
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicroporous carbon in the selective electro-oxidation of molecular biomarkers: uric acid, ascorbic acid, and dopamine†
Tidapa Rattanaumpaa,
Santi Maensirib and
Kamonwad Ngamchuea *a
*a
aSchool of Chemistry, Institute of Science, Suranaree University of Technology, 111 University Avenue, Suranaree, Muang, Nakhon Ratchasima, 30000, Thailand. E-mail: kamonwad@g.sut.ac.th; Tel: +66 (0) 44 224 637
bSchool of Physics, Institute of Science, Suranaree University of Technology, 111 University Avenue, Suranaree, Muang, Nakhon Ratchasima, 30000, Thailand
First published on 27th June 2022
Abstract
Herein, we demonstrate the superior electrocatalytic activities of microporous carbon in the oxidation of three molecular biomarkers, ascorbic acid (AA), dopamine (DA), and uric acid (UA), which are co-present in biological fluids. The voltammetric responses of AA, DA, and UA at the low-cost microporous carbon electrode show significantly better sensitivity and selectivity than other more expensive and commonly used electrode materials such as copper(II) oxide, copper(I) oxide, and carbon nanotube. Differential pulse voltammetry at the microporous carbon electrode allows the detection of AA, DA, and UA with linear ranges of 100–2000 μM (AA), 10–150 μM (DA), and 10–150 μM (UA), sensitivities of 6.8 ± 0.2 nA μM−1 (AA), 261.4 ± 3.4 nA μM−1 (DA), and 93.5 ± 2.0 nA μM−1 (UA), and detection limits of 23.1 μM (AA), 0.2 μM (DA), and 1.7 μM (UA). The method has been validated with a synthetic urine sample to yield ∼100% recoveries for all three analytes. The developed method has been further applied in the investigation of the peroxide scavenging activity of UA.
1 Introduction
Uric acid (UA), ascorbic acid (AA), and dopamine (DA) are co-present in biological fluids and play important roles in physiological processes and metabolism.1 Uric acid (UA) is the end product of purine metabolism, which participates in energy metabolism, signal transduction, plasma antioxidant activity, and the formation of nucleic acids, DNA, and RNA.2 At high concentrations of uric acid above its saturation level (6.8 mg dL−1 in serum), uric acid crystals can deposit in joints, nearby tissues, kidneys and throughout the body, leading to the development of gout inflammatory arthritis.2,3 The abnormal level of UA is an indicator of several diseases such as hyperuricemia,2,3 gout,2,3 lysis syndrome,4 acute kidney injury,5 metabolic syndrome,6 major depressive disorder,7 and schizophrenia.8Ascorbic acid (AA) or vitamin C is an essential nutrient for human body. It is an important enzyme cofactor and antioxidant in various physiological functions such as collagen synthesis, liver detoxification, and the immune system.9 Abnormal levels of AA are associated with cancer,10 neurodegenerative diseases,11 chronic inflammation diseases,12 hepatic disease,13 and oxidative stress.13
Dopamine (DA) is an important neurotransmitter and neuromodulator in the nervous system.14 Low concentration of DA is a sign of neurological disorders, schizophrenia, Parkinson’s disease, restless leg syndrome, and HIV infection.15 Despite their diagnostic implications, point-of-care testing of UA, AA, and DA has often been neglected. This is in part due to the lack of simple testing methods for these species. This work therefore aims to develop a fast, simple, and rapid sensor for the analysis of UA, AA, and DA.
Several analytical methods have been employed in the detection of UA, AA, and DA, including mass spectrometry,16 high-performance liquid chromatography,17 colorimetry,18 fluorescence,19 and biosensors.20 However, the application of these methods in point-of-care is often limited due to long analysis time, the difficulty of sample preparation, and expensive instruments. Electrochemical measurements allow rapid and highly sensitive detection of redox active analytes at low cost.21–25 The electroactivities of UA, AA, and DA enable them to be quantitatively determined via electrochemical methods.1 However, the similar redox properties of the three compounds give rise to signals at similar potentials. The chemically and electrochemically irreversible nature of their electrochemical processes also inevitably passivate the electrodes. The main challenges in the electrochemical detection of UA, AA, and DA are therefore to resolve their overlapping oxidation peaks, overcome the issue of electrode fouling, as well as to enhance the sensitivity of the measurement.
Several electrode materials have been developed and employed to achieve these goals, mainly noble metals (e.g., Pt, Pd), metal oxides (e.g., Cu2O, CuO), and carbon nanomaterials (e.g., carbon nanotube, graphene).26–36 Considering the cost and the analytical performance, our focus herein is thus on the copper- and carbon-based materials. In this work, the performances of copper(II) oxide (CuO), copper(I) oxide (Cu2O), carbon nanotube (CNT), and microporous carbon (MC) in the electrochemical detection of UA, AA, and DA are evaluated.
In particular, we will show the excellent performance of microporous carbon in enhancing the selectivity and sensitivity of UA, AA, and DA measurements. High conductivity, chemical stability, large surface area, and high microporosity are strong characteristics of microporous carbon which can greatly enhance the efficiency of electrochemical sensors.37,38 The selectivity of the measurement is induced by differences in diffusional rates of the analytes in the microporous structure. Possible interaction between the analytes and various functional groups on the microporous carbon surface such as –OH, –COOH, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –NH2 can further improve electron transfer and enhance the sensitivity and selectivity of the electrochemical sensor.18,39
O and –NH2 can further improve electron transfer and enhance the sensitivity and selectivity of the electrochemical sensor.18,39
In addition to the development of a highly selective and sensitive electrochemical sensor for UA, AA, and DA detection, we further use UA as a paradigmatic example to demonstrate the potential of the sensor in the investigation of reactive oxygen species (ROS) scavenging activity. Uric acid is the main antioxidant in human plasma which contributes up to approximately 50% of the total antioxidant activity, significantly higher than other antioxidants such as ascorbic acid (vitamin C) and vitamin E.40 Excessive formation of free radicals namely peroxyl radicals (RO2˙) and hydroxyl radical (OH˙) accelerates chronic diseases, DNA damage, and carcinogenesis.40,41 This work employs H2O2, which is the major source of OH˙ radicals, as a model system in the investigation of the scavenging properties.
2 Experimental
2.1 Chemical reagents
All chemical reagents were of analytical grade and used as received: sodium L-lactate (C3H5NaO3, ≥99.0%, Sigma-Aldrich), citric acid anhydrous (C6H8O7, 99.5%, QRëC), urea (CH4N2O, ≥99.0%, Sigma-Aldrich), calcium chloride (CaCl2, 94%, APS Ajax Finechem), sodium chloride (NaCl, ≥99.0%, Sigma-Aldrich), magnesium sulfate anhydrous (MgSO4, ≥98.0%, Tokyo Chemical Industry), sodium bicarbonate (Na2HCO3, 99.8%, QRëC), L-ascorbic acid (C6H8O6, ≥99.0%, Sigma-Aldrich), dopamine hydrochloride ((HO)2C6H3CH2CH2NH2·HCl, ≥97.5.0%, Sigma-Aldrich), uric acid (C5H4N4O3, ≥99.0%, Sigma-Aldrich), ethanol (C2H5OH, ≥99.9%, QRëC), hydrochloric acid (HCl, 37%, RCI Labscan), hydrogen peroxide (H2O2, 35%, ANaPURE), sodium phosphate monobasic (NaH2PO4, ≥99.0%, Sigma-Aldrich), sodium phosphate dibasic (Na2HPO4, ≥99.0%, Sigma-Aldrich), hexaammineruthenium(III) chloride (98%, Thermo Scientific), and Nafion (D-520 dispersion, 5% w/w in water and 1-propanol, Alfa Aesar). Microporous carbon was obtained from IRPC Public Company Limited, Thailand. Copper(II) oxide (CuO powder, <50 nm diameter) and copper(I) oxide (Cu2O powder, <5 μm diameter) were obtained from Sigma-Aldrich. Multi-walled carbon nanotube (20–40 nm diameter, 5–15 μm length, ≥95%) was purchased from Tokyo Chemical Industry. The commercially available electrode materials (CuO, Cu2O, multi-walled carbon nanotube, and microporous carbon) were used as received without further purification, modification, or activation.2.2 Electrochemical studies
All electrochemical experiments were carried out using a Metrohm 910 PSTAT Mini potentiostat (Metrohm, Netherlands) and a standard three-electrode system in a Faraday cage thermostated at 25 °C. A glassy carbon electrode (GCE, 3.0 mm diameter, ItalSens, Netherlands), a silver/silver chloride electrode (Ag/AgCl in saturated KCl, ItalSens, Netherlands), and a platinum coil (Redoxme AB, Sweden) were used as working, reference, and counter electrodes, respectively.In cyclic voltammetry measurements, the peak currents were determined using linear regression of the front baseline, which accounts for the capacitive charging background current.42 In differential pulse voltammetry (DPV) of the AA, DA, and UA mixtures where the regression front baseline was not possible, the peak currents were determined by selecting two baseline points on either side of the DPV peak. The two points were joined together to form a linear baseline.43 Note that the currents at this extrapolated linear baseline at a microporous carbon electrode are comparable to the response measured in a blank electrolyte (presented later in Section 3.4.4) and is thus a justifiable method for baseline correction for DPV measurements in the solution mixtures.
