
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic deoxygenation of palm oil over metal phosphides supported on palm fiber waste derived activated biochar for producing green diesel fuel
Napat Kaewtrakulchaiab,
Masayoshi Fujic and
Apiluck Eiad-Ua *a
*a
aCollege of Materials Innovation and Technology, King Mongkut's Institute of Technology, Bangkok 10520, Thailand. E-mail: apiluck.ei@kmitl.ac.th; Fax: +66–2–329–8625; Tel: +66–2–329–8300 ext. 3132
bKasetsart Agricultural and Agro-Industrial Product Improvement Institute, Kasetsart University, Bangkok 10900, Thailand
cAdvanced Ceramic Center, Nagoya Institute of Technology, Tajimi, Gifu, Japan
First published on 13th September 2022
Abstract
Palm oil conversion into green diesel by catalytic deoxygenation (DO) is one of the distinctive research topics in biorefinery towards a bio-circular-green economic model to reduce the greenhouse gas emissions. In this study, palm fiber waste was explored as an alternative precursor for the preparation of activated biochar as a support material. A new series of nickel phosphide (Ni–P) and iron phosphide (Fe–P) catalysts supported on palm fiber activated biochar (PFAC) was synthesized by wetness impregnation, and extensive characterization was performed by several techniques to understand the characteristics of the supported metal phosphide catalysts prior to palm oil deoxygenation for producing of green diesel (C15–C18 hydrocarbons). The PFAC support exhibited suitable physicochemical properties for catalyst preparation, such as high carbon content, and high porosity (SBET of 1039.64 m2 g−1 with VT of 0.572 cm3 g−1). The high porosity of the catalyst support (PFAC) significantly promotes the metal phosphide nanoparticle dispersion. The DO of palm oil was tested in a trickle bed down flow reactor under hydrogen atmosphere. The outstanding catalytic performance of supported Ni–P and Fe–P catalysts provided an impressive liquid hydrocarbon yield between 63.37 and 79.65% with the highest green diesel selectivity of 62.64%. Decarbonylation (DCO) and decarboxylation (DCO2) are the main pathways for the relative phosphide catalysts as presented by the high number of Cn−1 atoms (C15 and C17 hydrocarbons). In addition, metal phosphide/PFAC catalysts could achieve great potential application as a promising alternative catalyst for biofuel production via deoxygenation for large-scale operation owing to their excellent catalytic activity, simple preparation, and utilization of sustainable resources.
1. Introduction
The production of green chemicals and biofuels from renewable and environmentally sustainable resources, such as waste materials, agricultural byproducts and natural triglycerides from plants and animals, has attracted much scientific and industrial interest due to its greenness, carbon sequestration and CO2 emission reduction. This process could alleviate the global warming situation by replacing the use of fossil fuels.1,2 Presently, the consumption of petroleum in transportation and industrial sectors, particularly diesel fuel, is gradually increasing day by day owing to the fast growing economic demand and the ballooning of the world's population. The conversion of waste biomass to a green energy source has been recognized as a promising pathway to avoid using petroleum diesel in the future.3,4 Conventionally, biodiesel, well-developed from trans-esterification of natural triglycerides including plant-based oils, animal fats, and algae oils into fatty acid methyl esters (FAMEs), has become the most popular transportation biofuel. However, the drawback of biodiesel is the oxygen content, which deteriorates the relative fuel properties, such as high viscosity, low freezing point, and low thermal stability, which ultimately causes serious problems in engine systems, such as blockage of the injector and fuel filter, carbon deposition on pistons, ring sticking, cylinder head, etc4,5. To overcome these gaps, several technologies for conversion of triglycerides to efficient quality diesel-like fuel, which meet to the petroleum diesel quality need to be investigate conscientiously. Therefore, other attractive processes to convert the vegetable oils, and tallows to bio-hydrocarbons in the range of green diesel (C15–C18 paraffinic hydrocarbons) are significantly investigated. Technically, the catalytic deoxygenation essentially established for high quality of several green fuel products as though in petroleum fuels.6,7 Generally, catalytic deoxygenation for biofuel production is usually conducted in both hydrogen and hydrogen-free systems at 250–420 °C over moderate acidic micropore or mesopore heterogeneous catalysts under the presence of high pressure between 10–100 bar.8 The three parallel reactions in the deoxygenation for removal of oxygenated compounds are namely decarboxylation (DCO2), decarbonylation (DCO), and hydrodeoxygenation (HDO) that the contribution of each reaction pathway quite depends on types of relative reaction conditions, such as catalyst, reaction temperature, and hydrogen pressure.9Currently, catalytic deoxygenation for upgrading biomass feedstocks into green fuels was run over porous materials supported noble metals (e.g., palladium, platinum, and rhodium) catalysts, which is regularly employed as a commercial benchmark catalyst.10–12 However, the utilization of noble metals is less economically attractive than transition metal-based catalysts: nickel, cobalt, iron, molybdenum in commercialization scale. Furthermore, supported noble metal catalysts in metallic form show the poor stability in long-term catalytic run that should be urgently encountered. Over the last decade, the most important metal active phases for deoxygenation studied in several literatures are other bimetallic catalysts,13–15 metal sulfides,16,17 metal carbides,7,18,19 and metal phosphides,20–22 appears as alternative deoxygenation catalyst because of their very excellent catalytic performance, long life-time in use, and affordable price.14,15,17,20 Moreover, supported bimetallic catalysts showed an excellent catalytic behavior on biofuel processing, and several recent studies have done on these investigated, hence, this catalyst is still less effective for biofuels.13–15 Also, metal sulfide catalysts have a superior catalyst activity. However, the sulfur leaching in final hydrocarbon products is still a serious problem follows the fuel standardize. Thus, non-sulfide metal catalysts are the current research focus nowadays. Among these catalysts, phosphide metals, exhibited an excellent catalytic activity on the conversion of triglycerides into biofuels, such as bio-jet fuel, and green diesel due to its low activity for methanation that is a side reaction during deoxygenation, results in reduction of liquid hydrocarbon yield.6,16,17 Additionally, metal phosphide catalysts provide both metallic and acidic catalyst behaviors, corresponding to small metal positive charge leads to Lewis acid and Brønsted acid site from P–OH groups left from phosphate reduction, which is effective for oxy-compound removal.20,21 However, only the few endeavors have been reported in the utilization of metal phosphide catalyst for green diesel production from plant-based oil and animal fats. For example, Carmaro et al. demonstrated the high catalytic activity of zeolite supported nickel phosphide for producing green diesel from oleic acid.21 Pham et al. also showed the upgrading waste cooking oil into liquid hydrocarbons over activated carbon supported nickel phosphide. They revealed Ni2P phase presented a superior removal efficiency of oxygenated compounds, as it exhibited the great deoxygenation performance via decarboxylation and decarbonylation pathways.22 Kochaputi et al. reported the conversion of oleic acid to green diesel by deoxygenation using copper, nickel, and cobalt phosphide supported on silica, alumina, and zeolite.23 Rakmea et al. converted the palm oil feedstock into diesel-liked fuel over sodium mordenite supported nickel phosphide catalyst via hydrodeoxygenation.24 As mentioned above, supported metal phosphide catalysts have become the attractive candidate for green diesel production via deoxygenation of natural triglycerides.
In addition to deoxygenation catalysts, several recent studies have explored the utilization of high surface area materials, such as alumina, silica, zeolites, and porous carbons as metal catalyst support in the catalytic deoxygenation due to an enhancement of metal catalyst-nanoparticles dispersion and stabilization on the surface of support materials.25 The different support materials also display the differences in acidity and surface density of metal site, which significantly impacts the catalyst activity.26 To date, one of novel support materials is porous carbons developed from lignocellulosic biomass including plant-based materials and agricultural by-products. Due to this support shows some interests, such as low-cost, eco-friendly, and sustainability. Moreover, the differences in textural porosity and surface chemistry of relative porous carbon support can be facilely developed by adjusting the production parameters according to match contemplated applications. Moreover, the utilization of porous carbon as a catalyst supporter in deoxygenation inhibited a coke formation on catalyst surface during the reaction due to the neutral property of carbon materials. Also, porous carbon allows the excellent facility regarding the recovery of spent precious metal catalyst by direct burning the carbon support.27,28 These have been reported in recent studies as follows: Khalit and colleagues reported the usage of activated carbon from charcoal supported nickel-based catalysts in green biofuel production. The AC supported catalysts showed very effective deoxygenation activity, especially decarboxylation with hydrocarbons yields above 60% in the range of green diesel fractions (C15 and C17 hydrocarbons).29 Roy et al. studied the douglas fir derived biochar supported nickel and cobalt catalysts for hydrotreatment of carinata oil.30 Thangadurai also demonstrated the performance of activated carbon supported cobalt and iron oxide catalysts for the catalytic cracking of waste cooking oil to produce biohydrocarbons.31 Based on literature reviews, very few studies have done on investigation of utilizing waste biomass derived activated biochar supported metal phosphide catalysts in the deoxygenation of palm oil for green diesel production. This still caused a lack information on this field. To fill this knowledge gap, the approach to utilize waste biomass from palm industry as a support material for metal phosphide catalysts supplied to the production of palm oil-based green diesel offers an alternative concept in biorefinery has been explored.
Herein, we aimed to report the development of agricultural waste for producing activated biochar as a support material. The second objective of this work is to establish the alternative catalysts of activated biochar supported nickel phosphide (Ni–P) and iron phosphide (Fe–P) for producing green diesel (C15–C18 hydrocarbons) from deoxygenation of palm oil. The activated biochar support was successfully produced from palm fibers, which is one of relevant agricultural wastes collected from oil palm plantation in South-East Asia. Moreover, the development of oil palm residues to a promising catalyst for producing biofuel from oil palm feedstock effectively shows the sustainability concept of bio-refinery. The effects of experimental parameters (i.e. activation temperature and activating agent ratios) on the specific characteristic of palm fiber activated biochar were obviously investigated. The selected activated biochar was then applied as a support of phosphide catalysts in palm oil deoxygenation. Also, the catalytic activity of a series of synthesized phosphide catalysts was compared to the production of green diesel fuel. The selectivity to green diesel product was successfully controlled by tuning reaction conditions. However, the suitable reaction condition for producing green diesel by using these synthesized catalysts was conducted.
2. Experimental
2.1 Materials
Palm fibers was collected from consorted plantation in South of Thailand as a precursor of activated biochar processing. Nickel(II) nitrate hexahydrate; Ni(NO3)2·6H2O, (98%), iron nitrate nonahydrate; Fe(NO3)3·9H2O (98%), potassium hydroxide (KOH), phosphoric acid (H3PO4), and hydrochloric acid (HCl) were purchased from CARLO ERBA Reagents.Also, the palm oil, which is largely produced in Southeast Asia countries and sufficient to use for green diesel production, was supplied from local market in Bangkok, Thailand. The main composition of palm oil feedstock is described as follows: lauric acid (C12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0) 0.4%; myristic acid (C14:0) 0.8%; palmitic acid (C16:0) 37.8%; palmitoleic acid (C16:1) 0.2%; steraric acid (C18:0) 3.6%; oleic acid (C18:1) 45.8%; linoleic acid (C18:2) 11.1%; linolemic acid (18:3) 0.3%; arachidic acid (C20:0) 0.3%; and eicosenoic acid (C20:1) 0.1%. The main composition of palm oil is composed by palmitic acid and oleic acid. Thereby, palm oil appears as a potential resource for producing biofuel in diesel range hydrocarbons with huge commercial and industrial interests.
:0) 0.4%; myristic acid (C14:0) 0.8%; palmitic acid (C16:0) 37.8%; palmitoleic acid (C16:1) 0.2%; steraric acid (C18:0) 3.6%; oleic acid (C18:1) 45.8%; linoleic acid (C18:2) 11.1%; linolemic acid (18:3) 0.3%; arachidic acid (C20:0) 0.3%; and eicosenoic acid (C20:1) 0.1%. The main composition of palm oil is composed by palmitic acid and oleic acid. Thereby, palm oil appears as a potential resource for producing biofuel in diesel range hydrocarbons with huge commercial and industrial interests.
