions to the dramatic increase in environmental pollution.31–33 Lithium-ion (Li-ion) batteries are the most widespread energy storage systems used in portable electronic devices, electric vehicles (EVs), stationary energy storage and satellites.34–38 Nevertheless, as energy storage requirements expand, the energy density and specific capacity of commercial Li-ion batteries currently in use are proving to be insufficient for future applications. One of the possible candidates to replace Li-ion batteries are lithium-sulphur (Li–S) batteries thanks to their high theoretical specific capacity of 1675 mA h g−1, high energy density (2600 W h kg−1), natural abundance, and the low cost of sulphur.39,40 However, there are fundamental challenges regarding the sulphur cathode, including the low conductivity of sulphur and the final discharge product of lithium sulphide, the dissolution and diffusion of polysulphide intermediates in the electrolyte, and the large volumetric variations (∼80%) of the sulphur and polysulfides during cycling. During discharging, sulphur reacts with lithium ions, and the intermediates are higher (soluble; Li2Sx, 4 < x < 8) and lower (insoluble; Li2S2, Li2S) polysulfides. The reverse reaction is present during charging.41–43 The soluble polysulfides are involved in the “shuttle effect” where higher polysulfides migrate between the electrodes.44,45 To avoid these problems, various improvements were published,46–49 e.g. conductive additives with high stabilities50,51 and organic binders42,43 in the cathode material, separator modifications52 and insertion of an interlayer.53 Nevertheless, the search for functional materials for Li–S battery improvement remains urgently required.54,55 Improving the electrochemical behaviour of the sulphur cathode can be achieved with various carbon materials with high conductivity, tunability, and porosity.56 As such, several types of materials have been used to improve cyclic stability: functionalized carbon,57,58 metal oxides/sulphides/nitrides,59–61 covalent-organic frameworks62 and metal–organic frameworks,63–69 all of the mentioned materials have a high affinity for polar polysulphides.
To be employed as sulphur conductive additives in Li–S batteries, first of all, MOFs need to be electrochemically stable during cycling to ensure a permanent porous structure and thus confine sulphur and polysulfides inside. Second, the pore structures need to be well-designed, with suitably large pore size. Third and most importantly, these pores need to offer suitable chemical environments to effectively interact with sulphur and polysulfides.70–72 For example, ZIF-8 is one of the most widely studied materials in Li–S batteries application, showing a different specific capacity depending on the preparation of the composite used as a cathode in a range between 400–900 mA h g−1.73
Herein, we report the example of porphyrinic MOF self-assembled from porphyrin H2TCPP4− ligands, which was post-synthetically doped by Co2+/Ni2+ ions as adsorbent of carbon dioxide and the sulphur conductive additive for Li–S batteries. Accessible open metal sites of Co2+/Ni2+ ions increase adsorption of carbon dioxide from 22.8 wt% for undoped material (GaTCPP) to 26.5 wt% for Ni2+ analogue. The initial discharge capacity of the electrode with unmodified GaTCPP reaches the value of 667 mA h g−1 at 0.2C, and after fifty cycles, the capacity increases up to 699 mA h g−1. The doping by Ni2+ ions increases the conductivity of the electrode material, which increases the capacity, although the addition of Co2+ ions resulted in a capacity decrease and instability of the electrode. In addition, the cathode material with a pristine GaTCPP exhibited stable cycle performance, low fading rate per cycle (0.07% at 0.5C after 200 cycles), and high coulombic efficiency (94.6% at 0.5C during 200 cycles). The capacity fading rate per cycle during long-term cycling at 0.5C for GaTCPP(Co) was 0.14% and for the GaTCPP(Ni) electrode reached the value of 0.11%.
Experimental
Syntheses
Used chemicals Ga(NO3)3·xH2O (99.99%), Ni(NO3)2·6H2O (99.99%), Co(NO3)2·6H2O (99.99%), N,N′-dimethylformamide (DMF; 99.9%), acetone (99%), dimethylsulfoxide (DMSO; 99.9%), H6TCPP (4,4′,4′′,4′′′-(5,10,15,20-porphyrintetrayl)-tetrabenzoic acid, 98%), sulphur (99.5%), carbon Super P (99%), polyvinylidene fluoride (PVDF; Mw = 534.000 by GPC), N-methyl-2-pyrrolidone (NMP; 99.5%), 1,2-dimethoxyethane (DME; 99.5%), 1,3-dioxolane (DOL; 99.5%), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI; 99.99%), and lithium nitrate (LiNO3; 99.99%) were used in the synthesis and applications. Materials for synthesis/modification of GaTCPP and electrochemical measurements were obtained from Sigma-Aldrich, Timcal or PorphyChem companies and used without further purification.Syntheses of materials
Microporous material GaTCPP(AS) (AS – as-synthesized) with chemical formula {[Ga2(H2TCPP)(OH)2]·5DMF·2H2O}n was prepared by modification of the procedure described by Rhauderwiek et al.74 In the described synthesis, 180 mg (0.022 mmol) of H6TCPP (4, 4′,4′′,4′′′-(5,10,15,20-porphyrintetrayl)-tetrabenzoic acid) was dissolved in 8.7 cm3 of DMF (N,N′-dimethylformamide) and subsequently mixed with 1 M aqueous solution of Ga(NO3)3·H2O (0.9 mL, 0.7 mmol). The as-prepared mixture was sealed into a 45 mL Teflon-lined stainless-steel autoclave and heated at 120 °C for 48 h. Subsequently, the autoclave was cooled down to ambient temperature with a cooling rate of 5 °C min−1. The brown powdered material of GaTCPP(AS) was filtered off, two times washed with DMF, two times with acetone, and dried at 40 °C in an oven. A yield of 215 mg, 88.5% (based on H6TCPP) was obtained for GaTCPP(AS). Elemental analysis for GaTCPP(AS) {[Ga2(H2TCPP)(OH)2]·5DMF·2H2O}n; (Mw = 1069.33 g mol−1): CHN clcd: C 57.28%, H 3.68%, N 6.55%; exp.: C 57.92%, H 4.01%, N 6.83%.Post-synthetically modified materials with Co2+ or Ni2+ ions were obtained by soaking 200 mg GaTCPP(AS) in appropriate nitrate salt solutions in 10 mL of DMSO with concertation of 20 mg mL−1 at 90 °C for 72 hours under atmospheric conditions. After the reaction time, the reaction mixture was cooled down to ambient temperature and centrifugated with a 50% mixture of the suspension and acetone. The suspension was filtrated, washed with acetone and dried at 55 °C in an oven in an ambient atmosphere. Corresponding yields were 180 mg, 90% for GaTCPP(Co) and 150 mg, 75% for and GaTCPP(Ni) (both based on GaTCPP(AS)).
Preparation of electrode materials based on GaTCPP and cell assembly
Metal–organic framework GaTCPP and its modifications with Co2+ and Ni2+ ions were used as a matrix for sulphur in cathode materials. Sulphur, GaTCPP(AS)/(Co)/(Ni), and carbon Super P were placed into a zirconium oxide grinding jar and milled in a planetary ball mill at 500 rpm for 30 min to prepare the electrode materials. Binder PVDF was dissolved in NMP and the prepared mixture of the electrode material was added. The resulting mass ratio of the electrode components was 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:15:10 for sulphur, GaTCPP(AS)/(Co)/(Ni), Super P and PVDF, respectively. The electrode slurry was stirred on a magnetic stirrer for 24 h, coated on the aluminium current collector with a carbon surface modification and dried at 60 °C for 24 hours. The electrodes with a diameter of 18 mm were cut out, pressed with a pressure of 315 kg cm−2, dried under vacuum, and in the oven in a glove box at 60 °C for 24 hours. The weight of sulphur per area was controlled, around 2.2 mg cm−2. The electrochemical performance of the prepared cathodes was evaluated using an El-Cell® assembled with a glass fibre separator and lithium metal anode with a diameter of 18 mm. The composition of the electrolyte was as follows: DME and DOL (2:1 volume ratio) with 0.7 M of LiTFSI and 0.25 M of LiNO3 as an additive. The ratio of electrolyte/sulfur in the cell was 25 μL mg−1. The electrochemical test cells were assembled in the argon-filled glove box Jacomex.
:15:15:10 for sulphur, GaTCPP(AS)/(Co)/(Ni), Super P and PVDF, respectively. The electrode slurry was stirred on a magnetic stirrer for 24 h, coated on the aluminium current collector with a carbon surface modification and dried at 60 °C for 24 hours. The electrodes with a diameter of 18 mm were cut out, pressed with a pressure of 315 kg cm−2, dried under vacuum, and in the oven in a glove box at 60 °C for 24 hours. The weight of sulphur per area was controlled, around 2.2 mg cm−2. The electrochemical performance of the prepared cathodes was evaluated using an El-Cell® assembled with a glass fibre separator and lithium metal anode with a diameter of 18 mm. The composition of the electrolyte was as follows: DME and DOL (2:1 volume ratio) with 0.7 M of LiTFSI and 0.25 M of LiNO3 as an additive. The ratio of electrolyte/sulfur in the cell was 25 μL mg−1. The electrochemical test cells were assembled in the argon-filled glove box Jacomex.
Methods and characterization
The elemental analysis was performed using a CHNOS Elemental Analyzer Vario MICRO from Elementar Analysensysteme GmbH with a sample weight of approximately 3 mg.The infrared spectra of the samples were measured at laboratory temperature and recorded using an Avatar FTIR 6700 spectrometer in the range of wavenumbers 4000–400 cm−1 with 64 repetitions for a single spectrum, using the ATR (attenuated total reflectance) technique.
The thermal behaviour of prepared samples was studied by thermogravimetric analysis (TGA) and differential thermal analysis (DTA) with a sample weight of approximately 20 mg, using corundum crucibles. Samples were heated in the temperature range of 25–900 °C with a heating rate of 9 °C min−1 in a dynamic air atmosphere with a flow rate of 50 cm3 min−1, using a Netzsch 409-PC STA apparatus.
The powder X-ray diffraction (PXRD) patterns were obtained on a Rigaku MiniFlex 600 powder diffractometer using Cu Kα radiation (λ = 1.5406 Å) in 2 theta range 5–60°.