2.3 Electrode preparation and characterization
The GCE working electrode was cleaned by polishing with 1.0, 0.3 and 0.05 μm alumina powder (Buehler, USA) on soft lapping pads (Buehler, USA) prior to use. In the preparation of modified electrodes, 1.0 mg of microporous carbon, CuO, Cu2O, or carbon nanotube was dispersed in 1.0 mL ethanol and sonicated for 5 minutes. Afterwards, the suspension was drop-casted onto a clean GCE surface and dried in a vacuum oven (Faithful, DZ-A/BC II Series, Huanghua City, China) at 55 °C for 10 minutes.The porosity of microporous carbon was investigated using N2 adsorption/desorption isotherms. The specific surface area and pore sizes were calculated using the Brunauer–Emmett–Teller (BET) analysis at the pressures 5.8 × 10−7 ≤ P/P0 ≤ 1.00 at 77 K carried out using a 3Flex Physisorption – Surface Characterization Analyzer (Micromeritics, ATS Scientific Inc., Burlington, Canada). The morphology of microporous carbon was characterized using a field emission scanning electron microscope (FE-SEM, 3.00 kV, Zeiss AURIGA). Surface chemical analysis of microporous carbon was performed using Fourier transform infrared spectroscopy (ATR-FTIR, Bruker Tensor 27, Germany) in the spectral range of 400–4000 cm−1. The electroactive surface area of the electrodes were estimated by cyclic voltammetry measurements in 1.0 mM hexaammineruthenium(III) or RuHex in the presence of 0.10 M KCl supporting electrolyte at varied scan rates (10–400 mV s−1). The thickness and optical profile of the microporous carbon electrodes were measured using a laser confocal microscope (OLYMPUS OLS5100, Tokyo, Japan).
2.4 Application to synthetic urine samples
The developed method was validated by spiking and recovery tests (standard addition) of AA, DA, and UA in synthetic urine samples in the presence of 0.10 M HCl. Synthetic urine was prepared according to Jiang et al.44 The synthetic urine consists of 11.0 mM sodium L-lactate, 2.0 mM citric acid, 25.0 mM NaHCO3, 170.0 mM urea, 2.5 mM CaCl2, 90.0 mM NaCl, 2.0 mM MgSO4, 10.0 mM Na2SO4, 7.0 mM NaH2PO4, and 7.0 mM Na2HPO4. The samples were subjected to differential pulse voltammetry in the potential range of –0.2 to +0.9 V at an MC/GCE electrode at the scan rate of 10 mV s−1, pulse amplitude of 0.01 V, and pulse width of 50 ms. The obtained results were analyzed and reported as percentage recovery (the mean value of triplicate measurements ± standard deviation).3 Results and discussion
First, the electrochemical properties of AA, DA, and UA were investigated at different electrode materials: CuO, Cu2O, Nafion, carbon nanotube (CNT), and microporous carbon (MC). The electrode which showed the highest selectivity and sensitivity in the analysis of AA, DA, and UA was then chosen for further studies. The measuring conditions such as pH, scan rates, and pulse amplitude were optimized. The developed sensor was then validated in synthetic urine sample and applied in the investigation of peroxide scavenging properties.3.1 Effects of electrode material
The electrochemical behaviors of AA, DA, and UA were investigated at selected low-cost copper- and carbon-based electrode materials, including copper(II) oxide (CuO), copper(I) oxide (Cu2O), carbon nanotube (CNT), and microporous carbon (MC). The materials (10 μg) were immobilized on the glassy carbon electrode (GCE) and subjected to voltammetric measurements in 100 μM AA, 100 μM DA, and 100 μM UA in the presence of 0.10 M KCl electrolyte at the scan rate of 10 mV s−1.Overall, electrode modifications with CuO, Cu2O, CNT, and MC yielded higher peak currents and thus higher sensitivities than a bare GCE due to an increase in the electroactive surface area (Fig. 1). However, the voltammetric waveshapes and the selectivity of the measurements differ from material to material.
 | ||
| Fig. 1 CV of 100 μM AA, 100 μM DA, and 100 μM UA in 0.10 M KCl (pH ∼ 7) at different working electrodes: (a) GCE, (b) carbon nanotube (CNT/GCE), (c) copper(II) oxide (CuO/GCE), (d) copper(I) oxide (Cu2O/GCE), (e) microporous carbon (MC/GCE), (f) Nafion-coated microporous carbon (Nafion/MC/GCE). Scan rate: 10 mV s−1. E vs. Ag/AgCl (saturated KCl) reference electrode. | ||
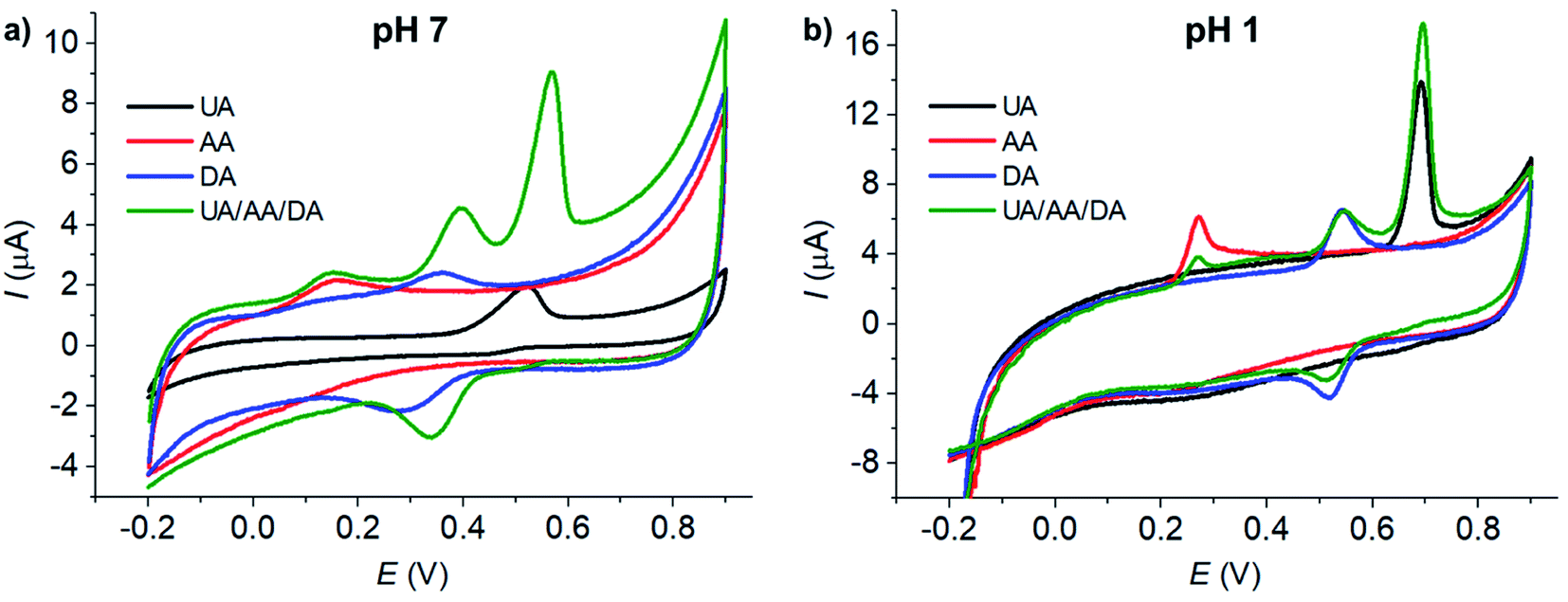
At bare GCE (Fig. 1a), CNT/GCE (Fig. 1b), and CuO/GCE (Fig. 1c), the voltammetric responses showed broad overlapping oxidation peaks of AA, DA, and UA, indicating poor selectivity of the electrodes. It is therefore not possible to determine the concentrations of individual AA, DA, and UA species due to the overlapping peak positions.
At Cu2O/GCE (Fig. 1d), three anodic peaks were observed in the oxidation of AA, DA, and UA at 0.21 V, 0.40 V, and 0.64 V, respectively. However, the peaks were still relatively broad and slightly overlapping, lowering the accuracy in the quantification of each species.
At MC/GCE (Fig. 1e), three well-resolved anodic peaks of AA, DA, and UA were observed at 0.14 V, 0.40 V, and 0.56 V, respectively. The occurrence of the AA, DA, and UA peaks at low overpotentials (cf. bare GCE) clearly indicated the electrocatalytic activities of the microporous carbon. The large separation of the peak potentials allows simultaneous selective detection of the individual concentrations of AA, DA, and UA.
The microporosity of the electrode material altered the mass transport behaviours of AA, DA, and UA from ‘planar/linear diffusion’ to ‘thin layer diffusion’, causing the shift in the peak potentials. As the electrochemical parameters such as formal potentials (Ef), standard electrochemical rate constants (k0) and transfer coefficients (β) of the three species are different, the voltammetric peaks shifted to different extents, which in this case inducing wider separation of the voltammetric peaks. In addition, the presence of various functional groups on the surface of the microporous carbon and the different interactions of the surface with AA, DA, and UA, which take part in the inner-sphere electron transfer processes, may also contribute to the improved selectivity of the electrode.1
In addition to nanomaterials, the use of conductive polymers as part of the protective and selective electrode component has been widely employed in electrochemical sensors.45 Nafion, in particular, has been demonstrated to show enhanced analytical performances towards the detection of molecular biomarkers such as serotonin,46 histamine,47 as well as uric acid, ascorbic acid, and dopamine.48,49 In an attempt to further improve the performance of microporous carbon electrodes, the effect of Nafion coating was next evaluated in Fig. 1f.