2.2 Synthesis of metal phosphide catalysts
Palm fiber activated biochar support (PFAC) was prepared via carbonization at 400 °C for 2 h under nitrogen atmosphere followed by chemical activation using potassium hydroxide (KOH) as an activating agent. The activation experiments were conducted at several conditions using activating agent ratios of 0.25 0.5, and 1.0 w/w, and different activation temperatures of 700, 750, and 800 °C under nitrogen flow of 100 mL min−1 for 1 h. After the activation process, the samples were completely cooled down to ambient temperature. Then, the samples were repeatedly washed with HCL, and distilled water, respectively. The washed samples were dried at 105 °C in an electrical oven for 24 h. Finally, the resulting PFAC were crushed and sieved to the uniform particle size of 0.4 mm using mesh 40 (U.S Standard Testing Sieve) for further characterization and utilization of catalyst support.Ni–P and Fe–P catalysts were synthesized by phosphate reduction technique using an incipient wetness impregnation at 1.0 metal/phosphorus molar ratio. Among 10 wt% metal loading content was constantly used for catalyst preparation. Initially, aqueous solutions of nickel and iron precursors were prepared followed by dropwise adding of concentrated phosphoric acid. Afterwards, the PFAC was then immersed in the prepared solution and subsequently dried at 80 °C for overnight. The dried samples were pyrolyzed at 500 °C for 2 h in nitrogen flow of 100 mL min−1 to obtain a metal-polyphosphate species. Prior to catalyst characterization and testing in deoxygenation, the prepared samples were completely reduced under a hydrogen atmosphere to obtain the metal phosphide/PFAC catalysts.
2.3 Characterization of PF activated biochar and catalysts
The chemical composition of raw PF and prepared PFACs including moisture, volatile matter, fixed carbon, and ash was determined by proximate analysis.32 The %Moisture, was calculated by drying process for 24 h at 105 °C follows the American Society for Testing Materials, ASTM D2867-99 method (ASTM, 2014).33 %Volatile matter was measured by carbonization according to an ASTM D5832-98 (ASTM, 2008).33 %Ash was analyzed by direct combustion follows ASTM D2866-94 (ASTM, 2011).33,34 In addition, a solid carbon remaining in the sample referred to as %Fixed carbon, is reported by subtracting moisture, volatile matter, and ash contents from 100%.32 Also, the elemental compositions (carbon: C, hydrogen: H, and nitrogen: N contents) of palm fibers and PF activated biochar were measured by the CHN elemental analyzer (Leco truespec CHN-628).The porosity of PFACs and synthesized catalysts were measured by nitrogen physisorption at −196 °C on the Quantachrome Autosorp iQ-MP-XR. The samples were pretreated at 300 °C for 3 h before the analysis begins. The determination of surface areas was computed by BET (Brunauer–Emmett–Teller) model. The pore size distribution was measured by the DFT (density functional theory) method, and the total pore volume (VT) was calculated by a condensation of liquid nitrogen at the relative pressure of 0.99 using the Quantachrome ASiQwin 2.0 software. In addition, the micropore volume (Vmi) was determined by the t-plot model. The mesopore volume (Vme) was calculated by subtraction of Vmi from VT.35
The species of metal phosphide catalysts were characterized on X-ray diffractometer (SmartLab, Rigaku, Japan) operated at 40 kV and 40 mA using Cu-Kα radiation (λ = 1.5406 Å). The 2θ-scanning range was from 15 to 80°, and the scanning rate was 0.5° s−1. The crystallite size of metal phosphide catalysts was measured following the Debye–Scherrer equation.
Surface morphology of PFAC support and synthesized catalysts were observed on a scanning electron microscope, SEM (Zeiss EVO50, Germany) operated at 20 kV, FE-SEM (Hitachi SU8030, Japan), and a transmission electron microscope, TEM (JEOL JEM-2100, Japan) operated at 200 keV, respectively.
The acidity of metal phosphide catalysts was carried out by using ammonia-temperature programmed desorption (NH3–TPD) technique (5% NH3/He) in a Quantachrome Chemisorption Analyzer ChemStar (TPX Series), equipped with a thermal conductivity detector.
Surface functional characteristics of PFAC support was characterized by using Fourier transform infrared spectroscopy (FTIR). The infrared adsorption spectrum is the wavenumber ranges from 4000–400 cm−1. The samples were put into infrared platform and impressed directly before to start characterization in transmittance mode on FTIR spectrometer, PerkinElmer UATR Two.
The study of surface chemical compounds of PFAC support, and metal phosphide catalysts were conducted by using X-ray photoelectron spectroscopy (XPS), Kratos AXIS supra (XPS) surface analytical with Al Kα radiation as the excitation source. The samples were situated on the carbon tape placed on the sample stub and substituted to high vacuum system before measurement. All binding energy spectra were deconvoluted by XPS Kratos ESCApe data processing software to fit the desired spectra, such as C 1s, O 1s (for carbon support), Ni 2p, Fe 3p (for studied catalysts), respectively.
2.4 Palm oil deoxygenation
The deoxygenation performance of Ni–P and Fe–P catalysts on green diesel production was investigated in the continuous down-flow reactor under different reaction conditions. 8 mL of Ni–P and Fe–P catalysts were used in the DO of palm oil follows the feedstock flow rate at liquid hourly space velocity (LHSV) of 1 h−1. The synthesized catalyst was packed into the middle of stainless-steel reactor tube, and then in situ reduced immediately under H2 flow of 50 mL min−1 at 650 °C for 3 h by using a ramping rate of 5 °C min−1 to ensure that the metal phosphide phase was achieved for the reaction. After the catalyst reduction, the system was cooled down to the desired reaction temperatures of 340, 360, 380, and 400 °C, and then pressurized with hydrogen of 50 bar using a back-pressure regulator under the controlled H2 flow of 100 mL min−1. The reaction test was evaluated after the start up time of 4 h. This is for reaching steady state of catalytic reaction. The fresh catalyst was used in all experiments to assure the catalyst performance because the catalyst deactivation can be occurred after each experimental run.36 In product analysis, the liquid product collection was periodically conducted at every 2 h intervals for analysis to obtain the average value. The 50 mg of liquid product compositions after separation of water phase was diluted with 1 ml of hexane, and 1 μL of prepared sample was characterized by offline gas chromatography equipped with a mass spectrometer detector (GCMS-QP2020, Shimadzu, Japan) using capillary column (DB-1HT, 30 m × 0.32 mm x 0.1 μm) and a flame ionization detector (FID). The temperature of injection port and detector was set at 370 °C. The analyzed mass spectra of C8–C20 hydrocarbons were matched with the National Institute of Standard and Testing (NIST) library. The conversion of palm oil, liquid hydrocarbon yield (HCs, C9–C24), and green diesel (C15–C18) selectivity were calculated by using the following equations (eqn. (1), (2) and (3)), respectively.
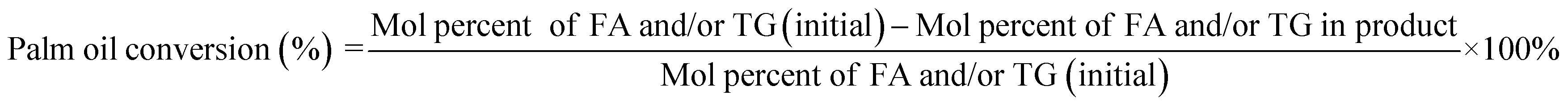 | (1) |
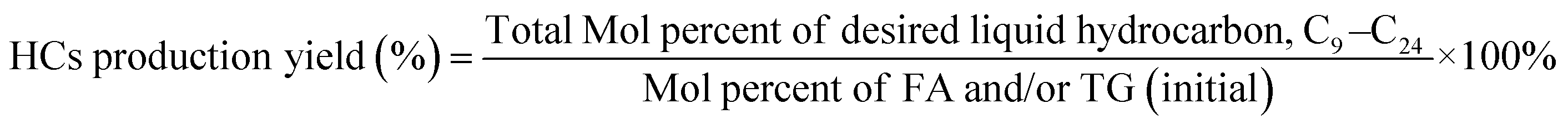 | (2) |
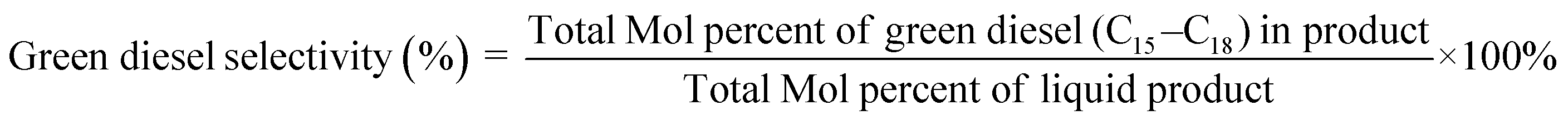 | (3) |
3. Results and discussion
3.1 Characterization of activated biochar support
| Samples | Proximate analysis (dried basis, %) | Ultimate analysis (dried basis, %) | |||||
|---|---|---|---|---|---|---|---|
| VM | FCa | A | C | H | N | Oa | |
| a Calculated by difference, M: moisture, VM: volatile matter, FC: fixed carbon, A: ash; (means ± SD; n = 3). | |||||||
| PF feedstock | 72.24 ± 2.47 | 21.63 ± 0.52 | 6.13 ± 0.18 | 46.73 ± 1.35 | 2.27 ± 0.43 | 1.36 ± 0.12 | 49.64 ± 3.32 |
| PF biochar | 29.26 ± 0.45 | 62.21 ± 3.96 | 8.53 ± 0.53 | 66.49 ± 4.12 | 1.64 ± 0.25 | 1.12 ± 0.06 | 30.75 ± 1.38 |
| PFAC700-0.25 | 20.43 ± 2.19 | 68.38 ± 5.74 | 9.19 ± 1.31 | 70.75 ± 5.04 | 1.04 ± 0.17 | 1.06 ± 0.02 | 27.15 ± 1.24 |
| PFAC700-0.5 | 20.62 ± 1.83 | 71.14 ± 7.12 | 8.24 ± 0.58 | 72.44 ± 2.35 | 1.02 ± 0.26 | 1.01 ± 0.07 | 25.53 ± 1.13 |
| PFAC700-1 | 21.26 ± 1.48 | 69.49 ± 4.65 | 9.25 ± 0.75 | 72.16 ± 1.83 | 0.98 ± 0.12 | 0.95 ± 0.03 | 25.91 ± 0.95 |
| PFAC750-0.25 | 17.72 ± 0.63 | 74.37 ± 2.24 | 7.91 ± 1.03 | 75.19 ± 2.14 | 0.97 ± 0.11 | 1.06 ± 0.08 | 22.78 ± 1.21 |
| PFAC750-0.5 | 18.89 ± 1.25 | 72.49 ± 1.28 | 8.62 ± 0.70 | 74.37 ± 1.21 | 0.92 ± 0.09 | 0.99 ± 0.05 | 23.72 ± 1.07 |
| PFAC750-1 | 17.04 ± 2.81 | 73.61 ± 2.49 | 9.35 ± 1.29 | 77.62 ± 1.09 | 0.93 ± 0.14 | 1.01 ± 0.09 | 20.44 ± 1.15 |
| PFAC800-0.25 | 12.53 ± 2.58 | 79.71 ± 3.85 | 7.76 ± 1.32 | 79.94 ± 4.73 | 0.83 ± 0.16 | 0.96 ± 0.03 | 18.27 ± 0.98 |
| PFAC800-0.5 | 15.86 ± 0.93 | 76.64 ± 5.12 | 7.50 ± 0.97 | 78.26 ± 2.07 | 0.89 ± 0.11 | 0.97 ± 0.06 | 19.89 ± 1.19 |
| PFAC800-1 | 13.02 ± 1.16 | 78.64 ± 6.27 | 8.34 ± 0.85 | 80.31 ± 1.42 | 0.92 ± 0.08 | 0.93 ± 0.04 | 17.97 ± 1.06 |
Nonetheless, the carbonization was firstly carried out at 400 °C for 2 h for pretreatment of a raw PF to develop the fixed carbon percent by releasing volatile and oxygenated compounds to produce a solid carbon before activation process. The value of carbon percentage and fixed carbon content significantly increased, when compared to proximate and ultimate analyzes of PF due to the thermal decomposition of lignocellulosic compounds that occurred a devolatilization. Besides, the percentage of oxygen was lower than that of PF feedstock suggesting that moisture and volatile matters are removed.39 Moreover, the ultimate and proximate analyzes of activation samples (PFAC) with different conditions of activation temperature and dosage of KOH activating agent are also demonstrated in Table 1.
