DOI:
10.1039/D2RA03226K
(Paper)
RSC Adv., 2022,
12, 34126-34141
Design, and synthesis of selectively anticancer 4-cyanophenyl substituted thiazol-2-ylhydrazones†
Received
23rd May 2022
, Accepted 18th November 2022
First published on 28th November 2022
Abstract
Cyclization of substituted thiosemicarbazones with α-bromo-4-cyanoacetophenone allows rapid single-step sustainable syntheses of 4-cyanophenyl-2-hydrazinylthiazoles libraries (30 examples, 66–79%). All show anticancer efficacy against HCT-116 and MCF-7 carcinoma cell lines with the majority being more active than cisplatin positive controls. The compounds 2-(2-(2-hydroxy-3-methylbenzylidene)hydrazinyl)-4-(4-cyanophenyl)thiazole (3f) and 2-(2-((pentafluorophenyl)methylene)-hydrazinyl)-4-(4-cyanophenyl)thiazole (3a′) show optimal GI50 values (1.0 ± 0.1 μM and 1.7 ± 0.3 μM) against MCF-7 breast cancer cells. Against colorectal carcinoma HCT-116 cells, (2-(2-(3-bromothiophen-2-yl)methylene)hydrazinyl)-4-(4-cyanophenyl)thiazole (3b′), 2-(2-(2-hydroxy-3-methylbenzylidene)hydrazinyl)-4-(4-cyanophenyl)thiazole (3f), 2-(2-(2,6-dichlorobenzylidene)hydrazinyl)-4-(4-cyanophenyl)thiazole (3n) and 2-(2-(1-(4-fluorophenyl)ethylidene)hydrazinyl)-4-(4-cyanophenyl)thiazole (3w) are the most active (GI50 values: 1.6 ± 0.2, 1.6 ± 0.1, 1.1 ± 0.5 and 1.5 ± 0.8 μM respectively). Control studies with MRC-5 cells indicate appreciable selectivity towards the cancer cells targeted. Significant (p < 0.005) growth inhibition and cytotoxicity effects for the thiazoles 3 were corroborated by cell count and clonogenic assays using the same cancer cell lines at 5 and 10 μM agent concentrations. Cell cycle, caspase activation and Western blot assays demonstrated that compounds 3b′ and 3f induce cancer cell death via caspase-dependent apoptosis. The combination of straight forward synthesis and high activity makes the thiazoles 3 an interesting lead for further development.
1. Introduction
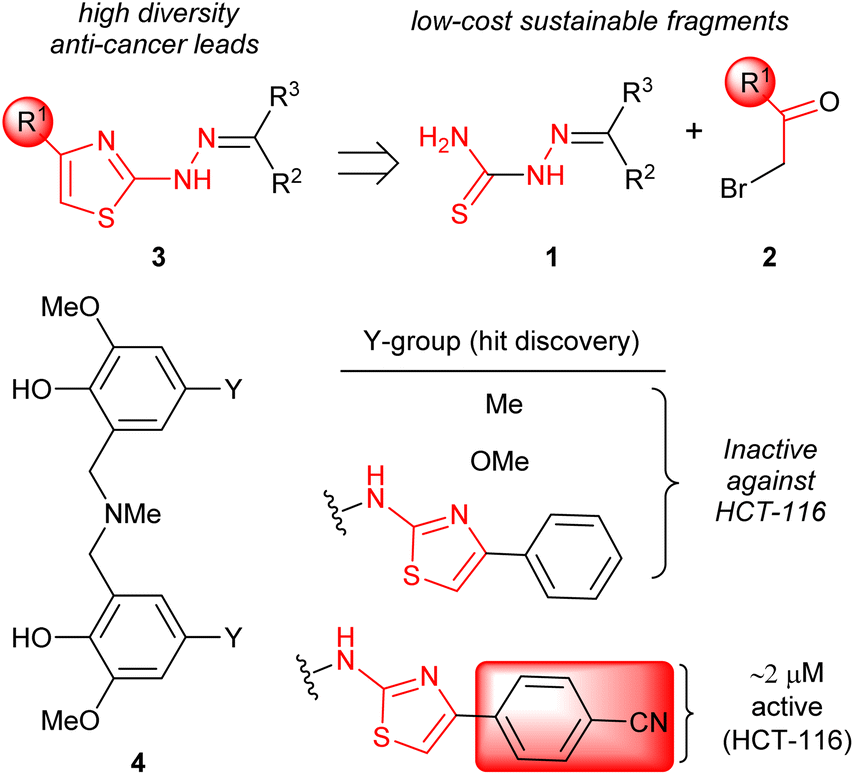
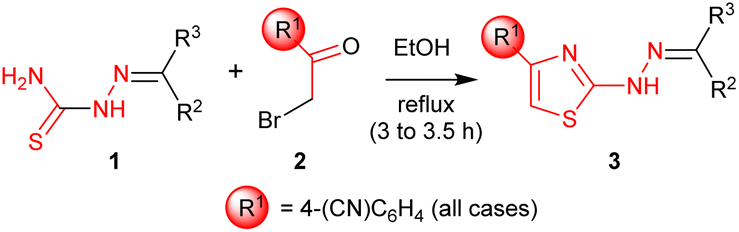
The attainment of affordable and sustainable cancer therapeutics is an emerging issue for both developing and other nations.1–3 Lead structures that can be realised in only a few synthetic steps, but still allow diverse structure–activity relationships to be defined are of high desirability in this area. Additionally, the use of only widely-available low-cost chemical building blocks from sustainable sources is also an enabling strategy. One such attractive fragment is the thiazole core (Scheme 1). Straightforward to prepare, their derivatives have already provided many disparate active lead compounds.4,5 The 4-substituted 2-hydrazinylthiazole species (3) are attractive, as these are easily prepared from ubiquitous thiosemicarbazones (1) and low cost α-bromoketones (2) (Scheme 1). In preliminary studies, associated with another programme,6 we had incorporated 2-hydrazinylthiazole units into the ligand (4). To our surprise the exemplar with a 4-cyanophenyl group (alone in the series) showed very high activity against the HCT-116 human colon cancer cell line, while all others had GI50 values > 100 μM (Scheme 1).
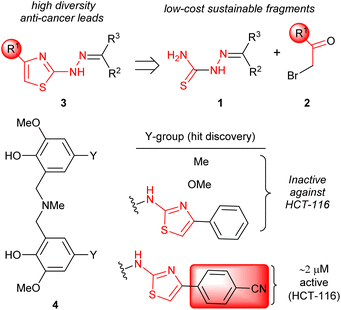 |
| | Scheme 1 Accessing 2-hydrazinylthiazoles (3) and initial hit discovery. | |
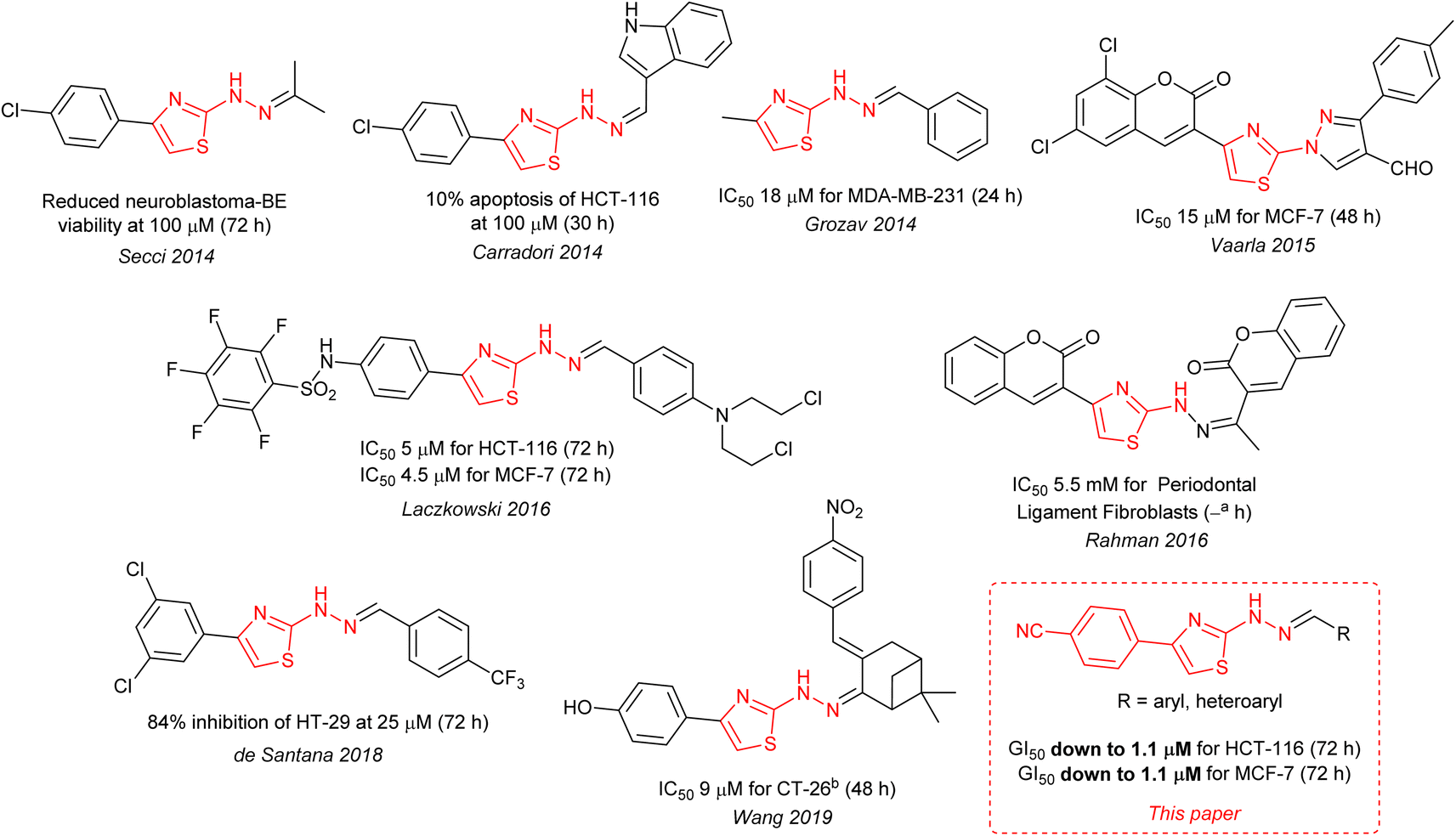
The ∼2 μM activity of the 4-cyanophenyl derivative of 4 is unusual. The anti-cancer activities of 2-hydrazinylthiazoles (3) are normally quite modest (see Fig. 1). In 2014 Secci and co-workers proposed that pharmacophores of type 3 are histone acetyltransferase (HAT) inhibitors that facilitate the onset of apoptosis in HCT-116.7,8
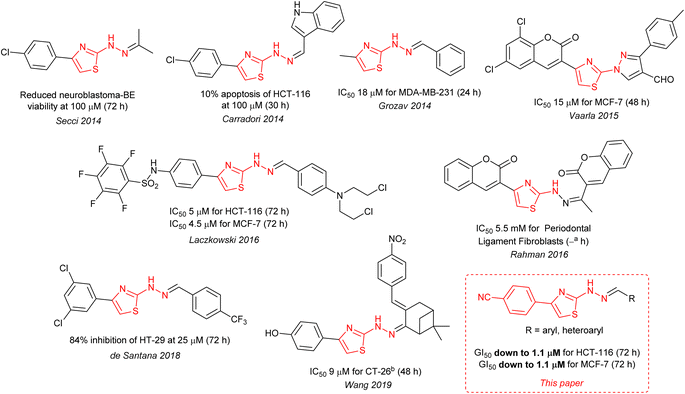 |
| | Fig. 1 Optimal literature8–12 cytotoxic activities for 2-hydrazinylthiazoles (3), and one closely related compound, compared to the present work. Notes: atime period not given; bmouse model, all other cell lines are human derived. | |
The Secci study activities are low (only 10% apoptosis at 100 μM).7,8 However, in the subsequent 8 years a significant range of additional thiazole derivatives (ca. 80 compounds) have been prepared by various groups, the most active of which are summarised in Fig. 1.9–13 Although some improved activities are present in this global library, over the original Secci lead, the most active compounds are still at best only equipotent to cisplatin and related positive controls. We noticed from both thiazole and related literature that incorporation of cyano functionality onto thiazole and other heterocycles, in many cases, enhances anticancer potency.14–16 Although the compounds 4 suggest a clear (anti-cancer) benefit from 4-cyanophenyl inclusion within the 2-hydrazinylthiazole derivative 4, this series was synthetically problematic to work with. Therefore, as part of our ongoing research,17 we set out to synthesise a range of more simply and easily accessed 2-hydrazinylthiazole species (3), all bearing a cyanoaryl moiety at position R1 (Scheme 1), to confirm the generality of the apparent activity effect detected.
2. Experimental
2.1. General
All reagents used in synthesis and biological evaluation were commercial (Merck, Fischer and Acros Organics) and were used as supplied. Initial thiosemicarbazones (1) are readily available either commercially and via simple one-step literature procedures (see ref. 17–25). Chromatographic silica gel 60 (220–240 mesh) was obtained from Merck. Thin layer chromatography was performed on foil-backed plates coated with Merck Silica gel 60 F254. Plates were developed using ultraviolet light and basic aqueous potassium permanganate. Uncorrected melting points were measured on a DMP-300 A&E Lab apparatus. Ultraviolet (UV) absorption spectra were recorded on Shimadzu Ultraviolet-1800 spectrophotometer in the range 200–800 nm. Infrared spectra were recorded on a Bruker OPUS FT-IR spectrometer by attenuated total reflection (diamond-ATR) on solid films. Proton and 13C NMR spectra were recorded on Bruker DPX-400 and 500 MHz spectrometers at ambient temperatures; where necessary HSQC and HMBC techniques were used to confirm structural connectivities. Mass spectra were recorded on Bruker Micro TOF-ESI positive targeted mode. Elemental (CHN) analyses were conducted on a CE-440 Elemental Analyzer.
2.2. Synthesis of 2-(2-benzylidenehydrazinyl)-4-(4-cyanophenyl) thiazoles (3a–d′)
Syntheses of (3a–d′) were achieved by refluxing equimolar mixtures of the required thiosemicarbazone (1) (1.00 mmol) and α-bromo-4-cyanoacetophenone (1.00 mmol) in absolute ethanol (20 mL) for 3 to 3.5 hours. The progress of reaction was monitored by TLC using acetone/n-hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), if necessary, reflux times were extended to 5 h; the Rf values for individual compounds are given below. On completion the reaction mixture was allowed to cool to room temperature and poured on crushed ice (ca. 20 g). The precipitated solids were filtered off, washed with copious water and dried under vacuum to afford 3a–d′. Spectral data as described below. Purity estimates are based on expected %C values from elemental analyses.
:3), if necessary, reflux times were extended to 5 h; the Rf values for individual compounds are given below. On completion the reaction mixture was allowed to cool to room temperature and poured on crushed ice (ca. 20 g). The precipitated solids were filtered off, washed with copious water and dried under vacuum to afford 3a–d′. Spectral data as described below. Purity estimates are based on expected %C values from elemental analyses.