At Nafion/MC/GCE, the anodic peaks of DA and UA were observed at 0.40, and 0.56 V, respectively. The peak currents of DA increased, while that of UA decreased and the signal of AA completely disappeared in the presence of Nafion coating. The pKa values of AA, DA, and UA are 4.17,9 8.87,50,51 and 5.40,52 respectively. At pH ∼7 under the measuring conditions, AA and UA were thus negatively charged, while DA was neutral. The electrostatic repulsion between the sulfonic groups of Nafion and the negatively charged AA and UA lowered the voltammetric responses of the two species. Under neutral conditions, Nafion coating may thus be suitable for selective analysis of DA the presence of AA and UA interferences. Other electrically charged coating materials may also be selected to suit the choice of the analyte of interest, considering the pKa of the analytes and the coating material. However, if the analysis of all the three compounds is required under neutral condition, it is preferable to use MC/GCE electrodes without Nafion coating. The effects of pH on the electrochemical responses of AA, DA, and UA at Nafion/MC/GCE electrodes have also been investigated, and the results are presented in Section S1 (ESI†).
3.2 Characterization of microporous carbon
Fig. 2a shows the SEM image of microporous carbon. A large number of pores in the range of 2–34 nm were observed throughout the sample. Due to the limited resolution of the SEM (ca. 0.5–2 nm), only larger pores were observed, and the size distribution measured using SEM was biased towards the more easily observable larger pores. The nitrogen adsorption–desorption isotherm of microporous carbon was further used to determine the pore sizes and surface area of the microporous carbon. The microporous carbon exhibited type I isotherm (Fig. 2b), confirming the characteristic microporous structure.53 The specific surface area of the microporous carbon was determined by the Brunauer–Emmett–Teller (BET) analysis in Fig. 2b (inlay) to be 1270 m2 g−1.54 The pore sizes were determined from the desorption loop from the amount of N2 adsorbate lost in the desorption step.55 The Brunauer–Emmett–Teller (BET), Barrett–Joyner–Halenda (BJH), and Dollimore–Heal (DH) models predicted the average pore diameters of 1.89 nm, 2.93 nm, and 2.95 nm, respectively. The latter two models, however, used Kelvin equation of pore filling which is only applicable to mesopores and small macropores.55 From the average diameters and the morphology observed in the SEM images, the results suggested that the material consisted of a mixture of micropores and mesopores.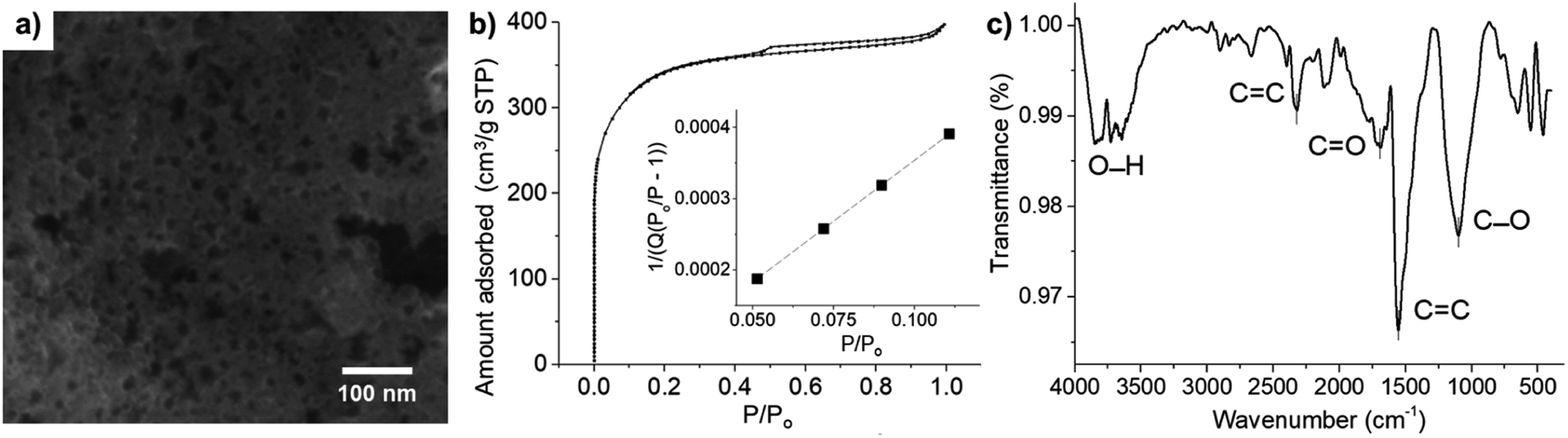 | ||
| Fig. 2 (a) SEM image, (b) N2 adsorption–desorption isotherm, (inlay) a plot of 1/[Q(P0/P − 1)] against the relative pressure (P/P0) for the determination of BET surface area, where Q is the amount of N2 adsorbed on microporous carbon (cm3 g−1 STP), and (c) FT-IR spectra of microporous carbon. | ||
FT-IR analysis was used to identify functional groups on the surface of microporous carbon (Fig. 2c). The broad band at 3700 cm−1 was attributed to the O–H stretching. The stretching band of the carbonyl CO group was observed at 1692 cm−1. The bands at 1554 cm−1 and 2320 cm−1 corresponded to CC stretching.56 The peak at 1098 cm−1 can be assigned to C–O stretching vibration. The C–C stretching was also observed in the range of 400–700 cm−1.57–59 There were thus several functional groups on the surface of microporous carbon which may interact with AA, DA, and UA through H-bonding and electrostatic attraction.
3.3 Cyclic voltammetry of AA, DA, and UA at MC/GCE
 | ||
| Fig. 3 CV of (red) 100 μM AA, (blue)100 μM DA, (black)100 μM UA and (green) the mixture of 100 μM AA, 100 μM DA, and 100 μM UA at MC/GCE at the scan rate of 10 mV s−1 in different electrolytes: (a) 0.10 M KCl and (b) 0.10 M HCl. E vs. Ag/AgCl (saturated KCl) reference electrode. | ||
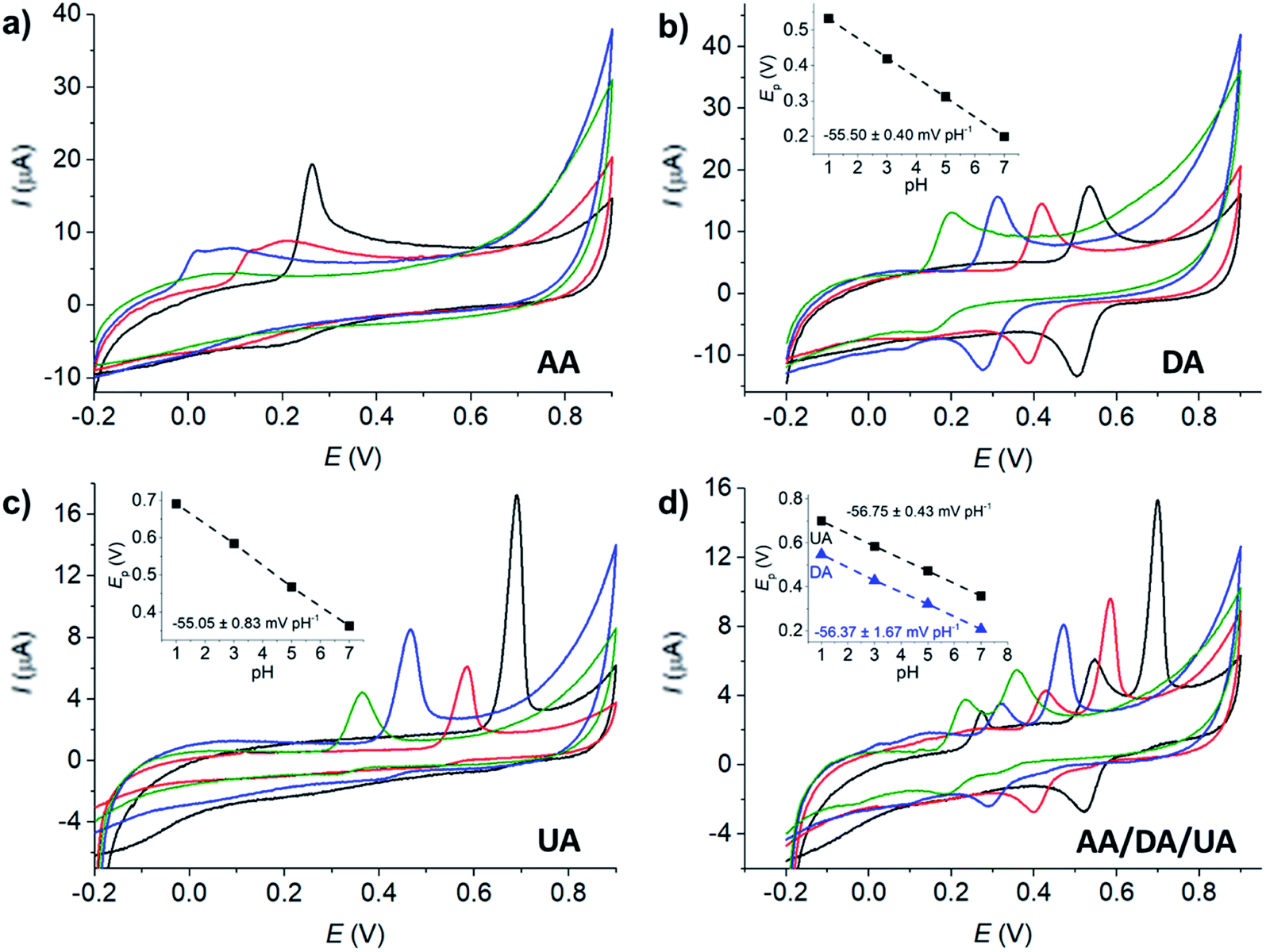
The voltammetric peaks of AA, DA, and UA became better resolved in acidic solution (0.10 M HCl, pH = 1, Fig. 3b), possibly due to the change in the protonation states of the analytes and the surface function groups of microporous carbon altering the interaction between the two components. The effects of the solution pH will be further investigated next.
 | ||
| Fig. 4 CV of (a) 100 μM AA, (b) 100 μM DA, (c) 100 μM UA, and (d) 100 μM AA, DA, and UA mixture at MC/GCE at the scan rate of 10 mV s−1 in aqueous buffers of various pH: (black) pH 1.0, (red) pH 3.0, (blue) pH 5.0, (green) pH 7.0. E vs. Ag/AgCl (saturated KCl) reference electrode. | ||
The effects of pH on the voltammetric responses of the mixture of 100 μM AA, DA, and UA were next investigated at MC/GCE electrodes (Fig. 4d). In the solution mixtures, the anodic peak potentials (Ep) of DA and UA negatively shifted with pH with the slopes of −56.75 ± 0.02 mV pH−1 and −56.37 ± 0.03 mV pH−1, respectively. The slopes of Ep vs. pH of DA and UA measured in the solution mixtures were not significantly different from when measured on their own, clearly indicating that the oxidation of AA, DA, or UA is independent of the other two species.