The morphology of the MOF materials was investigated using transmission electron microscopy (TEM) and energy-dispersive X-ray spectroscopy (EDS) on a JEOL JEM 2100F UHR, equipped with an EDS analyser. The surface characterization and elemental mapping of the electrode materials were performed by scanning electron microscope TESCAN VEGA 3, equipped with an EDS analyser. The surface areas and pore volumes of the samples were measured by argon sorption at −186 °C using a Quantachrome AUTOSORB-1-MP automated gas sorption system. Prior to the measurements, the samples were degassed in a vacuum at 100 °C for 2 hours followed by an increase in temperature to 140 °C, which was held for 14 hours. The total surface area was calculated via the Brunauer Emmett Teller (BET) equation, and the micropore volume was obtained using the DFT method (NLDFT kernel). Adsorptions of carbon dioxide at 0 °C and 20 °C were measured using a Quantachrome AUTOSORB-iQ-C combined volumetric and dynamic sorption system. Before measurements, the samples were activated in a vacuum at 100 °C for 2 hours followed by an increase in temperature to 140 °C which was held for 14 hours. The composition of MOF materials was analyzed via X-ray photoelectron spectroscopy (XPS) with a Kratos Axis Supra with an Al Kα X-ray source.
All electrochemical measurements were performed using a BioLogic VMP3 potentiostat. Cyclic voltammetry (CV) was measured in a potential window from 1.8 V to 3.0 V (vs. Li/Li+) and the scan rate was set to 0.1 mV s−1. Galvanostatic cycling was carried out within the potential range of 1.8–2.8 V (vs. Li/Li+). Electrochemical impedance spectroscopy (EIS) was measured in the frequency range of 1 MHz to 100 mHz with an amplitude of 10 mV.
Results and discussion
Crystal structure
The MOF material GaTCPP(AS) crystallizes in the orthorhombic crystal system in the space group Cmmm, with cell parameters a = 32.9509(40) Å, b = 6.6990(16) Å, and c = 16.5555(19) Å, with two formula units in the cell. In the compound GaTCPP(AS), all four benzoic moieties –COOH groups of the H6TCPP ligand are deprotonated and coordinated to Ga(III) ions (see Fig. 1a). The terminal benzoate groups are rotated 90° relative to the macrocycle ring, and the distance between the parallel neighbouring porphyrin rings along the b crystallographic axis is 6.69 Å. The Ga3+ ions are six-coordinated, with the six oxygen atoms originating from the four independent H2TCPP4− ligands, the remaining two oxygens atoms come from bringing μ-OH groups. Coordination of GaO6 octahedrons using μ-OH bridges creates a polymer network of trans-corner-sharing polyhedral along the b crystallographic axis (see Fig. 1b). Furthermore, the coordination of H2TCPP4− ligand to eight Ga(III) ions leads to the formation of the final three-dimensional open porous framework (see Fig. 1c) containing three mutually crossing pores propagating along all crystallographic axes. Two types of cavities are formed each along the b and c crystallographic axis with the size of approximately 8.1 × 11.4 Å2 and 4.4 × 8.8 Å2, respectively. From the crystallographic point of view, the crystal structure of GaTCPP(AS) is isostructural with Al-PMOF.75 A high similarity could be found with MIL-60,76 in which V3+ ions are present instead of Ga3+ ions and the macrocyclic porphyrin ligand is replaced by the rigid tetradentate ligand 1,2,4,5-benzenetetracarboxylic acid.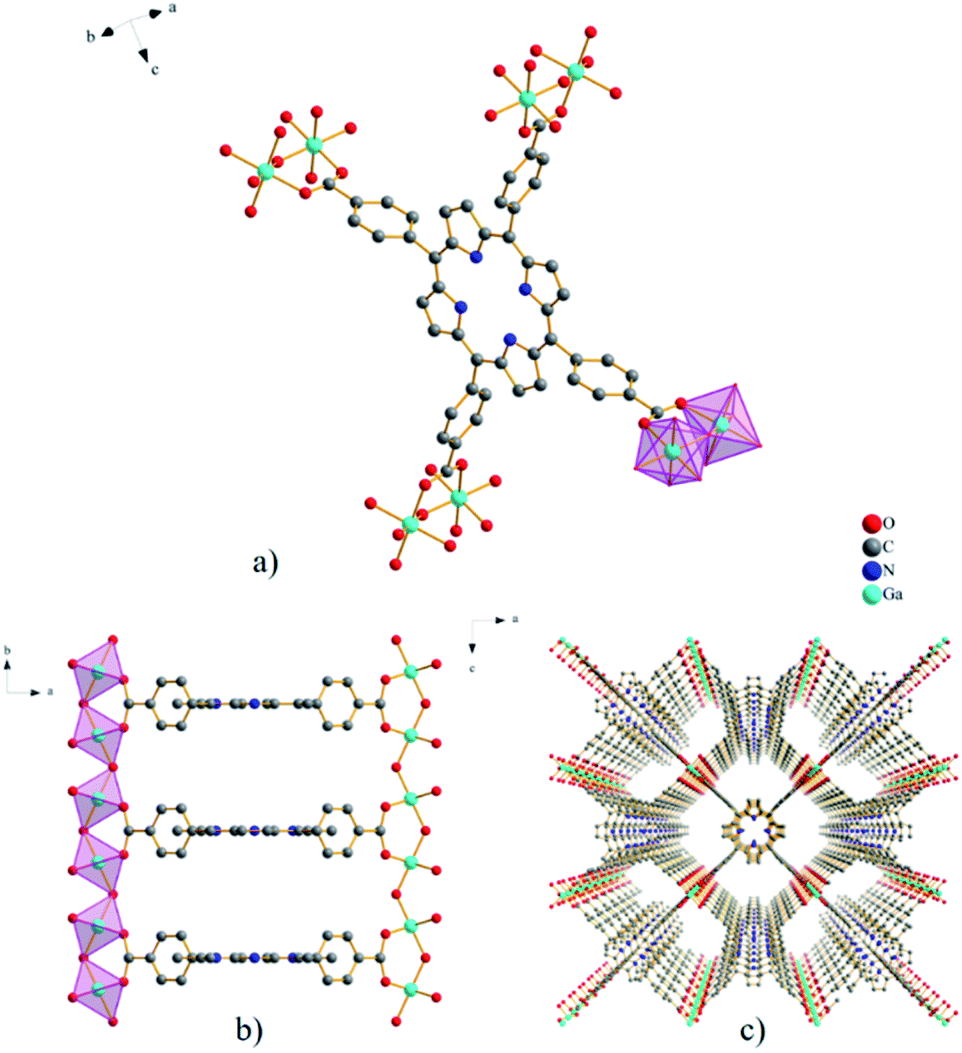 | ||
| Fig. 1 (a) Coordination environment of H2TCPP4− linker in GaTCPP(AS) with a view of GaO6 octahedra binged by carboxylate groups. (b) A view along the c-axis showing trans-corner-sharing polymeric chains propagated along the b-axis. (c) The final 3D porous framework along the b-axis with the corresponding 1D channel (8.1 × 11.4 Å2). | ||
After successful synthesis of GaTCPP(AS), the material was post-synthetically modified with Co2+ and Ni2+ ions. Mentioned ions were bound to the central porphyrin ring of the TCPP ligand throughout the modification process. Two pyrrole aromatic scaffolds were deprotonated during the coordination process to compensate for the positive charge of included metal ions.
Characterization and textural properties
The infrared spectra of all the prepared materials are shown in Fig. 2a, and the wavenumbers of the most important vibrations are summarized in Table 1. Since similarities between GaTCPP(AS), GaTCPP(Co), and GaTCPP(Ni) samples were observed in the infrared spectra, only the IR spectrum of GaTCPP(AS) is described as an example. The spectrum of the as-synthetized sample exhibits a broad band in the range from 3500 to 3000 cm−1, which was attributed to the O–H stretching vibrations of crystallization water molecules and OH ligands. Moreover, in this area, the ν(N–H) vibration of the pyrrole NH bond at 3308 cm−1 was observed. The characteristic stretching vibrations of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups were observed by a medium absorption band around 1549 cm−1. The asymmetric (νas) and symmetric (νs) stretching vibrations of the coordinated carboxylate groups were observed at 1588 and 1417 cm−1, respectively. Deformation vibration of –COO− group was assigned to the δ(COO−) and was observed at 769 cm−1. The tetrapyrrole core was also confirmed by stretching vibration ν(C–N–C) at 1362 cm−1, further characteristic vibrations were found at 959, 799, 703 and 509 cm−1 for δ(C–H), γ(C–H)1,4-subst., γ(C–H)out of plane and δ(CC)skeleton, respectively. The presence of DMF in the channel system of as-synthethized sample was confirmed by the appearance of ν(CO) stretching vibrations at 1708 cm−1, and ν(CH)aliph. stretching vibration in the area under 3000 cm−1. Observed shifted ν(CH)aliph. stretching vibration at higher vibrations for GaTCPP(Co)/(Ni) were attributed to vibrations of DMSO used during the process of modification, also the presence of ν(SO) vibration at 1032 cm−1. The absence of ν(N–H) vibration in IR spectra of post-synthetically modified materials confirmed the successful incorporation of Co2+/Ni2+ ions within the framework of GaTCPP.
C groups were observed by a medium absorption band around 1549 cm−1. The asymmetric (νas) and symmetric (νs) stretching vibrations of the coordinated carboxylate groups were observed at 1588 and 1417 cm−1, respectively. Deformation vibration of –COO− group was assigned to the δ(COO−) and was observed at 769 cm−1. The tetrapyrrole core was also confirmed by stretching vibration ν(C–N–C) at 1362 cm−1, further characteristic vibrations were found at 959, 799, 703 and 509 cm−1 for δ(C–H), γ(C–H)1,4-subst., γ(C–H)out of plane and δ(CC)skeleton, respectively. The presence of DMF in the channel system of as-synthethized sample was confirmed by the appearance of ν(CO) stretching vibrations at 1708 cm−1, and ν(CH)aliph. stretching vibration in the area under 3000 cm−1. Observed shifted ν(CH)aliph. stretching vibration at higher vibrations for GaTCPP(Co)/(Ni) were attributed to vibrations of DMSO used during the process of modification, also the presence of ν(SO) vibration at 1032 cm−1. The absence of ν(N–H) vibration in IR spectra of post-synthetically modified materials confirmed the successful incorporation of Co2+/Ni2+ ions within the framework of GaTCPP.