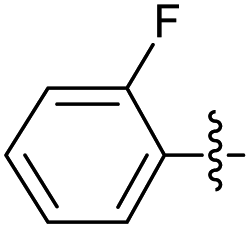
2.2.1 2-(2-(2-Fluorobenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3a). Off-yellow solid (69% yield), mp 230–232 °C, Rf 0.43 (acetone/n-hexane, 1:3), λmax 339 nm, IR (ATR) νmax 3257 (N–H), 3116 (C–H), 2218 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1606 (thiazole), 1562 (C
N), 1606 (thiazole), 1562 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1487, 1435, 1357, 1330 (CC), 1053–717 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, CDCl3) δ (ppm) 8.14 (s, 1H, HCN), 7.97–7.89 (m, 3H, ArH), 7.73 (d, J = 8.3 Hz, 2H, ArH), 7.41 (d, J = 6.7 Hz, 1H, ArH), 7.23 (app. t, J = 7.7 Hz, 1H, ArH), 7.12 (app. t, J = 9.3 Hz, 1H, ArH), 7.05 (s, 1H, thiazole-H), 13C NMR (100 MHz, CDCl3) δ 168.9 (thiazole C2), 161.4 (d, 1JCF = 216.8 Hz, Aripso), 146.3 (thiazole C4), 138.5 (CN), 136.2, 132.9, 132.1, 126.7, 126.4, 124.6, 121.1 (Ar), 118.6 (CN), 116.0 (d, 2JCF = 20.9 Hz, Ar), 112.1 (Aripso), 106.1 (thiazole C5), not all signals could be observed due to JCF couplings and low compound solubility, 19F NMR (375 MHz, CDCl3) δ −120.03, HRMS m/z 323.0757 [M + H]+ calcd for C17H11FN4S: 323.0766. Anal. for 3a, as isolated, C17H11FN4S: C, 66.34; H, 3.44; N, 17.38. Found: C, 67.39; H, 3.70; N, 17.43, corresponding to 98% purity.
N), 1487, 1435, 1357, 1330 (CC), 1053–717 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, CDCl3) δ (ppm) 8.14 (s, 1H, HCN), 7.97–7.89 (m, 3H, ArH), 7.73 (d, J = 8.3 Hz, 2H, ArH), 7.41 (d, J = 6.7 Hz, 1H, ArH), 7.23 (app. t, J = 7.7 Hz, 1H, ArH), 7.12 (app. t, J = 9.3 Hz, 1H, ArH), 7.05 (s, 1H, thiazole-H), 13C NMR (100 MHz, CDCl3) δ 168.9 (thiazole C2), 161.4 (d, 1JCF = 216.8 Hz, Aripso), 146.3 (thiazole C4), 138.5 (CN), 136.2, 132.9, 132.1, 126.7, 126.4, 124.6, 121.1 (Ar), 118.6 (CN), 116.0 (d, 2JCF = 20.9 Hz, Ar), 112.1 (Aripso), 106.1 (thiazole C5), not all signals could be observed due to JCF couplings and low compound solubility, 19F NMR (375 MHz, CDCl3) δ −120.03, HRMS m/z 323.0757 [M + H]+ calcd for C17H11FN4S: 323.0766. Anal. for 3a, as isolated, C17H11FN4S: C, 66.34; H, 3.44; N, 17.38. Found: C, 67.39; H, 3.70; N, 17.43, corresponding to 98% purity.
2.2.2 2-(2-(3-Fluorobenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3b). Yellow solid (79% yield), mp 177–178 °C, Rf 0.44 (acetone/n-hexane, 1:3), λmax 337 nm, IR (ATR) νmax 3278 (N–H), 3126 (C–H) 2218 (CN), 1606 (thiazole), 1573 (CN), 1487, 1415, 1355, 1257 (CC), 1051–717 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H, NH), 8.05 (d, J = 2.5 Hz, 2H, ArH), 8.02 (s, 1H, HCN), 7.86 (d, J = 8.5 Hz, 2H, ArH), 7.65 (s, 1H, thiazole-H), 7.52–7.42 (m, 3H, ArH), 7.21 (app. tt, J = 9.2, 2.5 Hz, 1H, ArH), 13C NMR (100 MHz, DMSO-d6) δ 168.9 (thiazole C2), 162.9 (d, 1JCF = 243.6 Hz, Aripso), 149.4 (thiazole C4), 140.6, 139.2 (CN), 137.3 (d, 3JCF = 8.1 Hz, Ar), 133.2, 131.3 (d, 3JCF = 8.4 Hz, Ar), 126.6, 123.1 (d, 4JCF = 2.9 Hz, Ar), 119.5 (CN), 116.5 (d, 2JCF = 21.6 Hz, Ar), 112.7 (d, 2JCF = 22.7 Hz, Ar), 110.1 (Aripso), 108.3 (thiazole C5), 19F NMR (375 MHz, DMSO-d6) δ −112.80 (s, 1F), HRMS m/z 323.0759 [M + H]+ calcd for C17H11FN4S: 323.0766. Anal. for 3b, as isolated, C17H11FN4S: C, 63.34; H, 3.44; N, 17.38; found: C, 67.44; H, 3.59; N, 17.51%, corresponding to 95% purity.
2.2.3 2-(2-(1-(3-Bromophenyl)ethylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3c). Off-white solid (70% yield), mp 224–226 °C, Rf 0.56 (acetone/n-hexane, 1:3), λmax = 336 nm, IR (ATR) νmax 3305 (N–H), 3061, 2927 (C–H), 2223 (CN), 1604–1421 (thiazole), 1558 (CN), 1479, 1342, 1280, 1261 (CC), 1053–727 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, CDCl3) δ 7.94 (app d, J = 5.0 Hz, 2H, ArH), 7.91 (s, 1H, Ar–H), 7.72 (app d, J = 4.6 Hz, 2H, ArH), 7.69 (s, 1H, thiazole-H) 7.53 (d, J = 7.9 Hz, 1H, ArH), 7.31 (d, 1H, Ar–H, J = 7.9 Hz), 7.10 (s, 1H, ArH), 2.29 (s, 3H, CH3), 13C NMR (100 MHz, CDCl3) δ 169.1 (thiazole C2), 149.2 (thiazole C4), 139.4 (HCN), 138.5, 132.6, 132.1, 130.0, 129.0, 126.3, 124.5, 122.8 (Ar), 118.9 (CN), 111.1 (Aripso), 107.2 (thiazole C5), 12.9 (CH3), HRMS m/z 397.0117 [M + H]+ calcd for C18H13BrN4S 397.0122. Anal. for 3c, as isolated, calcd for C18H13BrN4S: C, 54.42; H, 3.30; N, 14.10. Found: C, 54.23; H, 3.07; N, 13.92%, corresponding to >99% purity.
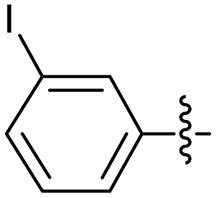
2.2.4 2-(2-(3-Iodobenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3d). Off-yellow solid (67% yield), mp 197–198 °C, Rf 0.41 (acetone/n-hexane, 1:3), λmax 341 nm, IR (ATR) νmax 3259 (N–H) overlapped by br 3600–2600 (N–HBr) 3041 (C–H), 2221 (CN), 1618 (thiazole), 1566 (CN), 1494, 1415, 1359, 1265 (CC), 1051–680 (characteristic of thiazole) cm−1, 1H NMR (500 MHz, DMSO-d6) δ 12.36 (s, 1H, NH), 8.03 (d, J = 8.5 Hz, 2H, Ar–H), 8.00 (s, 1H, HCN), 7.99 (s, 1H, ArH), 7.86 (d, J = 8.5 Hz, 2H, ArH), 7.72 (d, J = 8.0 Hz, 1H, ArH), 7.66 (d, J = 8.0 Hz, 1H, ArH), 7.65 (s, 1H, thiazole-H), 7.22 (app. t, J = 8.0 Hz, 1H, ArH), 13C NMR (125 MHz, DMSO-d6) δ 168.8 (thiazole C2), 149.3 (thiazole C4), 140.4, 139.1 (CN), 138.2, 137.1, 134.9, 133.2, 131.4, 128.9, 126.6, 126.1 (Aripso), 119.5 (CN), 110.1 (Aripso), 108.3 (thiazole C5), HRMS m/z 430.9841 [M + H]+ calcd for C17H11IN4S: 430.9827. Anal. for 3d·1/10HBr, as isolated, C17H11.1Br0.1IN4S: C, 46.58; H, 2.55; N, 12.78. Found: C, 46.43; H, 2.39; N, 12.74, corresponding to >99% for this salt.
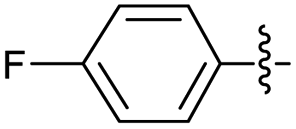
2.2.5 2-(2-(1-(4-Fluorophenyl)ethylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3e). Off-yellow solid (76% yield), mp 267–269 °C, Rf 0.62 (acetone/n-hexane, 1:3), λmax 332 nm, IR (ATR) νmax 3348 (N–H), 3116, 3041 (C–H), 2220 (CN), 1606–1408 (thiazole), 1558 (CN), 1508, 1346, 1280, 1213 (CC), 1053–702 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 11.35 (s, 1H, NH), 8.06 (d, J = 8.5 Hz, 2H, ArH), 7.87 (d, J = 8.5 Hz, 2H, Ar–H), 7.82 (dd, J = 9.0, 5.5 Hz, 2H, ArH), 7.65 (s, 1H, thiazole-H), 7.26 (app t, J = 9.0 Hz, 2H, ArH), 2.33 (s, 3H, CH3), 13C NMR (100 MHz, DMSO-d6) δ 170.6 (thiazole C2), 163.0 (d, 1JCF = 246.1 Aripso), 149.5 (thiazole C4), 146.4, 139.4 (imine CN), 134.8 (d, 4JCF = 2.9 Hz, Ar), 133.2, 128.3 (d, 3JCF = 8.4 Hz, Ar), 126.6 (Ar), 119.5 (CN), 115.9 (d, 2JCF = 21.6 Hz, Ar), 110.0 (Aripso), 108.5 (thiazole C5), 14.6 (CH3), 19F NMR (375 MHz, DMSO-d6) δ −113.09, HRMS m/z 337.0922 [M + H]+ calcd for C18H13FN4S 337.0923. Anal. for 3e, as isolated, calcd for C18H13FN4S: C, 65.27; H, 3.90; N, 16.66. Found: C, 67.67; H, 3.97; N, 16.86%, corresponding to >99% purity.
2.2.6 2-(2-(2-Hydroxy-3-methylbenzylidene) hydrazinyl)-4-(4-cyanophenyl) thiazole (3f). Light orange solid (73% yield) mp 273–275 °C; Rf 0.34 (acetone/n-hexane, 1:3), λmax 352 nm, IR (ATR) νmax 3298 (N–H), 3163 (O–H), 3165, 3041 (C–H), 2216 (CN), 1604–1431 (thiazole), 1573 (CN), 1483, 1357, 1249, 1147 (CC), 1083 (C–O), 1051–702 (characteristic of thiazole) cm−1, 1H NMR (500 MHz, DMSO-d6) δ 12.38 (s, 1H, NH), 10.28 (s, 1H, OH), 8.30 (s, 1H, HCN), 8.04 (d, J = 8.5 Hz, 2H, ArH), 7.87 (d, J = 8.5 Hz, 2H, ArH), 7.67 (s, 1H, thiazole-H), 7.30 (d, J = 7.8 Hz, 1H, ArH), 7.18 (d, J = 7.1 Hz, 1H, ArH), 6.85 (app t, J = 7.5 Hz, 1H, ArH), 2.23 (s, 3H, CH3), 13C NMR (125 MHz, DMSO-d6) δ 167.9 (thiazole C2), 155.1 (Aripso), 149.6 (thiazole C4), 144.9, 139.0 (CN), 133.2, 132.4, 127.8, 126.7, 125.5, 119.9, 119.4 (Ar), 118.6 (CN), 110.3 (Aripso), 107.7 (thiazole C5), 16.1 (CH3), HRMS m/z 335.0958 [M + H]+ calcd for C18H14N4OS: 335.0966. Anal. for 3f, as isolated, calcd for C18H14N4OS: C, 64.65; H, 4.22; N, 16.75. Found: C, 64.18; H, 4.21; N, 16.55%, corresponding to >99% purity.
2.2.7 2-(2-(5-Chloro-2-hydroxybenzylidene) hydrazinyl)-4-(4-cyanophenyl) thiazole (3g). Off-white solid (78% yield), mp 277–278 °C, Rf 0.33 (acetone/n-hexane, 1:3), λmax 344 nm, IR (ATR) νmax 3211 (O–H), 3192 (C–H), 3109 (N–H), 2229 (CN), 1608 (thiazole), 1575 (CN), 1479, 1382, 1327, 1288 (CC), 1182 (C–O), 1056–740 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.26 (br s, 1H, OH), 10.34 (br s, 1H, NH), 8.29 (s, 1H, HCN), 8.03 (d, J = 8.5 Hz, 2H, ArH), 7.86 (d, J = 8.5 Hz, 2H, ArH), 7.63 (s, 1H, thiazole-H), 7.62 (d, J = 2.8 Hz, 1H, ArH), 7.24 (dd, J = 8.7, 2.8 Hz, 1H, ArH), 6.93 (d, J = 8.7 Hz, 1H, ArH), 13C NMR (100 MHz, DMSO-d6) δ 168.7 (thiazole C2), 155.1 (Aripso), 149.2 (thiazole C4), 139.1 (CN), 138.0, 133.2, 130.4, 126.6, 125.1, 123.7, 122.6 (Ar), 119.4 (CN), 118.4 (Ar), 110.1 (Aripso), 108.0 (thiazole C5), HRMS m/z 355.0428 [M + H]+ calcd for C17H11ClN4OS: 355.0420. Anal. for 3g·1/3H2O, as isolated, C17H11.66N4O1.33S: C, 56.60; H, 3.26; N, 15.53. Found: C, 57.04; H, 2.93; N, 15.18%, corresponding to 99% purity for solvate.
2.2.8 2-(2-(5-Bromo-2-hydroxybenzylidene) hydrazinyl)-4-(4-cyanophenyl) thiazole (3h). Off-white solid (70% yield), mp 274–275 °C, Rf 0.32 (acetone/n-hexane, 1:3), λmax 352 nm, IR (ATR) νmax 3286 (br, O–H), 3115 (N–H), 3115 (C–H stretching), 2214 (CN), 1604 (thiazole), 1581 (CN), 1473, 1377, 1282, 1180 (CC), 1155 (C–O), 1053–698 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H, NH), 10.38 (s, 1H, OH), 8.28 (s, 1H, HCN), 8.04 (d, J = 8.4 Hz, 2H, ArH), 7.87 (d, J = 8.4 Hz, 2H, ArH), 7.76 (d, J = 2.6 Hz, 1H, ArH), 7.65 (s, 1H, thiazole-H), 7.36 (dd, J = 8.8, 2.6 Hz, 1H, ArH), 6.88 (d, J = 8.8 Hz, 1H, ArH), 13C NMR (100 MHz, DMSO-d6) δ 168.7 (thiazole C2), 155.5 (Aripso), 149.4 (thiazole C4), 139.2 (CN), 137.7, 133.2, 133.2, 127.9, 126.6, 123.2, 119.5 (Ar), 118.9 (CN), 111.3 (Aripso), 110.1 (Aripso), 108.1 (thiazole C5), HRMS m/z 398.9903 [M + H]+ calcd for C17H11BrN4OS: 398.9915. Anal. for 3p, as isolated, calcd for C17H11BrN4OS: C, 51.14; H, 2.78; N, 14.03. Found: C, 51.03; H, 2.65; N, 13.73%, corresponding to >99% purity.
2.2.9 2-(2-(2,5-Dimethylbenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3i). Off-yellow solid (75% yield) mp 187–188 °C, Rf 0.54 (acetone/n-hexane, 1:3), λmax 349 nm, IR (ATR) νmax 3302 (N–H), 2949 (C–H), 2233 (CN), 1604–1384 (thiazole), 1575 (CN), 1481, 1348, 1271, 1204 (CC), 1049–700 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.4 Hz, 2H, ArH), 7.72 (s, 1H, thiazole-H), 7.69 (d, 2H, J = 8.4 Hz, ArH), 7.32 (s, 1H, HCN), 7.09 (s, 1H, ArH), 7.09–7.01 (m, 2H, ArH), 2.36 (s, 3H, CH3), 2.12 (s, 3H, CH3), 13C NMR (100 MHz, CDCl3) δ 170.3 (thiazole C2), 148.9 (thiazole C4), 142.4, 138.5 (CN), 135.7, 133.6, 132.7, 131.2, 131.0, 130.5, 127.5, 126.4 (Ar), 118.8 (CN), 111.2 (Aripso), 106.8 (thiazole C5), 20.9 (CH3), 19.3 (CH3), HRMS m/z 333.1167 [M + Na]+ calcd for C19H16N4S: 333.1174, Anal. for 3i, as isolated, calcd for C19H16N4S: C, 68.65; H, 4.85; N, 16.85. Found: C, 68.54; H, 4.61; N, 16.69%, corresponding to >99% purity.