The anodic peak currents (Ip) increased as the solutions became more acidic. The peaks were also sharper and narrower in acidic conditions, allowing better separation of the oxidation peaks of AA, DA and UA. The pH of 1.0 was thus chosen for further analyses.
The redox process of DA (Fig. 4b) shows a clear backward cathodic peak which suggests that the reaction is chemically reversible. The absence of backward cathodic peaks in the oxidation of AA (Fig. 4a) and UA (Fig. 4c) indicates the chemical irreversibility of the processes due to further chemical reactions following the electron transfer steps.61 The results are similar to those previously reported in the literatures.21,26
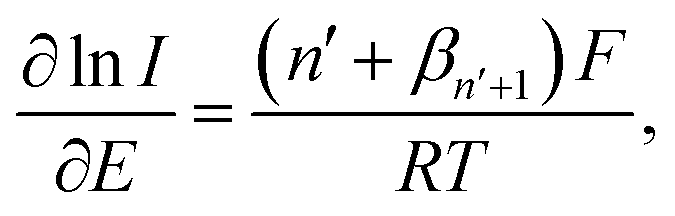 | (1) |
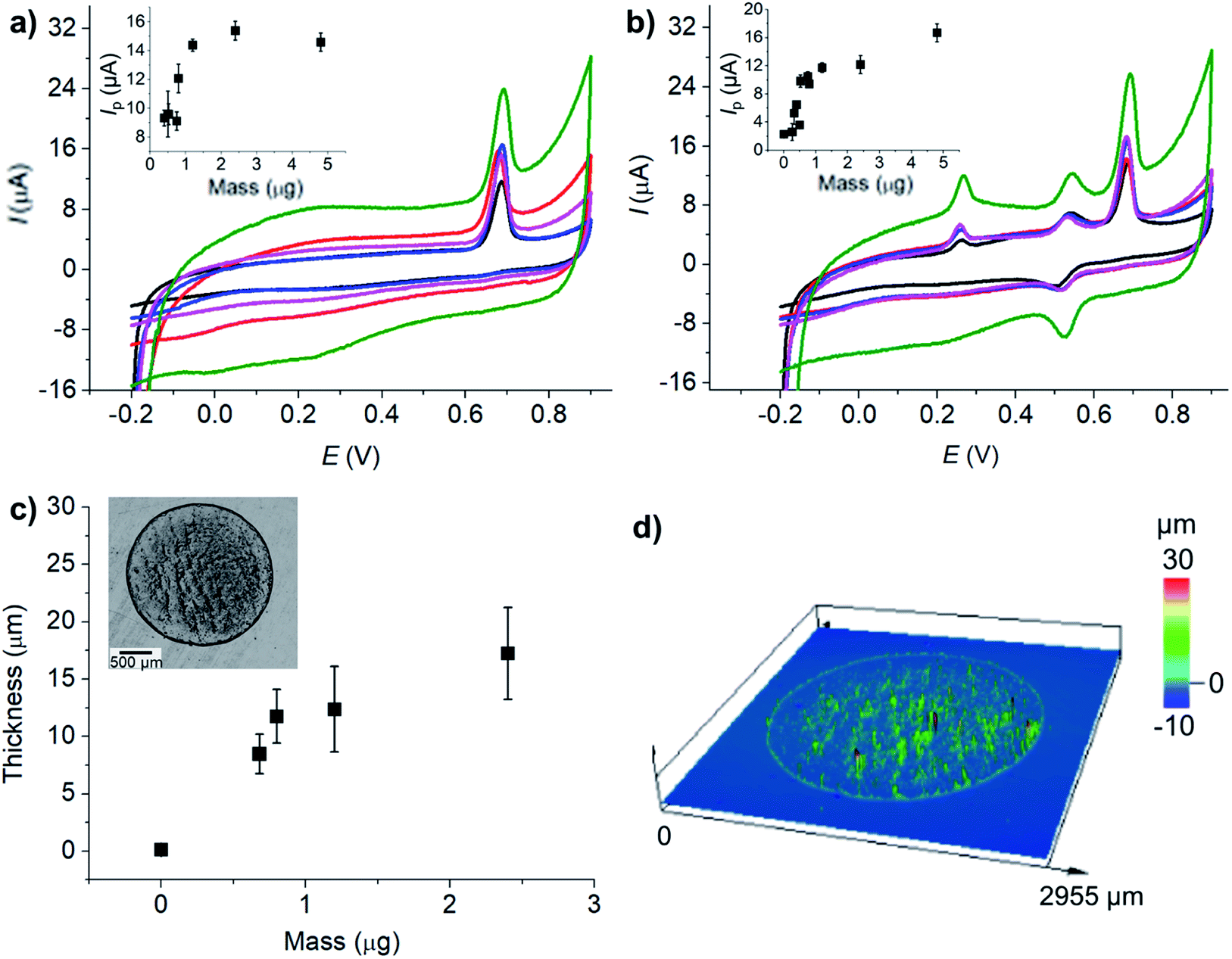
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), R is the molar gas constant (8.314 J K−1 mol−1), T is the absolute temperature (298 K), n′ is the number of electron transfer before the rate determining step (RDS), βn′+1 is the anodic transfer coefficient of the RDS. The line of best fit of lnI vs. E was determined using currents in the range of 15–50% of the peak current to eliminate the mass transport effect.54
485 C mol−1), R is the molar gas constant (8.314 J K−1 mol−1), T is the absolute temperature (298 K), n′ is the number of electron transfer before the rate determining step (RDS), βn′+1 is the anodic transfer coefficient of the RDS. The line of best fit of lnI vs. E was determined using currents in the range of 15–50% of the peak current to eliminate the mass transport effect.54Analysis of the voltammograms at the slow scan rate of 10 mV s−1 yielded the n′ + βn′+1 values at a bare GCE of 0.14 ± 0.05, 0.17 ± 0.02, and 0.46 ± 0.10 for AA, DA, and UA, respectively. At an MC/GCE, n′ + βn′+1 were determined to be 0.53 ± 0.01, 0.34 ± 0.05, and 0.69 ± 0.01 for AA, DA, and UA, respectively. The results thus indicated that AA, DA, and UA have similar mechanisms with the first e− transfer being the rate determining step (n′ = 0).