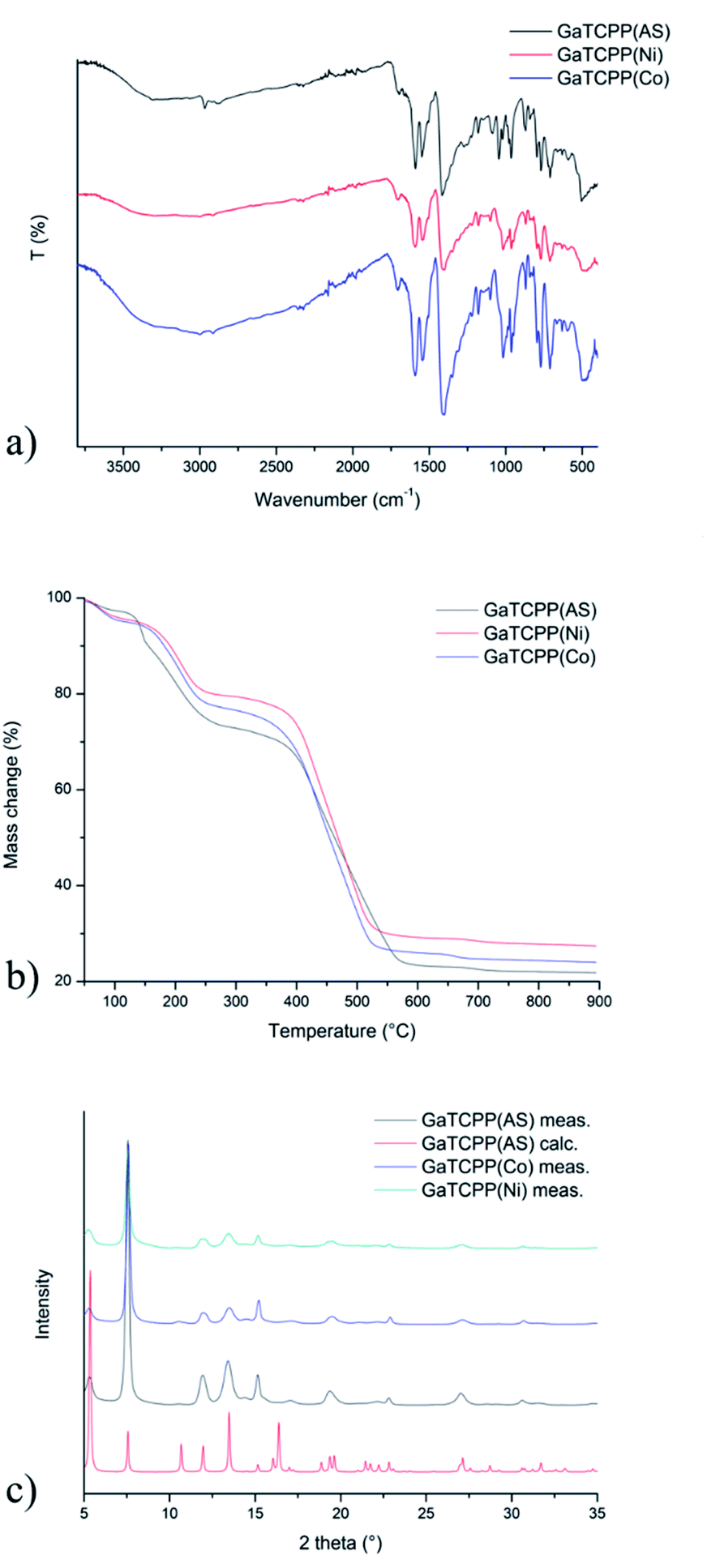 | ||
| Fig. 2 (a) Infrared spectra of GaTCPP(AS) (black line), GaTCPP(Co) (blue line), GaTCPP(Ni) (red line). (b) Thermogravimetric curve of materials: GaTCPP(AS/Co/Ni). (c) The comparison of PXRD patterns of GaTCPP(Co) (red line) and GaTCPP(Ni) (blue line) samples and the calculated pattern from single X-ray diffraction data for GaTCPP(AS) (black line). | ||
| Vibration | Wavenumber | ||
|---|---|---|---|
| GaTCPP(AS) [cm−1] | GaTCPP(Co) [cm−1] | GaTCPP(Ni) [cm−1] | |
| ν(OH)H2O | 3401 | 3373 | 3378 |
| ν(NH)pyrrole | 3314 | — | — |
| ν(CH)aliph. DMF | 2971/2867 | 3002/2906 | 2999/2903 |
| ν(CO)DMF |
1706 | 1708 | 1707 |
| νas(COO) | 1585 | 1585 | 1585 |
| ν(CC) |
1546 | 1546 | 1546 |
| νs(COO) | 1419 | 1417 | 1417 |
| ν(SO) |
— | 1032 | 1032 |
| γ(C–H)1,4-subst. | 796 | 795 | 796 |
| γ(C–H)out of plane | 709 | 709 | 709 |
| δ(CC)skeleton |
504 | 506 | 506 |
The thermogravimetric curves of the prepared materials, GaTCPP(AS) (black line in Fig. 2b), GaTCPP(Co) (blue line in Fig. 2b), and GaTCPP(Ni) (red line in Fig. 2b), display thermal stability of prepared materials. In the TG curves, we can observe three characteristic steps of weight loss. The first small weight loss, about 3.3 wt%, is observed up to 120 °C and can be attributed to the release of lattice water molecules from the channel system. The second weight loss observed between 120 °C and 300 °C can be attributed to the removal of DMF (∼25 wt%) or DMSO (from ion-modified samples) from the pores. The materials were thermally stable up to 300 °C. Above this temperature, the decomposition of the framework (∼50 wt%) started at around 350 °C. The residual solid product represented 20 wt%, 24 wt%, and 27 wt%, corresponding to the gallium oxide (calculated 18 wt%) and the mixture of Ga2O3 with cobalt/nickel oxides. An increasing percentage of residual masses confirms the presence of metal oxides of corresponding ions in the modified samples. The amounts of released solvent molecules differ slightly due to sample storage conditions before measurements and the synthetic procedure for preparing GaTCPP(Co) and GaTCPP(Ni) (possible presence of DMSO).
The crystallinity and bulk composition of all prepared materials were studied by powder X-ray diffraction (PXRD), and obtained PXRD patterns are depicted in Fig. 2c. Fig. 2c shows the comparison of the experimental and calculated PXRD patterns from the single-crystal X-ray data for GaTCPP(AS).74 Both patterns of GaTCPP(Co) (red curve) and GaTCPP(Ni) (blue curve) are almost identical, evidencing the phase purity of the prepared samples. The differences in intensities of diffraction lines can be attributed to the variation in the preferred orientation of the crystallites in the analysed powdered materials and the presence of solvent molecules in frameworks. Moreover, it can be concluded from the results obtained using PXRD measurements, that the modification of GaTCPP(AS) with Co2+/Ni2+ ions does not degrade the quality of GaTCPP framework.
The particle size, morphology, and successful incorporation/distribution of metal ions in the prepared samples were investigated by TEM measurements. Fig. 3 shows TEM images of the GaTCPP(Co) and GaTCPP(Ni) samples, which showed that the crystals of the compounds have a rod-like shape with different sizes, approximately from 60 to 300 nm for both materials and tend to agglomerate. The EDS mapping was used to confirm the elemental composition and distribution of Co2+/Ni2+ in prepared materials (yellow dots in Fig. 3a-Co and b-Ni). According to the obtained results from EDS mapping, we can conclude the presence of the desired ions and their regular distribution in the samples. SEM images of GaTCPP(AS), GaTCPP(Co), and GaTCPP(Ni) are available in ESI (see Fig. S1†).
 | ||
| Fig. 3 TEM images of the modified materials: (a) GaTCPP(Co) and (b) GaTCPP(Ni) with elemental mapping of Co2+/Ni2+ (yellow), Ga3+ (green) and N (red) using EDS analysis. | ||
In order to further investigate the composition of GaTCPP MOF materials and the chemical state of the present metals, XPS characterization was carried out. The wide-scan XPS spectra of GaTCPP(AS), GaTCPP(Co), and GaTCPP(Ni) are shown in Fig. 4. The peak located at 20 eV is not possible to unequivocally determine, it may be attributed to C 2s, Ga 3d, or O 2s. All observed peaks are slightly shifted to lower binding energy for modified GaTCPP(Ni) compared to GaTCPP(AS) and GaTCPP(Co). The XPS peak of Ga shows the presence of Ga in the MOF materials as it was observed in the TEM image. The peaks corresponding to Ga 3p3/2 and Ga 3p1/2 are observed at 103 and 106 eV for GaTCPP(Ni), and at 105 and 109 eV for GaTCPP(AS)/(Co), respectively. Ga 3s is found at 158 eV for GaTCPP(Ni) and 161 eV for GaTCPP(AS)/(Co). The Auger peak shapes Ga LMM can be seen around 423 eV. The peaks related to Ga 2p3/2 and Ga 2p1/2 are accessible at 1115 eV and 1142 for GaTCPP(Ni), and at 1119 eV and 1146 eV for GaTCPP(AS)/(Co), respectively. The presence of carbon (C 1s) can be observed at 281 eV for GaTCPP(Ni) and 285 eV for GaTCPP(AS)/(Co). The peak corresponding to N 1s is found at 396 eV for GaTCPP(Ni) and 399 eV for GaTCPP(AS)/(Co). Oxygen O 1s is seen in GaTCPP(Ni) spectrum at 528 eV and GaTCPP(AS)/(Co) spectra at 533 eV. The Auger peak O KLL is located around 978 eV. The presence of cobalt in GaTCPP(Co) is confirmed by a peak at 779 eV corresponding to Co 2p3/2. Nickel in GaTCPP(Ni) is determined by a peak at 852 eV, which is attributed to Ni 2p3/2. The atomic and mass concentration of the elements in GaTCPP materials is summarized in Table S1 in ESI.†
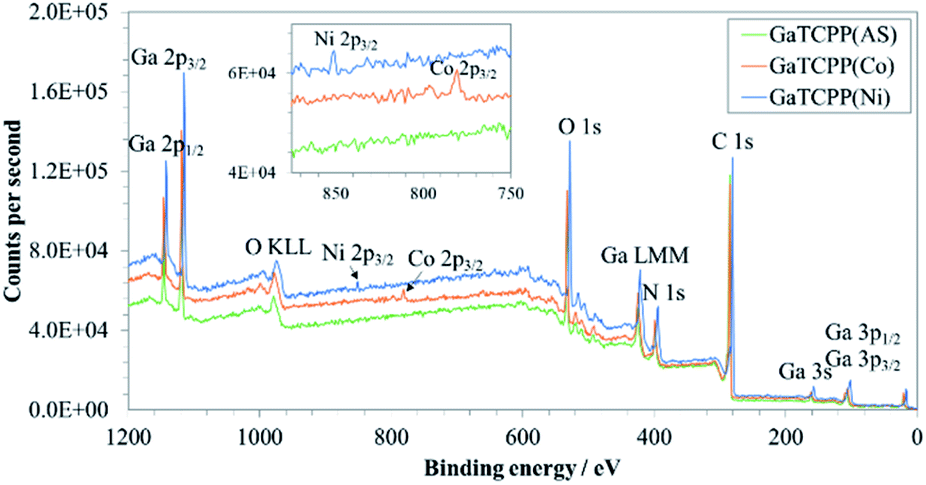 | ||
| Fig. 4 Wide-scan XPS spectra of GaTCPP(AS), GaTCPP(Co), and GaTCPP(Ni). | ||
After pressing, the surface of the sulphur-containing cathodes with the GaTCPP samples, and its modifications with Co2+ and Ni2+, were also studied by SEM and EDS analyses (see Fig. 5). The SEM images are depicted at a view field of 41.5 μm, and elemental mapping was probed on the area of 400 μm × 400 μm. The mapped elements on the cathode surface were S (green colour), C (red colour), and Ga (blue colour), respectively. The elements Co2+ a Ni2+ were not analysed in the electrode by EDS, as they were previously analysed in the pure material. The electrode materials containing GaTCPP(AS) and GaTCPP(Ni) (Fig. 5a and c) are homogenous with uniform distribution of the elements. However, the electrode prepared with GaTCPP(Co) (Fig. 5b) shows less homogeneity. The sulphur particles are aggregated, and this phenomenon is not visible in samples GaTCPP(AS) and GaTCPP(Ni).