2.2.10 2-(2-(2,5-Dimethoxybenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3j). Beige solid (72% yield), mp 158–159 °C, Rf 0.52 (acetone/n-hexane, 1:3), λmax 347 nm, IR (ATR) νmax 3263 (N–H), 3109, 2953 (C–H), 3100–2600 br (N–HBr salt and C–H overlapped), 2221 (CN), 1604–1427 (thiazole), 1564 (CN), 1496, 1278 (CC), 1083 (C–O), 1051–702 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 8.34 (s, 1H, HCN), 8.03 (d, J = 8.4 Hz, 2H, ArH), 7.86 (d, J = 8.4 Hz, 2H, ArH), 7.63 (s, 1H, thiazole-H), 7.31 (d, J = 3.1 Hz, 1H, ArH), 7.03 (d, J = 9.0 Hz, 1H, ArH), 6.96 (dd, J = 9.0, 3.1 Hz, 1H, ArH), 3.80 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 13C NMR (100 MHz, DMSO-d6) δ 169.0 (thiazole C2), 153.7 (Aripso), 152.1 (Aripso), 149.3 (thiazole C4), 139.2 (CN), 137.4, 133.1, 126.6, 123.4 (Ar), 119.5 (CN), 116.7, 113.7, 110.0 (Ar), 109.8 (Aripso), 108.0 (thiazole C5), 56.6 (CH3), 55.8 (CH3), HRMS m/z 365.1058 [M + H]+ calcd for C19H16N4O2S: 365.1072. Anal. for 3j·1/4HBr, as isolated, calcd for C19H16.25Br0.25N4O2S: C, 59.33; H, 4.26; N, 14.57. Found: C, 59.54; H, 4.32; N, 14.16%, corresponding to >99% purity for this salt.
2.2.11 2-(2-(4-Methoxy-2-methylbenzylidene) hydrazinyl)-4-(4-cyanophenyl) thiazole (3k). Beige solid (65% yield), mp 240–242 °C, Rf 0.51 (acetone/n-hexane, 1:3), λmax 343 nm, IR (ATR) νmax 3269 (N–H), 3120, 2918 (C–H), 2223 (CN), 1606 (thiazole), 1566 (CN), 1504, 1448, 1411, 1359 (CC), 1170 (C–O), 1051–709 (characteristic of thiazole vibrations), 1H NMR (500 MHz, DMSO-d6) δ 12.04 (s, 1H, NH), 8.23 (s, 1H, HCN), 8.03 (d, J = 8.6 Hz, 2H, ArH), 7.86 (d, J = 8.6 Hz, 2H, ArH), 7.64 (d, J = 8.5 Hz, 1H, ArH), 7.59 (s, 1H, thiazole-H), 6.88–6.80 (m, 2H, ArH), 3.78 (s, 3H, OCH3), 2.43 (s, 3H, CH3), 13C NMR (125 MHz, DMSO-d6) δ 169.2 (thiazole C2), 160.3 (Aripso), 149.3 (thiazole C4), 141.6, 139.3 (CN), 138.4, 133.1, 128.5, 126.6, 125.4, 119.4 (CN), 116.5 (Ar), 112.6 (Aripso), 109.9 (Ar), 107.7 (thiazole C5), 55.6 (OCH3), 20.4 (CH3), HRMS m/z 349.1119 [M + H]+ calcd for C19H16N4OS: 349.1123. Anal. for 3k, as isolated, calcd for C19H16N4OS: C, 65.50; H, 4.63; N, 16.08. Found: C, 65.51; H, 4.56; N, 16.14%, corresponding to >99% purity.
2.2.12 2-(2-(2,4-Dimethoxybenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3l). Brick red solid (71% yield), mp 237–238 °C, Rf 0.53 (acetone/n-hexane, 1:3), λmax 345 nm, IR (ATR) νmax 3277 (N–H), 2972 (C–H), 2223 (CN), 1598 (thiazole), 1568 (CN), 1471, 1452, 1354, 1276 (CC), 1168 (C–O), 1051–707 (characteristic of thiazole) cm−1, 1H NMR (500 MHz, DMSO-d6) δ 12.03 (s, 1H, NH), 8.29 (s, 1H, HCN), 8.03 (d, J = 8.6 Hz, 2H, ArH), 7.86 (d, J = 8.6 Hz, 2H, ArH), 7.71 (d, J = 9.3 Hz, 1H, ArH), 7.59 (s, 1H, thiazole-H), 6.65–6.60 (m, 2H, ArH), 3.85 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 13C NMR (125 MHz, DMSO-d6) δ 169.1 (thiazole C2), 162.4 (Aripso), 158.9 (Aripso), 149.3 (thiazole C4), 139.3 (CN), 137.9, 133.1, 126.6, 126.5 (Ar), 119.5 (CN), 115.6, 109.9 (Aripso), 107.6 (thiazole C5), 106.9 (Ar), 98.7 (Ar), 56.2 (CH3), 55.9 (CH3), HRMS m/z 365.1062, [M + Na]+ calcd for C19H16N4O2S: 365.1072. Anal. for 3l, as isolated, calcd for C19H16N4O2S: C, 62.62; H, 4.43; N, 15.37. Found: C, 62.27; H,4.29; N, 15.09%, corresponding to >99% purity.
2.2.13 2-(2-(2,4-Bis-(trifluoromethyl) benzylidene) hydrazinyl)-4-(4-cyanophenyl) thiazole (3m). Off-yellow solid (66% yield), mp 253–255 °C, Rf 0.55 (acetone/n-hexane, 1:3), λmax 335 nm, IR (ATR) νmax 3317 (N–H), 3101 (C–H), 2218 (CN), 1504 (thiazole), 1562 (CN), 1344, 1269 (CC), 1083–742 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.78 (s, 1H, NH), 8.37 (d, J = 2.3 Hz, 1H, HCN), 8.29 (d, 1H, J = 8.5 Hz, ArH), 8.09 (d, J = 8.5 Hz, 1H, ArH), 8.03 (s, 1H, ArH), 8.01 (d, J = 8.5 Hz, 2H, ArH), 7.85 (d, J = 8.5 Hz, 2H, Ar–H), 7.72 (s, 1H, thiazole-H), 13C NMR (100 MHz, DMSO-d6) δ 168.2 (thiazole C2), 149.4 (thiazole C4), 138.9 (CN), 136.7, 135.2, 133.2, 132.8, 130.0, 128.8, 127.7, 126.6, 125.2 (Aripso), 123.6, 122.5, 119.4 (CN), 110.2 (Aripso), 109.1 (thiazole C5); not all signals could be observed due to multiple JCF couplings and low compound solubility, 19F NMR (375 MHz, DMSO-d6) δ −57.77 (s, 3F), −61.44 (s, 3F), HRMS m/z 441.0606 [M + H]+ calcd for C19H10F6N4S 441.0608. Anal. for 3h·1/2EtOH, as isolated, calcd for C20.33H14F6Cl2N4O0.66S: C, 51.84; H, 2.83; N, 12.09. Found: C, 51.60; H, 2.00; N, 11.46%, corresponding to >99% purity for this solvate. The residual solvent was easily removed under prolonged drying.
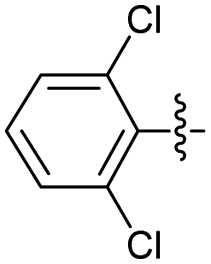
2.2.14 2-(2-(2,6-Dichlorobenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3n). Off yellow solid (68% yield), mp 250–251 °C, Rf 0.45 (acetone/n-hexane, 1:3), λmax 331 nm, IR (ATR) νmax 3278 (N–H), 3115 (C–H), 2216 (CN), 1604–1408 (thiazole), 1581 (CN), 1562, 1483, 1433, 1361 (CC), 1051–705 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.53 (s, 1H, NH), 8.29 (s, 1H, HCN), 8.03 (d, J = 8.5 Hz, 2H, ArH), 7.87 (d, J = 8.5 Hz, 2H, ArH), 7.65 (s, 1H, thiazole-H), 7.55 (d, J = 8.2 Hz, 2H, ArH), 7.39 (app. t, J = 7.5 Hz, 1H, ArH), 13C NMR (100 MHz, DMSO-d6) δ 168.8 (thiazole C2), 149.4 (thiazole C4), 139.1 (CN), 136.8 (Ar), 134.0 (Aripso), 133.2, 130.9, 130.0, 129.9, 126.6 (Ar), 119.5 (CN), 110.1 (Aripso), 108.6 (thiazole C5), HRMS m/z 373.0072 [M + Na]+ calcd for C17H10Cl2N4S 373.0081, Anal. for 3n, as isolated, calcd for C17H10Cl2N4S: C, 54.70; H, 2.70; N, 15.01. Found: C, 54.56; H, 2.59; N, 14.76%, corresponding to >99% purity.
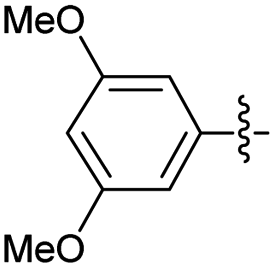
2.2.15 2-(2-(3,5-Dimethoxybenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3o). Off-yellow solid (75% yield), mp 235–237 °C, Rf 0.50 (acetone/n-hexane, 1:3), λmax 335 nm, IR (ATR) νmax 3304 (N–H), 3113, 2939 (C–H) overlapped by br (O–H), 2216 (CN), 1573 (thiazole), 1463 (CN), 1361, 1298 (CC), 1051–702 (characteristic of thiazole) cm−1, 1H NMR (500 MHz, DMSO-d6) δ 12.29 (s, 1H, NH), 8.04 (d, J = 8.6 Hz, 2H, ArH), 7.98 (s, 1H, HCN), 7.87 (d, J = 8.6 Hz, 2H, ArH), 7.65 (s, 1H, thiazole-H), 6.83 (d, J = 2.3 Hz, 2H, ArH), 6.53 (t, J = 2.3 Hz, 1H, ArH), 3.79 (s, 6H, OCH3), 13C NMR (125 MHz, DMSO-d6) δ 168.9 (thiazole C2), 161.2 (Aripso), 149.4 (thiazole C4), 141.9, 139.2 (CN), 136.7, 133.2, 126.6 (Ar), 119.5 (CN), 110.1 (Aripso), 108.2 (thiazole C5), 104.7 (Ar), 101.9 (Ar), 55.7 (CH3), HRMS m/z 365.1062 [M + Na]+ calcd for C19H16N4O2S: 365.1072. Anal. for 3o·1/2H2O, as isolated, C19H16.25Br0.25N4O2S: C, 59.33; H, 4.26; N, 14.57. Found: C, 59.54; H, 4.32; N, 14.16%, corresponding to >99% purity for this solvate.
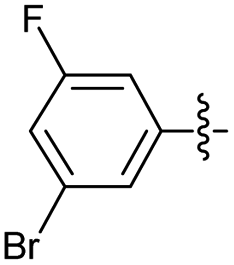
2.2.16 2-(2-(3-Bromo-5-fluorobenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3p). Off-yellow solid (70% yield), mp 246–247 °C, Rf 0.44 (acetone/n-hexane, 1:3), λmax 342 nm, IR (ATR) νmax 3300–2600 br (N–H and N–HBr), 3076 (C–H), 2225 (CN), 1629 (thiazole), 1595 (CN), 1568, 1494, 1400, 1265 (CC), 1053–756 (characteristic of thiazole) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 8.03 (s, 1H, HCN), 8.02 (d, J = 8.5 Hz, 2H, ArH), 7.86 (d, J = 8.5 Hz, 2H, ArH), 7.68 (app. s, 2H, thiazole and ArH), 7.50 (app. dd, J = 16.6, 9.0 Hz, 2H, ArH), 13C NMR (100 MHz, DMSO-d6) δ 168.7 (thiazole C2), 164.8 (d, 1JCF = 248.3 Hz, Aripso), 149.3 (thiazole C4), 139.0 (d, 3JCF = 8.8 Hz, Ar), 133.2 (CN), 132.8, 128.8, 126.6 (Ar), 125.5 (d, 4JCF = 2.9 Hz, Ar), 122.9 (d, 3JCF = 10.6 Hz, Ar), 119.6 (CN), 119.4 (d, 4JCF = 5.1 Hz, Ar) 112.3 (d, 2JCF = 23.1 Hz, Ar), 110.1 (Aripso), 108.6 (thiazole C5), 19F NMR (375 MHz, DMSO-d6) δ −110.20 (s, F), HRMS m/z 400.9866 [M + H]+ calcd for C17H10BrFN4S: 400.9872. Anal. for 3p·1/6HBr, as isolated, calcd for C17H10.16Br1.16FN4S: C, 49.24; H, 2.47; Br, 22.47; N, 13.51. Found: C, 49.57; H, 2.27; N, 13.10%, corresponding to 99% purity for this salt.
2.2.17 2-(2-(3-Fluoro-5-(trifluoromethyl)benzylidene) hydrazinyl)-4-(4-cyanophenyl) thiazole (3q). Off-yellow solid (78% yield), mp 233–235 °C, Rf 0.52 (acetone/n-hexane, 1:3), λmax 342 nm, IR (ATR) νmax 3286 (N–H), 3138 (C–H) 2218 (CN), 1604 (thiazole), 1558 (CN), 1471, 1450, 1367, 1340 (CC), 1055–705 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.59 (s, 1H, NH), 8.11 (s, 1H, HCN), 8.03 (d, J = 8.3 Hz, 2H, ArH), 7.86 (m, 3H, ArH), 7.77 (d, J = 9.7 Hz, 1H, ArH), 7.69 (s, 1H, thiazole-H), 7.66 (d, J = 8.7 Hz, 1H, ArH), 13C NMR (100 MHz, DMSO-d6) δ 168.7 (thiazole C2), 162.8 (d, 1JCF = 246.1 Hz, Aripso), 149.4 (thiazole C4), 139.3–138.4 (m, –CF3), 133.2, 126.6, 124.9, 122.3 (CN), 119.3 (d, 3JCF = 10.6 Hz, Ar), 116.8 (d, 2JCF = 22.7 Hz, Ar), 113.4 (d, 2JCF = 28.2 Hz, Ar), 110.1 (Aripso), 108.7 (thiazole C5), 19F NMR (375 MHz, DMSO-d6) δ (ppm) −61.48 (s, 1F, CF), −110.14 (s, 3F, CF3), HRMS m/z 391.0637 [M + H]+ calcd for C18H10F4N4S: 391.0640. Anal. for 3q, as isolated, calcd for C18H10F4N4S: C, 55.38; H, 2.58; N, 14.35. Found: C, 57.33; H, 2.56; N, 14.62%, corresponding to 97% purity.
2.2.18 2-(2-(3-Bromo-5-nitrobenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3r). Yellow solid (71% yield), mp 276–278 °C, Rf 0.42 (acetone/n-hexane, 1:3), λmax 335 nm, IR (ATR) νmax 3284 (N–H), 3089 (C–H), 2214 (CN), 1604 (thiazole), 1556 (CN), 1525 (NO), 1404 (CC), 1342 (NO), 1286 (CC), 1053–731 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.67 (s, 1H, NH), 8.46 (s, 1H, ArH), 8.32 (s, 1H, ArH), 8.25 (s, 1H, ArH), 8.13 (s, 1H, HCN), 8.04 (d, J = 8.4 Hz, 2H, ArH), 7.87 (d, J = 8.4 Hz, 2H, ArH), 7.71 (s, 1H, thiazole-H), 13C NMR (100 MHz, DMSO-d6) δ 168.5 (thiazole C2), 149.5 (thiazole C4, Aripso overlap), 139.1 (HCN), 138.5, 138.2, 135.0, 133.2, 126.6, 126.2, 122.9 (Ar), 119.8 (CN), 110.2 (Aripso), 108.8 (thiazole C5), HRMS m/z 427.9817 [M + H]+ calcd for C17H10BrN5O2S: 427.9817. Anal. for 3r, as isolated, C17H10BrN5O2S: C, 47.68; H, 2.35; N, 16.35. Found: C, 47.41; H, 2.15; N, 16.14%, corresponding to 99% purity.