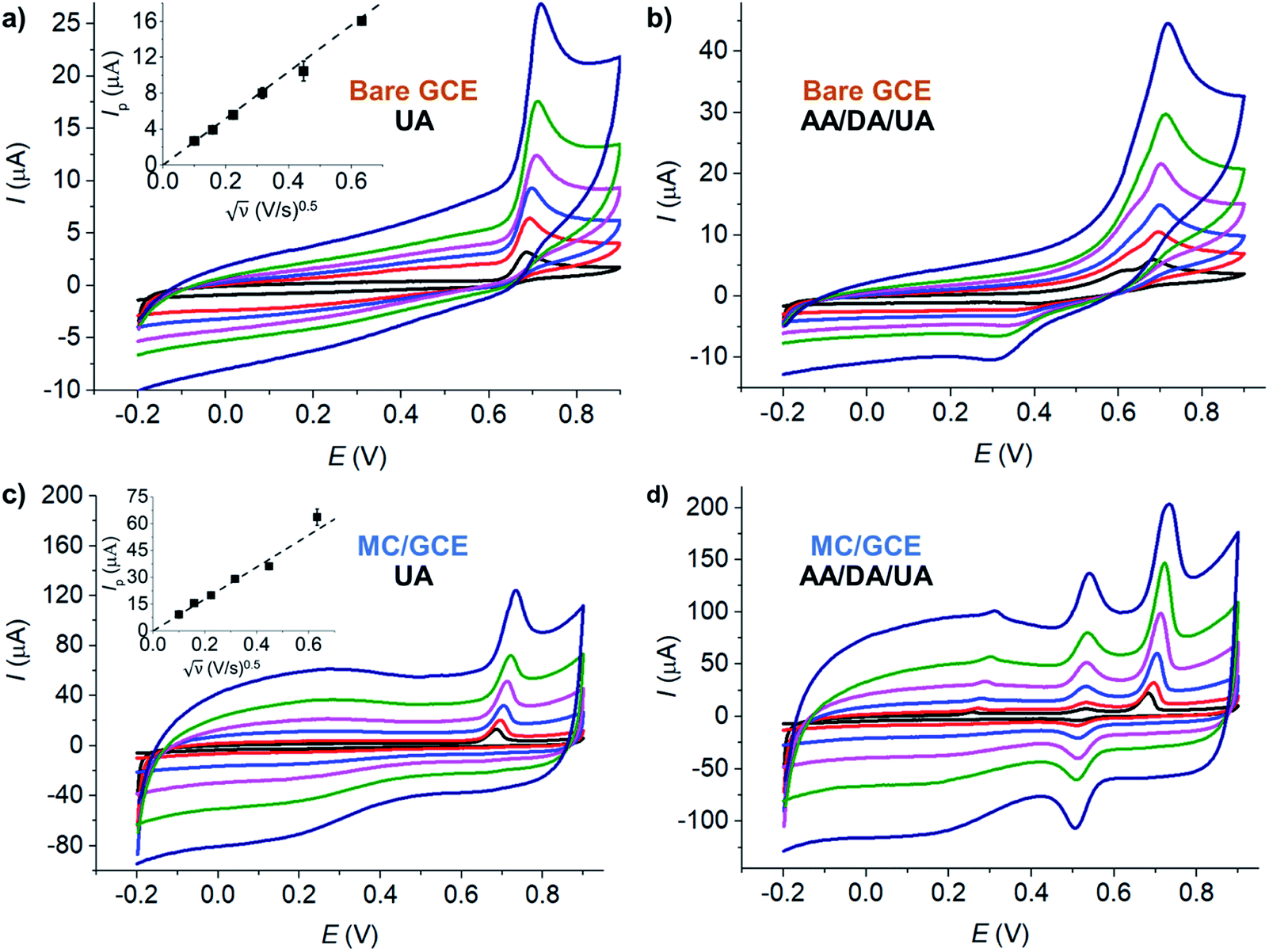 | ||
| Fig. 5 CV at bare GCE at varied scan rates of (a) 100 μM UA and (b) 100 μM AA, DA, UA. CV at MC/GCE at varied scan rates of (c) 100 μM UA and (d) 100 μM AA, DA, UA. (black) 10 mV s−1, (red) 25 mV s−1, (blue) 50 mV s−1, (pink) 100 mV s−1, (green) 200 mV s−1, and (dark blue) 400 mV s−1. All measurements were performed in the presence of 0.10 M HCl at pH 1.0. E vs. Ag/AgCl (saturated KCl) reference electrode. | ||
The diffusion coefficient of UA was analyzed at a bare GCE (electrode surface area = 7.068 × 10−6 m2). The electrode size has been checked by length measurements under a microscope to yield the diameter of 3.00 ± 0.01 mm. The GCE has also been characterized using hexaammineruthenium(III) or RuHex, a standard outer-sphere redox probe, refer to Section S2 in the ESI.† The scan rates were varied between 10 and 400 mV s−1. The temperature was thermostated at 25 °C (298 K). The electrolyte was 0.10 M HCl. The diffusion coefficient of UA was determined by eqn (2) to be 5.09 × 10−9 ± 0.02 × 10−9 m2 s−1, in the same order of magnitude as the value previously reported in the literature (3.2 × 10−5 m2 s−1).64
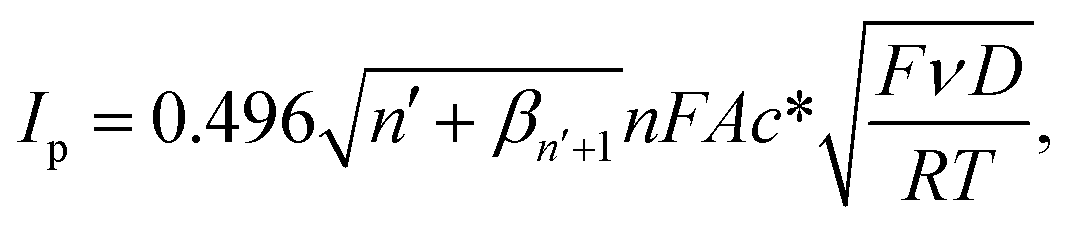 | (2) |
485 C mol−1), A is the electrode surface area (m2), c* is the bulk concentration of the redox analyte, R is the molar gas constant (8.314 J K−1 mol−1), T is the absolute temperature (K), and D is the analyte diffusion coefficient (m2 s−1).65
At MC/GCE electrodes, the anodic peak currents of UA also increased linearly with square root of scan rates, indicating a diffusion-controlled process (Fig. 5c). The simultaneous oxidation of AA, DA, and UA gave well-defined and widely separated peaks at MC/GCE electrodes (Fig. 5d). The three anodic peaks of AA, DA, and UA were better resolved at slow scan rates. The scan rate of 10 mV s−1 was thus chosen as the optimal condition for the simultaneous detection of AA, DA, and UA.
 | ||
| Fig. 6 (a) CV of 100 μM UA, and (b) CV of 100 μM UA, AA, and DA at MC/GCE prepared using different amounts of microporous carbon: (black) 0.75 μg, (red) 0.8 μg, (blue) 1.2 μg, (pink) 2.4 μg, (green) 4.8 μg. Scan rate: 10 mV s−1. Inlays: the plots of the anodic peak currents (Ip) vs. the amount of microporous carbon. E vs. Ag/AgCl (saturated KCl) reference electrode. (c) Average thickness of the dropcasted microporous carbon at different amounts (inlay: confocal microscopic image of the MC (1.2 μg)/GCE surface). (d) Optical profile of the MC (1.2 μg)/GCE surface. | ||
The electroactive surface areas of the electrodes modified with different amounts of microporous carbon were further determined using a standard outer-sphere redox probe, hexaammineruthenium(III). The results which are presented in Fig. S3 (ESI†) showed that the electroactive surface area increased with the amount of microporous carbon in a similar trend to that observed for the responses of AA, DA, and UA in Fig. 6.
The thickness and optical profile of the microporous carbon layers were next measured and presented in Fig. 6c and d. The thickness of the microporous carbon layers increased with the amount dropcasted. The thick layers of microporous carbon hinder mass transport of the analytes to the electrode surface due to slow diffusion within the microporous structures.66 An increase in the amount of microporous carbon also caused the capacitative background currents to be significantly large. Importantly, the peak positions of AA, DA, and UA were not significantly altered by the amount of microporous carbon. The amount of 1.2 μg of microporous carbon was thus chosen as the optimal condition for the analysis of AA, DA, and UA.
At MC/GCE electrodes, the quantification of AA had the linear range of 1–1200 μM, the sensitivity of 0.018 μA μM−1, and the LOD of 5.1 μM. DA measurements at MC/GCE yielded the linear range of 1–140 μM, the sensitivity of 0.0465 μA μM−1, and the LOD of 1.4 μM. For UA at MC/GCE, the linear range, sensitivity, and LOD were 1–60 μM, 0.16 μA μM−1, and 1.8 μM, respectively. The results thus clearly demonstrated the improved sensitivities and LOD at the microporous carbon electrodes. Chronoamperometry of AA, DA, and UA in 0.10 M HCl is also presented in Fig. S4, ESI.†
3.4 Differential pulse voltammetry of AA, DA, and UA at MC/GCE
Next, differential pulse voltammetry (DPV) was employed to enhance the analytical performances of MC/GCE electrodes in the determination of AA, DA, and UA.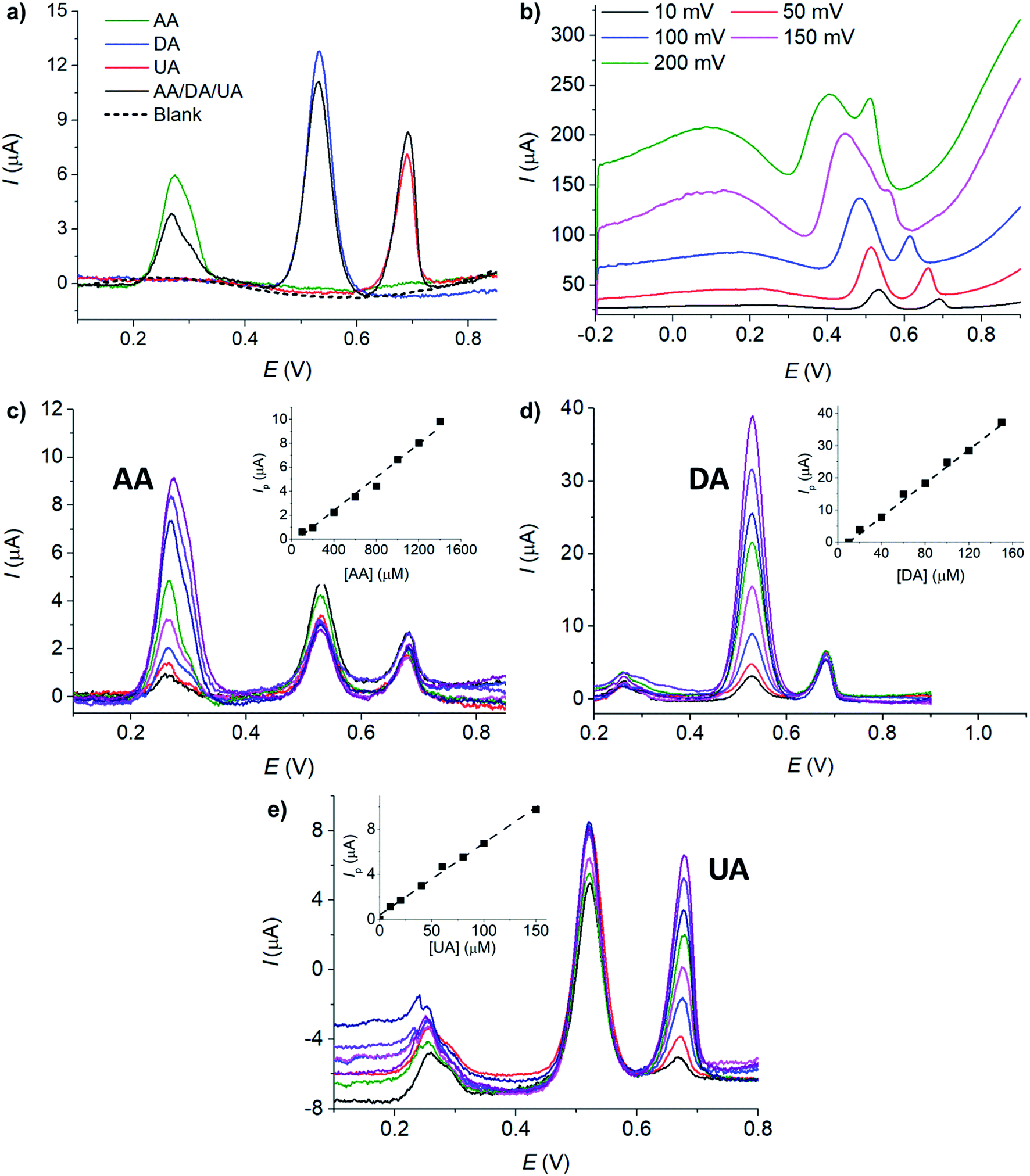 | ||
| Fig. 7 (a) DPV of AA, DA, UA, the mixture of AA, DA, and UA, and a blank electrolyte. (b) DPV of the mixture of AA, DA, and UA at different pulse amplitudes (10–200 mV). (c) DPV of varied [AA] in the presence of 20 μM DA and 20 μM UA. (d) DPV of varied [DA] in the presence of 500 μM AA and 50 μM UA, (e) DPV of varied [UA] in the presence of 300 μM AA and 20 μM DA. Electrolyte: 0.10 M HCl. Electrode: MC/GCE. Scan rate: 10 mV s−1. Otherwise stated: pulse amplitude = 10 mV and pulse width = 50 ms. E vs. Ag/AgCl (saturated KCl) reference electrode. | ||
| Ascorbic acid | Dopamine | Uric acid | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Electrode | Linear range (μM) | LOD (μM) | Sensitivity (μA μM−1) | Ref. | Electrode | Linear range (μM) | LOD (μM) | Sensitivity (μA μM−1) | Ref. | Electrode | Linear range (μM) | LOD (μM) | Sensitivity (μA μM−1) | Ref. |
| Graphene/Pt | 0.15–34.4 | 0.15 | 0.3457 | 26 | Graphene/Pt | 0.03–8.13 | 0.03 | 0.9695 | 26 | Graphene/Pt | 0.05–11.85 | 0.05 | 0.4119 | 26 |
| Reduced graphene oxide | 0.7–100 | 0.7 | 0.4342 | 27 | Reduced graphene oxide | 0.1–400 | 0.1 | 0.3304 | 27 | Reduced graphene oxide | 2–600 | 1 | 0.0153 | 27 |
| MOF-5/3D kenaf stem-derived porous carbon | 0.7–11.5 | 0.24 | 0.00686 | 28 | Graphene coated by polydopamine/multi-walled carbon nanotubes | 7–297 | 1 | 223.7 | 35 | Graphene coated by polydopamine/multi-walled carbon nanotubes | 20–320 | 15 | 209.6 | 35 |
| Nitrogen doped graphene | 5.0–1300 | 2.2 | 0.09311 | 29 | Nitrogen doped graphene | 0.5–170 | 0.25 | 0.2049 | 29 | Nitrogen doped graphene | 0.1–20 | 0.045 | 0.1320 | 29 |