 | ||
| Fig. 5 SEM images and elemental mapping (S – green colour, C – red colour and Ga – blue colour) of the prepared electrode materials based on sulphur with: (a) GaTCPP(AS), (b) GaTCPP(Co) and (c) GaTCPP(Ni). | ||
Gas adsorption
The sorption experiments using Ar at −186 °C and CO2 at different temperatures (0 °C and 20 °C) as adsorptive gases were performed for the surface characterization of all prepared samples (see Table 2 and Fig. 6). Before adsorption measurements, the samples were activated for 2 hours at 100 °C followed by 14 hours at 140 °C under a vacuum (temperatures were selected due to the gradual removal of solvent molecules H2O and DMF (DMSO in doped materials) based on TG analysis). The calculated surface area (SBET) was obtained using the BET (Brunauer–Emmet–Teller) equation from the argon adsorption isotherms. The pore volume was calculated using the DFT method (NLDFT kernel). The shape of the adsorption isotherms for all three samples GaTCPP(AS), GaTCPP(Co) and GaTCPP(Ni) can be characterized as type I by IUPAC,77 which is characteristic for microporous materials (see Fig. 6). The surface area of the non-modified material GaTCPP(AS) was estimated to be 1010 m2 g−1 and the pore volume of 0.74 cm3 g−1. The obtained surface area is comparable to the formerly published value of 1150 m2 g−1 by Rhauderwiek et al.74 However, in this publication N2 was applied as adsorbate, and a measurement temperature of −196 °C was used. The calculated BET surface area is smaller than the isostructural analogue Al-PMOF (1400 m2 g−1).75 Evaluation of the argon sorption data for modified materials using the BET equation gave higher specific surface area values of 1177 and 1188 m2 g−1 for materials GaTCPP(Co) and GaTCPP(Ni), with the pore volumes being 0.730 and 0.876 cm3 g−1, respectively.| Material | Adsorbate | |||||
|---|---|---|---|---|---|---|
| Ar | CO2 | |||||
| −186 °C | 20 °C; 1 bar | 0 °C; 1 bar | ||||
| SBET (m2 g−1) | Vp (cm3 g−1) | |||||
| wt% | mmol g−1 | wt% | mmol g−1 | |||
| GaTCPP(AS) | 1010 | 0.74 | 19.0 | 4.31 | 22.8 | 5.17 |
| GaTCPP(Co) | 1177 | 0.73 | 15.8 | 3.59 | 23.1 | 5.26 |
| GaTCPP(Ni) | 1188 | 0.88 | 25.5 | 5.77 | 26.5 | 6.00 |
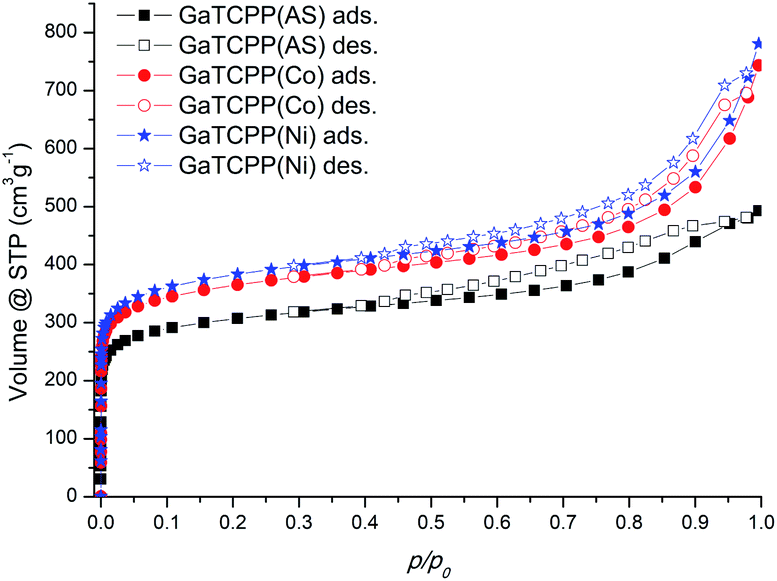 | ||
| Fig. 6 Argon adsorption/desorption isotherms at −186 °C of the prepared materials. | ||
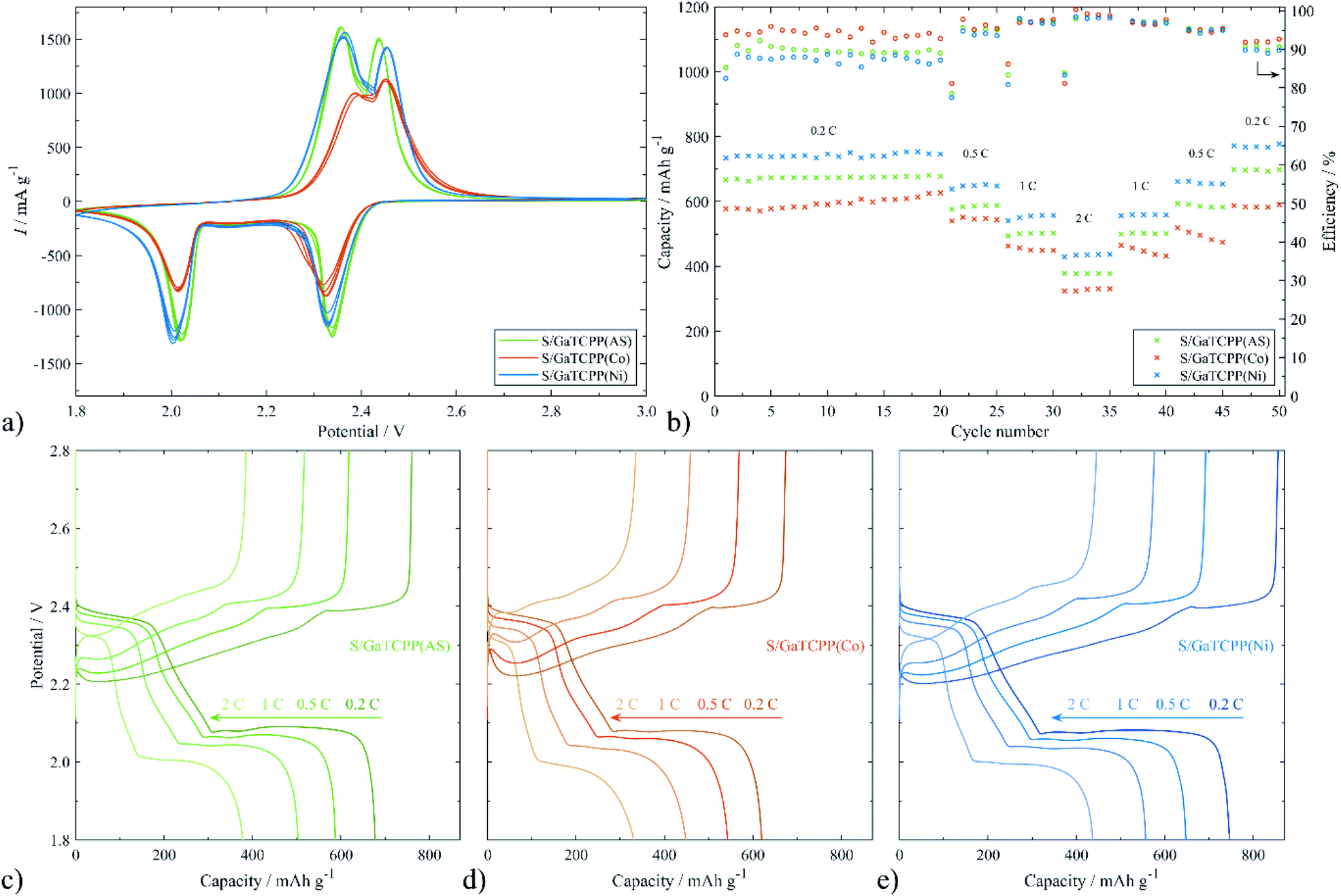 | ||
| Fig. 7 Electrochemical performance of the electrodes based on GaTCPP: (a) cyclic voltammograms at a scan rate of 0.1 mV s−1; (b) galvanostatic cycling at different C-rates; charge and discharge curves of the cell with the (c) S/GaTCPP(AS), (d) S/GaTCPP(Co) and (e) S/GaTCPP(Ni) cathode. | ||
Carbon dioxide measurements were performed at two temperatures, 0 °C and 20 °C (see Fig. S2a and b in ESI†). Carbon dioxide adsorption isotherms at 20 °C (see Fig. S2a in ESI† and Table 2) showed that the as-synthetized material GaTCPP(AS) adsorbed 103 cm3 g−1 of CO2 up to 1 bar, which corresponds to the storage capacity of 19.0 wt% (4.31 mmol g−1) CO2. The modified materials GaTCPP(Co) and GaTCPP(Ni) adsorbed 86 cm3 g−1 and 138 cm3 g−1 CO2 up to 1 bar, corresponding to 15.8 wt% (3.59 mmol g−1) and 25.5 wt% (5.77 mmol g−1) CO2, respectively. As the adsorption phenomenon is an exothermic process, the adsorbed amount of CO2 increases with decreasing temperature. The carbon dioxide adsorption measurements at 0 °C (see Fig. S2b in ESI† and Table 2) showed that materials adsorbed 115, 117, and 134 cm3 g−1 of CO2, corresponding to the maximal uptake of 22.8, 23.1, and 26.5 wt% (5.17, 5.26 and 6 mmol g−1) at 1 bar, respectively for GaTCPP(AS), GaTCPP(Co) and GaTCPP(Ni). Differences in the influence of doped materials, with the lowering of the temperature from 20 °C to 0 °C should be caused by the difference in binding energy or accessibility of binding sites. One of the most significant effects on increasing the capacity and selectivity of adsorptive, especially carbon dioxide over other gases are open metal sites (OMS). In the activation process, excess solvents are removed from the metal sites of the lattice, leading to the formation of open metal sites (OMSs). Doping with metal ions (Co2+/Ni2+) increases the number of OMSs in the framework, leading to the increased CO2 adsorption capacity of modified materials. Since the characteristic coordination number of the incorporated ions is six, we assume that they are coordinated in the cavity of the TCPP ligand by the 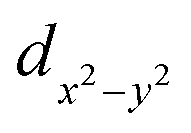 orbital (in the equatorial plane), while coordinated solvent molecules occupy the
orbital (in the equatorial plane), while coordinated solvent molecules occupy the 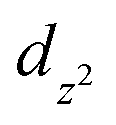 orbital (axial position). Activation of materials results in removing solvents and the formation of a free orbital, which represents OMS. In fact, these metal centres form the skeleton's surface as active sites for the capture of CO2 molecules and their binding by dipole–quadrupole interactions.78 The increased CO2 sorption capacity of GaTPCC(Ni) compared to GaTCPP(Co) can be explained by the presence of highly polarizing Ni2+ adsorption sites, leading to the dense packing of carbon dioxide within the framework and large binding enthalpies.73
orbital (axial position). Activation of materials results in removing solvents and the formation of a free orbital, which represents OMS. In fact, these metal centres form the skeleton's surface as active sites for the capture of CO2 molecules and their binding by dipole–quadrupole interactions.78 The increased CO2 sorption capacity of GaTPCC(Ni) compared to GaTCPP(Co) can be explained by the presence of highly polarizing Ni2+ adsorption sites, leading to the dense packing of carbon dioxide within the framework and large binding enthalpies.73
Electrochemical characterization
Encouraged by the unique MOF structures, the electrode materials based on GaTCPP and its modifications with Co2+ and Ni2+ ions are considered to be an appropriate candidate for the capture and confinement of sulphur. Afterwards, the CV profiles at a scan rate of 0.1 mV s−1 were measured and collected in Fig. 7a. All the electrodes show two reduction peaks around 2.01 and 2.33 V, respectively. The observed peaks are in agreement with the reduction of S8 to higher polysulphides (2.33 V) and lower polysulphides (2.01 V). Moreover, two oxidation peaks at around 2.36 and 2.45 V can also be found, corresponding to the reverse process, the oxidation of lower polysulphides to sulphur. It should be noted that for the S/GaTCPP(AS) and S/GaTCPP(Ni) electrodes, the intensity of the peaks is apparently increased compared to the S/GaTCPP(Co) electrode. Additionally, the peaks of the S/GaTCPP(AS) and S/GaTCPP(Ni) electrodes are well overlapped with the number of cycles, and their shape is sharper, indicating mitigation of polarization and enhancement of the redox kinetics