According to proximate analysis, PFAC samples have a fixed carbon content between 68.38 and 79.71%. It was found that the fixed carbon content essentially gets raised by increasing activation temperature (700–850 °C), while the activating agent ratio slightly impacted on the percentage of fixed carbon. This is due to the high temperature in PFAC manufacturing is significantly released the volatile matters via devolatilization, and this also partially converted the oxygenated compounds into solid carbon via graphitization, simultaneously.39,40 On the other hand, volatile content also reduced with increasing activation temperature in the range from 12.53–21.26%. This finding consisted with the reducing oxygen content in ultimate analyzes of PFAC, which results in a higher hydrophobicity. One of crucial parameters in the proximate analysis of carbon material derived from biomass is ash content since the ash might cause an influence on a significant role in specific applications such as hydrophilicity and some catalytic effects.33,39,41 The prepared activated biochar derived from palm fiber contains low ash content (7.50–9.35%).
The ultimate analyzes showed that PFACs was approximately 70.75–80.31% C, which is quite high when compared to a commercial activated carbon (85–90% C).42 Noticeably, the percentage of carbon in activated carbon is always seemingly proportional with its surface area value, which the majority property of activated carbon sample is a porosity and surface area since the pore development is mainly occurred by partial oxidation of carbon atoms during activation process, while the high content of inorganic substances (ash composition) may suppress the porosity enhancement results in reducing specific surface area and porosity, directly.33,41 Hence, the high carbon content of PFACs may be desirable for the excellent pore formation. The highest surface area approximately 1039.64 m2 g−1 was obtained from PFAC750–0.25 with %C of 75.19%. Table 1 also shows the N and H contents of PFAC samples. The small amount of N was found nearly less than 1% (0.93–1.06%). This can be left from the nitrogen content in plant structure and partially chemisorbed during activation stage.41 Hydrogen content was also observed between 0.83 and 1.04%, which is normally a chemical H bonding with carbon atom in carbon structure.43 Additionally, PFAC samples have O content 17.97–27.15%. It was gradually decreased from 49.64% O in a raw PF. This is due to the releasing of volatiles and oxygenated compounds in the plant-based biomass during high-temperature processing, which the results are in good agreement with the study of Mahamad et al.44 and Zubrik et al.38 According to the finding results, it was found suggested that both chemical contents and elemental compositions of PFACs were strongly influenced by the high temperature during activation process. These analyzes have a good agreement with several literatures, which reported the similar observations that increasing activation temperature significantly obtained the product with higher carbon percentage and fixed carbon content due to the influence of thermal decomposition reactions during activation occurred simultaneously. In addition, the enough activating agent may decrease in fixed carbon content.37,38,43,45
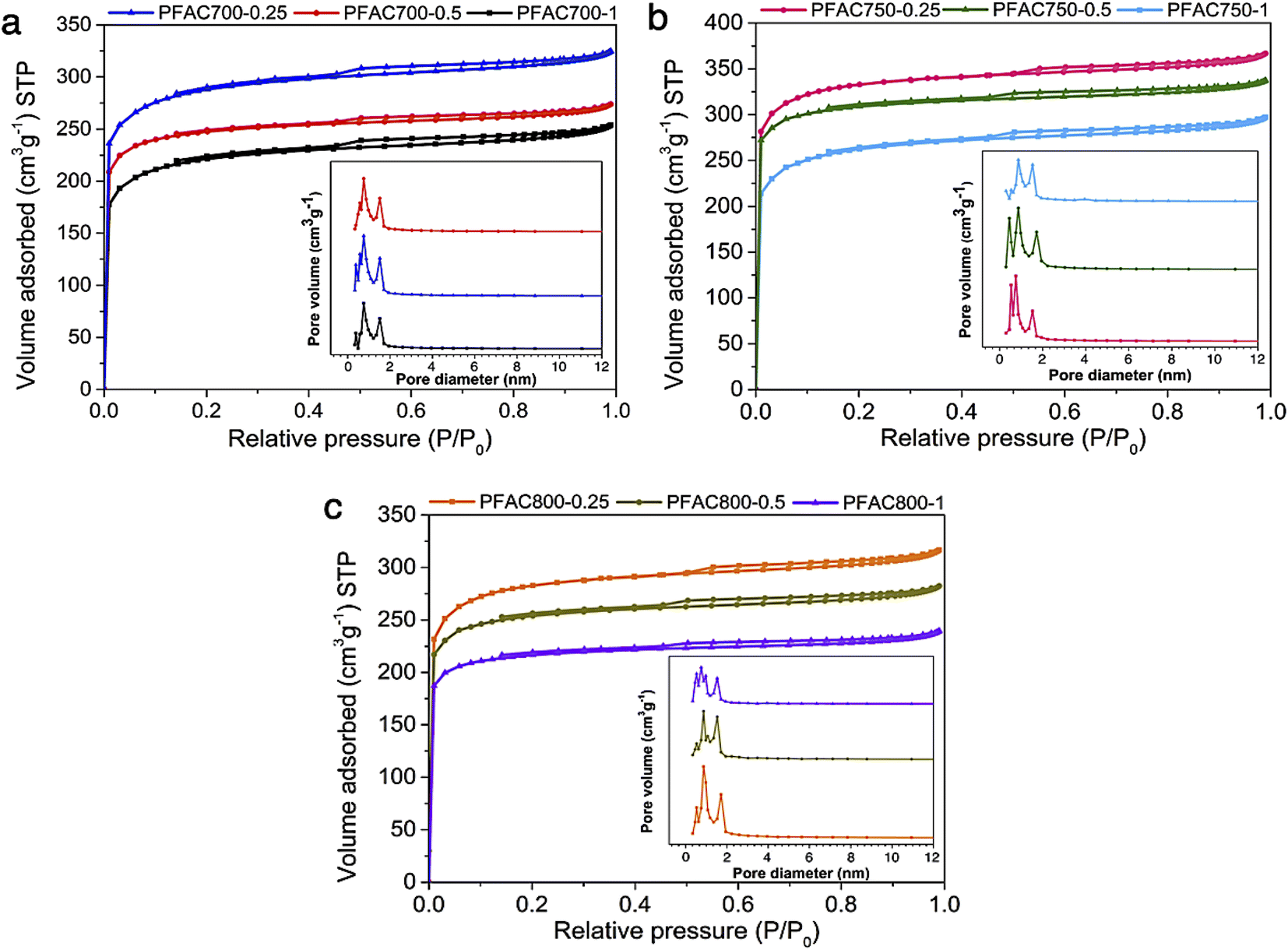 | ||
| Fig. 1 N2 adsorption/desorption isotherms and pore size distribution of PFAC samples obtained at (a) 700 °C, (b) 750 °C, and (c) 800 °C with different KOH ratio. | ||
| Samples | SBET (m2 g−1) | Vtotal (cm3 g−1) | Vmicro (%) | Vmeso (%) | DAVG (nm) |
|---|---|---|---|---|---|
| a The highest porosity of PFAC that was applied as a metal catalyst support in palm oil deoxygenation. | |||||
| PF biochar | 178.35 | 0.104 | 35.52 | 64.48 | 3.13 |
| PFAC700-0.25 | 893.85 | 0.505 | 57.36 | 42.64 | 2.55 |
| PFAC700-0.5 | 717.26 | 0.398 | 61.09 | 38.91 | 1.98 |
| PFAC700-1 | 629.39 | 0.346 | 58.23 | 41.77 | 2.06 |
| PFAC750–0.25a | 1039.64 | 0.572 | 71.66 | 28.34 | 1.82 |
| PFAC750-0.5 | 937.31 | 0.486 | 60.31 | 39.69 | 1.99 |
| PFAC750-1 | 830.25 | 0.441 | 61.75 | 38.25 | 1.97 |
| PFAC800-0.25 | 975.21 | 0.517 | 68.63 | 31.37 | 1.89 |
| PFAC800-0.5 | 813.98 | 0.438 | 56.68 | 43.32 | 2.04 |
| PFAC800-1 | 705.33 | 0.392 | 63.69 | 36.31 | 1.98 |
The typical PFAC samples have total pore volume (Vtotal) of 0.346–0.572 cm3 g−1−1. However, PFACs also have a micropore volume of 56.68–71.66%, and mesopore volume (Vmeso) of 28.34–42.64%, respectively. The surface area (SBET) of all PFACs is higher than the PF biochar without activation. Technically, the activation process significantly develops the surface area of carbon materials. An increase in surface area and porosity enhancement depends on the activation parameters, such as activation temperature, dosage of activating agents, and types of activating agents.33,39,48 In this study, KOH was used as an activating agent, which is one of effective chemicals for well development of superior surface area and porosity in the porous carbon production.48,49
According to activation temperature, the effect of activation temperature (700, 750, and 800 °C) on SBET of PFACs are shown in Table 3. The pore characteristics of PFACs are significantly influenced. The increasing surface area and porosity are generally correlated with the increasing activation temperature to reach the highest porosity and SBET. However, the porosity and surface area might be decreased at the exceed activation temperature stage due to the excessive energy for activation reaction leads to the pore ablation and micropore sintering.48 Also, the KOH dosage is the crucial factor to maximize the SBET of PFACs. Furthermore, increase in KOH until excess exhibited the highest SBET and even their porosity. More excess activating agent gradually decreased in SBET because the optimum porous structures of PFACs, which completely formed at the excess KOH activation stage, was broken leading to the reduction of SBET and porosity.50 The PFAC750–0.25 has a superior pore structure than the others with the highest SBET of 1039.64 m2 g−1, and Vtotal of 0.572 cm3 g−1, respectively. Nonetheless, the activation using the condition of KOH ratio of 0.125 at 750 °C was also tested to assure that the condition of 750 °C using 0.25 KOH ratio is the real optimum condition to obtain PFAC with highest porosity. The results showed that PFAC750–0.125 has a SBET of 831.45 m2 g−1, and Vtotal of 0.446 cm3 g−1. In addition, the PFAC750–0.25 exhibited the special properties, such as high surface area, and porosity, high carbon content for its feasibility to be utilized as a support material. Therefore, the PFAC750–0.25 was selected as a supporter of metal phosphides on the producing green diesel via palm oil deoxygenation.
| Wavenumber (cm−1) | Functional group | Description |
|---|---|---|
| 3680–3000 | O–H stretching | Hydroxyl or carboxyl groups, alcohol from cellulose or phenols from lignin |
| 2925 | C–H stretching | Aliphatic |
| 1700 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching O stretching |
Carbonyl, ester or carboxyl from cellulose and lignin |
| 1600 | CC stretching |
Aromatic skeletal present in lignin |
| 1290–950 | C–O stretching | Ester from hemicellulose |
| 860–724 | C–H bending | Aromatic |
| <500 | Metal-O stretching | Inorganic compound (ash) |
 | ||
| Fig. 2 SEM micrographs of PF biochar and PFAC. | ||
As shown in the SEM micrographs, the observation revealed that the increase of activation temperature and KOH dosage significantly caused a changing with the appearance of PFAC surface morphology in terms of enhance the extensive external pore cavities, which are the entry way into internal pores (mesopores an micropores). However, the observation on the PFACs surface morphology is correlated with their porous structure analyzes. This suggests that the PFACs with high surface area can be observed the appearance of various pores on its external surface, as compared with Table 2.