| Cu4(OH)6SO4 nanorods | 17–6000 | 6.4 | 0.01753 | 30 | CuO/nanorice | 1–150 | 0.42 | 32.8 | 36 | CuO/nanorice | 1–160 | 1.2 | 9.97 | 36 |
| Poly[N-(ferrocenyl formacyl) pyrrole]@multi-walled carbon nanotubes | 200–400 | 40.0 | 0.000542 | 31 | Poly[N-(ferrocenyl formacyl) pyrrole]@multi-walled carbon nanotubes | 2–16 | 1.1 | 0.01951 | 31 | Poly[N-(ferrocenyl formacyl) pyrrole]@multi-walled carbon nanotubes | 2–16 | 0.73 | 0.02793 | 31 |
| Pristine graphene | 9.00–2314 | 6.45 | 0.06674 | 32 | Pristine graphene | 5.00–710 | 2.00 | 0.1125 | 32 | Pristine graphene | 6.00–1330 | 4.82 | 0.1029 | 32 |
| Hierarchical nanoporous PtTi | 450–1000 | 17.5 | 27.50 | 33 | Hierarchical nanoporous PtTi | 25–50 | 2.8 | 21.05 | 33 | Hierarchical nanoporous PtTi | 120–230 | 5.7 | 26.34 | 33 |
| Reduced graphene oxide/Au nanoplates | 240–1500 | 51 | 0.0117 | 34 | Reduced graphene oxide/Au nanoplates | 6.8–41 | 1.4 | 1.8 | 34 | Reduced graphene oxide/Au nanoplates | 8.8–53 | 1.6 | 3.6 | 34 |
| This work | 100–2000 | 23.1 | 6.8 | This work | 10–150 | 0.2 | 261.4 | This work | 10–150 | 1.7 | 93.5 | |||
3.5 Validation in synthetic urine
The developed sensor was next validated in synthetic urine sample prepared according to Jiang et al.,44 refer to Section 2.4. Fig. S6 in the ESI† showed that the oxidation peaks of AA, DA, and UA in urine sample in the presence of 0.10 M HCl were not significantly altered compared with the responses in a standard 0.10 M HCl solution. Next, the % recovery was evaluated. The concentrations of 500 μM AA, 2.0 μM DA, and 200 μM UA were spiked into synthetic urine and subjected to DPV measurements at MC/GCE under the optimized conditions (1.2 μg MC, scan rate: 10 mV s−1, pulse amplitude 10 mV, pulse width 50 ms). The employed concentrations were chosen to be within the normal levels of AA, DA, and UA found in human urine (AA: 458–9602 μM,67 DA: 0.3–3.13 μM,68 UA: 149–416 μM).69 The percentage recoveries determined by standard addition methods were 96%, 102%, and 101% for AA, DA, and UA, respectively, suggesting high accuracy of the method and excellent tolerance to possible interferences presented in the synthetic urine sample.3.6 Application of the developed sensor in the evaluation of peroxide scavenging activity of uric acid
As uric acid is an important antioxidant in human plasma, we next applied our developed sensor in the investigation of the scavenging properties of uric acid towards reactive oxygen species (ROS) by using hydrogen peroxide (H2O2) as a model system.70The solutions of 100 μM or 200 μM UA were mixed with various concentrations of H2O2 (0.0–50.0 mM H2O2) and kept in a dark place at room temperature for 24 hours. UA concentrations were then measured electrochemically by the developed method.
The % of UA consumed in the reaction with H2O2 was calculated by eqn (3):
| % UA consumed = [(Ip,control − Ip,sample)/Ip,control] × 100, | (3) |
Guelcin et al.70 suggested that the ‘% UA consumed’ parameter can be used in the estimation of peroxide scavenging activity of UA. Fig. 8 showed that higher percentages of UA were consumed at higher H2O2 concentrations. Higher percentages of UA were consumed when the initial concentration of UA was lower. When 100 μM UA was used, the peroxide scavenging activity approached the limiting value of 46.7 ± 0.8% at the [H2O2]/[UA] ratio of ca. 350-fold, close to the value reported by Gülçin et al. in spectrophotometric measurements (41.1 ± 2.9%).70 With the developed simple electrochemical method, it was thus possible to determine the peroxide scavenging activity of UA. The advantage of the electrochemical method over the previously used spectrophotometric technique70 is that there is no interference between the responses of UA and H2O2 (Fig. S8, ESI†).
 | ||
| Fig. 8 % UA consumed after (black) 100 μM UA or (red) 100 μM UA were incubated with 0.0–50.0 mM H2O2 for 24 hours. | ||
4 Conclusions
The electrochemical behaviors of AA, DA, and UA were investigated at various electrode materials including Cu2O, CuO, carbon nanotube, and microporous carbon. The superior sensitivity and selectivity of microporous carbon in the simultaneous analysis of AA, DA, and UA were clearly demonstrated. The highly porous structure of the carbon material greatly increased the electroactive surface area, enhancing the sensitivity of the measurement. The selectivity of the electrochemical detection was significantly improved by the microporosity and the presence of various surface functional groups on microporous carbon. The synergistic effect of microporous carbon enables it to be a suitable electrode material for the analysis of AA, DA, and UA, better than its alternatives such as carbon nanotubes and copper oxides. Excellent sensitivities and limits of detection with ∼100% recoveries were achieved. The sensor was capable of detecting AA, DA, and UA simultaneously and selectively in the presence of various interferences in synthetic urine. Furthermore, we have shown that the developed electrochemical sensor can be applied in the evaluation of peroxide scavenging activity using UA as a paradigmatic example. Overall, the developed sensor shows strong potentials for applications which require fast measurements of AA, DA, and/or UA in biological fluids.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by Office of the Permanent Secretary, Ministry of Higher Education, Science, Research and Innovation (OPS MHESI), Thailand Science Research and Innovation (TSRI), and Suranaree University of Technology [Research Grant for New Scholar (RGNS) Grant No. RGNS 63-118]. Rattanaumpa T. acknowledges funding from the Royal Thai government under the Development and Promotion of Science and Technology Talents Project.References
- K. Ngamchuea, K. Chaisiwamongkhol, C. Batchelor-McAuley and R. G. Compton, Chemical analysis in saliva and the search for salivary biomarkers–a tutorial review, Analyst, 2018, 143(1), 81–99 RSC.
- J. Maiuolo, F. Oppedisano, S. Gratteri, C. Muscoli and V. Mollace, Regulation of uric acid metabolism and excretion, Int. J. Cardiol., 2016, 213, 8–14 CrossRef PubMed.
- M. A. Martillo, L. Nazzal and D. B. Crittenden, The crystallization of monosodium urate, Curr. Rheumatol. Rep., 2014, 16(2), 400 CrossRef PubMed.