of polysulphides compared with the S/GaTCPP(Co) cathode.The rate performance of the S/GaTCPP(AS), S/GaTCPP(Co), and S/GaTCPP(Ni) electrodes at different current densities is illustrated in Fig. 7b. The C-rate value increases from 0.2 to 2C and back to 0.2C as displayed in the figure. The initial discharge capacities at 0.2C of the S/GaTCPP(AS), S/GaTCPP(Co), and S/GaTCPP(Ni) electrodes are 666.5, 578.2, and 733.8 mA h g−1, respectively. The electrode with pristine, unmodified GaTCPP(AS) exhibits highly stable cycle performance with sufficient discharge capacities even at a high C-rate, the discharge capacity at 2C was 378.0 mA h g−1. The electrode containing modified GaTCPP with Co achieved lower discharge capacities, and the cycle performance was less stable than that containing the unmodified material. The highest discharge capacities, despite multi-current cycling, were observed for the S/GaTCPP(Ni) electrode. The discharge capacity at 2C acquires the value of 439.6 mA h g−1, which demonstrates good cycle performance and reversibility. The average value of coulombic efficiency at different C-rates of the S/GaTCPP(AS), S/GaTCPP(Co), and S/GaTCPP(Ni) electrodes was as follows 92.9, 95.3, and 91.7% respectively. The comparison of charge/discharge curves at the last cycles of each C-rate is depicted in Fig. 7c–e. The charge/discharge voltage plateaus correspond well with the redox peaks in the CVs. Both plateaus are visibly suppressed for the S/GaTCPP(Co) electrode indicating a higher impact of the shuttle effect during cycling than in the other samples. The most obvious difference in the shape of the charge/discharge curves was observed from cycling at 2C. The half-capacity during discharging and charging at 0.2C for the S/GaTCPP(AS) and S/GaTCPP(Ni) electrodes were located at 2.00 and 2.40 V, respectively. However, the value of half-capacity for the S/GaTCPP(Co) electrode was shifted to 1.99 and 2.42 V, respectively. The shift of the half-capacity to lower potential during discharging and higher potential during charging may indicate lower stability of the electrode material and enhanced material degradation.
Comparison literature of MOF based on 1,4-BDC (benzenedicaboxylate) linker was applied as cathode support in a Li–S battery where the initial discharge capacity at 0.2C was 392 mA h g−1 with 46% retention after 100 cycles.79 Also, HKUST-1 was studied as a host for sulphur in ref. 80, the discharge capacity at 0.1C achieved a value of around ∼550 mA h g−1 and at 1C ∼300 mA h g−1. After 100 cycles, the discharge capacity decreased to 300 mA h g−1 at 0.1C. It can be noted that the structure of GaTCPP is more suitable for capturing sulphur and polysulphides during cycling, which results in higher and more stable capacity than mentioned MOF materials reported in the literature.
To further study the electrochemical properties of the electrode materials improved by GaTCPP and its modifications with Co2+ and Ni2+ ions in Li–S batteries, 200 cycles of charge/discharge test were carried out at 0.5C. As shown in Fig. 8, the initial discharge capacity for the S/GaTCPP(AS) electrode was 600.6 mA h g−1, and after 200 cycles the capacity retention reached the value of 86.1% which corresponds with the fading rate of 0.07% per cycle. Coulombic efficiency during long-cycling at 0.5C was 94.6%. For comparison, the S/GaTCPP(Co) electrode shows the discharge capacity in the 200th cycle of 374.0 mA h g−1 with a capacity retention of 71.4% and fading rate of 0.14% per cycle respectively. Coulombic efficiency during cycling was decreasing due to the instability of the electrode material and the average value was around 88.4%. Finally, the initial discharge capacity of the S/GaTCPP(Ni) electrode reached the value of 609.9 mA h g−1, and the capacity retention after 200 cycles was 78.5% with a fading rate of 0.11% per cycle. Beyond, the specific capacity of the S/GaTCPP(Ni) electrode was higher than S/GaTCPP(AS) up to the 85th cycle, then the capacity was comparable up to the 100th cycle and it was followed by a gentle decrease of capacity for the S/GaTCPP(Ni) electrode which resulted in the highest capacity of the S/GaTCPP(AS) electrode after 200 cycles. Coulombic efficiency was lower compared to the S/GaTCPP(AS) electrode and reached the value of 88.3%. In addition, discharge profiles during cycling at 0.5C for all electrodes are presented in Fig. 8b–d. The most significant decrease in high voltage plateau and the reduced amount of higher polysulphides after 200 cycles is observed for the S/GaTCPP(Co) electrode indicating the weakest ability to trap polysulphides. The S/GaTCPP(Ni) electrode achieved the highest capacity in the high voltage plateau, the stability of the high voltage plateau was noticeably higher than that of S/GaTCPP(Co). The modification of GaTCPP with Ni2+ ions showed enhanced polysulfide trapping than modification with Co2+ ions. The S/GaTCPP electrode exhibit the most stable cycle performance and efficient capture of polysulphides, as indicated by the high stability of the high voltage plateau throughout the galvanostatic cycling. To sum up, the S/GaTCPP(AS) and S/GaTCPP(Ni) electrodes exhibit high and comparable discharge capacities during long-cycling at 0.5C. Moreover, coulombic efficiency of the S/GaTCPP(Ni) electrode decreases with cycle number, while it is stable at a high value for the S/GaTCPP(AS) electrode. However, the obvious difference between capacity retention and coulombic efficiency is the higher stability of the S/GaTCPP(AS) electrode and efficient polysulphide confinement in the GaTCPP material structure. Chen et al.81 presented MOF Ce-UiO-66-BPDC (benzenebiphenyldicaboxylate) as a cathode host of the Li–S battery. The initial discharge capacity at 0.1C was 757 mA h g−1, the decrease of capacity in the first 5 cycles was rapid and after 100 cycles the capacity retention was only around 20%. Also, MOF-5 was applied as a matrix for sulphur, which had an initial discharge capacity that was higher compared to our materials (874 mA h g−1 at 0.5C), although after fifty cycles the capacity retention was unsatisfactory and exhibit the value of 31.1%.82 The inferior cycling stability of the S/GaTCPP(Co) electrode compared to the S/GaTCPP(Ni) electrode may be due to weaker coordination between Co2+ and polysulfides than Ni2+ and polysulfides. The length of the high-voltage plateau does not shrink significantly for the S/GaTCPP(Ni) electrode, which is in agreement with improved coordination between Ni2+ and polysulfides. Moreover, the results are in accordance with the order of stability constant of complexes by bivalent ions of the first–row transition metals Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+(II).83 The comparison of parameters obtained from multi-current and long-term cycling for all electrodes based on GaTCPP structure is summarized in Table 3.