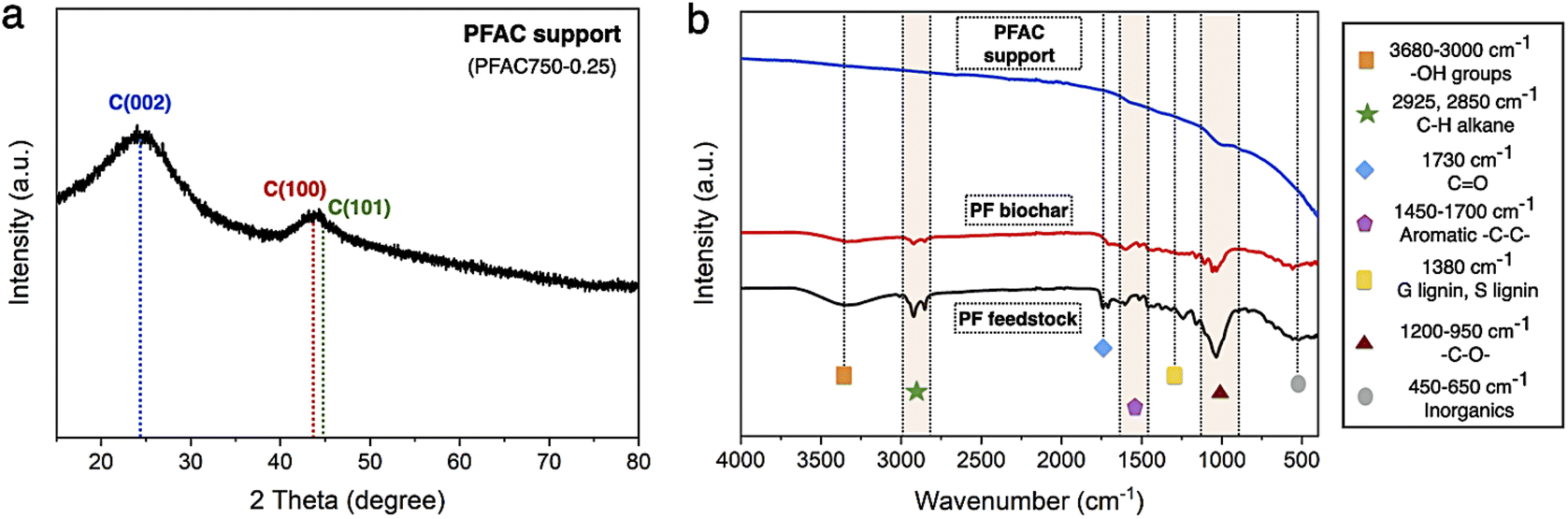 | ||
| Fig. 3 Characterization of PFAC support (a) XRD pattern, and (b) surface functional group analyzes. | ||
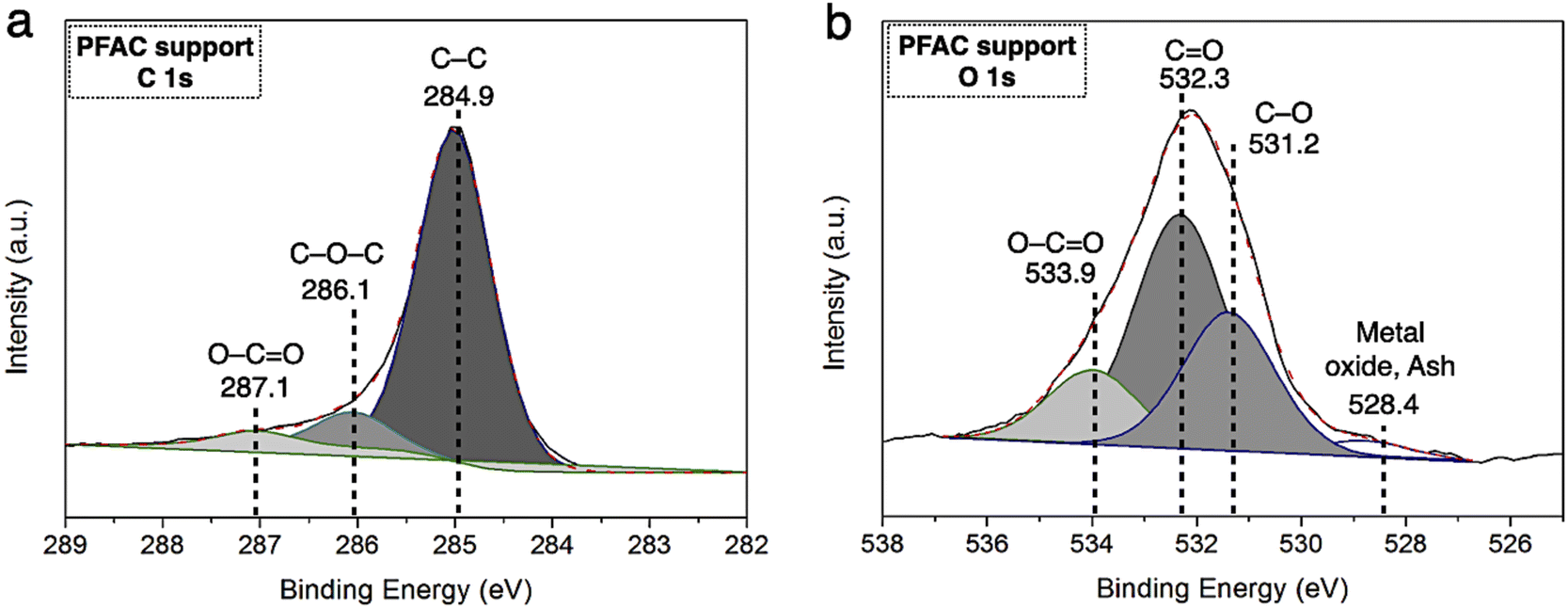 | ||
| Fig. 4 XPS spectra of PFAC support (a) C 1s spectra, and (b) O 1s spectra. | ||
In Fig. 3b, the FTIR spectra of PF feedstock, PF biochar, and PFAC support were determined by the infrared absorption spectra using direct transmittance in the wavenumber ranges from 4000–400 cm−1, where the surface functionalities of lignocellulose materials are explained in Table 2. FTIR analyzes shows the appearance of several chemical functions. The polycyclic aromatics from lignin in plant-based biomass are including –OH stretching of hydroxyl and carbonyl groups at 3680–3000 cm−1, and aromatic –CC– rings between 1600 and 1512 cm−1.51 Moreover, aliphatic C–H groups of polysaccharides in cellulose and hemicellulose was at the vibration bands between 2925 and 2850 cm−1.52 However, the stretching bands of lignin composition is also observed at 1380–1240 cm−1 and 1460 cm−1. In addition, the band of CO, and –C–O– vibrations correspond to cellulose and hemicellulose structure was at 1550–1730 cm−1, and 1200 and 950 cm−1, respectively. Some inorganic molecules or metal compounds, which known as ash could be necessary observed at 500 cm−1 of the intense band.51,52 Finding results exhibited the similar FTIR spectra frameworks of PF feedstock, PF biochar, and PFAC support, as displayed in Fig. 3b. In FTIR spectra of PFAC support, the symmetrical stretching of organic functional groups, which greatly disappeared from the observed spectra because the surface functional groups were successfully decomposed. The decrease in the observed FTIR intensity was occurred by thermal decomposition of lignocellulosic components at high temperature during PFAC manufacturing.53 Moreover, the FTIR spectra of PMFC cannot observed on the relative intense bands below 500 cm−1, indicating that PFAC support consisted of a low ash content.
The resolutions of C 1s and O 1s spectrum of PFAC support (PFAC750-0.25), which are obtained by XPS analysis, are displayed in Fig. 4. In Fig. 4a, C 1s spectrum can be revolved into three component peaks which represented the peak intensity of graphitic carbon (–CC–, 284.75 eV), the group of carbon in alcohol and/or ether linkage (C–O–C, 286.23 eV) and carbon in carbonyl group (O–CO, 288.79 eV), respectively. The high intensity peaks centered at 284.75 eV, suggesting that PFAC support has a high proportion of graphitic carbon on its surface.39,45 Also, it can be indicated that PFAC support composed of high C content. This could be larger content than PF feedstock and PF biochar. These results are consistence with the ultimate analyzes (Table 1). Besides, O 1s spectrum (Fig. 4b) exhibited the four relevant spectra representing C–O in phenols and ethers groups at 531.2 eV. Also, the organic CO in carboxylic acid and/or ester group was centered 532.3 eV.45 The O–CO of organic molecule in carbon support was demonstrated at 534.0 eV, and metal oxide or inorganic molecules ascribes to ash content in carbon support was at 528.4 eV.45,54 It was found that PFAC support (PFAC750-0.25) has a low shoulder peak, as seen in O 1s spectrum. This can be implied that PFAC support contains a low ash content. This result is consisted with the information of chemical composition from proximate analysis, as shown in Table 1.