- A. E. Mirrakhimov, P. Voore, M. Khan and A. M. Ali, Tumor lysis syndrome: A clinical review, World J. Crit. Care Med., 2015, 4(2), 130–138 CrossRef PubMed.
- B. Lisowska-Myjak, Serum and urinary biomarkers of acute kidney injury, Blood Purif., 2010, 29(4), 357–365 CrossRef CAS PubMed.
- A. López-Suárez, J. Elvira-González, A. Bascuñana-Quirell, J. Rosal-Obrador, A. Michán-Doña, J. Escribano-Serrano and E. Benítez-Rodríguez, Concentraciones séricas de uratos y excreción urinaria de ácido úrico en individuos con síndrome metabólico, Med. Clin., 2006, 126(9), 321–324 CrossRef PubMed.
- P. Zheng, J.-j. Chen, T. Huang, M.-j. Wang, Y. Wang, M.-x. Dong, Y.-j. Huang, L.-k. Zhou and P. Xie, A novel urinary metabolite signature for diagnosing major depressive disorder, J. Proteome Res., 2013, 12(12), 5904–5911 CrossRef CAS PubMed.
- H.-L. Cai, H.-D. Li, X.-Z. Yan, B. Sun, Q. Zhang, M. Yan, W.-Y. Zhang, P. Jiang, R.-H. Zhu and Y.-P. Liu, Metabolomic analysis of biochemical changes in the plasma and urine of first-episode neuroleptic-naive schizophrenia patients after treatment with risperidone, J. Proteome Res., 2012, 11(8), 4338–4350 CrossRef CAS PubMed.
- Z. Shad, A. Arsalan, R. Barto, M. F. Khan and I. Ahmed, Physicochemical, Biochemical and Antioxidant Properties of Ascorbic Acid, Baqai J. Health Sci., 2011, 14(2), 33–40 Search PubMed.
- J. Du, J. J. Cullen and G. R. Buettner, Ascorbic acid: chemistry, biology and the treatment of cancer, Biochim. Biophys. Acta, Rev. Cancer, 2012, 1826(2), 443–457 CrossRef CAS PubMed.
- M. Moretti, D. B. Fraga and A. L. S. Rodrigues, Preventive and therapeutic potential of ascorbic acid in neurodegenerative diseases, CNS Neurosci. Ther., 2017, 23(12), 921–929 CrossRef CAS PubMed.
- A. Sorice, E. Guerriero, F. Capone, G. Colonna, G. Castello and S. Costantini, Ascorbic acid: its role in immune system and chronic inflammation diseases, Mini-Rev. Med. Chem., 2014, 14(5), 444–452 CrossRef CAS PubMed.
- K. Q. De Andrade, F. A. Moura, J. M. Dos Santos, O. R. P. De Araújo, J. C. de Farias Santos and M. O. F. Goulart, Oxidative stress and inflammation in hepatic diseases: therapeutic possibilities of N-acetylcysteine, Int. J. Mol. Sci., 2015, 16(12), 30269–30308 CrossRef PubMed.
- A. A. Ensafi, M. Taei and T. Khayamian, A differential pulse voltammetric method for simultaneous determination of ascorbic acid, dopamine, and uric acid using poly (3-(5-chloro-2-hydroxyphenylazo)-4,5-dihydroxynaphthalene-2, 7-disulfonic acid) film modified glassy carbon electrode, J. Electroanal. Chem., 2009, 633(1), 212–220 CrossRef CAS.
- A. Pandikumar, G. T. S. How, T. P. See, F. S. Omar, S. Jayabal, K. Z. Kamali, N. Yusoff, A. Jamil, R. Ramaraj and S. A. John, Graphene and its nanocomposite material based electrochemical sensor platform for dopamine, RSC Adv., 2014, 4(108), 63296–63323 RSC.
- S. Chen, H. Zheng, J. Wang, J. Hou, Q. He, H. Liu, C. Xiong, X. Kong and Z. Nie, Carbon nanodots as a matrix for the analysis of low-molecular-weight molecules in both positive-and negative-ion matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and quantification of glucose and uric acid in real samples, Anal. Chem., 2013, 85(14), 6646–6652 CrossRef CAS PubMed.
- N. Ye, T. Gao and J. Li, Hollow fiber-supported graphene oxide molecularly imprinted polymers for the determination of dopamine using HPLC-PDA, Anal. Methods, 2014, 6(18), 7518–7524 RSC.
- E. Sohouli, E. M. Khosrowshahi, P. Radi, E. Naghian, M. Rahimi-Nasrabadi and F. Ahmadi, Electrochemical sensor based on modified methylcellulose by graphene oxide and Fe3O4 nanoparticles: Application in the analysis of uric acid content in urine, J. Electroanal. Chem., 2020, 877, 114503 CrossRef CAS.
- T. S. Thanh, P. T. Qui, N. T. T. Tu, T. T. T. Toan, T. T. B. Hoa, L. V. T. Son, D. M. Nguyen, T. N. Tuyen and D. Q. Khieu, Electrochemical determination of uric acid in urine by using zeolite imidazolate framework-11 modified electrode, J. Nanomater., 2021, 2021, 9914062 Search PubMed.
- M. Li, W. Guo, H. Li, W. Dai and B. Yang, Electrochemical biosensor based on one-dimensional MgO nanostructures for the simultaneous determination of ascorbic acid, dopamine, and uric acid, Sens. Actuators, B, 2014, 204, 629–636 CrossRef CAS.
- K. Ngamchuea, C. Batchelor-McAuley and R. G. Compton, Understanding electroanalytical measurements in authentic human saliva leading to the detection of salivary uric acid, Sens. Actuators, B, 2018, 262, 404–410 CrossRef CAS.
- K. Ngamchuea, S. Wannapaiboon, P. Nongkhunsan, P. Hirunsit and I. Fongkaew, Structural and Electrochemical Analysis of Copper-Creatinine Complexes: Application in Creatinine Detection, J. Electrochem. Soc., 2022, 169(2), 020567 CrossRef.
- K. Ngamchuea, C. Lin, C. Batchelor-McAuley and R. G. Compton, Supported microwires for electroanalysis: sensitive amperometric detection of reduced glutathione, Anal. Chem., 2017, 89(6), 3780–3786 CrossRef CAS PubMed.
- K. Chaisiwamongkhol, K. Ngamchuea, C. Batchelor-McAuley and R. G. Compton, Electrochemical detection and quantification of gingerol species in ginger (Zingiber officinale) using multiwalled carbon nanotube modified electrodes, Analyst, 2016, 141(22), 6321–6328 RSC.
- K. Ngamchuea, B. Tharat, P. Hirunsit and S. Suthirakun, Electrochemical oxidation of resorcinol: mechanistic insights from experimental and computational studies, RSC Adv., 2020, 10(47), 28454–28463 RSC.
- C. L. Sun, H. H. Lee, J. M. Yang and C. C. Wu, The simultaneous electrochemical detection of ascorbic acid, dopamine, and uric acid using graphene/size-selected Pt nanocomposites, Biosens. Bioelectron., 2011, 26(8), 3450–3455 CrossRef CAS PubMed.
- H. Wang, F. Ren, C. Wang, B. Yang, D. Bin, K. Zhang and Y. Du, Simultaneous determination of dopamine, uric acid and ascorbic acid using a glassy carbon electrode modified with reduced graphene oxide, RSC Adv., 2014, 4(51), 26895–26901 RSC.
- Y. Song, C. Gong, D. Su, Y. Shen, Y. Song and L. Wang, A novel ascorbic acid electrochemical sensor based on spherical MOF-5 arrayed on a three-dimensional porous carbon electrode, Anal. Methods, 2016, 8(10), 2290–2296 RSC.
- Z.-H. Sheng, X.-Q. Zheng, J.-Y. Xu, W.-J. Bao, F.-B. Wang and X.-H. Xia, Electrochemical sensor based on nitrogen doped graphene: simultaneous determination of ascorbic acid, dopamine and uric acid, Biosens. Bioelectron., 2012, 34(1), 125–131 CrossRef CAS PubMed.
- C. Xia and W. Ning, A novel bio-electrochemical ascorbic acid sensor modified with Cu4 (OH) 6SO4 nanorods, Analyst, 2011, 136(2), 288–292 RSC.
- S. Zhang, F. Xu, Z. Q. Liu, Y. S. Chen and Y. L. Luo, Novel electrochemical sensors from poly[N-(ferrocenyl formacyl) pyrrole]@multi-walled carbon nanotubes nanocomposites for simultaneous determination of ascorbic acid, dopamine and uric acid, Nanotechnology, 2019, 31(8), 085503 CrossRef PubMed.
- S. Qi, B. Zhao, H. Tang and X. Jiang, Determination of ascorbic acid, dopamine, and uric acid by a novel electrochemical sensor based on pristine graphene, Electrochim. Acta, 2015, 161, 395–402 CrossRef CAS.
- D. Zhao, G. Yu, K. Tian and C. Xu, A highly sensitive and stable electrochemical sensor for simultaneous detection towards ascorbic acid, dopamine, and uric acid based on the hierarchical nanoporous PtTi alloy, Biosens. Bioelectron., 2016, 82, 119–126 CrossRef CAS PubMed.
- C. Wang, J. Du, H. Wang, C. e. Zou, F. Jiang, P. Yang and Y. Du, A facile electrochemical sensor based on reduced graphene oxide and Au nanoplates modified glassy carbon electrode for simultaneous detection of ascorbic acid, dopamine and uric acid, Sens. Actuators, B, 2014, 204, 302–309 CrossRef CAS.