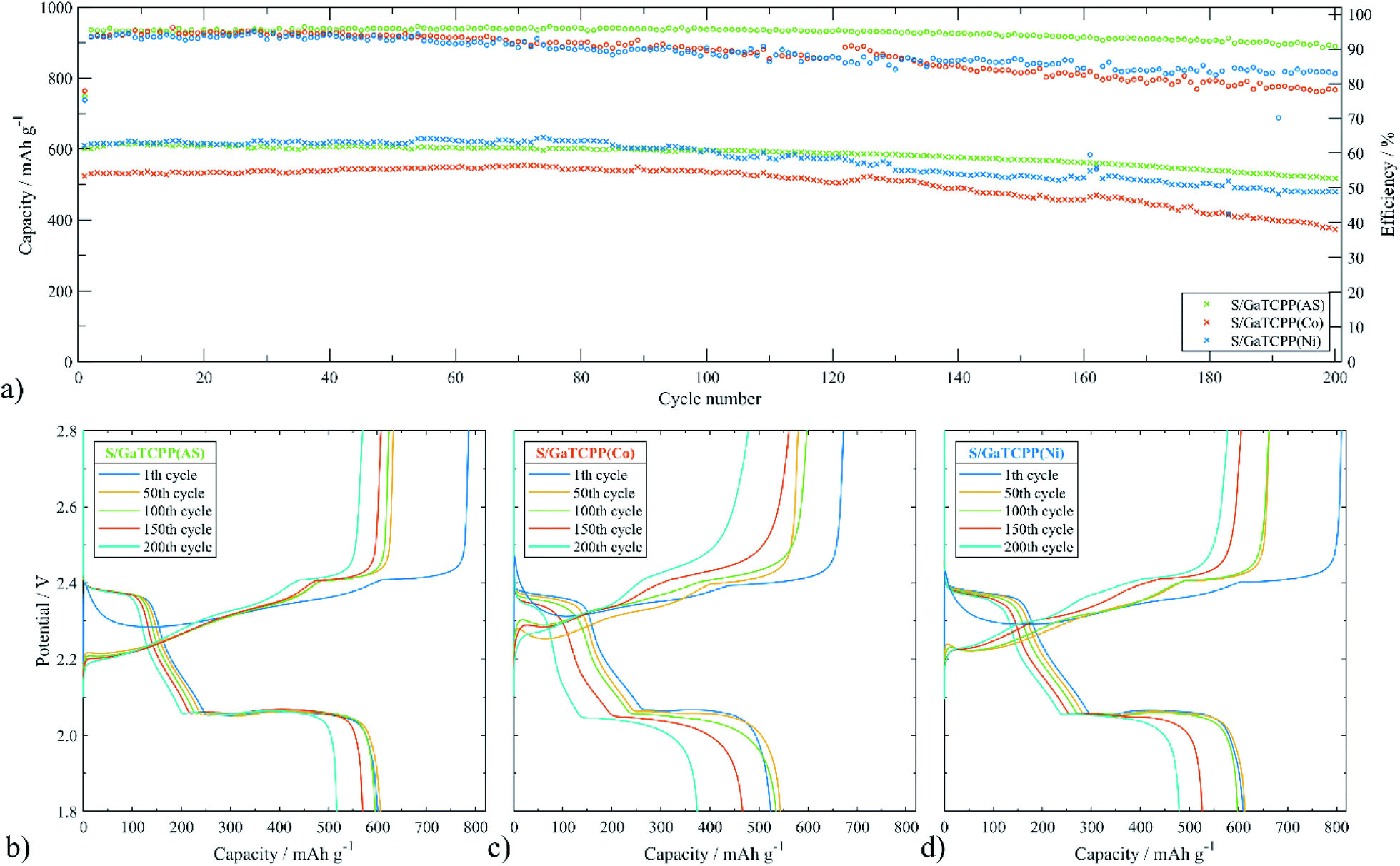 | ||
| Fig. 8 (a) Cycling performance of the cell with S/GaTCPP(AS), S/GaTCPP(Co), and S/GaTCPP(Ni) electrodes at 0.5C and corresponding charge/discharge profiles of the (b) S/GaTCPP(AS), (c) S/GaTCPP(Co) and (d) S/GaTCPP(Ni) electrode. | ||
| Parameter | S/GaTCPP(AS) | S/GaTCPP(Co) | S/GaTCPP(Ni) |
|---|---|---|---|
| Capacity [mA h g−1] in 1st cycle at 0.2C | 666.5 | 578.2 | 733.8 |
| Capacity [mA h g−1] in 20th cycle at 0.2C | 677.4 | 626.1 | 746.6 |
| Capacity [mA h g−1] in 25th cycle at 0.5C | 587.4 | 543.2 | 648.0 |
| Capacity [mA h g−1] in 30th cycle at 1C | 502.9 | 448.6 | 557.1 |
| Capacity [mA h g−1] in 35th cycle at 2C | 378.0 | 331.0 | 436.9 |
| Capacity [mA h g−1] in 50th cycle at 0.2C | 699.2 | 590.6 | 778.0 |
| Coulombic efficiency [%] – various C-rates | 92.9 | 95.3 | 91.7 |
| Capacity [mA h g−1] in 1st cycle at 0.5C | 600.6 | 523.8 | 609.9 |
| Capacity [mA h g−1] in 100th cycle at 0.5C | 597.9 | 534.8 | 597.9 |
| Capacity [mA h g−1] in 200th cycle at 0.5C | 516.9 | 374.0 | 478.9 |
| Capacity retention after 200 cycles [%] | 86.1 | 71.4 | 78.5 |
| Coulombic efficiency [%] – 0.5C | 94.6 | 88.4 | 88.3 |
In order to investigate the effect of various GaTCPP MOF materials on the electrochemical behaviour of the electrode, electrochemical impedance spectroscopy (EIS) was performed before cycling and after cycling at 2C 35 cycles. The EIS spectra of fresh and aged Li–S cells are shown in Fig. 9. All EIS curves consist of two semi-circles and a straight line in low frequencies. The applied equivalent circuit for the simulation of the EIS spectra was proposed in ref. 84 and depicted in Fig. 9. The electrolyte resistance (Re) represents the contribution of ohmic resistance. The first loop is expressed by Rint||CPEint representing interphase contact resistance and the capacitance of the sulphur cathode bulk. The second loop is represented by Rct||CPEdl, the charge transfer resistance, and the capacitance of the double layer on the electrode. The last part of the spectra is described as Rdiff||CPEdiff with Warburg element (W) and reflects the diffusion process. The electrolyte resistance was shifted to a slightly higher resistance for the S/GaTCPP electrode (4.5 Ω compared to 2.2 Ω for S/GaTCPP(Co) and 2.1 Ω S/GaTCPP(Ni)). After cycling, the Re of the S/GaTCPP electrode decreased to 2.9 Ω and raised to 4.4 Ω for S/GaTCPP(Co) and S/GaTCPP(Ni). The decrease of the electrolyte resistance after cycling for the S/GaTCPP may be associated with improved contact of sulphur with the conductive support for the GaTCPP than in its modified forms. The interphase contact resistance is associated with the position of sulphur in the porous structure of the cathode. The Rint was comparable for the fresh cells (5.0 Ω for S/GaTCPP, 5.9 Ω for S/GaTCPP(Co), and 4.6 Ω for S/GaTCPP(Ni)). After cycling, the Rint decreased for the S/GaTCPP (3.0 Ω) and S/GaTCPP(Ni) (0.6 Ω) but increased for the S/GaTCPP(Co) (6.8 Ω). This phenomenon may be related to the improved position of sulphur in GaTCPP and GaTCPP(Ni). The reaction kinetics influence the charge transfer resistance. Insignificant differences were observed for the fresh cells for charge transfer resistance (0.7 Ω for S/GaTCPP, 0.5 Ω for S/GaTCPP(Co), and 0.8 Ω for S/GaTCPP(Ni)). The cycled cells showed higher charge transfer resistance, the highest value showed the S/GaTCPP(Co) electrode (16.7 Ω), lower was observed for the S/GaTCPP (13.8 Ω), and the lowest was for the S/GaTCPP(Ni) (9.4 Ω). It may be assumed that the reaction kinetics was improved for the S/GaTCPP and S/GaTCPP(Ni) electrodes.
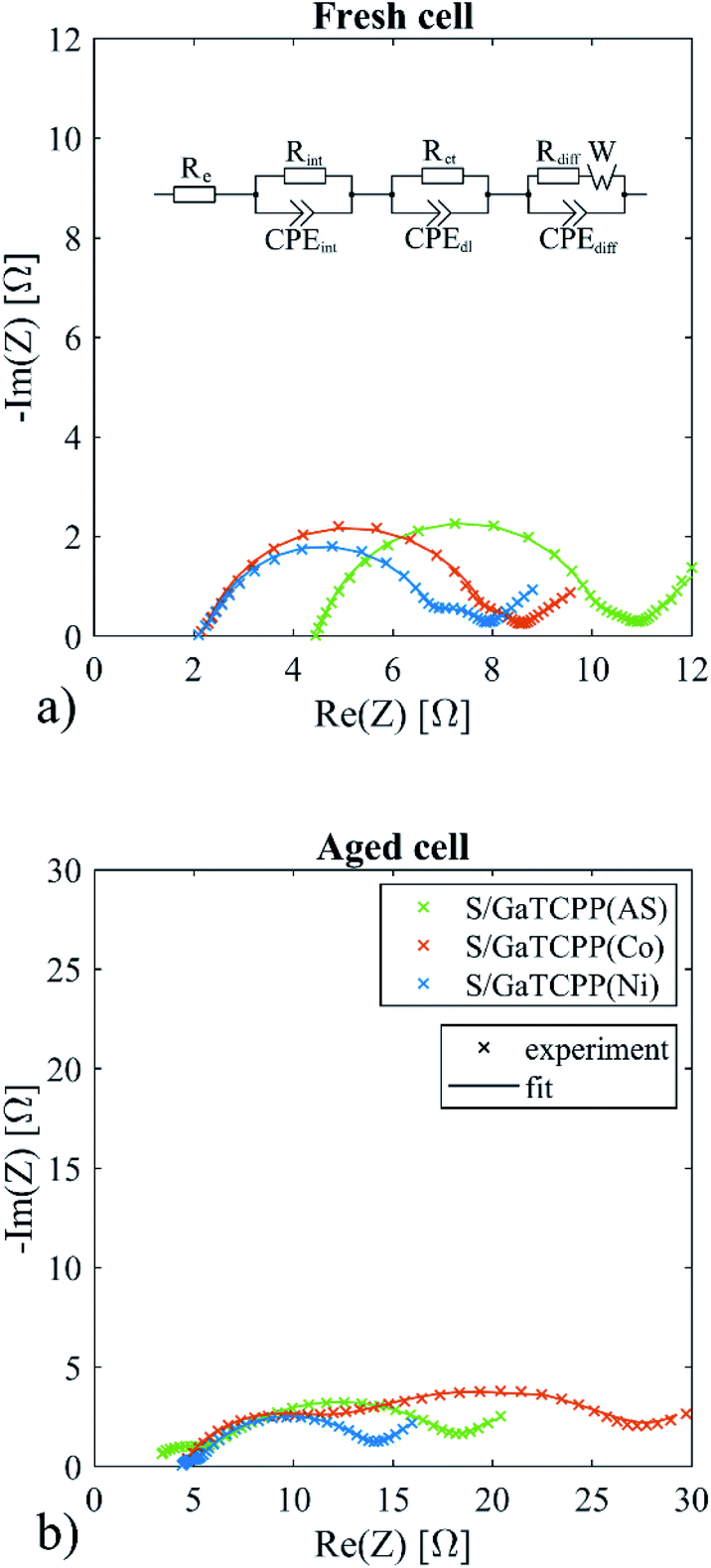 | ||
| Fig. 9 The equivalent circuit and EIS spectra of the S/GaTCPP(AS), S/GaTCPP(Co), and S/GaTCPP(Ni) electrodes (a) before cycling and (b) after 35 cycles. | ||
The comparison of the electrochemical performance of presented GaTCPP-based electrodes in Li–S batteries with the state-of-the-art is presented in Table 4. The initial discharge capacity of GaTCPP-based electrode materials is mostly lower than the compared MOF materials, but their stability is worse and their capacity retention at the end of cycling is lower compared to our electrode materials.