3.2 Catalyst characterization
Fig. 5a shows the N2 adsorption–desorption isotherm of reduced metal phosphide catalysts before testing on palm oil deoxygenation. The sorption isotherms of both Ni–P/PFAC and Fe–P/PFAC catalysts obviously has a mixing of Type I and Type IV characteristics according to the IUPAC classification, implying that the obtained catalysts show a combination of microporous and mesoporous structures.54,55 The adsorbed volumes on supported Fe–P and Ni–P are lower than the bare PFAC support due to the instead of metal nanoparticles on the carbon surface. This could be decrease in the pore volume (seen in Tables 3 and 4). Nonetheless, the reduction of catalysts before characterization and reaction testing may decrease in total pore volume because of the pore collapse by thermal decomposition during high temperature operation.56,57 The surface area of Ni–P/PFAC and Fe–P/PFAC catalysts are 718.65 m2 g−1 and 850.92 m2 g−1, respectively (Table 4). Also, the pore size distributions of corresponding catalysts are displayed in Fig. 5b. The size distributions are in the range of 0.5–2.5 nm, which the main pore size distribution was in micropores range (<2 nm), and the narrow mesopore size distribution occurred at 1.9–2.5 nm. However, Table 3 also presented that a higher proportion of micropore volume than mesopore were observed from the obtained catalysts. The high porosity and surface area of synthesized catalysts can be implied that very well dispersion of metal nanoparticles was achieved on the PFAC surface. This indicates the essential characteristics of metal catalyst for catalytic deoxygenation resulting in excellent catalyst activity.58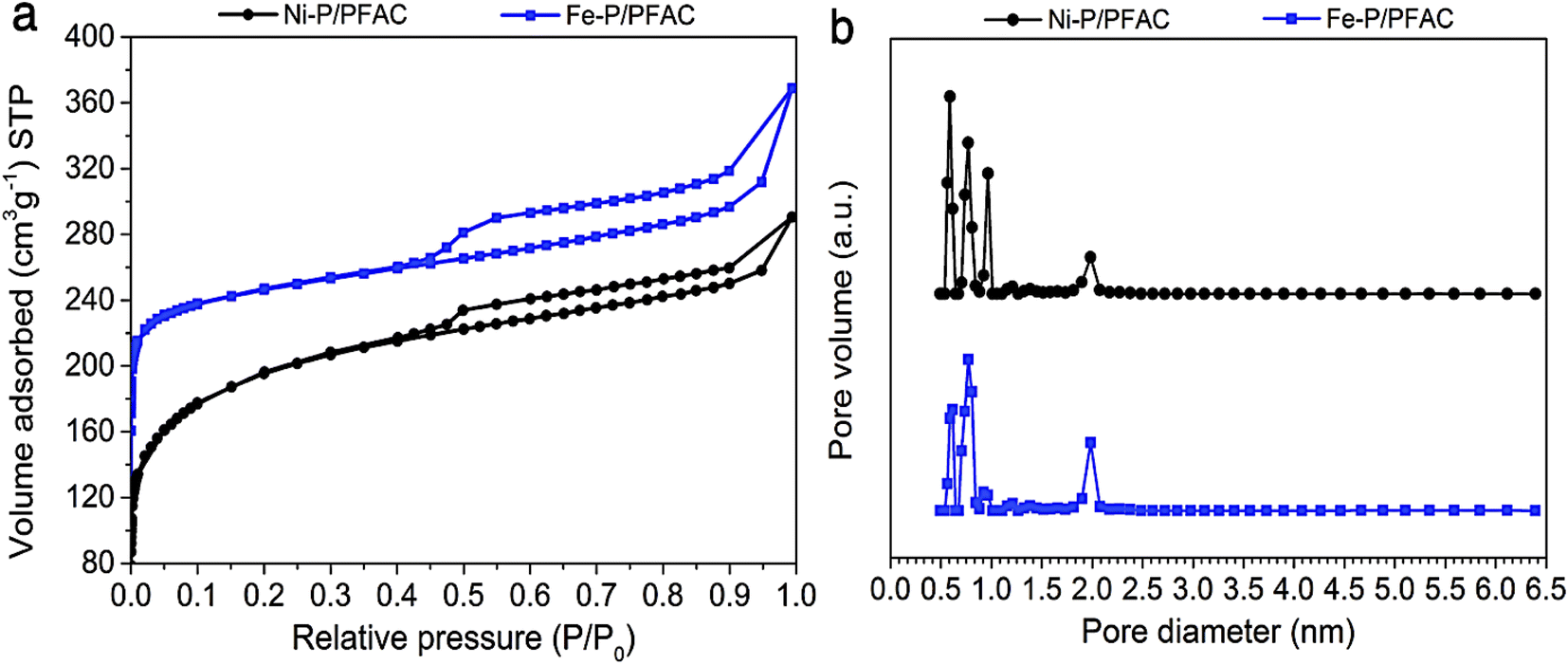 | ||
| Fig. 5 Porosity analyzes of studied catalysts (a) N2 adsorption–desorption isotherms, and (b) Pore size distribution curves. | ||
| Catalysts | SBET (m2 g−1) | VT (cm3 g−1) | Vmic (%) | Vmes (%) | Crystallite size (nm) | NH3 uptake (μmol g−1) |
|---|---|---|---|---|---|---|
| Ni-P/PFAC | 718.65 | 0.437 | 68.29 | 31.71 | 10.58 | 119.54 |
| Fe-P/PFAC | 850.92 | 0.511 | 61.45 | 38.55 | 6.13 | 144.72 |
According to morphology observation of catalysts, the SEM, TEM, and particle size distribution of both supported Ni–P and Fe–P are displayed in Fig. 6. In the same implication on SEM micrograph, the clusters of spherical shape of both Ni–P and Fe–P nanoparticles were obtained with well distribution on carbon support, as seen in Fig. 6a and b, respectively. This result implies that the active sites is significantly increased resulting in the improvement of catalyst performance. Additionally, the observation on dark spots ascribes to Ni–P and Fe–P is also shown in TEM image (Fig. 6c and d). This indicated that Ni–P and Fe–P nanoparticles are well dispersed on both the surface and inside of PFAC support. However, the low scattering contrast of the dark spots referred to the corresponding Ni–P and Fe–P were observed. It was implied that metal phosphides nanoparticles probably locate both inside and outside pores of PFAC supports. The porosity and surface area of PFAC support were gradually decreased after the deposition of metal phosphide, indicating that the metal phosphide nanoparticles might cause the pore filling.59,60
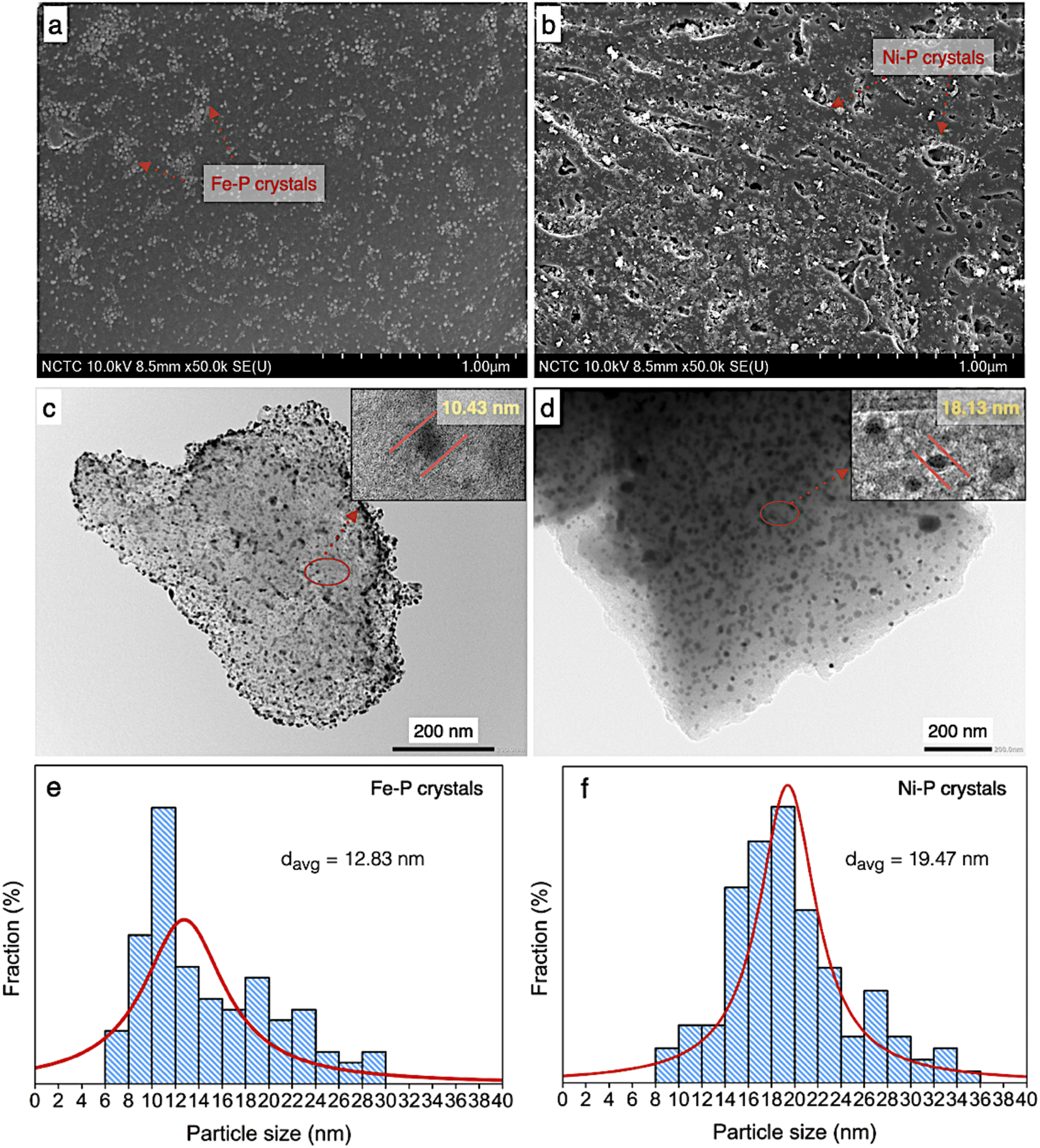 | ||
| Fig. 6 Physical morphology of PFAC supported Ni–P and Fe–P catalysts; FESEM images of (a) Fe–P/PFAC, (b) Ni–P/PFAC, TEM images of (c) Fe–P/PFAC and, (d) Ni–P/PFAC, and particle size distribution (e and f). | ||
According to TEM micrographs, the Fe–P nanoparticles seemingly better disperse on the PFAC in comparison to Ni–P. The average particles sizes and particle size distribution (Fig. 6e and f) are also correlates with TEM results. Fe–P and Ni–P nanoparticles has an average size of 12.83 and 19.47 nm, respectively. Moreover, the particle size distribution of Fe–P was observed between 6 and 30 nm, which the main distribution is at 6–14 nm. In contrast, the particle size distribution of Ni–P was found with shifting on the right axis within 8–36 nm. The distribution of Ni–P size was mainly between 14 and 24 nm. Besides, the crystallite sizes from XRD analysis (Table 4), which calculated by using Debye–Scherrer equation, showed that Fe–P and Ni–P nanoparticles is approximately 6.13 and 10.58 nm, respectively. Remarkably, well dispersion of metal phosphide crystals results in high active surface area of obtained catalysts is a promising characteristic for enhancement of catalyst activity. This can be developed from porous carbon materials derive from agricultural waste.61
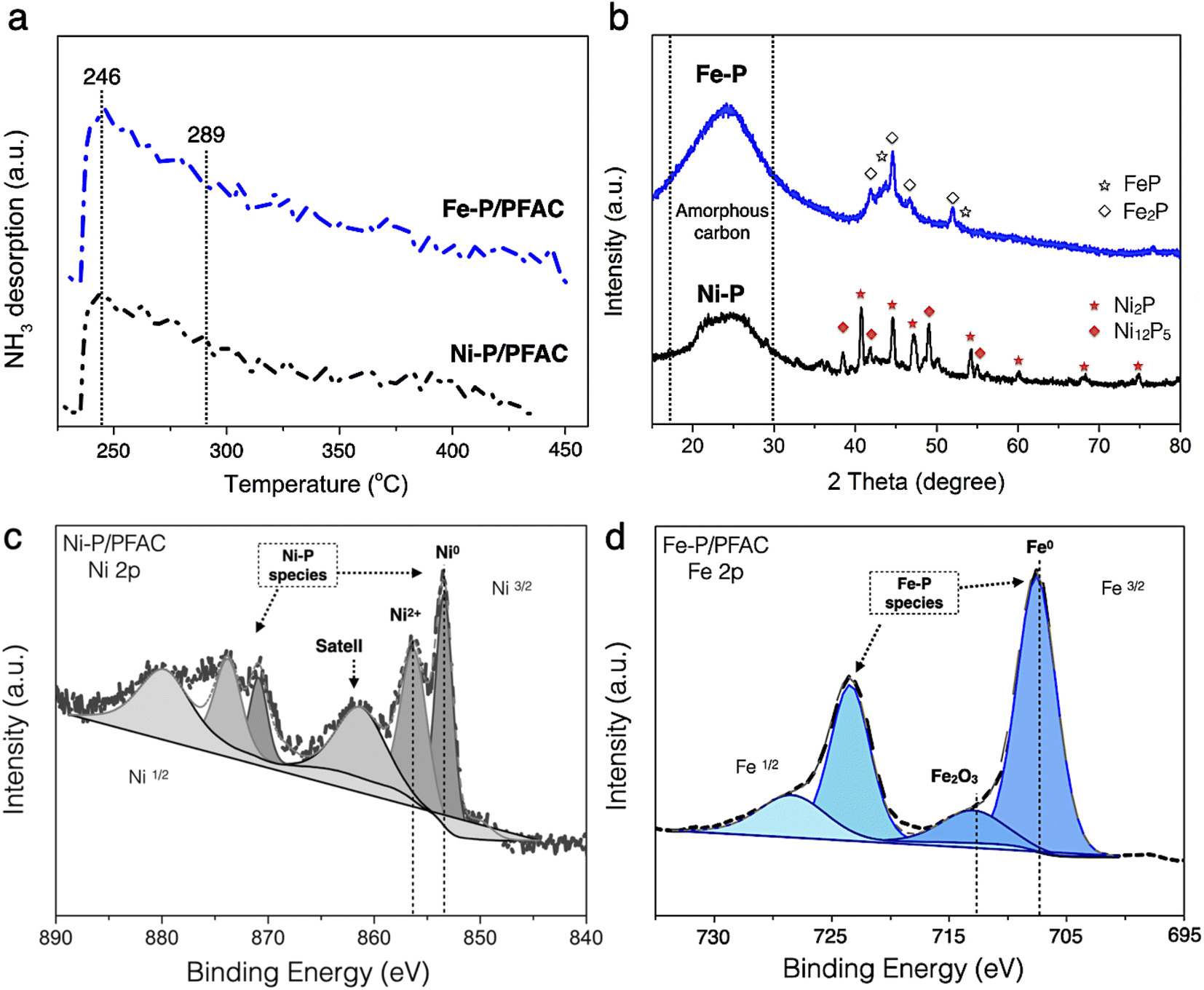
The acidity of obtained catalysts after reduction process is shown in the NH3-TPD profiles, as seen in Fig. 7a. The amount of desorbed NH3 is reported in Table 4. The profiles of desorbed NH3 of corresponding catalysts have a dominant desorbed peak at approximately 246 °C that correlated with the characteristic of NH3-desorption at the low temperature between 100 and 250 °C corresponds to weak acidity, the Brønsted acid site, particularly conducted from the surface of M−OH and P–OH groups in unreduced or phosphates species of catalysts.62 This might occur during preparation for the catalyst characterizations. However, the strong shoulder peak also observed at the temperature around 289 °C corresponds to moderate and strong acid sites (the Lewis acid), which is specifically attributed to the characteristic of some Mδ+ charges of Ni–P and Fe–P nanoparticles. This characteristic could be due to a greater electron mobility in the metal phosphide structure.63 Consuelo Alvarez-Galval et al.64 also reported that the intense NH3 desorption peak of reduced metal phosphides was found in the ranges of moderate and strong acid sites (>250 °C). As the observation, the NH3 desorption peak of supported Fe–P catalyst showed a strong intense and broader shoulder peaks at high temperature than Ni–P/PFAC catalyst, which also showed the larger acid sites and stronger acidic were obtained from Fe–P/PFAC. Also, the total desorbed NH3 amount of Fe–P/PFAC and Ni–P/PFAC catalysts were obtained approximately 144.72 and 119.54 μmol g−1, respectively. This consequence is due to the better dispersion and smaller size of Fe–P crystals resulted in the increasing NH3 adsorption on Fe–P surface.64,65 Moreover, strong acidity catalysts significantly exhibited an excellent activity in deoxygenation of oxygenated compounds since the high acidity is favored to the cleavage of C–C and C–O bonds. In addition, the hydrocracking reaction for efficient production of light hydrocarbons is also represented in the similar contribution.66,67