- C. Wang, J. Li, K. Shi, Q. Wang, X. Zhao, Z. Xiong, X. Zou and Y. Wang, Graphene coated by polydopamine/multi-walled carbon nanotubes modified electrode for highly selective detection of dopamine and uric acid in the presence of ascorbic acid, J. Electroanal. Chem., 2016, 770, 56–61 CrossRef CAS.
- K. Krishnamoorthy, V. Sudha, S. M. Senthil Kumar and R. Thangamuthu, Simultaneous determination of dopamine and uric acid using copper oxide nano-rice modified electrode, J. Alloys Compd., 2018, 748, 338–347 CrossRef CAS.
- S. M. Manocha, Porous carbons, Sadhana, 2003, 28(1–2), 335–348 CrossRef CAS.
- M. Xu, D. Li, Y. Yan, T. Guo, H. Pang and H. Xue, Porous high specific surface area-activated carbon with co-doping N, S and P for high-performance supercapacitors, RSC Adv., 2017, 7(69), 43780–43788 RSC.
- F. Momtazan, A. Vafaei, M. Ghaedi, A. M. Ghaedi, D. Emadzadeh, W.-J. Lau and M. M. Baneshi, Application of copper sulfide nanoparticles loaded activated carbon for simultaneous adsorption of ternary dyes: Response surface methodology, Korean J. Chem. Eng., 2018, 35(5), 1108–1118 CrossRef CAS.
- A. Amić, Z. Marković, J. M. Dimitrić Marković, B. Lučić, V. Stepanić and D. Amić, The 2H+/2e− free radical scavenging mechanisms of uric acid: thermodynamics of NH bond cleavage, Comput. Theor. Chem., 2016, 1077, 2–10 CrossRef.
- B. N. Ames, R. Cathcart, E. Schwiers and P. Hochstein, Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis, Proc. Natl. Acad. Sci., 1981, 78(11), 6858–6862 CrossRef CAS PubMed.
- C. Batchelor-McAuley, M. Yang, E. M. Hall and R. G. Compton, Correction factors for the analysis of voltammetric peak currents measured using staircase voltammetry, J. Electroanal. Chem., 2015, 758, 1–6 CrossRef CAS.
- A. P. Brown and F. C. Anson, Cyclic and differential pulse voltammetric behavior of reactants confined to the electrode surface, Anal. Chem., 1977, 49(11), 1589–1595 CrossRef CAS.
- G. Jiang, J. Wang, Y. Yang, G. Zhang, Y. Liu, H. Lin, G. Zhang, Y. Li and X. Fan, Fluorescent turn-on sensing of bacterial lipopolysaccharide in artificial urine sample with sensitivity down to nanomolar by tetraphenylethylene based aggregation induced emission molecule, Biosens. Bioelectron., 2016, 85, 62–67 CrossRef CAS PubMed.
- M. H. Naveen, N. G. Gurudatt and Y.-B. Shim, Applications of conducting polymer composites to electrochemical sensors: A review, Appl. Mater. Today, 2017, 9, 419–433 CrossRef.
- K. Khoshnevisan, H. Maleki, E. Honarvarfard, H. Baharifar, M. Gholami, F. Faridbod, B. Larijani, R. Faridi Majidi and M. R. Khorramizadeh, Nanomaterial based electrochemical sensing of the biomarker serotonin: a comprehensive review, Microchim. Acta, 2019, 186(1), 1–21 CrossRef CAS PubMed.
- J. Liu and Y. Cao, An electrochemical sensor based on an anti-fouling membrane for the determination of histamine in fish samples, Anal. Methods, 2021, 13(5), 685–694 RSC.
- N. F. Atta, A. Galal, S. M. Ali and D. M. El-Said, Improved host–guest electrochemical sensing of dopamine in the presence of ascorbic and uric acids in a β-cyclodextrin/Nafion®/polymer nanocomposite, Anal. Methods, 2014, 6(15), 5962–5971 RSC.
- Z. Xu, M.-q. Zhang, H.-q. Zou, J.-s. Liu, D.-z. Wang, J. Wang and L.-d. Wang, Non-enzymatic electrochemical detection of uric acid with electrodeposited Nafion film, J. Electroanal. Chem., 2019, 841, 129–134 CrossRef CAS.
- N. Tukimin, J. Abdullah and Y. Sulaiman, Electrodeposition of poly(3,4-ethylenedioxythiophene)/reduced graphene oxide/manganese dioxide for simultaneous detection of uric acid, dopamine and ascorbic acid, J. Electroanal. Chem., 2018, 820, 74–81 CrossRef CAS.
- H. Jeong and S. Jeon, Determination of dopamine in the presence of ascorbic acid by nafion and single-walled carbon nanotube film modified on carbon fiber microelectrode, Sensors, 2008, 8(11), 6924–6935 CrossRef CAS PubMed.
- C. L. Benn, P. Dua, R. Gurrell, P. Loudon, A. Pike, R. I. Storer and C. Vangjeli, Physiology of hyperuricemia and urate-lowering treatments, Front. Med., 2018, 160 CrossRef PubMed.
- M. Kaya, Ö. Şahin and C. Saka, Preparation and TG/DTG, FT-IR, SEM, BET surface area, iodine number and methylene blue number analysis of activated carbon from pistachio shells by chemical activation, Int. J. Chem. React. Eng., 2018, 16(2), 20170060 CAS.
- K. Kaewket, S. Maensiri and K. Ngamchuea, Adsorptive stripping voltammetry at microporous carbon: Determination and adsorption characteristics of environmental contaminants, Colloid Interface Sci. Commun., 2020, 38, 100310 CrossRef CAS.
- D. Dollimore and G. Heal, An improved method for the calculation of pore size distribution from adsorption data, J. Appl. Chem., 1964, 14(3), 109–114 CrossRef CAS.
- N. Mojoudi, N. Mirghaffari, M. Soleimani, H. Shariatmadari, C. Belver and J. Bedia, Phenol adsorption on high microporous activated carbons prepared from oily sludge: equilibrium, kinetic and thermodynamic studies, Sci. Rep., 2019, 9(1), 19352 CrossRef CAS PubMed.
- Y. Zhao, C.-W. Cho, L. Cui, W. Wei, J. Cai, G. Wu and Y.-S. Yun, Adsorptive removal of endocrine-disrupting compounds and a pharmaceutical using activated charcoal from aqueous solution: kinetics, equilibrium, and mechanism studies, Environ. Sci. Pollut. Res., 2019, 26(33), 33897–33905 CrossRef CAS PubMed.
- J. Shu, S. Cheng, H. Xia, L. Zhang, J. Peng, C. Li and S. Zhang, Copper loaded on activated carbon as an efficient adsorbent for removal of methylene blue, RSC Adv., 2017, 7(24), 14395–14405 RSC.
- R. H. Hesas, A. Arami-Niya, W. M. A. W. Daud and J. Sahu, Preparation and characterization of activated carbon from apple waste by microwave-assisted phosphoric acid activation: application in methylene blue adsorption, BioResources, 2013, 8(2), 2950–2966 Search PubMed.
- M. Noroozifar, M. Khorasani-Motlagh, F. Z. Jahromi and S. Rostami, Sensitive and selective determination of uric acid in real samples by modified glassy carbon electrode with holmium fluoride nanoparticles/multi-walled carbon nanotube as a new biosensor, Sens. Actuators, B, 2013, 188, 65–72 CrossRef CAS.
- G. Dryhurst, Electrochemical oxidation of uric acid and xanthine at the pyrolytic graphite electrode mechanistic interpretation of electrochemistry, J. Electrochem. Soc., 1972, 119(12), 1659–1664 CrossRef CAS.
- R. Guidelli, R. G. Compton, J. M. Feliu, E. Gileadi, J. Lipkowski, W. Schmickler and S. Trasatti, Defining the transfer coefficient in electrochemistry: An assessment (IUPAC Technical Report), Pure Appl. Chem., 2014, 86(2), 245–258 CrossRef CAS.
- D. Li, C. Lin, C. Batchelor-McAuley, L. Chen and R. G. Compton, Tafel analysis in practice, J. Electroanal. Chem., 2018, 826, 117–124 CrossRef CAS.
- R. Gupta and V. Ganesan, Gold nanoparticles impregnated mesoporous silica spheres for simultaneous and selective determination of uric acid and ascorbic acid, Sens. Actuators, B, 2015, 219, 139–145 CrossRef CAS.
- R. G. Compton and C. E. Banks, Understanding voltammetry, World Scientific, 2018 Search PubMed.
- K. Ngamchuea, K. Tschulik, S. Eloul and R. G. Compton, In situ detection of particle aggregation on electrode surfaces, ChemPhysChem, 2015, 16(11), 2338–2347 CrossRef CAS PubMed.
- M. L. Brigden, D. Edgell, M. McPherson, A. Leadbeater and G. Hoag, High incidence of significant urinary ascorbic acid concentrations in a west coast population-implications for routine urinalysis, Clin. Chem., 1992, 38(3), 426–431 CrossRef CAS.
- S. Dalirirad and A. J. Steckl, Lateral flow assay using aptamer-based sensing for on-site detection of dopamine in urine, Anal. Biochem., 2020, 596, 113637 CrossRef CAS PubMed.
- J. A. Buledi, S. Ameen, S. A. Memon, A. Fatima, A. R. Solangi, A. Mallah, F. Karimi, S. Malakmohammadi, S. Agarwal and V. K. Gupta, An improved non-enzymatic electrochemical sensor amplified with CuO nanostructures for sensitive determination of uric acid, Open Chem., 2021, 19(1), 481–491 CrossRef CAS.
- I. Guelcin, M. Oktay, E. Koeksal, H. Serbetci, S. Beydemir and O. Kuefrevioglu, Antioxidant and radical scavenging activities of uric acid, Asian J. Chem., 2008, 20(3), 2079–2090 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03126d |
| This journal is © The Royal Society of Chemistry 2022 |