| MOF | Initial capacity [mA h g−1] | Number of cycles | Final capacity [mA h g−1] | C-Rate | S content [wt%] | Capacity retention [%] | References |
|---|---|---|---|---|---|---|---|
| MOF-5 | 874 | 50 | 272 | 0.5C | 58 | 31.1 | 82 |
| ZIF-67 | 1225 | 100 | 422 | 0.1C | 75 | 34.5 | 85 |
| Ce-UiO-66-BPDC | ∼700 | 50 | ∼300 | 0.1C | 25 | 42.3 | 81 |
| Mg-1,4-BDC | 392 | 100 | 180 | 0.2C | 69 | 46.0 | 79 |
| HKUST-1 | 431 | 300 | 286 | 0.5C | 30 | 66.4 | 65 |
| GaTCPP(Co) | 524 | 200 | 374 | 0.5C | 60 | 71.4 | This work |
| GaTCPP(Ni) | 610 | 200 | 479 | 0.5C | 60 | 78.5 | This work |
| GaTCPP | 601 | 200 | 517 | 0.5C | 60 | 86.1 | This work |
Conclusions
In conclusion, GaTCPP metal–organic framework was synthesized, post-synthetically doped, fully characterized (IR, TG, PXRD, TEM, EDS, SEM) and tested as materials for carbon dioxide capture and as a matrix for sulphur in Li–S battery. In the way to improve the performance of the cathode, the GaTCPP material was doped with Co2+/Ni2+ ions. The presence of Co2+/Ni2+ ions was confirmed by XPS analysis. It was demonstrated that post-synthetic doping increases the amount of accessible open metal sites (OMS), which has an impact on the increase of adsorbed gases (Ar, CO2). Argon adsorption measurements revealed that doped material increase their SBET surface area from 1010 m2 g−1 (GaTCPP(AS)) to 1177 m2 g−1 and 1188 m2 g−1 for GaTCPP(Co) and GaTCPP(Ni), respectively. Also, a significant change was observed in CO2 adsorption at 0 °C and 1 bar, where the maximum capacities observed on GaTCPP(Ni) was 26.5 wt%, for GaTCPP(Co) was 23.1 wt%, compared to undoped material GaTCPP(AS), which adsorbed 22.8 wt%. It should be stated, post-synthetic doping has a positive effect on sorption properties, increasing the adsorption capacity of argon and carbon dioxide (at 0 °C). However, these results are temperature dependent, as at 20 °C, doping has a negative effect on CO2 sorption for the Co2+ doped material, while for the Ni2+ doped material, a positive effect. It can be concluded that GaTCPP electrode materials are suitable as a matrix for sulphur in Li–S batteries. The modification of GaTCPP with Co2+ ions resulted in the lowest ability to trap and confine polysulfides for the S/GaTCPP(Co) electrode. The conductivity and stability of the electrode material were improved by the modification of GaTCPP with Ni2+ ions. The best cycle performance at different C-rates showed the S/GaTCPP(Ni) electrode, slightly lower discharge capacities were obtained for the S/GaTCPP(AS) electrode. However, in terms of long-term cycling, at the beginning of cycling the highest discharge capacity was observed for the S/GaTCPP(Ni) electrode, although the S/GaTCPP(AS) electrode showed high capacity retention, which resulted in the highest capacity after 200 cycles. The S/GaTCPP(AS) electrode showed the best potential to capture the polysulphides and suppress the polysulfide shuttle during long-term cycling. The results acquired could provide the roadmap for the application of MOF materials with porphyrin structure with Ga3+ ions and the impact of doped MOF with Ni2+ and Co2+ ions on the electrochemical properties of the Li–S battery.Author contributions
N. Király – conceptualization, data curation, formal analysis, investigation, validation, visualization, writing – original draft. D. Capková – data curation, formal analysis, investigation, validation, visualization, writing – original draft. M. Almáši – data curation, investigation, formal analysis, validation, writing – review & editing. T. Kazda – data curation, validation, methodology, supervision, writing – review & editing. O. Čech – data curation, validation. P. Čudek – data curation, validation. A. Straková Fedorková – funding acquisition, project administration, supervision, writing – review & editing. M. Lisnichuk – data curation, validation. V. Meynen – methodology, supervision, writing – review & editing. V. Zeleňák – methodology, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Scientific Grant Agency of the Slovak Republic (VEGA) under project 1/0865/21; by the Cultural and Educational Grant Agency of Ministry of Education, Science, Research and Sport of the Slovak Republic (KEGA) project no. 006TUKE-4/2021; by the Slovak Research and Development Agency under contracts APVV-18-0197, APVV-20-138; projects from P. J. Safarik University no. VVGS-2020-1667, VVGS-2022-2123; iCoTS No. 313011V334; Ministry of Education, Youth and Sports of the Czech Republic under project no. LTT19001 and specific graduate research of the Brno University of Technology no. FEKT-S-20-6206. V. M. acknowledges the Research Foundation—Flanders (FWO) for project K801621N. Author N. K. and D. C. thanks Erasmus+ for funding their internships at the Laboratory of Adsorption & Catalysis (LADCA) at the University of Antwerp, Belgium and the Brno University of Technology, Czech Republic, respectively. The authors (V. Z., M. A.) thank the Ministry of Education, Science, Research and Sport of the Slovak Republic and the Accreditation Commission for the financial support of the TRIANGEL team in the frame of the scheme “Top Research Teams in Slovakia”.References
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- S. P. Shet, S. Shanmuga Priya, K. Sudhakar and M. Tahir, Int. J. Hydrogen Energy, 2021, 46, 11782–11803 CrossRef CAS.
- M. Fakhraei Ghazvini, M. Vahedi, S. Najafi Nobar and F. Sabouri, J. Environ. Chem. Eng., 2021, 9, 104790 CrossRef CAS.
- M. Almáši, N. Király, V. Zeleňák, M. Vilková and S. Bourrelly, RSC Adv., 2021, 11, 20137–20150 RSC.
- V. Zeleňák and I. Saldan, Nanomaterials, 2021, 11, 1638 CrossRef PubMed.
- M. Almáši, V. Zeleňák, R. Gyepes, Ľ. Zauška and S. Bourrelly, RSC Adv., 2020, 10, 32323–32334 RSC.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- T. A. Goetjen, J. Liu, Y. Wu, J. Sui, X. Zhang, J. T. Hupp and O. K. Farha, Chem. Commun., 2020, 56, 10409–10418 RSC.
- M. Almáši, V. Zeleňák, M. V. Opanasenko and J. Čejka, Catal. Lett., 2018, 148, 2263–2273 CrossRef.
- J. Cao, X. Li and H. Tian, Curr. Med. Chem., 2020, 27, 5949–5969 CrossRef CAS PubMed.
- Y. Wang, J. Yan, N. Wen, H. Xiong, S. Cai, Q. He, Y. Hu, D. Peng, Z. Liu and Y. Liu, Biomaterials, 2020, 230, 119619 CrossRef CAS PubMed.
- M. Almáši, J. Coord. Chem., 2021, 74, 2111–2127 CrossRef.
- Y. Y. Enakieva, E. A. Zhigileva, A. N. Fitch, V. V. Chernyshev, I. A. Stenina, A. B. Yaroslavtsev, A. A. Sinelshchikova, K. A. Kovalenko, Y. G. Gorbunova and A. Y. Tsivadze, Dalton Trans., 2021, 50, 6549–6560 RSC.
- J. Cao, W. Ma, K. Lyu, L. Zhuang, H. Cong and H. Deng, Chem. Sci., 2020, 11, 3978–3985 RSC.
- X. Chen and G. Li, Inorg. Chem. Front., 2020, 7, 3765–3784 RSC.
- K. Lee, J. Park, I. Song and S. M. Yoon, Bull. Korean Chem. Soc., 2021, 42, 1170–1183 CrossRef CAS.
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716–8789 CrossRef CAS PubMed.
- N. Király, V. Zeleňák, A. Zeleňáková, A. Berkutova, M. Almáši, R. Gyepes and E. Čižmár, Acta Phys. Pol., A, 2020, 137, 770–772 CrossRef.
- N. Király, V. Zeleňák, N. Lenártová, A. Zeleňáková, E. Čižmár, M. Almáši, V. Meynen, A. Hovan and R. Gyepes, ACS Omega, 2021, 6, 24637–24649 CrossRef PubMed.
- W.-Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 RSC.