 | ||
| Fig. 7 Characterization on of PFAC supported Ni–P and Fe–P catalysts (a) temperature programed desorption (NH3-TPD), (b) XRD pattern of studied catalysts, (c) XPS spectra of Ni–P/PFAC and, (d) XPS spectra of Fe–P/PFAC. | ||
Fig. 7b shows the XRD diffraction pattern of obtained catalysts at the 2θ angle 15–80°. The broad peak of diffraction between 15–30° could be ascribed to amorphous carbon of PFAC support, as illustrated in Fig. 3a. Additionally, average crystallite size of metal phosphide was calculated by the Scherrer equation using XRD peak intensity of relative catalyst, as listed in Table 4. The average crystallite size of Fe–P and Ni–P was about 6.13 and 10.58 nm, respectively. The average domain sizes of corresponding catalysts were correlated to the particle sizes from the TEM analysis (Fig. 6). The XRD patterns of both supported metal phosphide catalysts were a sharp peak with high intensity suggesting that the crystalline phase was obtained. The supported Ni–P catalyst was observed the intense peak at 2θ angle of 35–75°. The intense peaks at 2θ of 40.7, 44.6, 47.4, 54.2, 59.9, 69.7, and 74.6° were mainly Ni2P crystal (PDF 65-1989), also, low intensity peaks at 2θ of 38.3, 42.2, 49.9, and 55.0° corresponds to Ni12P5 (PDF 22-1190). The transformation of Ni2P to Ni12P5 could be occurred during pyrolysis and reduction process in the catalyst synthesis.68 According to supported Fe–P catalyst, the XRD pattern was an overlapped diffraction lines between 2θ angle of 40–55°, which ascribed to the Fe2P (PDF 85-1725) (2θ = 41.8, 44.5, 46.9, and 52.1°), and FeP (PDF 81-1173) (2θ = 43.9 and 53.7°) phases.64 This also indicates that the Fe2P could be transform to FeP during catalyst preparation. The high temperature and long resident time in the preparation might influence on the ratio of metal/P by partial releasing phosphorus as a volatile species such as phosphine (PH3) resulted in the phase transformation.69
The surface chemistry of corresponding catalysts in a reduced form were observed by XPS, as displayed in Fig. 7c and d. The XPS spectra were reported at binding energies of Ni 2p3/2 and Fe 2p3/2 core levels, respectively. The binding energy of Ni 2p was separated in two main contributions at 853.7 eV, attributed to metallic nickel (Niδ+ species) in metal phosphide, and approximately 856.4 eV, which was accompanied by a broad satellite peak is assigned to Ni2+ in unreduced PO3−/PO4−, respectively.68–70 Noted that, the Ni2+ in unreduced probably formed by re-oxidation with air during the catalyst preparation for XPS analysis.70 According to supported Fe–P catalyst, the Fe 2p3/2 spectra are also shown in two main contributions. The sharp peak at binding energy of 707 eV is corresponded to the binding energy of metallic iron (Feδ+) in iron phosphide crystals (FeP and Fe2P). However, the peak centered at binding energy around 712.6 eV is ascribed to a satellite peak of the oxide layer (Fe2O3) on iron surface.62,71 Regarding to XPS results, it can be implied that both phosphate and metal oxide species is probably formed by oxidation of catalyst surface during XPS sample preparation. Also, the formation of unreduced species might represent by an incomplete reduction process.70,71 This result is a good agreement with the results of XPS analysis reported by Lui et al.,70 and Dong et al.71
3.3 Test of catalyst performance in deoxygenation of palm oil
In DO of palm oil, the relative supported metal phosphide catalysts were evaluated on its catalytic performance for producing of green diesel fuel due to the individual properties of each catalyst, with higher porosity, greater acidity, could be ascribed to the high efficiency alternative catalyst. The palm oil conversion was completely obtained in the trickle bed down-flow reactor at the deoxygenation temperatures of 340–400 °C under the H2 pressure of 50 bar using H2 flow of 100 mL min−1, and the palm oil was fed at the liquid hourly space velocity (LHSV) of 1 h−1 by using HPLC pump, as seen in schematic process diagram on Fig. 8. However, the purpose of reaction pathway according to the product distribution during the conversion of palm oil over metal phosphide was clearly illustrated in Fig. 9. Firstly, the unsaturated triglyceride in palm oil were hydrogenated to saturated triglyceride and were then degraded to fatty acid molecules (FFAs) and propane gas as a by-product by hydrogenolysis reaction. Before the deoxygenation stage, the FFAs was probably transformed to larger FFAs molecules via oligomerization, which could be assured by the appearance of long-chain hydrocarbon products after deoxygenation.72 The green diesel fuel and other hydrocarbons were obtained from the deoxygenation of FFAs through three main reactions (i.e. HDO, DCO, DCO2), simultaneously. HDO reaction required an excess H2 consumption to produce hydrocarbons with the same number of carbon atoms in feedstock by the cleavage of C–O bonding as well as the water as a by-product. For DCO reaction, H2 was partially used to form hydrocarbon molecules with CO and water as a by-product, while DCO2 reaction does not required H2 because the hydrocarbons were produced by C–C bond cleavage with releasing CO2 resulted in odd number of carbon atom hydrocarbons.73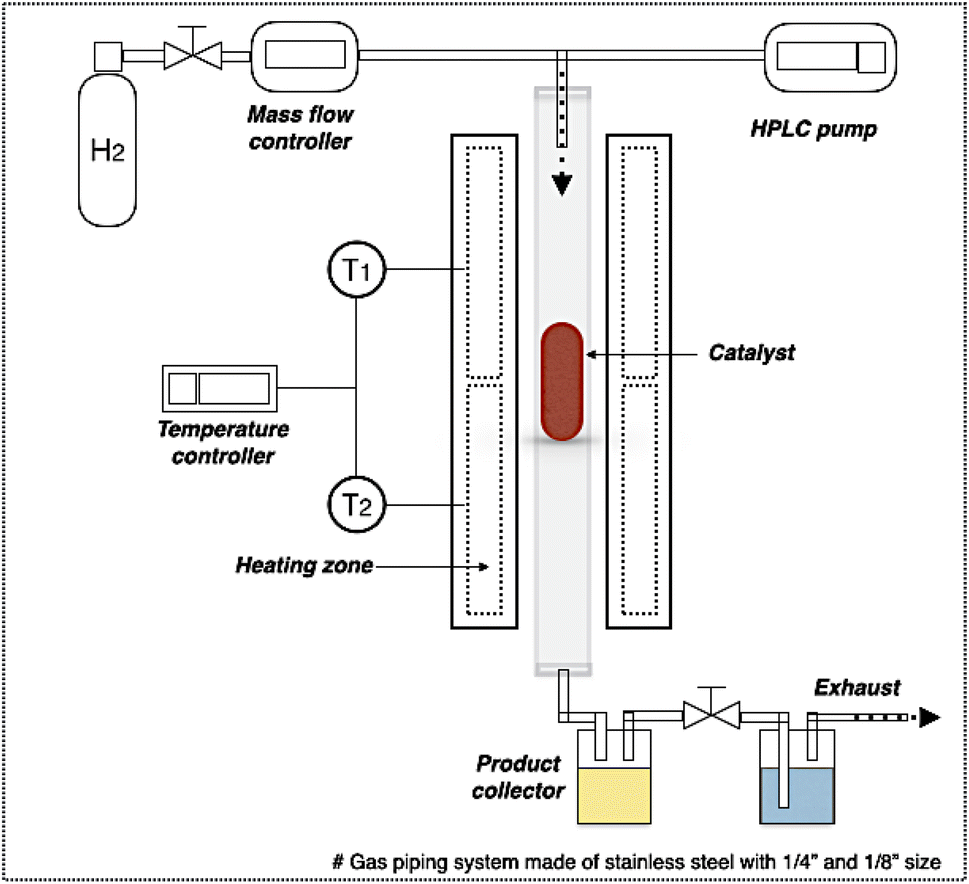 | ||
| Fig. 8 Schematic diagram of deoxygenation unit. | ||
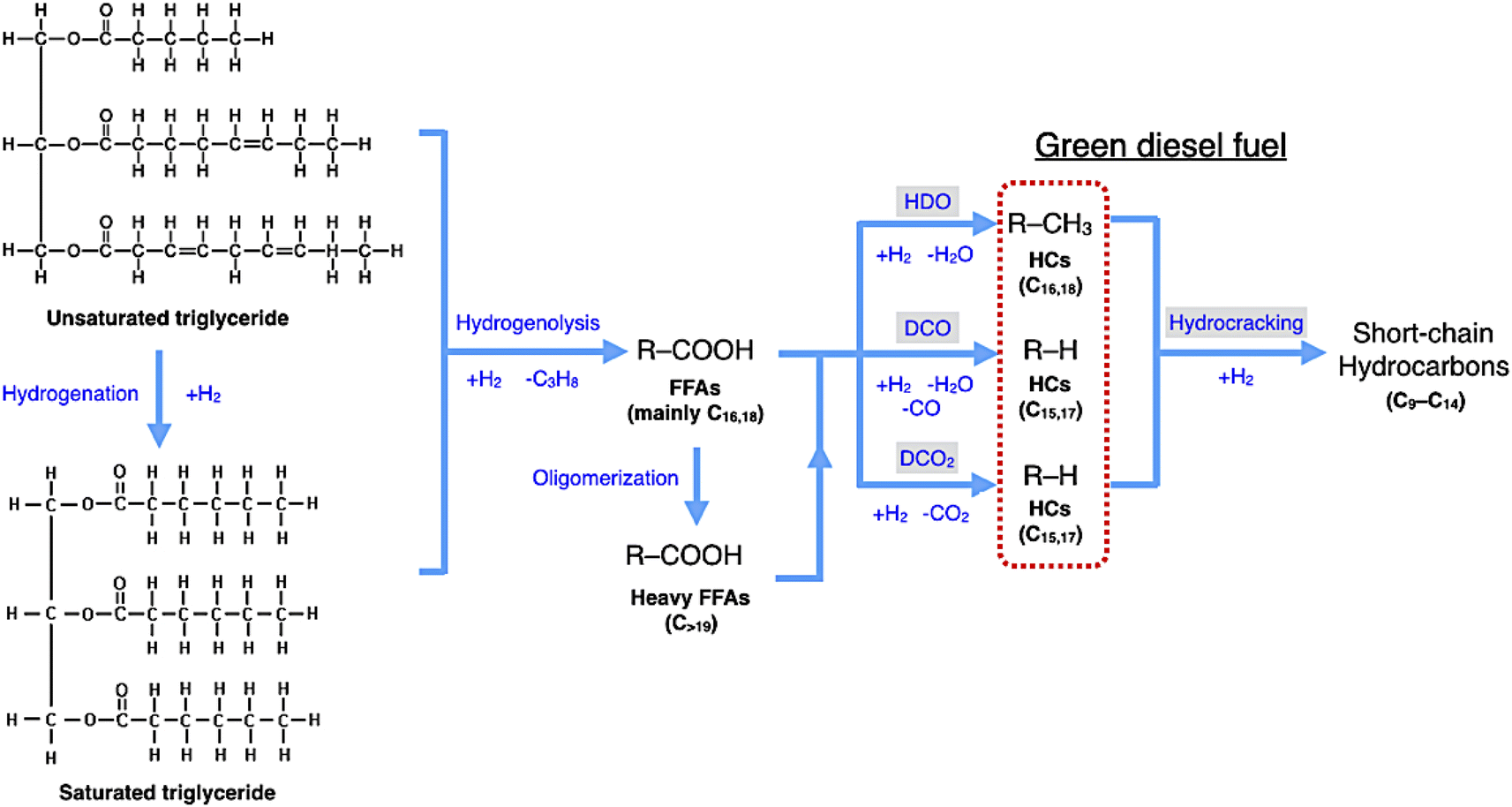 | ||
| Fig. 9 Reaction pathway of palm oil conversion to green diesel. | ||