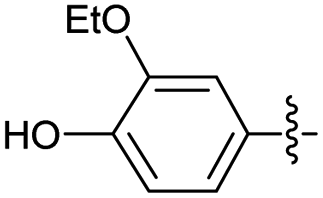
2.2.19 2-(2-(3-Ethoxy-4-hydroxybenzylidene) hydrazinyl)-4-(4-cyanophenyl) thiazole (3s). Brick red solid (76% yield), mp 223–225 °C, Rf 0.36 (acetone/n-hexane, 1:3), λmax 339 nm, IR (ATR) 3338 νmax (O–H), 3290 (N–H), 3122, 2991 (C–H), 2231 (CN), 1606 (thiazole), 1570 (CN), 1516, 1408, 1352, 1267 (CC), 1174 (C–O), 1047–711 (characteristic of thiazole) cm−1, 1H NMR (500 MHz, DMSO-d6) δ 12.04 (s, 1H, NH), 8.03 (d, J = 8.5 Hz, 2H, ArH), 7.95 (s, 1H, HCN), 7.86 (d, J = 8.5 Hz, 2H, ArH), 7.59 (s, 1H, thiazole-H), 7.22 (d, J = 2.0 Hz, 1H, ArH), 7.08 (dd, 1H, Ar–H, J = 8.2, 2.0 Hz, 1H, ArH), 6.85 (d, J = 8.2 Hz, 1H, ArH), 4.07 (q, J = 7.0 Hz, 2H, OCH2CH3), 1.37 (t, J = 7.0 Hz, 3H, OCH2CH3), 13C NMR (125 MHz, DMSO-d6) δ 169.2 (thiazole C2), 149.1 (Aripso), 147.6 (thiazole C4), 142.9 (Aripso), 139.3 (CN), 133.1, 126.6, 126.2, 120.9 (Ar), 119.5 (CN), 116.2 (Ar), 111.1 (Aripso), 109.9 (thiazole C5), 107.6 (Ar), 64.3 (CH2), 15.2 (CH3), HRMS m/z 365.1063 [M + H]+ calcd for C19H16N4O2S: 365.1072. Anal. for 3a′·1/5EtOH, as isolated, calcd for C19.4H17.2N4O2.2S: C, 62.36; H, 4.64; N, 15.00. Found: C, 61.58; H, 4.23; N, 14.65%, corresponding to 99% purity. The residual solvent could be removed only under prolonged drying.
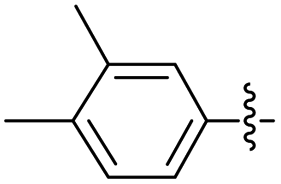
2.2.20 2-(2-(3,4-Dimethylbenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3t). Off-yellow solid (73% yield), mp 192–194 °C, Rf 0.46 (acetone/n-hexane, 1:3), λmax 335 nm, IR (ATR) νmax 3294 (N–H), 3115, 2924 (C–H), 2227 (CN), 1604 (thiazole), 1575 (CN), 1504, 1454, 1409, 1386 (CC), 1051–709 (characteristic of thiazole) cm−1, 1H NMR (500 MHz, DMSO-d6) δ 12.14 (s, 1H, NH), 8.03 (d, J = 8.4 Hz, 2H, ArH), 7.99 (s, 1H, HCN), 7.86 (d, J = 8.4 Hz, 2H, ArH), 7.62 (s, 1H, thiazole-H), 7.42 (s, 1H, ArH), 7.38 (d, J = 7.7 Hz, 1H, ArH), 7.19 (d, J = 7.8 Hz, 1H, ArH), 2.26 (s, 3H, CH3), 2.24 (s, 3H, CH3), 13C NMR (125 MHz, DMSO-d6) δ 169.0 (thiazole C2), 149.3 (thiazole C4), 142.5, 139.3 (CN), 138.4, 137.2, 133.2, 132.4, 130.4, 127.8, 126.6, 124.4 (Ar), 119.5 (CN), 110.0 (Aripso), 107.9 (thiazole C5), 19.9 (2× overlapping CH3), HRMS m/z 333.1167 [M + H]+ calcd for C19H16N4S: 333.1174. Anal. for 3t, as isolated, C19H16N4S: C, 68.65; H, 4.85; N, 16.85. Found: C, 68.33; H, 4.69; N, 16.52%, corresponding to >99% purity.
2.2.21 2-(2-(3,4-Dichlorobenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3u). Off-yellow solid (74% yield), mp 262–263 °C, Rf 0.48 (acetone/n-hexane, 1:3), λmax = 343 nm, IR (ATR) νmax 3113 (N–H), 3100–2600 br (N–HBr salt and C–H overlapped), 2225 (CN), 1629–1473 (thiazole), 1608 (CN), 1556, 1373, 1271, 1222 (CC), 1056–715 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 8.05–7.99 (m, 3H, ArH), 7.91 (d, J = 8.4 Hz, 2H, ArH), 7.86 (s, 1H, HCN), 7.84 (s, 1H, ArH), 7.67–7.61 (m, 3H, overlapping thiazole-H and ArH), 13C NMR (100 MHz, DMSO-d6) δ 168.7 (thiazole C2), 149.3 (thiazole C4), 139.3 (HCN), 135.6, 133.1, 132.8, 132.1, 132.1, 131.8, 131.5, 128.8, 126.6 (Ar), 119.4 (CN), 110.1 (Aripso), 108.4 (thiazole C5), HRMS m/z 373.0161 [M + H]+ calcd for C17H10Cl2N4S: 373.0081. Anal. analysis calcd (%) for 3u·1/2HBr, as isolated, calcd for C17H10.5Br0.5Cl2N4S2: C, 49.35; H, 2.56; N, 13.54; found: C, 49.28; H, 2.30; N, 13.77%, corresponding to >99% purity for this salt.
2.2.22 2-(2-(2,4,6-Trimethylbenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3v). Beige solid (68% yield), mp 221–223 °C, Rf 0.56 (acetone/n-hexane, 1:3), λmax 340 nm, IR (ATR) νmax 3298 (N–H), 2910 (C–H), 2218 (CN), 1604 (thiazole), 1571 (CN), 1479, 1411, 1382, 1332 (CC), 1049–717 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.08 (s, 1H, NH), 8.41 (s, 1H, HCN), 8.03 (d, J = 8.5 Hz, 2H, ArH), 7.86 (d, J = 8.5 Hz, 2H, ArH), 7.59 (s, 1H, thiazole-H), 6.92 (s, 2H, ArH), 2.42 (s, 6H, CH3), 2.25 (s, 3H, CH3), 13C NMR (100 MHz, DMSO-d6) δ 169.2 (thiazole C2), 149.4 (thiazole C4), 142.4, 139.3 (CN), 138.2, 137.3, 133.1, 130.1, 128.2, 126.6 (Ar), 119.5 (CN), 110.0 (Aripso), 107.7 (thiazole C5), 21.9 (CH3), 21.2 (CH3), HRMS m/z 347.1324 [M + H]+ calcd for C20H18N4S: 347.1330. Anal. for 3v, as isolated, calcd for C20H18N4S: C, 69.34; H, 5.24; N, 16.17. Found: C, 69.14; H, 5.11; N, 16.02%, corresponding to >99% purity.
2.2.23 2-(2-(2,4,6-Trifluorobenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3w). Yellow solid (72% yield), mp 270–271 °C, Rf 0.43 (acetone/n-hexane, 1:3), λmax 331 nm, IR (ATR) νmax 3273 (N–H), 3115 (C–H), 2216 (CN), 1606 (thiazole), 1573 (CN), 1490, 1427, 1344, 1288 (CC), 1035–738 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.40 (s, 1H, NH), 8.07 (s, 1H, HCN), 8.03 (d, J = 8.5 Hz, 2H, ArH), 7.86 (d, J = 8.5 Hz, 2H, ArH), 7.65 (s, 1H, thiazole-H), 7.29 (app. t, J = 9.0 Hz, 2H, ArH), 13C NMR (100 MHz, DMSO-d6) δ 168.6 (thiazole C2), 149.4 (thiazole C4), 133.2, 131.2, 126.6 (Ar), 119.4 (CN), 110.1 (Aripso), 108.6 (thiazole C5), 102.0 (Ar), not all signals could be observed due to JCF couplings and low compound solubility, 19F NMR (375 MHz, DMSO-d6) δ (ppm) −106.34 (app. d, J = 8.1 Hz, 1F), −109.73 (app. t, J = 8.1 Hz, 2F), HRMS m/z 359.0577 [M + H]+ calcd for C17H9F3N4S: 359.0578. Anal. for 3w, as isolated, C17H9F3N4S: C, 56.98; H, 2.53; N, 15.64. Found: C, 56.88; H, 2.4; N, 15.42%, corresponding to >99% purity.
2.2.24 2-(2-(2,4,6-Trihydroxybenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3x). Brick red solid (74% yield), mp 264–265 °C, Rf: 0.24 (acetone/n-hexane, 1:3), λmax 343 nm, IR (ATR) νmax 3113 (N–H) overlapped by br 3600–2600 (O–H, N–HBr and C–H), 2223 (CN), 1604 (thiazole), 1473 (CN), 1346, 1255 (CC), 1147 (C–O), 1047 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H, OH), 8.49 (s, 1H, HCN), 8.02 (d, J = 8.5 Hz, 2H, ArH), 7.86 (d, J = 8.5 Hz, 2H, ArH), 7.59 (s, 1H, thiazole-H), 5.89 (s, 2H, ArH), 13C NMR (100 MHz, DMSO-d6) δ 167.8 (thiazole C2), 161.6 (Aripso), 159.3 (Aripso), 149.4 (thiazole C4), 143.2, 139.0 (CN), 133.2, 126.7 (Ar), 119.4 (CN), 110.2 (Aripso), 106.9 (thiazole C5), 99.5 (Ar), 94.9 (Ar), HRMS m/z 353.0700 [M + H]+ calcd for C17H12N4O3S. Anal. for 3o·1/2EtOH·1/5HBr, as isolated, C18H15.2Br0.2N4O3.5S: C, 55.21; H, 3.91; N, 14.31. Found: C, 55.47; H, 3.48; N, 14.04%, corresponding to >99% purity for this composition. The ethanol was removed on prolonged drying, but not the HBr.
2.2.25 2-(2-(3,4,5-Trimethoxybenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3y). Orange solid (77% yield), mp 197–199 °C, Rf 0.50 (acetone/n-hexane, 1:3), λmax 347 nm, IR (ATR) νmax 3500–3000 br (N–H and N–HBr), 2940 (C–H), 2227 (CN), 1600 (thiazole), 1565 (CN), 1500, 1445, 1348, 1273 (CC), 1137 (C–O stretching), 1020–700 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 8.04 (s, 1H, HCN), 8.01 (d, J = 8.5 Hz, 2H, ArH), 7.86 (d, J = 8.5 Hz, 2H, ArH), 7.64 (s, 1H, thiazole-H), 6.98 (s, 2H, ArH), 3.83 (s, 6H, OCH3), 3.70 (s, 3H, OCH3), 13C NMR (100 MHz, DMSO-d6) δ 169.0 (thiazole C2), 153.7 (Aripso), 149.2 (thiazole C4), 146.4, 142.1 (Aripso), 139.2 (CN), 133.2, 132.9, 130.3, 128.8, 126.6 (Ar), 119.5 (CN), 110.0 (Aripso), 108.0 (thiazole C5), 104.0 (Ar), 60.6 (CH3), 56.4 (CH3), 56.3 (CH3), HRMS m/z 395.1177 [M + H]+ calcd for C20H18N4O3S: 395.1178. Anal. for 3y·1/5HBr, as isolated, calcd for C20H18.2Br0.2N4O3S: C, 58.50; H, 4.47; N, 13.64. Found: C, 59.19; H, 4.16; N, 13.16%, corresponding to 99% purity.
2.2.26 2-(2-(2,4,5-Trimethoxybenzylidene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3z). Dark grey solid (78% yield), mp 213–214 °C, Rf 0.49 (acetone/n-hexane, 1:3), λmax 353 nm, IR (ATR) νmax 3109 (N–H), 2935 (C–H) overlapped by (N–HBr), 2223 (CN), 1604 (thiazole), 1571 (CN), 1478, 1359, 1278, 1205 (CC), 1134 (C–O), 1024–738 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H, HCN), 7.90 (d, J = 8.5 Hz, 2H, ArH), 7.70 (d, J = 8.5 Hz, 2H, ArH), 7.40 (s, 1H, thiazole-H), 6.98 (s, 1H, ArH), 6.48 (s, 1H, ArH), 3.96 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 3.82 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ 168.9 (thiazole C2), 153.8 (Aripso), 152.7 (Aripso), 145.1 (Aripso), 143.7 (thiazole C4), 142.4, 135.7 (CN), 132.8, 126.3 (Ar), 118.6 (CN), 113.0, 112.0 (Ar), 108.2 (Aripso), 105.3 (thiazole C5), 96.5 (Ar), 56.4 (CH3), 56.3 (CH3), 56.1 (CH3), HRMS m/z 395.1171 [M + H]+ calcd for C20H18N4O3S: 395.1178 Anal. calcd (%) for 3z·1/5HBr, as isolated, C20H18.2Br0.2N4O3S: C, 58.50; H, 4.47; N, 13.64. Found: C, 58.55; H, 4.29; N, 13.48%, corresponding to >99% purity for this salt.
2.2.27 2-(2-((Pentafluorophenyl)methylene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3a′). Yellow solid (67% yield), mp 279–280 °C, Rf 0.40 (acetone/n-hexane, 1:3), λmax 335 nm, IR (ATR) νmax 3315 (N–H), 3130 (C–H), 2216 (CN), 1571 (CN), 1517, 1489, 1409, 1361 (CC), 1056–705 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.66 (s, 1H, NH), 8.05 (d, J = 8.4 Hz, 2H, ArH), 8.03 (s, 1H, HCN), 7.88 (d, J = 8.4 Hz, 2H, ArH), 7.71 (s, 1H, thiazole-H); 13C NMR (100 MHz, DMSO-d6) δ 142.1, 137.8 (CN), 133.2, 129.4, 126.6 (Ar), 119.4 (CN), 110.2 (Aripso), not all signals could be observed due to multiple JCF couplings and low compound solubility, 19F NMR (375 MHz, DMSO-d6) δ −143.17 (app. dd, 2F, J = 22.6, 7.3 Hz), −154.78 (app. t, 1F, J = 22.6 Hz), −162.85 (app. td, 2F, J = 22.6, 7.3 Hz), HRMS m/z 395.0383 [M + H]+ calcd for C17H7F5N4S: 395.0390. Anal. for 3a′, as isolated, calcd for C17H7F5N4S: C, 51.78; H, 1.79; N, 14.21. Found: C, 51.66; H, 1.74; N, 13.92, corresponding to >99 purity.