- S. Huh, S.-J. Kim and Y. Kim, CrystEngComm, 2016, 18, 345–368 RSC.
- M. C. Das, S. Xiang, Z. Zhang and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 10510–10520 CrossRef CAS PubMed.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- X. S. Wang, M. Chrzanowski, L. Wojtas, Y. S. Chen and S. Ma, Chem.–Eur. J., 2013, 19, 3297–3301 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincă, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- J. A. Botas, G. Calleja, M. Sánchez-Sánchez and M. G. Orcajo, Langmuir, 2010, 26, 5300–5303 CrossRef CAS PubMed.
- H. Yue, Z. Shi, Q. Wang, Z. Cao, H. Dong, Y. Qiao, Y. Yin and S. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 17067–17074 CrossRef CAS PubMed.
- M. Almáši, in Current development in MOFs for hydrogen storage: a mechanistic investigation in metal-organic framework-based nanomaterials for energy conversion and storage, Elsevier, 2022, pp. 1–31 Search PubMed.
- T. Ghanbari, F. Abnisa and W. M. Wan Daud, Sci. Total Environ., 2020, 707, 135090 CrossRef CAS PubMed.
- S. Wang, Y. Liang, T. Dai, Y. Liu, Z. Sui, X. Tian and Q. Chen, J. Colloid Interface Sci., 2021, 591, 264–272 CrossRef CAS PubMed.
- L. Huang, J. Li, B. Liu, Y. Li, S. Shen, S. Deng, C. Lu, W. Zhang, Y. Xia, G. Pan, X. Wang, Q. Xiong, X. Xia and J. Tu, Adv. Funct. Mater., 2020, 30, 1910375 CrossRef CAS.
- Y. Hu, W. Chen, T. Lei, Y. Jiao, J. Huang, A. Hu, C. Gong, C. Yan, X. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 2000082 CrossRef CAS.
- Y. Zheng, S. Zheng, H. Xue and H. Pang, J. Mater. Chem. A, 2019, 7, 3469–3491 RSC.
- V. Knap, L. K. Vestergaard and D.-I. Stroe, Energies, 2020, 13, 4097 CrossRef CAS.
- V. Knap and D.-I. Stroe, J. Power Sources, 2021, 498, 229913 CrossRef CAS.
- L. Chladil, D. Kunický, T. Kazda, P. Vanýsek, O. Čech and P. Bača, Journal of Energy Storage, 2021, 41, 102907 CrossRef.
- L. Chladil, D. Kunický, P. Vanýsek and O. Čech, ECS Trans., 2018, 87, 107–114 CrossRef CAS.
- C. Ma, Z. Zheng, X. Jia, X. Liu, J. Wang, W. Qiao and L. Ling, J. Power Sources, 2021, 486, 229358 CrossRef CAS.
- Z. Y. Wang, L. Wang, S. Liu, G. R. Li and X. P. Gao, Adv. Funct. Mater., 2019, 29, 1901051 CrossRef.
- T. Kazda, M. Krbal, M. Pouzar, J. Vondrák, A. F. Straková, M. Slávik, T. Wagner and J. M. Macak, J. Power Sources, 2016, 331, 293–298 CrossRef CAS.
- T. Kazda, D. Capková, K. Jaššo, A. Fedorková Straková, E. Shembel, A. Markevich and M. Sedlaříková, Materials, 2021, 14, 5578 CrossRef CAS PubMed.
- D. Capkova, T. Kazda, P. Čudek and A. Strakova Fedorkova, ECS Trans., 2020, 99, 161–167 CrossRef CAS.
- J. Aguilera-Sigalat and D. Bradshaw, Coord. Chem. Rev., 2016, 307, 267–291 CrossRef CAS.
- K. Vellingiri, A. Deep and K.-H. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 29835–29857 CrossRef CAS PubMed.
- M. Jana, R. Xu, X.-B. Cheng, J. S. Yeon, J. M. Park, J.-Q. Huang, Q. Zhang and H. S. Park, Energy Environ. Sci., 2020, 13, 1049–1075 RSC.
- Y. Hu, W. Chen, T. Lei, Y. Jiao, J. Huang, A. Hu, C. Gong, C. Yan, X. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 2000082 CrossRef CAS.
- S. Dörfler, H. Althues, P. Härtel, T. Abendroth, B. Schumm and S. Kaskel, Joule, 2020, 4, 539–554 CrossRef.
- Z. Wang, Z. Wang, L. Yang, H. Wang, Y. Song, L. Han, K. Yang, J. Hu, H. Chen and F. Pan, Nano Energy, 2018, 49, 580–587 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- T. Kazda, P. Čudek, J. Vondrák, M. Sedlaříková, J. Tichý, M. Slávik, G. Fafilek and O. Čech, J. Solid State Electrochem., 2017, 22, 537–546 CrossRef.
- S. Bai, X. Liu, K. Zhu, S. Wu and H. Zhou, Nat. Energy, 2016, 1, 16094 CrossRef CAS.
- Y.-S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- X.-C. Xie, K.-J. Huang and X. Wu, J. Mater. Chem. A, 2018, 6, 6754–6771 RSC.
- Y. Zheng, S. Zheng, H. Xue and H. Pang, J. Mater. Chem. A, 2019, 7, 3469–3491 RSC.
- M. Rana, B. Luo, M. R. Kaiser, I. Gentle and R. Knibbe, J. Energy Chem., 2020, 42, 195–209 CrossRef.
- Z. Du, X. Chen, W. Hu, C. Chuang, S. Xie, A. Hu, W. Yan, X. Kong, X. Wu, H. Ji and L.-J. Wan, J. Am. Chem. Soc., 2019, 141, 3977–3985 CrossRef CAS PubMed.
- D. Capková, T. Kazda, A. Straková Fedorková, P. Čudek and R. Oriňaková, ECS Trans., 2019, 95, 19–26 CrossRef.
- Z. Xiao, Z. Yang, L. Wang, H. Nie, M. Zhong, Q. Lai, X. Xu, L. Zhang and S. Huang, Adv. Mater., 2015, 27, 2891–2898 CrossRef CAS PubMed.
- Z. W. Seh, J. H. Yu, W. Li, P.-C. Hsu, H. Wang, Y. Sun, H. Yao, Q. Zhang and Y. Cui, Nat. Commun., 2014, 5, 5017 CrossRef CAS PubMed.
- W. Dong, D. Wang, X. Li, Y. Yao, X. Zhao, Z. Wang, H.-E. Wang, Y. Li, L. Chen, D. Qian and B.-L. Su, J. Energy Chem., 2020, 48, 259–266 CrossRef.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- D. Capková, M. Almáši, T. Kazda, O. Čech, N. Király, P. Čudek, A. S. Fedorková and V. Hornebecq, Electrochim. Acta, 2020, 354, 136640 CrossRef.
- G. Chen, Y. Li, W. Zhong, F. Zheng, J. Hu, X. Ji, W. Liu, C. Yang, Z. Lin and M. Liu, Energy Storage Mater., 2020, 25, 547–554 CrossRef.
- Z. Wang, B. Wang, Y. Yang, Y. Cui, Z. Wang, B. Chen and G. Qian, ACS Appl. Mater. Interfaces, 2015, 7, 20999–21004 CrossRef CAS PubMed.
- X.-C. Xie, K.-J. Huang and X. Wu, J. Mater. Chem. A, 2018, 6, 6754–6771 RSC.
- D. Skoda, T. Kazda, L. Munster, B. Hanulikova, A. Styskalik, P. Eloy, D. P. Debecker, P. Vyroubal, L. Simonikova and I. Kuritka, J. Mater. Sci., 2019, 54, 14102–14122 CrossRef CAS.
- D. Capková, T. Kazda, O. Čech, N. Király, T. Zelenka, P. Čudek, A. Sharma, V. Hornebecq, A. Straková Fedorková and M. Almáši, Journal of Energy Storage, 2022, 51, 104419 CrossRef.
- D. Capkova, T. Kazda, M. Almasi, O. Cech, K. Jasso, M. Macak, J. Macko, P. Čudek and A. Straková Fedorková, Proc. Int. Astronaut. Congr., 2021, C3, 177587 Search PubMed.
- J. Niu, A. Kushima, M. Li, Z. Wang, W. Li, C. Wang and J. Li, J. Mater. Chem. A, 2014, 2, 19788–19796 RSC.
- J. Zhou, R. Li, X. Fan, Y. Chen, R. Han, W. Li, J. Zheng, B. Wang and X. Li, Energy Environ. Sci., 2014, 7, 2715 RSC.
- Z. Wang, B. Wang, Y. Yang, Y. Cui, Z. Wang, B. Chen and G. Qian, ACS Appl. Mater. Interfaces, 2015, 7, 20999–21004 CrossRef CAS PubMed.
- A. Benítez, J. Amaro-Gahete, D. Esquivel, F. J. Romero-Salguero, J. Morales and Á. Caballero, Nanomaterials, 2020, 10, 424 CrossRef PubMed.
- T. Rhauderwiek, S. Waitschat, S. Wuttke, H. Reinsch, T. Bein and N. Stock, Inorg. Chem., 2016, 55, 5312–5319 CrossRef CAS PubMed.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- K. Barthelet, D. Riou, M. Nogues and G. Férey, Inorg. Chem., 2003, 42, 1739–1743 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- T. Ghanbari, F. Abnisa and W. M. Wan Daud, Sci. Total Environ., 2020, 707, 135090 CrossRef CAS PubMed.
- T. Dhawa, S. Chattopadhyay, G. De and S. Mahanty, ACS Omega, 2017, 2, 6481–6491 CrossRef CAS PubMed.
- K. Xi, S. Cao, X. Peng, C. Ducati, R. Vasant Kumar and A. K. Cheetham, Chem. Commun., 2013, 49, 2192 RSC.
- X. Chen, M. Zhang, J. Zhu, J. Wang, Z. Jiao and Y. Li, J. Alloys Compd., 2022, 901, 163649 CrossRef CAS.
- W. Bao, Z. Zhang, C. Zhou, Y. Lai and J. Li, J. Power Sources, 2014, 248, 570–576 CrossRef CAS.
- H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192–3210 RSC.
- D. Capkova, V. Knap, A. Strakova Fedorkova and D.-I. Stroe, J. Energy Chem., 2022, 72, 318–325 CrossRef.
- X. Ge, C. Li, Z. Li and L. Yin, Electrochim. Acta, 2018, 281, 700–709 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03301a |
| This journal is © The Royal Society of Chemistry 2022 |