Additionally, Fig. 10 represented palm oil conversion, yield of liquid hydrocarbon product, and selectivity towards green diesel obtained from different reaction temperatures of 340–400 °C by using Ni–P/PFAC and Fe–P/PFAC catalysts. According to the deoxygenation over Ni–P/PFAC, the reaction at 340 and 360 °C revealed the conversion about 97.36 and 99.69%, while the complete conversions (100%) were obtained from 380 and 400 °C, respectively (Fig. 10a). The incomplete conversion at the reaction temperature below 350 °C in a continuous flow system was consistent with the report from previous study that an incomplete conversion was significantly obtained at low-temperature operation because some intermediates in oxygenated compounds, such as palmitic acid, and stearic acid in the liquid hydrocarbon product could not undergo further to produce hydrocarbons by HDO, DCO, and DCO2 reactions.73 The evidence implied that supported Ni–P catalyst requires a higher reaction temperature in conversion of vegetable oils for biofuel production.70,71,73 In contrast, 100% conversion of palm oil was obtained at the reaction temperatures of 340–400 °C in the deoxygenation over Fe–P/PFAC, as seen in Fig. 10a. This finding implies that iron phosphide has a superior catalyst activity in deoxygenation. However, there are no recent studies reported the catalytic performance of iron phosphide in producing green diesel.
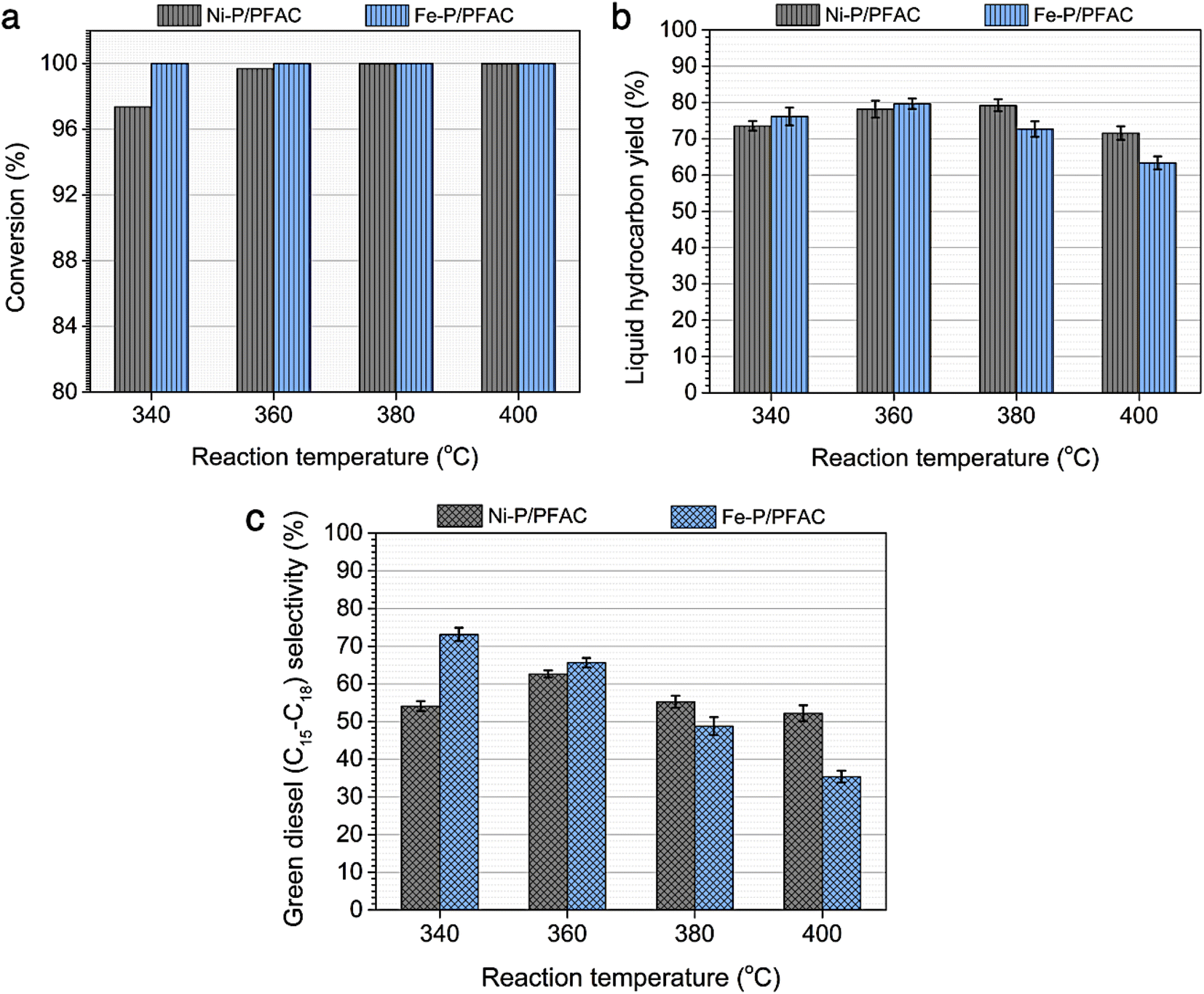 | ||
| Fig. 10 Performance study of Ni–P/PFAC and Fe–P/PFAC catalysts in palm oil deoxygenation for green diesel production at different reaction temperatures (a) palm oil conversion, (b) liquid hydrocarbon yield, and (c) green diesel selectivity. | ||
In addition, liquid hydrocarbon yield obtained from the corresponding catalysts are summarized in Fig. 10b. The study of catalytic activity on liquid hydrocarbon from palm oil were compared between Ni–P/PFAC and Fe–P/PFAC catalysts, which obtained at the different testing condition in DO with 100% conversion. The highest liquid hydrocarbon yield of palm oil deoxygenation over Fe–P/PFAC catalyst is approximately 79.65% (obtained at 360 °C). While the total liquid hydrocarbon yield obtained by using Ni–P/PFAC catalyst is about 79.21% (obtained at 380 °C). The liquid hydrocarbon yield was continuously decreased with increasing reaction temperature, due to the decomposition of palm oil and intermediated to gaseous hydrocarbon occurred at high-temperature condition by hydrocracking reaction.74 However, the finding results show the beneficial role of Fe–P catalyst at lower-temperature of operating condition compared to Ni–P catalyst. The green diesel (n-C15–C18) selectivity towards carried out from the Ni–P/PFAC and Fe–P/PFAC catalysts at varied deoxygenation temperatures are illustrated in Fig. 10c. According to the deoxygenation over supported Ni–P catalyst, the green diesel selectivity is approximately 52.2–62.6%, while the highest green diesel selectivity (62.6%) was obtained at the reaction temperature of 360 °C. The green diesel fraction was significantly increased by adjusting reaction temperature from 340 to 360 °C. This correlated with the complete palm oil conversion, as seen in Fig. 10a. Additionally, when the reaction temperature was shifted to 380 and 400 °C, the decrease in selectivity of green diesel was obtained. This is due to the hydrocracking of long-chain hydrocarbon significantly occurred at high temperature condition.74
However, the superior catalytic activity of supported Fe–P catalyst compared to supported Ni–P catalyst is certainly due to strong acidic characteristic and well dispersion resulted in higher surface of active sites. The highest green diesel selectivity (73.12%) was obtained at lower deoxygenation temperature, compared to Ni–P supported on carbon catalyst. This could be summarized that Fe–P/PFAC catalyst is effective for the deoxygenation at low temperature conditions to maximize the green diesel fraction. In contrast, the selectivity of green diesel was gradually decreased at the reaction temperature greater than 360 °C owning to the transformation of long-chain hydrocarbons to shorter-chain hydrocarbons at high-temperature operation during palm oil deoxygenation over supported Fe–P catalyst. This result shows the potential of support Fe–P catalyst that can be used as a promising alternative for hydrocracking in the production of light hydrocarbon, such as bio-gasoline and bio-jet fuel.64,74,75
Fig. 11 shows the hydrocarbon composition based on carbon number in green diesel fuels. As we known that, palm oil feed stock is mainly composed by unsaturated triglycerides, likes palmitic acid (C16:0, 37.8%) and oleic acid (C18:1, 45.8%).74 Therefore, the major hydrocarbon products should be C16 and C18 hydrocarbons followed the chemical structure of oil feedstock. However, the product distribution obtained from supported Ni–P and Fe–P catalysts is mainly C15 and C17 hydrocarbons. The amount of C15 and C17 hydrocarbons is approximately two times greater than C16 and C18 hydrocarbons. This evidence suggests that the deoxygenation of palm oil over the corresponding catalysts is transformed though the decarbonylation and decarboxylation as a major reaction. However, the hydrodeoxygenation also existed in the reaction route simultaneously, that is ensured by the formation of C16 and C18 hydrocarbons as a minor product.75,76 In deoxygenation over Ni–P/PFAC catalyst, the major products including C15 and C17 hydrocarbons was catalyzed by DCO/DCO2 due to the presence of Ni2P as a dominant active phase. These results are in good correlation with the study from Zhou et al.76 that reveal the Ni2P is favorable for decarbonylation through the scission of C–C bonding resulted in the hydrocarbon products with one less carbon atom (Cn−1) as the main product. Moreover, the Ni12P5 species exhibited both DCO and HDO simultaneously showing that the hydrocarbon products with similar carbon number of triglycerides in palm oil feedstock (Cn) is also observed. Furthermore, the hydrocarbon product obtained by using supported Fe–P catalyst exhibited the same product distribution (C15 and C17 hydrocarbons). This result implies that the DCO/DCO2 reactions are mainly crucial roles in deoxygenation of palm oil over supported Fe–P catalyst since the high acidity of Fe–P catalyst is important for catalyzing C–O and C–C cleavage to conduct Cn−1 hydrocarbons.75,77 Additionally, recent study also ensured that the DCO/DCO2 pathways was the favor reaction in deoxygenation over Fe phosphide catalyst.77 One remarkable, the observation from XRD analyzes demonstrated that Fe–P/PFAC catalyst particularly consists of the Fe2P active phase. This finding can be implied that Fe2P species show the similar catalytic behavior with Ni2P phase.