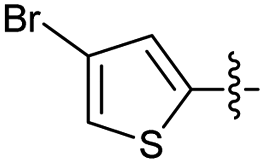
2.2.28 2-(2-((3-Bromothiophen-2-yl)methylene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3b′). Beige solid (73% yield), mp 223–225 °C, Rf 0.45 (acetone/n-hexane, 1:3), λmax 350 nm, IR (ATR) νmax 3174 (N–H), 3099 (C–H), 2227 (CN), 1608–1411 (thiazole), 1573 (CN), 1489, 1446, 1350, 1271 (CC), 1053–700 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6) δ 12.39 (s, 1H, NH), 8.22 (s, 1H, HCN), 8.03 (d, J = 8.5 Hz, 2H, Ar–H), 7.86 (d, J = 8.5 Hz, 2H, Ar–H), 7.70 (d, J = 5.4 Hz, 1H, thiophene-H), 7.65 (s, 1H, thiazole-H), 7.17 (d, J = 5.4 Hz, 1H, thiophene-H), 13C NMR (100 MHz, DMSO-d6) δ 168.3 (thiazole C2), 149.3 (thiazole C4), 139.1 (HCN), 135.4, 134.1, 133.2 (Ar), 131.2 (thiophene C2), 129.2 (thiophene C5), 126.6 (thiophene C4), 119.4 (CN), 112.1 (Aripso), 110.1 (thiophene C3), 108.4 (thiazole C5), HRMS m/z 388.9527, [M + H]+ calcd for C15H9BrN4S2 388.9530. Anal. for 3b′·1/6EtOH, as isolated, calcd for C15H10.33BrN4O0.67S2: C, 44.90; H, 2.60; N, 13.96. Found: C, 44.93; H, 2.35; N, 13.52%, corresponding to >99% purity for this solvate. The residual solvent was easily removed under drying.
2.2.29 2-(2-((4-Bromothiophen-2-yl)methylene)hydrazinyl)-4-(4-cyanophenyl) thiazole (3c′). Yellow solid (75% yield), mp 240–242 °C, Rf 0.48 (acetone/n-hexane, 1:3), λmax 348 nm, IR (ATR) νmax 3307 (N–H), 3107 (C–H), 2218 (CN), 1604 (thiazole), 1571 (CN), 1517, 1483, 1406, 1348 (CC), 1053–698 (characteristic of thiazole) cm−1, 1H NMR (500 MHz, DMSO-d6) δ 12.36 (s, 1H, NH), 8.19 (s, 1H, thiophene-H), 8.03 (d, J = 8.5 Hz, 2H, ArH), 7.87 (d, J = 8.5 Hz, 2H, ArH), 7.69 (s, 1H, thiophene-H), 7.65 (s, 1H, thiazole-H), 7.42 (s, 1H, HCN), 13C NMR (125 MHz, DMSO-d6) δ 168.4 (thiazole C2), 149.4 (thiazole C4), 141.0 (thiophene C2), 139.1 (CN), 135.9, 133.2 (Ar), 131.2 (thiophene C3), 126.6 (thiophene C5), 125.4 (Ar), 119.4 (CN), 110.1 (Aripso), 109.7 (thiopheneipso), 108.4 (thiazole C5), HRMS m/z 388.953 [M + H]+ calcd for C15H9BrN4S2: 388.9530. Anal. for 3c′, as isolated, C15H9BrN4S2: C, 46.28; H, 2.33; N, 14.39. Found: C, 46.16; H, 2.22; N, 14.13, corresponding to >99% purity.
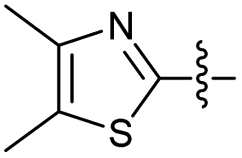
2.2.30 2-(2-((4,5-Dimethylthiazol-2-yl) methylene) hydrazinyl)-4-(4-cyanophenyl) thiazole (3d′). Turmeric yellow solid (69% yield), mp 262–263 °C, Rf 0.37 (acetone/n-hexane, 1:3), λmax 367 nm, IR (ATR) νmax 3174 (N–H), 3070, 2920 (C–H), 2223 (CN), 1606–1433 (thiazole), 1577 (CN), 1471, 1346, 1278, 1251 (CC), 1055–738 (characteristic of thiazole) cm−1, 1H NMR (400 MHz, DMSO-d6): δ 12.50 (s, 1H, NH), 8.08 (s, 1H, HCN), 8.01 (d, J = 8.4 Hz, 2H, ArH), 7.85 (d, J = 8.4 Hz, 2H, ArH), 7.64 (s, 1H, thiazole-H), 2.35 (s, 3H, CH3), 2.25 (s, 3H, CH3), 13C NMR (100 MHz, DMSO-d6) δ 149.7 (thiazole C4), 136.8 (HCN), 133.2, 130.0, 128.3, 126.6 (Ar), 119.4 (CN), 108.7 (thiazole C5), 14.9 (thiazole, C4–CH3), 11.7 (thiazole, C5–CH3), not all signals could be observed due to low compound solubility, HRMS m/z 340.0684 [M + H]+ calcd for C16H13N5S2: 340.0690. Anal. 3d′·1/6EtOH, as isolated, calcd for C16.33H14N5O0.17S2: C, 56.52; H, 4.07; N, 20.18. Found: C, 56.17; H, 3.71; N, 20.18%, corresponding to >99% purity for this solvate. The residual solvent could be removed only under prolonged drying.
2.3. Biological methods
2.3.1 Cell culture. The carcinoma cell lines used in anticancer biological experiments were from the American Type Tissue Cell Culture Collection (ATCC), cultivated in nutritional medium (RPMI 1640) supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) glutamine. Cell lines were passaged twice weekly and cultured at 37 °C in a 5% CO2 maintained incubator to maintain logarithmic growth. To decrease genotypic/phenotypic drift, ≤30 passaged cells used in the experiments. All compounds were dried prior to biological screening. Milligram quantities of cisplatin and compounds 3a–d′ were weighed using an ultra-micro balance (accurate to ±0.02 mg). Fresh stock solutions (10 mM) of trialled compounds and cisplatin controls were prepared in DMSO for each experiment, and all experiments were performed in triplicate. Colon cancer cell line (HCT-116), breast cancer cell line (MCF-7) and non-carcinogenic MRC-5 fibroblasts were used in the investigation.
2.3.2 MTT cell viability assay. MTT assays were used to evaluate the cell viability of HCT-116, MRC-5, and MCF7 cells, as described by Mosmann in 1983.26 In 96 well plates cells were seeded (180 μL per well). To allow exponential growth, cells were seeded at 3 × 103 cells per well and incubated for 24 h at 37 °C/5% CO2 prior to test agent introduction. From the stock solution, serial dilutions were made in nutrient medium to achieve final concentrations of 0.05 μM, 0.1 μM, 0.5 μM, 1 μM, 5 μM, 10 μM, 20 μM, and 50 μM, respectively. The T0 plate was read at the time of test agent addition, and the other plates after 72 h exposure to test agents. All plates were incubated for another 2.5 h following addition of 50 μL MTT solution (5 mg mL−1 PBS). The supernatant was removed from each well, and 150 μL DMSO was added. Depending on the number of viable cells, a colour change of varying intensity (purple colour) was detected. At a wavelength of 550 nm, absorbance was measured with a 2104 EnVision® multilabel plate reader. GI50 values (concentration required to achieve 50% growth inhibition) were calculated by interpolation, using standard procedures as detailed previously.6
2.3.3 Cell counts. While MTT assay is commonly used to assess cell viability and proliferation there are several drawbacks to this approach, such as false positive results caused by chemicals that may reduce the MTT (irrespective of any cellular process). Therefore a cell count test was used to confirm our MTT results. Cells were harvested and counted in the same way as for the MTT experiment, seeded at a density of 2 × 104 in 6-well plates. After being treated with test compounds at concentrations of 5 and 10 μM, the cells were grown for 72 h. A haemocytometer was used to count the cells.
2.3.4 Clonogenic assay. Clonogenic assays were carried out by seeding 400 cells per well in 6-well plates with 2 mL medium and allowing them to attach for 24 h. A haemocytometer was used to count the cells. Cells were treated with 3 at 5 and 10 μM concentrations, as guided by the initial MTT studies. The medium alone was used as a control. After 24 h of compound exposure, the medium was aspirated and washed with 1 mL of cold PBS. Fresh medium (2 mL) was added, and the cells were incubated at 37 °C. Daily inspection was conducted until colonies of ≥50 cells established in the control wells. Colonies were washed in PBS before being fixed in 100 percent methanol (0.5 mL) for 15 minutes. Finally, 0.7 mL of 0.05 percent methylene blue was used for staining (water:methanol 1:1). The number of cell colonies were counted.
2.3.5 Cell cycle assay. Cell cycle analyses were performed by the method reported by Nicoletti.27 In a 10 cm Petri dishes cells were seeded with 10 mL medium, density of 7 × 105 (control, 24 h) and 4 × 105 (48 and 72 h) cells per dish. After 24 h incubation at 37 °C cells were treated with desired concentrations of the synthesized compounds (5 and 10 μM). The medium and flocculent cells were transferred to a 15 mL Falcon tube after treatment period. The harvesting of cells was achieved by adding 0.5 mL of 1× trypsin EDTA solution. After cells had detached, they were added to the falcon tube and content were centrifuged at 1200 rpm for 5 min at 4 °C in a Beckman Coulter Allegro 33. Supernatant was removed and pellet was broken down ensuing 1 mL PBS addition and the tubes were vortexed and centrifuged twice and the cells were suspended in 0.7 mL cold hypotonic fluorochrome solution. In a labelled (FACS) fluorescence activated cell sorter tube cells were transferred and stored overnight at 4 °C in dark. To attain single cell suspensions the cells were vortexed. A total of 10000 events were recorded for each sample which were counted using a FC500 Beckman Coulter flow cytometer and the results were analysed by using WEASEL.6
2.3.6 Caspase-3/7 activity assay. Caspase-3/7 activity was determined by Apo-ONE® homogeneous caspase-3/7 assay (Promega).6 In 96 well opaque white cell culture plates, 3 × 103 per well cells were seeded and incubated at 37 °C for 24 h followed by addition of compounds 3b′ and 3f (i.e. 5 and 10 μM for 72 h exposure). Apo-ONE® caspase-3/7 reagent was added to each well, gently mixed at 300–500 rpm for 30 seconds followed by incubation for 30 min at room temperature. The EnVision multilabel plate reader (PerkinElmer) was used to measure the well fluorescence at wavelengths between 499 nm and 521 nm.
2.3.7 Western blot assay. Western blotting was carried out using method reported.28 Suitable 10 cm2 dishes cells were seeded at a density of 1–2 × 106 per dish and allowed to attach for 24 h prior to exposure to 5 μM and 10 μ test compounds. Cell lysates were prepared after the desired treatment period, protein concentrations were determined using the Bradford assay.29 SDS PAGE was used to separate the proteins (50 μg each sample) which were then transferred to a nitrocellulose membrane. Primary antibodies to PARP-1, and GAPDH were from Promega UK. Immunoblotting was used to detect the proteins, as previously described.30 ImageJ software was used for densitometric analyses.
3. Results and discussion
3.1. Synthesis of 4-substituted 2-hydrazinylthiazole species (3)
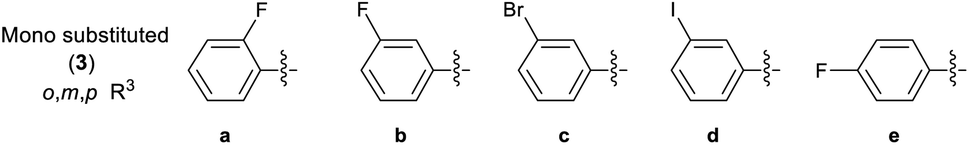
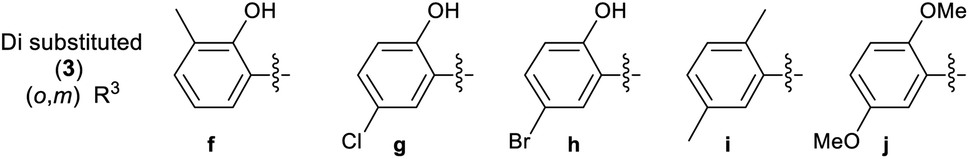
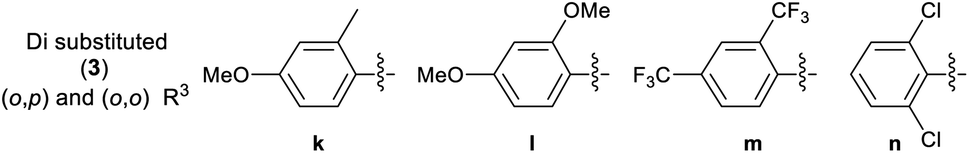
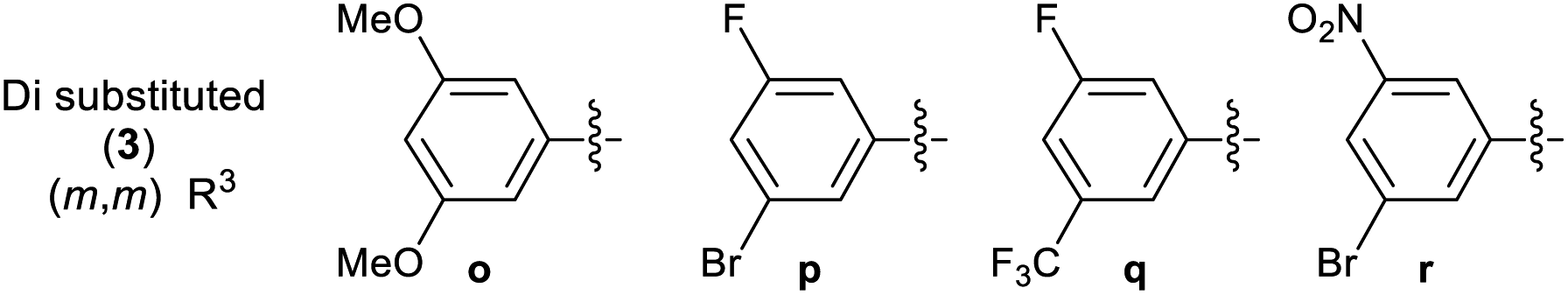
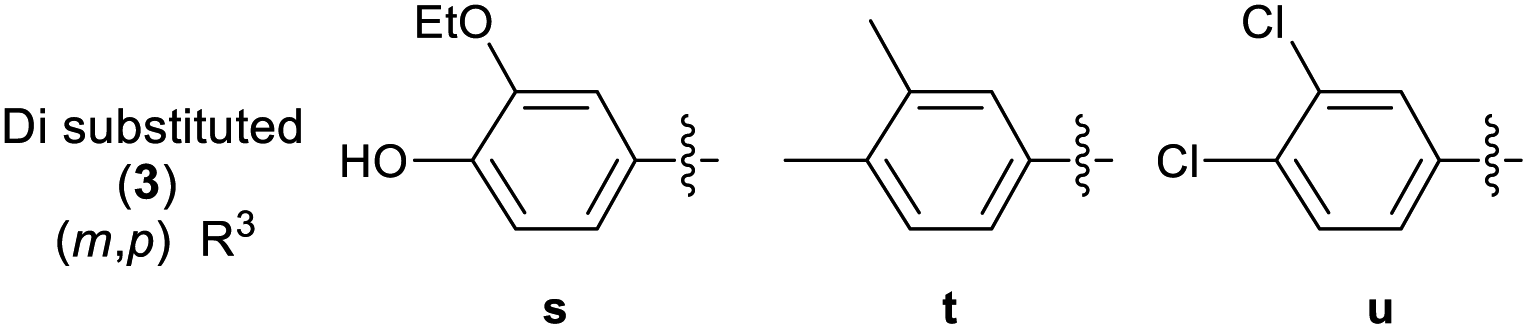
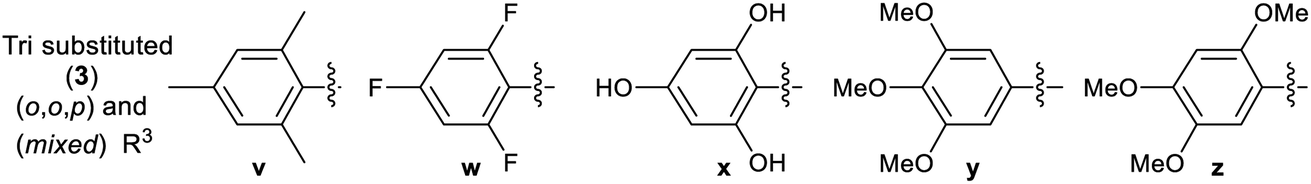
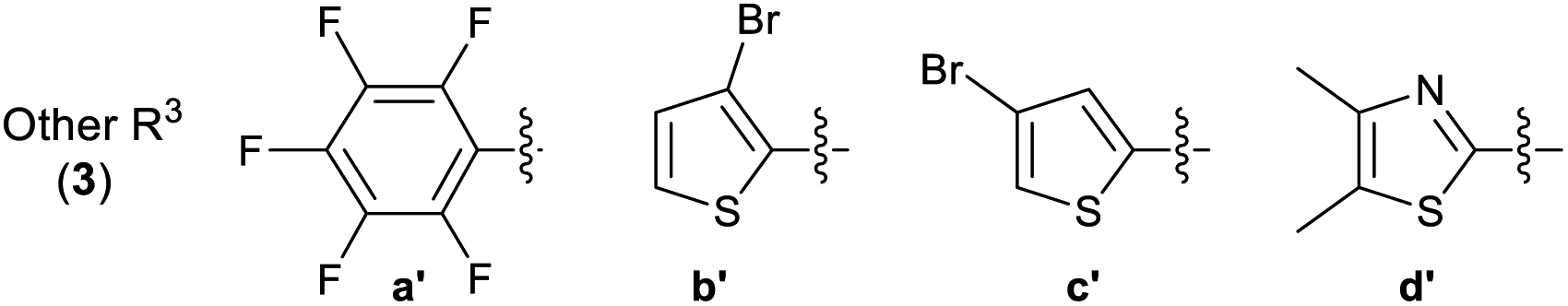
A library of 30 compounds was prepared using α-bromo-4-cyanoacetophenone (2, R1 = 4-C6H4CN in all cases) from a range of readily available thiosemicarbazones (1a–d′).17–25 Reflux of the reaction components in ethanol for 3–3.5 h afforded modest to good yields of 3a–d′ (Table 1) all of which could be isolated by simple filtration of directly precipitated products.
Table 1 4-Cyanophenyl-2-hydrazinylthiazole species (3) prepared in this study
Across the library of 30 thiazoles (3a–d′) the average purity was 98% with a standard deviation of 2%, based on elemental analysis data. Combustion analyses also revealed that traces of water/EtOH or low levels of product protonation by the HBr reaction by-products that fully accounted for the minor impurities. The HBr/H2O impurities can be seen by IR (where they cause a shallow broad νOH feature to develop 3600–2500 cm−1 due to extensive hydrogen bonding) but are best detected particularly by CHN combustion analysis. They are not easily quantified by NMR. All compounds 3 were of the desired formulation, except for 3u, where the hemi-HBr salt was the major product on the basis of combustion analysis. The latter compound was not further purified due to its low biological activity in preliminary studies (GI50 value 12.8 ± 0.7 μM and 27.2 ± 0.5 μM against MCF-7 and HCT-116 carcinoma cell lines, see later). It proved challenging to obtain accurate CHN combustion data on fluorine-containing compounds 3r, 3t and 3w, and this may reflect their resistance to combustion. The data presented represents the lower limit on their purity, which is still >95%. Issues associated with attaining high accuracy CHN analyses of F-containing organic compounds are well known.31 All compounds were extensively dried before use in biological assay.
Multinuclear NMR studies are also in line with isolated 3 analogues having purities of 95–98+% and confirm that proposed connectivity has been isolated. The observation of appropriate HRMS [M + H]+ also support the isolation of the desired targets. In all cases the presence of an azomethine linkage is confirmed by a 13C NMR signal in the range of 138.5–147.7 ppm. The presence of the hydrazone is evidenced by the presence of characteristic amine (3348–3174 cm−1) and imine (1629–1408 cm−1) stretches in the solid state IR spectra. In the latter case the band is sometimes overlapped by thiazole skeletal vibrations. 13C NMR spectroscopy is diagnostic of the substitution pattern in the thiazole: C2 appears in the range 167.8–170.6 ppm, C4 at 149.3–155.2 ppm while C5 appears at 105.3–109.9 ppm. Although well suited to aqueous biological studies at millimolar and lower concentrations, compounds 3 are poorly soluble in many organic solvents. Fully assigned 13C NMR spectra could be attained in most cases, but those 3 containing extensively fluorine-coupled carbons proved problematic, even for long spectral accumulation times. The 1H NMR spectra of 3a–d′ are unremarkable, but indicate the presence of the connectivity proposed in Table 1. By performing our analyses in DMSO-d6 we could slow NH exchange allowing this to also be observed at ca. 12.5 ppm. The studies proved that compounds with similar core exist in trans form,32,33 hence synthesized compounds should also exist in trans configuration.
3.2. Biological evaluation
3.2.1 MTT assayed anti-cancer activity. The growth inhibitory potential of 3a–d′ against the HCT-116 and MCF-7 cancer cell lines was determined by MTT assays. Potential growth inhibitory selectivity for carcinoma cell lines over normal cells was also probed using MRC-5 fibroblasts. Results of MTT assays were corroborated by cell count assays. Further, clonogenic assays were performed to determine cancer cell survival and retention of proliferative capacity after a brief challenge with test agent. The effects of the agents 3 on cell cycle perturbation and interrogation of apoptotic cell death were examined by flow cytometry, caspase activation and western blot assays. Growth inhibitory potential of 3a–d′ was compared with cisplatin as a positive control (Table 2). Using standard MTT methods,6 GI50 values (the concentration of test agent that inhibits cell growth by 50%) were calculated. The selectivity of compounds 3 towards cancer cell lines was determined by comparing their activity against normal human lung fibroblast (MRC-5) cells. Consequently, selectivity indices (SIs) were calculated for both cisplatin and the tested compounds 3.
Table 2 Activity of 2-(2-benzylidenehydrazinyl)-4-(4-cyanophenyl) thiazole (3a–d′) against MCF-7 and HCT-116 cancer cell lines and normal MRC-5 fibroblasts, as determined by MTT in vitro studiesa,b and SAR analysisc,d
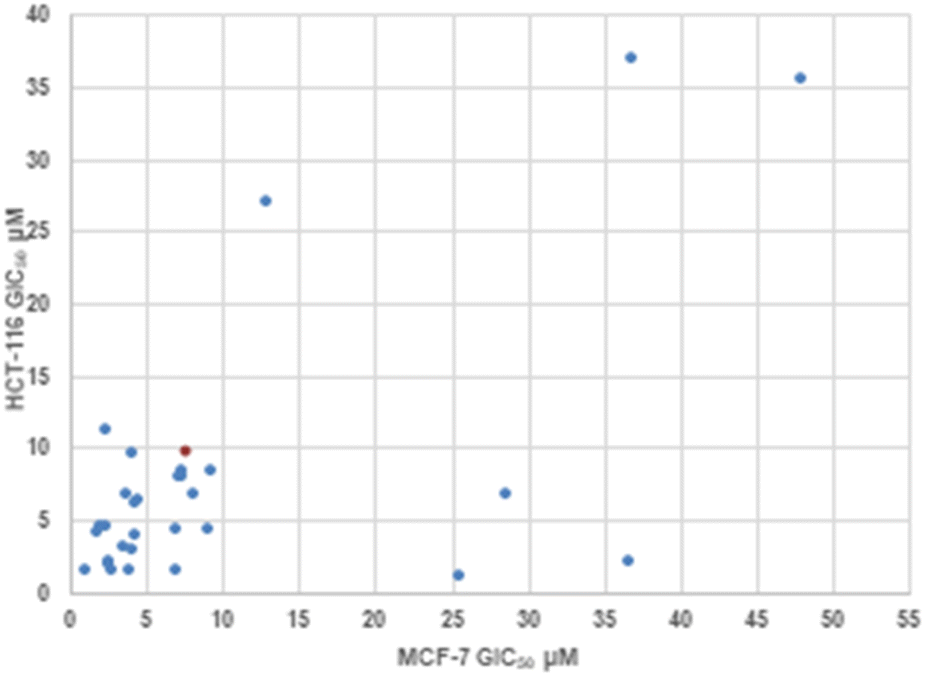
3.2.2 Effect of substituent R3 on the anti-cancer activity of 3 (SAR). Pleasingly from the array of compounds prepared, the majority of compounds were highly active in both cancer cell lines tested (Table 1 and Fig. 2). In fact, the majority of compound series 3 analogues are more active than the cisplatin positive control as cisplatin GI50 values of 7.5 ± 0.7 and 9.8 ± 0.2 μM against MCF-7 and HCT-116 cells respectively were recorded. The GI50 value of 9.7 μM against MRC-5 fibroblasts demonstrates the limited therapeutic window of cisplatin; cisplatin exhibited poor SIs of 0.9 and 1.3 for HCT-116 and MCF-7 respectively. The results (Table 2) indicated that only two compounds (3w and 3o) have appreciably greater activity against the MRC-5 cell line when compared with MCF-7 and HCT-116. Most other compounds are more selective towards carcinoma cell lines (3i is marginal). Compound 3f substituted with 2-hydroxy-3-methyl functionality was the most potent dual growth inhibitor from the library with GI50 values of 1.0 ± 0.1 μM and 1.6 ± 0.1 μM against MCF-7 and HCT-116 cells respectively, and due to its low molecular weight led to satisfactory ligand efficiency values for both cell lines. Compound 3f also demonstrated good selectivity with SI > 10 over MRC-5 fibroblasts. The clogP for this compound was calculated as 3.85, indicating the potential for good oral bioavailability.34,35 Pentafluorosubstituted compound 3a′ shows comparable potency to 3f against MCF-7 cells but with increased lipophilicity and maintaining a MRC-5 SI of 6.8. Compound 3m bearing two trifluoromethyl groups at positions 2 and 4 is also a potent growth inhibitor with GI50 2.0 ± 0.8 μM but with unfavourable lipophilicity. Compound 3n (R3 = 2,6-Cl2C6H3) was most active against HCT-116 showing a GI50 value 1.1 ± 0.5 μM but with a calculated lipophilicity value of 4.97, compound 3n presents a less favourable lead than 3f against HTC-116 cells with a comparable MRC-5 SI. An interesting trend can be observed in the 3-halogen substituted compounds 3b–d. An inverse relationship to size can be seen for potency against MCF-7 and HCT-116 cells. 3-Fluorosubstituted compound 3b shows poor activity against MCF-7 cells (36.6 μM) but highly potent against HCT-116 cells at 2.2 μM. Inversely, 3-iodosubstituted compound 3d shows good potency against MCF-7 cells (GI50 2.3 μM); but a 5-fold reduction in potency against HCT-116 cells, indicating that the size of the substituent in the meta-position has an influence on activity and that smaller substituents in this position may negatively impact MCF-7 potency. Fluorine substitution in the para-position was equally poorly active against MCF-7 cells with a 3-fold decrease in activity against HCT-116 cells. Compounds 3b′–d′ replace the aryl ring with heterocyclic isosteres. Compound 3b′ demonstrates good anti-cancer potency against both cancer cell lines with SI values of 5.0 and 12.2 respectively. 2-Bromothiophene compound 3c′ shows comparable potency to 3b′ against MCF-7 cells but shows comparable cytotoxicity resulting in a poor selective index. The 3-bromothiophene containing compound 3d′ displays modest growth inhibition against HCT-116 cells, with an improved SI of 5.1. Considering the methoxy substituted compounds 3j, 3l, 3o, 3y and 3z respectively it can be seen that the substitution pattern of the aromatic ring has a significant effect on the potency of the compound. For example, the introduction of a 4-position methoxy group to compound 3o to yield 3y increases the activity against MCF-7 and HCT-116 cells decreasing GI50 values from 47.8 ± 1.1 μM and 35.6 ± 1.1 μM to 2.7 ± 0.1 and 1.6 μM respectively. The same methoxy addition to compound 3j provides 3z which benefits from a 3.5-fold increase in activity against HCT-116 cells with no significant change in activity against fibroblasts, resulting in an improved SI from 3.3 to 11.5. Interestingly, 2,4-methoxy substituted compound 3l shows reduced potency against both cancer cell lines along with no significant activity against MRC-5. However, the need to add additional methoxy groups to improve the selectivity results in decreased ligand efficiency for these compounds. The use of electron-donating methoxy groups reduces fibroblast toxicity compared to other electron donating groups. Compounds 3i and 3j have similar overall molar refractivity 102.8 cm3 mol−1 and 105.5 cm3 mol−1 respectively; with comparable anticancer activity – but significantly different activity toward MRC-5 cells. Substitution with additional methoxy groups results in decreased in lipophilicity in compounds 3y and 3z showing good drug-like properties commensurate with oral bioavailability in line with compliance to Lipinski and Veber guidelines with modest ligand efficiency improved lipophilic efficient than 3f.35–37 Clearly, substitution on the aromatic ring in the R3 position can strongly influence the overall activity of these compounds; however, a wider scope library would be needed to fully investigate the nuances of the structure–activity relationships. As the electronic effects of substituents are not correlated to electron withdrawing or donating substituents as can be seen with the comparable potencies of 3a′ (pentafluoro) and 3y (3,4,5-methoxy) in both cell lines.
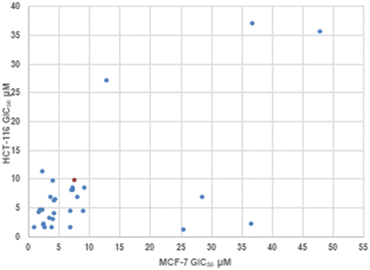 |
| | Fig. 2 Correlation plot between MCF-7 and HCT-116 GIC50 values. Cisplatin control shown in orange. | |
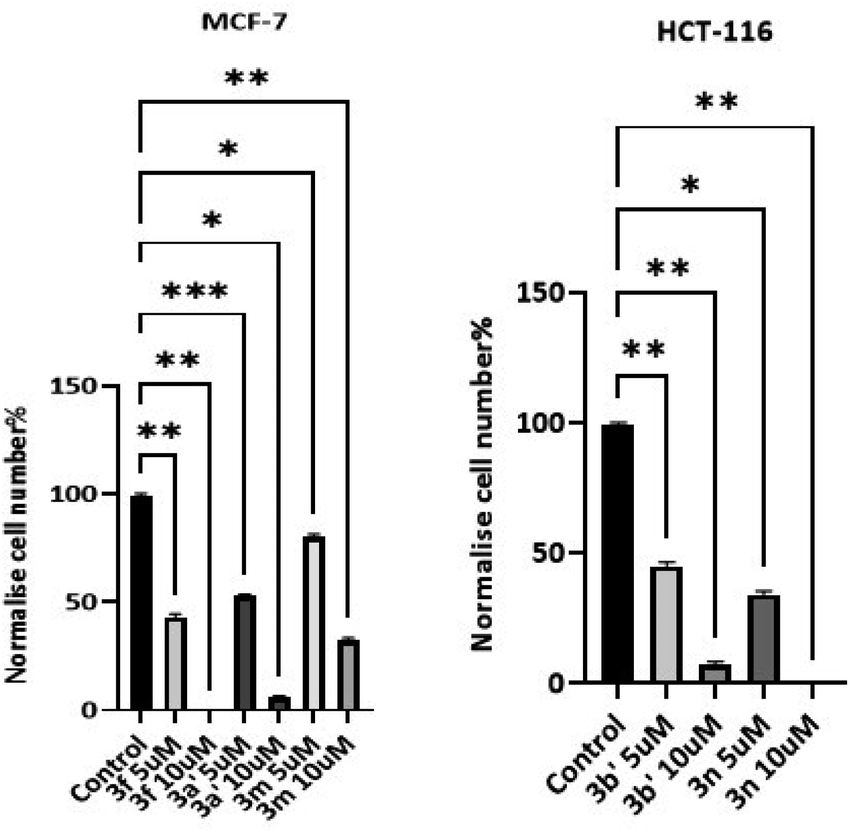
3.2.3 Cell count studies. To corroborate MTT studies, compounds with potent in vitro anticancer activity (3f, 3m, 3n, 3a′ and 3b′) were selected for cell count experiments (Fig. 3).
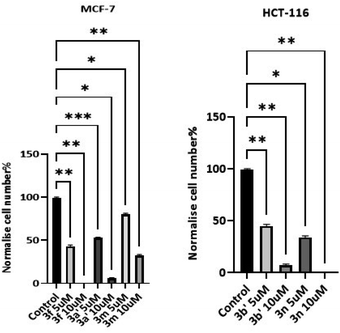 |
| | Fig. 3 Cell count assay for selected compounds against MCF-7 and HCT-116 cells. Cells were seeded at a density of 2 × 104 per well; counts were performed following 72 h exposure to test agent. Data represented as % control, mean ± SD for 2 independent experiments performed in triplicate. Significance is reported as *p < 0.01, **p < 0.004, *p < 0.02, **p < 0.006, *p < 0.01 and *p < 0.02 compared to control. The compounds screened and the concentrations used are shown on the x-axis. | |
These investigations show compound 3n (R3 = 2,6-dichlorophenyl) inhibited HCT-116 growth significantly, (50–55% of the untreated control sample at 5 μM and 100% at 10 μM). Additionally, 3b′ (R3 = 3-bromothiophene) exhibited 50–55% inhibition at 5 μM and 90–95% at 10 μM against HCT-116. Compound 3f (R3 = 2-hydroxy-3-methylphenyl) inhibited MCF-7 by 100% at 10 μM and 55–60% at 5 μM. At a concentration of 10 μM, compounds 3a′ (R3 = 1,2,3,4,5-pentafluorophenyl) exhibited 90–95% growth inhibition and 3h (R3 = 2,4-bis(trifluoromethyl)phenyl) showed 65–70% inhibitory potential.
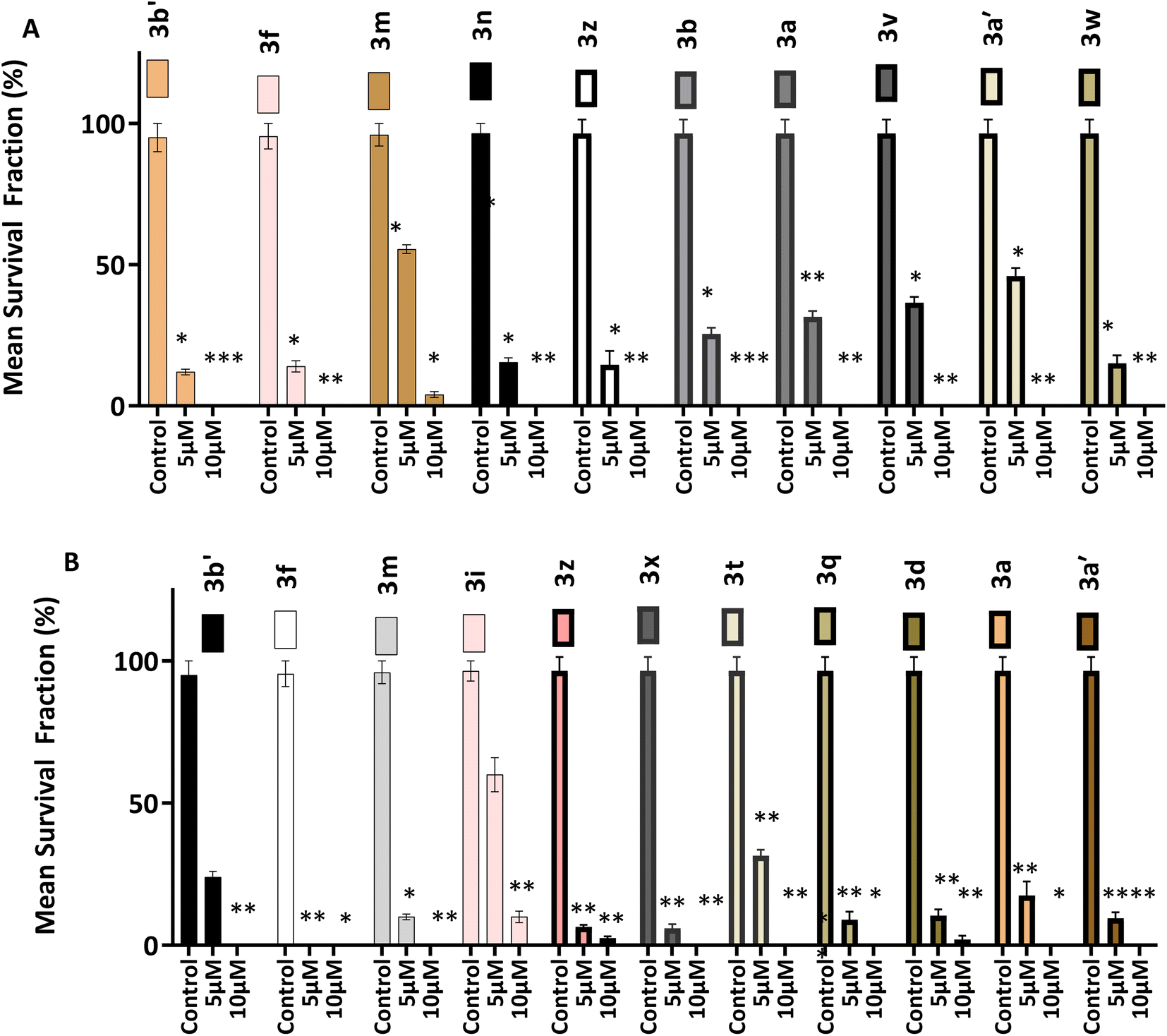
3.2.4 Clonogenic assays. The ability of single cells (HCT 116 and MCF-7) to survive brief challenge (24 h) by 3 and survive to produce progeny colonies was determined by clonogenic assay. Selected 3 analogues were interrogated at 5 and 10 μM. Fig. 4 shows results of clonogenic assays. The number of colonies formed after brief exposure to test agents are represented as survival fractions (in % values) of control colony numbers. After 24 h treatment compounds 3 strongly suppressed cell colony formation at 5 and 10 μM concentrations. Compounds 3a, 3b, 3f, 3n, 3v, 3w, 3z, 3a′ and 3b′ are presented in Fig. 3A, all compounds prevent (100%) colony formation at 10 μM concentration. At 5 μM between 5 and 50% inhibition of colony formation is seen. Fig. 3 also demonstrates compounds 3 suppress HCT-116 colony formation. Compound 3f was the most effective, completely inhibiting colony formation at both 5 and 10 μM. At 10 μM compounds 3a, 3q, 3t, 3a′ and 3b′ completely prevented colony formation while at 5 μM only 5–30% inhibition was observed.
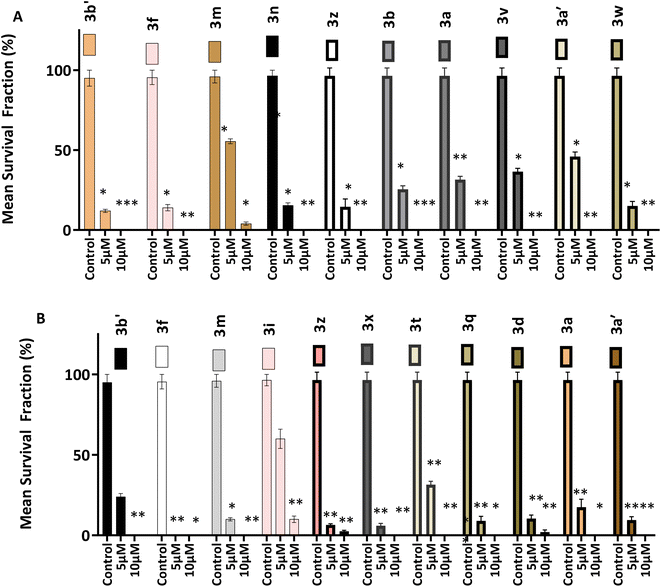 |
| | Fig. 4 Effect of selected compounds on (A) HCT-116 and (B) MCF-7, colony formation. Bar graphs (A and B) both show the mean survival fraction of % control represented of mean ± SD of five independent trials (n = 2 per trial). Significance is reported as *p < 0.05, **p < 0.005 compared to control. | |
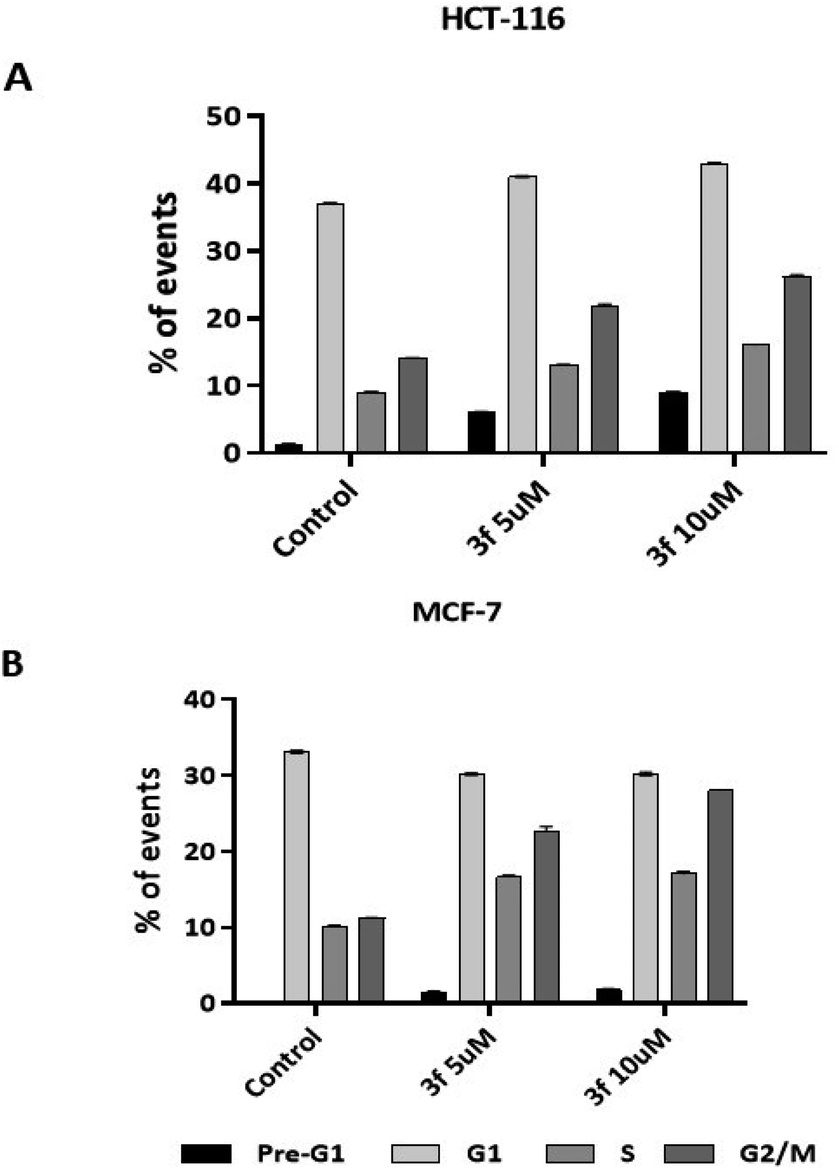
3.2.5 Cell cycle analysis. To shed light on the mechanism of action of this class of compound, 3f was investigated as a representative example in the cell-cycle assay. After 24 h treatment of HCT-116 and MCF-7 with compound 3f (5 and 10 μM) both cell lines showed increases in the portion of cells in the G2/M phase (Fig. 5). The observed G2/M arrest indicates DNA damage and cell cycle checkpoint attempts to initiate repair. Pre-G1 events (5–8%) were detected in HCT-116 populations exposed to 3f, an indication of caspase-excision of DNA characteristic of apoptosis. Consistent with this the MCF-7 cell line showed negligible pre-G1 cell populations; MCF-7 cells lack the caspase 3 proteases responsible for DNA excision and protein cleavage.27 Here the G2/M (MCF-7) cell accumulation also indicates DNA damage and attempt at repair. For HCT-116 the highest accumulation of cells in the G2/M phase at 5 and 10 μM concentrations was 22% and 26% compared to control (14%). The G2/M phase arrest is somewhat more obvious in MCF-7 cells, with the maximum accumulation of events observed in G2/M phase at 5 and 10 μM concentration being 23% and 28% compared to the control (11%). Our studies clearly showed that increase in agent 3f concentration resulted in significant (p < 0.01) increase in G2/M.
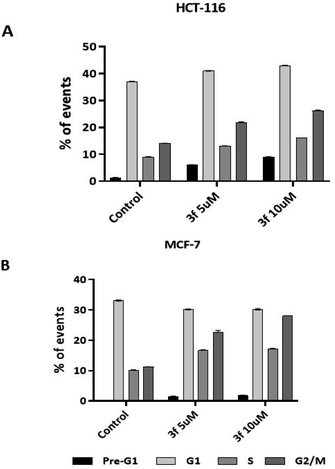 |
| | Fig. 5 Cell-cycle histograms summarising the effect of 3f on cell-cycle progression of HCT-116 (A) and MCF-7 (B) cells after 24 h of treatment at concentrations of 5 and 10 μM concentrations; 20000 events were counted. Number of internal repeats three times. Primary flow cytometry traces are given in the ESI.† | |
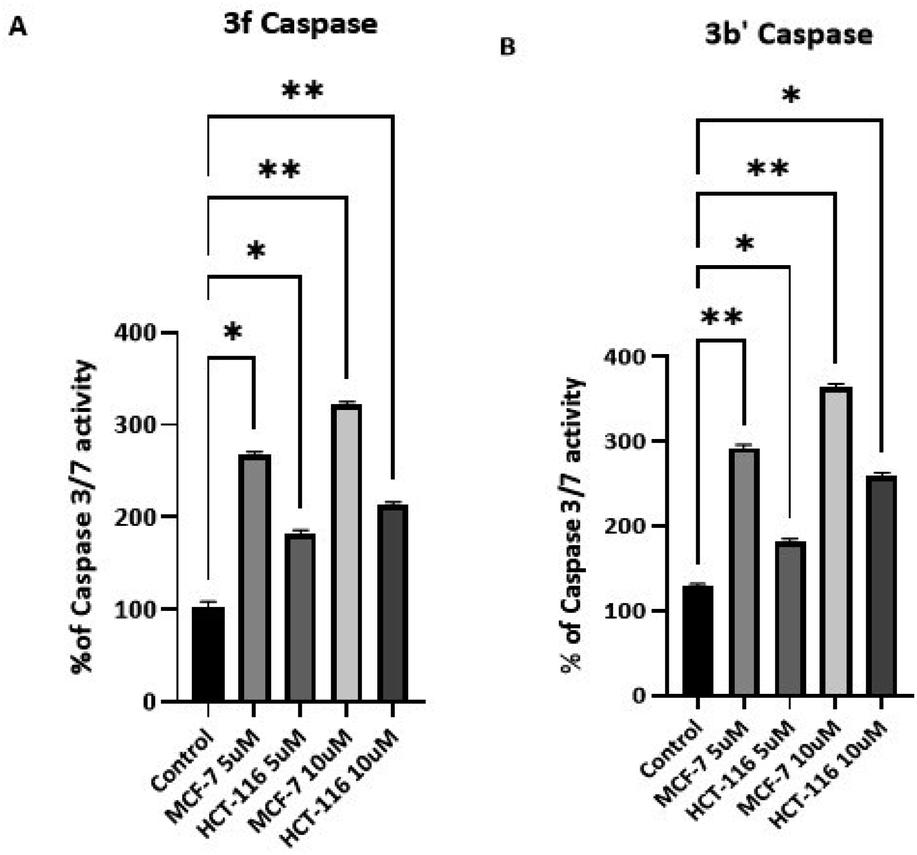
3.2.6 Caspase activation. To confirm that compounds 3 cause apoptosis in HCT-116 and MCF-7 cells, the activation of caspases 3 and 7 was studied following exposure of cells (72 h) to 5 and 10 μM 3b′ and 3f (Fig. 6). Significant dose-dependent caspase activation was observed after 72 h exposure of the cells to these agents (Fig. 5) which is consistent with apoptosis-induction. These data suggest that 3b′ and 3f exert their cytotoxic activity via induction of caspase-dependent apoptosis.
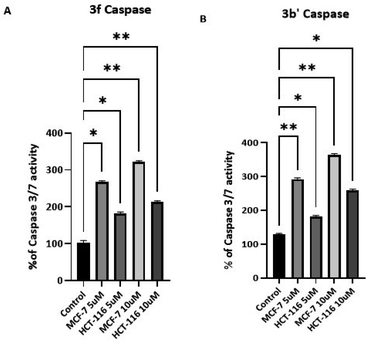 |
| | Fig. 6 Caspase-3/7 activity of HCT-116 (A) and MCF-7 (B) cells after 72 h of treatment with 3b′ and 3f at concentrations of 5 and 10 μM. Each experiment was repeated twice with five internal repeats. Bars and error bars refer to the mean ± SD. *p < 0.01 vs. control, ***p < 0.0005 vs. control respectively. | |
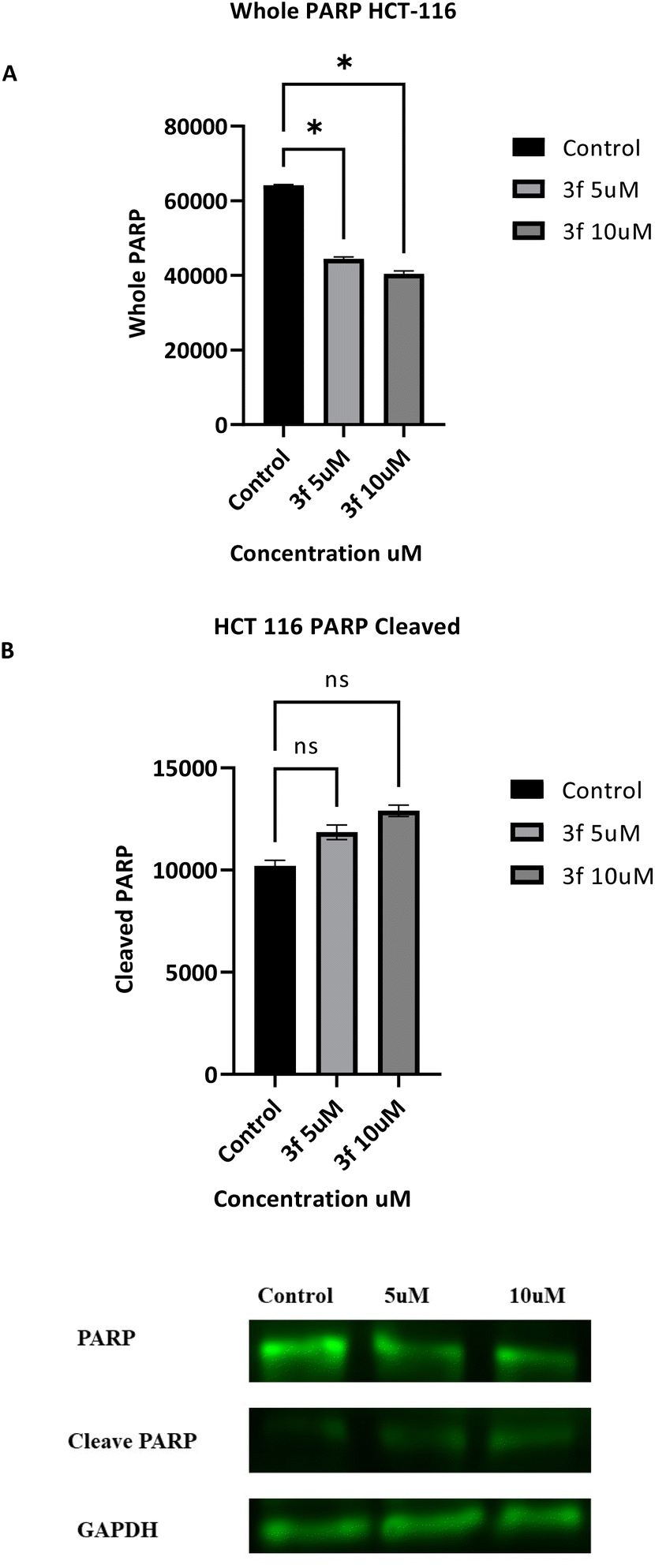
3.2.7 Western blot assay. Confirmation of apoptosis-induction by 3f was further investigated by monitoring the expression levels of key apoptotic proteins. Poly (ADP-ribose) polymerase-1 (PARP-1) is a substrate of the caspase-3 protein, present in HCT-116 cells and involved in a number of cellular processes. Its cleavage is indicative of apoptosis in HCT-116 cell lines.30 Therefore, HCT-116 cells were treated with 3f (5 and 10 μM, 24 h). The results obtained (Fig. 7) demonstrate that 3f induces concentration-dependent PARP-1 cleavage, accompanied by down-regulation of whole PARP, suggesting apoptosis induction. The presence of cleaved PARP is also indicative of caspase activation, consistent with the caspase 3/7 results above.
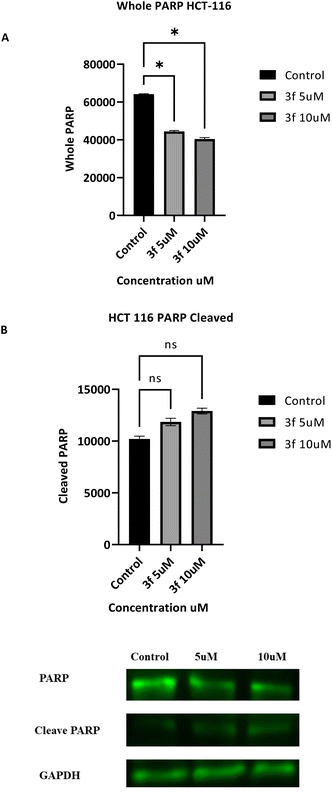 |
| | Fig. 7 Effect of compound 3f on expression of whole and cleaved PARP in HCT 116 cells. (A and B) Data are mean ± SD of three independent experiments (ANOVA followed by Dunnett's test), significance is reported as *p < 0.05. (C) Representative western blots of HCT 116 lysates following treatment of cells with 3f (5 and 10 μM) are given. The data are the mean SD of three independent experiments (ANOVA followed by Dunnett's test). Significance is reported as: *p < 0.05. | |
4. Conclusions
A library of 30 novel 2-(2-benzylidenehydrazinyl)-4-(4-cyanophenyl)thiazole derivatives (3a–d′) were synthesised. These demonstrate potent and selective antitumour activity against two cell lines representative of colorectal (HCT-116) and breast (MCF-7) cancer phenotypes. Compound 3f (R3 = 2-hydroxy-3-methylphenyl) was found to be the most general efficient growth inhibitor with GI50 value 1.0 ± 0.4 μM against MCF-7. Compound 3n (R3 = 2,6-Cl2C6H3) exhibited highest inhibition potential (GI50 1.1 ± 0.5 μM) against HCT-116. The selectivity of compounds towards the cancer cell lines over non-cancer cells was estimated by equivalent exposure to MRC-5 (normal fibroblasts). Only one compound 3w (R3 = 2,4,6-C6H3F3) was more active against MRC-5, while all other compounds were more active against the carcinoma cell lines (often appreciable so e.g. >5× for 3b′, >10× for 3f). Cell cycle, caspase activation and western blot assays established that compounds 3b′ (R3 = 3-bromothiophene) and 3f (R3 = 2-(OH)-3-MeC6H3) induce cancer cell death via caspase-dependent apoptosis and that these compounds are significantly more active than previously reported related thiazoles. It is too premature to speculate in depth on the exact mechanism of action of the compounds 3. However, we note that very recently a related group of thiazoles were described that are potent kinase inhibitors.40 It is possible that our own compounds operate similarly as preliminary experiments also showed ERK1/2 inhibition for 3f. In conclusion, these synthetically accessible molecules are worthy of further preclinical evaluation including further examination against broad-spectrum cancer phenotypes and elucidation of their mechanism of action.
Author contributions
Tashfeen Akhtar planned the synthesis. Hasnain Mehmood synthesized the compounds and carried out all spectral analysis. Mustapha Musa carried out all the biological assays. Simon Woodward led the writing of the manuscript. Tracey Bradshaw and Andrew Nortcliffe contributed in biological results analysis and interpretation. Shahdat Hossan assisted in biological assays. Muhammad Haroon provided critical review and feedback.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
HM is thankful to the Higher Education Commission (HEC) of Pakistan for a fellowship under the International Research Support Initiative (IRSIP) Program. MM is grateful to the University of Nottingham for the award of a Vice Chancellor's Scholarship for Research Excellence. We thank the referees for their valuable insights and suggestions for this paper.
References
- Green Approaches in Medicinal Chemistry for Sustainable Drug Design, ed. B. Banik, Elsevier, Amsterdam, 2020, p. 1021 Search PubMed.
- M. Milanesi, A. Runfola and S. Guercini, J. Clean. Prod., 2020, 261, 121204 CrossRef.
- M. Fridlender, Y. Kapulnik and H. Koltai, Front. Plant Sci., 2015, 6, 799 Search PubMed.
- P. C. Sharma, K. K. Bansal, A. Sharma, D. Sharma and A. Deep, Eur. J. Med. Chem., 2020, 188, 112016 CrossRef CAS PubMed.
- S. Jain, S. Pattnaik, K. Pathak, S. Kumar, D. Pathak, S. Jain and A. Vaidya, Mini-Rev. Med. Chem., 2018, 18, 640 CrossRef CAS PubMed.
- M. Abid, R. Nouch, T. D. Bradshaw, W. Lewis and S. Woodward, Eur. J. Inorg. Chem., 2019, 2774 CrossRef CAS.
- D. Secci, S. Carradori, B. Bizzarri, A. Bolasco, P. Ballario, Z. Patramani, P. Fragapane, S. Vernarecci, C. Canzonetta and P. Filetici, Bioorg. Med. Chem., 2014, 22, 1680 CrossRef CAS PubMed.
- S. Carradori, D. Rotili, C. De Monte, A. Lenoci, M. D'Ascenzio, V. Rodriguez, P. Filetici, M. Miceli, A. Nebbioso and L. Altucci, Eur. J. Med. Chem., 2014, 80, 569 CrossRef CAS PubMed.
- K. Z. Łączkowski, M. Świtalska, A. Baranowska-Łączkowska, T. Plech, A. Paneth, K. Misiura, J. Wietrzyk, B. Czaplińska, A. Mrozek-Wilczkiewicz, K. Malarz, R. Musiol and I. Grela, J. Mol. Struct., 2016, 1119, 139 CrossRef.
- F. S. A. Rahman, S. K. Yusufzai, H. Osman and D. Mohamad, J. Phys. Sci., 2016, 27, 77 Search PubMed.
- K. Vaarla, R. K. Kesharwani, K. Santosh, R. R. Vedula, S. Kotamraju and M. K. Toopurani, Bioorg. Med. Chem. Lett., 2015, 25, 5797 CrossRef CAS PubMed.
- T. I. de Santana, M. de Oliveira Barbosa, P. A. T. de Moraes Gomes, A. C. N. da Cruz, T. G. da Silva and A. C. L. Leite, Eur. J. Med. Chem., 2018, 144, 874 CrossRef PubMed.
- Y. Wang, C. Wu, Q. Zhang, Y. Shan, W. Gu and S. Wang, Bioorg. Chem., 2019, 84, 468 CrossRef CAS PubMed.
- R. Raveesha, A. Anusuya, A. Raghu, K. Y. Kumar, M. D. Kumar, S. B. Prasad and M. Prashanth, Comput. Toxicol., 2022, 21, 100202 CrossRef CAS.
- A. Rauf, M. K. Kashif, B. A. Saeed, N. A. Al-Masoudi and S. Hameed, J. Mol. Struct., 2019, 1198, 126866 CrossRef CAS.
- M. C. Al-Matarneh, R.-M. Amărandi, I. I. Mangalagiu and R. Danac, Molecules, 2021, 26, 2066 CrossRef CAS PubMed.
- H. Mehmood, M. Haroon, T. Akhtar, S. Woodward and H. Andleeb, J. Mol. Struct., 2022, 1250, 131807 CrossRef CAS.
- S. Bargujar, S. Chandra, R. Chauhan, H. K. Rajor and J. Bhardwaj, Appl. Organomet. Chem., 2018, 32, e4149 CrossRef.
- M. T. Scotti, L. Scotti, H. M. Ishiki, L. M. Peron, L. de Rezende and A. T. do Amaral, Chemom. Intell. Lab. Syst., 2016, 154, 137 CrossRef CAS.
- G. Springett, S. M. Sebti and N. J. Lawrence, PCT Patent, WO2015157242A1, 2015, p. 55.
- H. Hammoud, K. Elhabazi, R. E. Quillet, I. Bertin, V. Utard, E. Laboureyras, J.-J. Bourguignon, F. Bihel, G. Simonnet and F. Simonin, ACS Chem. Neurosci., 2018, 9, 2599 CrossRef CAS PubMed.
- A. D. Khalaji, K. Fejfarova and M. Dusek, J. Cryst. Miner., 2013, 19, 1 Search PubMed.
- B. Donarska, M. Świtalska, W. Płaziński, J. Wietrzyk and K. Z. Łączkowski, Bioorg. Chem., 2021, 110, 104819 CrossRef CAS PubMed.
- B. N. Sağlık, D. Osmaniye, S. Levent, U. A. Çevik, B. K. Çavuşoğlu, Y. Özkay and Z. A. Kaplancıklı, Eur. J. Med. Chem., 2021, 209, 112918 CrossRef PubMed.
- A. Mignot, M. Miocque, P. Binet, J. Rapin, P. Rinjard, M. Roux, M. Cals and J. Ekindjian, Eur. J. Med. Chem., 1980, 15, 33 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed.
- I. Nicoletti, G. Migliorati, M. Pagliacci, F. Grignani and C. Riccardi, J. Immunol. Methods, 1991, 139, 271 CrossRef CAS PubMed.
- M. K. B. Break, M. S. Hossan, Y. Khoo, M. E. Qazzaz, M. Z. Al-Hayali, S. C. Chow, C. Wiart, T. D. Bradshaw, H. Collins and T.-J. Khoo, Fitoterapia, 2018, 125, 161 CrossRef PubMed.
- E. Harlow and D. Lane, Cold Spring Harbor Protocols, 2006, 2006 DOI:10.1101/pdb.prot4283.
- T. Mahmood and P.-C. Yang, N. Am. J. Med. Sci., 2012, 4, 429 CrossRef PubMed.
- V. Fadeeva, V. Tikhova and O. Nikulicheva, J. Anal. Chem., 2008, 63, 1094 CrossRef CAS.
- M. Haroon, T. Akhtar, M. Yousuf, M. N. Tahir, L. Rasheed, S. S. Zahra and M. Ashfaq, BMC Chem., 2022, 16, 1 CrossRef PubMed.
- H. Mehmood, M. Khalid, M. Haroon, T. Akhtar, M. Ashfaq, M. N. Tahir, M. U. Khan, M. Imran, A. A. C. Braga and S. Woodward, J. Mol. Struct., 2021, 1245, 131043 CrossRef CAS.
- M. J. Waring, Expert Opin. Drug Discovery, 2010, 5, 235 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615 CrossRef CAS PubMed.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881 CrossRef CAS PubMed.
- Molinspiration Cheminformatics free web services, https://www.molinspiration.com, Slovensky Grob, Slovakia Search PubMed.
- T. Sander, J. Freyss, M. von Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460 CrossRef CAS PubMed.
- A. Y. Khormi, T. A. Farghaly, A. Bayazeed, Y. O. Al-Ghamdi, H. G. Adulwahab and M. R. Shaaban, RSC Adv., 2022, 12, 27355 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of prepared compounds and supporting biological information. See DOI: https://doi.org/10.1039/d2ra03226k |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Md Shahadat Hossanc,
Tracey D. Bradshaw
*b,
Md Shahadat Hossanc,
Tracey D. Bradshaw