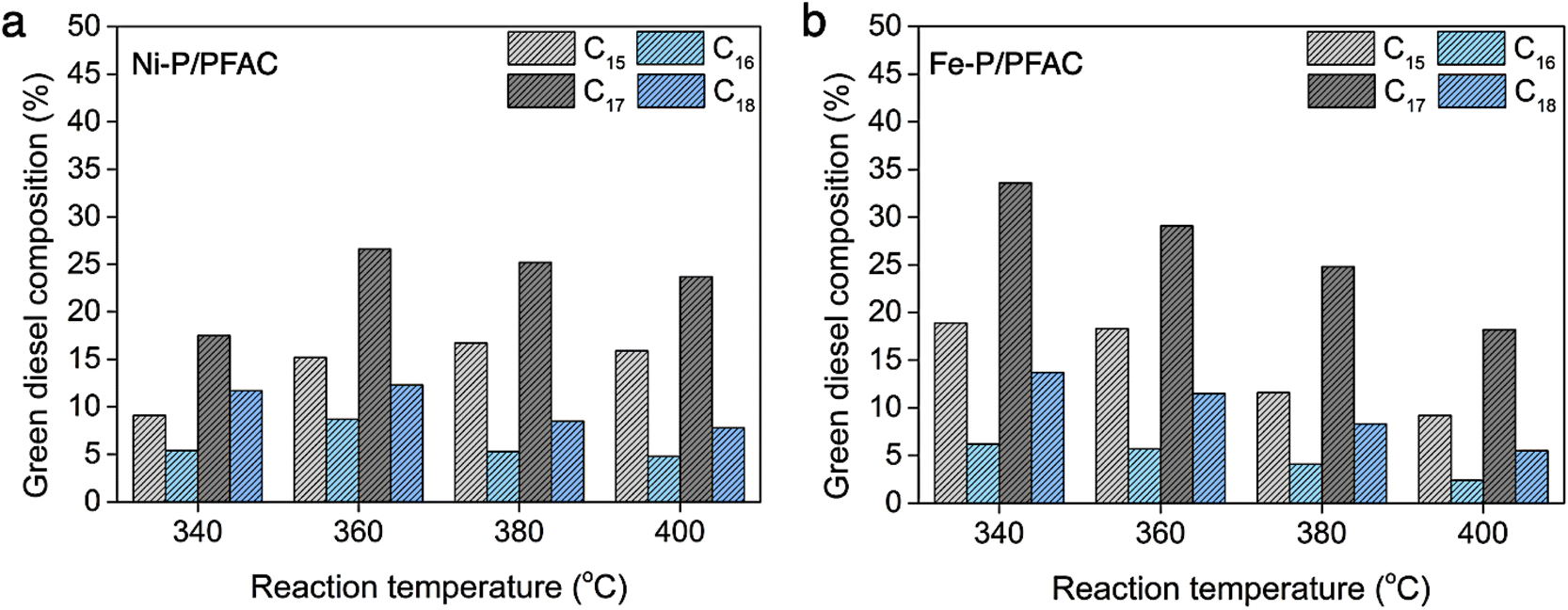 | ||
| Fig. 11 Green diesel composition based on carbon number (a) obtained by using Ni-P/PFAC, and (b) obtained by using Fe-P/PFAC. | ||
Besides, the comparison of current study with several literatures on the green diesel production over different catalysts and experimental conditions are listed in Table 5. In this study, the optimum condition of green diesel production from palm oil by catalytic deoxygenation is at reaction temperature of 340 °C over Fe–P/PFAC catalyst, providing the highest green diesel selectivity. This result could be implied that supported iron phosphide catalysts is a promising for the conversion of triglycerides into renewable diesel fuel due to lower cost than those noble metals, and excellent catalyst activity for production of biofuels.
| Catalyst | Reactant | Condition | GD selectivity (%) | Ref. |
|---|---|---|---|---|
| 30 wt% Ni–Co/MWCNT | Jatropha curcas oil | 350 °C, T = 1 h, 5% loading 10 mbar (Batch reactor) | 64 | 9 |
| 20 wt% NiO–ZnO | By-product (palm oil refining process) | 350 °C, T = 2 h, 5% loading, N2 atmosphere (Batch reactor) | 86 | 10 |
| 20 wt% MoP/SBA-15 | Methyl ester | 290 °C, H2 = 30 bar, WHSV = 20.1 h−1 (fixed bed, micro reactor) | <90% | 69 |
| 5 wt% Pd–Fe/Al2O3 | Palm oil | 400 °C, T = 2 h, H2 = 60 bar, 1% loading (High pressure batch reactor) | 62.2 | 75 |
| 5 wt% Ni–Co/SBA-15 | Palm oil | 350 °C, T = 2 h, 10% loading (without H2, Batch reactor) | 78 | 55 |
| Fe–P/activated biochar (10 wt% of FeP) | Palm oil | 340 °C, H2 = 50 bar, LHSV = 1 h−1 of palm oil feed (continuous-flow reactor) | 73.1 | This work |
4. Conclusions
Palm fiber waste derived activated biochar (PFAC) was successfully prepared by KOH activation process. The PFAC is efficiently applied as a catalyst support due to its special characteristics, such as high specific surface area and good mechanical strength. The synthesis of supported Ni–P and Fe–P catalysts was done by the wetness impregnation technique. The application of activated biochar as a support material could enhance metal phosphide dispersion resulted in higher catalyst activity. Green diesel fuel was completely produced over PFAC supported Ni–P and Fe–P catalysts in palm oil deoxygenation. This study shows that palm fiber waste derived activated biochar is suitable to be used as a support material for Ni–P and Fe–P catalysts in the catalytic deoxygenation of palm oil for producing green diesel fuel. The highest selectivity towards green diesel (73.1%) was obtained by using supported Fe–P catalysts at the deoxygenation temperature of 340 °C with a complete of palm oil conversion. The second highest green diesel selectivity (62.6%) was obtained by using supported Ni–P catalysts obtained at the reaction temperature of 360 °C. Interestingly, the liquid hydrocarbon product is mainly composed of C15 and C17 hydrocarbons indicating that high proportion Ni2P and Fe2P pure phases on supported metal phosphide catalysts are favored to DCO/DCO2 reactions. One remarkable result is the hydrocracking of long-chain hydrocarbon could be occurred at the condition of high-reaction temperature. Although, the deoxygenation performance of PFAC supported metal phosphide catalysts is not superior when compared to those supported noble metal catalysts (i.e. Pt-, Pd-, Rh-based catalysts), the synthesized catalyst exhibited better green diesel selectivity than 5 wt% Pd–Fe/Al2O3 (seen in Table 5). Hence, the further improvement of deoxygenation activity for biofuel production is still challenge on the catalyst selection, synthesis condition, and performance testing. From findings, this study provides the beneficial knowledges, which full fill the information gaps reference to the potential utilization of waste biomass derived porous carbon applied as a support material for DO catalysts. Nonetheless, the development of desired product contributions in the yield and selectivity of green diesel could be achieved by optimizing and find-tuning experimental variables, such as types of catalysts, loading content, and reaction conditions in palm oil deoxygenation for further study.Authors contributions
Napat Kaewtrakulchai: conceptualization, formal analysis, investigation, data curation, writing–original draft Masayoshi Fuji: conceptualization Apiluck Eiad-ua: writing–review and editing, conceptualization, supervision funding. This research received no external fundingConflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this publication.Acknowledgements
The authors acknowledge the post-doctoral research fund (KREF146407), and the College of Materials Innovation and Technology, King Mongkut's Institute of Technology Ladkrabang, Bangkok, for their laboratory, and facility financial support.References
- C. A. Scaldaferri and V. M. D. Pasa, Production of jet fuel and green diesel range biohydrocarbons by hydroprocessing of soybean oil over niobium phosphate catalyst, Fuel, 2019, 245, 458–466 CrossRef CAS.
- P. Vignesh, et al., Biodiesel and green diesel generation: an overview, Oil Gas Sci. Technol., 2021, 76 Search PubMed.
- H. I. Mahdi, et al., Catalytic deoxygenation of palm oil and its residue in green diesel production: A current technological review, Chem. Eng. Res. Des., 2021, 174, 158–187 CrossRef CAS.
- O. D. Onukwuli, C. Esonye and A. U. Ofoefule, Combustion exhaust release impact, diesel engine performance, and optimization studies of green diesel-petrodiesel blend in a high compression ratio direct-injection compression-ignition engine, Adv. Mech. Eng., 2021, 13(5), 1–25 Search PubMed.
- G. Di Vito Nolfi, K. Gallucci and L. Rossi, Green Diesel Production by Catalytic Hydrodeoxygenation of Vegetables Oils, Int. J. Environ. Res. Public Health, 2021, 18(24), 1–28 Search PubMed.
- A. Afshar Taromi and S. Kaliaguine, Green diesel production via continuous hydrotreatment of triglycerides over mesostructured γ-alumina supported NiMo/CoMo catalysts, Fuel Process. Technol., 2018, 171, 20–30 CrossRef CAS.
- F. Wang, et al., Activated carbon supported molybdenum and tungsten carbides for hydrotreatment of fatty acids into green diesel, Fuel, 2018, 228, 103–111 CrossRef CAS.
- M. Ameen, et al., Process optimization of green diesel selectivity and understanding of reaction intermediates, Renewable Energy, 2020, 149, 1092–1106 CrossRef CAS.
- N. Asikin-Mijan, et al., Production of green diesel via cleaner catalytic deoxygenation of Jatropha curcas oil, J. Cleaner Prod., 2017, 167, 1048–1059 CrossRef CAS.
- K. B. Baharudin, et al., Renewable diesel via solventless and hydrogen-free catalytic deoxygenation of palm fatty acid distillate, J. Cleaner Prod., 2020, 274 Search PubMed.
- M. Jin, et al., Coproduction of Value-Added Lube Base Oil and Green Diesel from Natural Triglycerides via a Simple Two-Step Process, Ind. Eng. Chem. Res., 2020, 59(19), 8946–8954 CrossRef CAS.
- H. Liu, et al., Catalytic Hydrodeoxygenation of Methyl Stearate and Microbial Lipids to Diesel-Range Alkanes over Pd/HPA-SiO2 Catalysts, Ind. Eng. Chem. Res., 2020, 59(39), 17440–17450 CrossRef CAS.
- M. Ameen, et al., Catalytic hydrodeoxygenation of rubber seed oil over sonochemically synthesized Ni-Mo/gamma-Al2O3 catalyst for green diesel production, Ultrason. Sonochem., 2019, 51, 90–102 CrossRef CAS PubMed.
- M. S. Gamal, et al., Effective catalytic deoxygenation of palm fatty acid distillate for green diesel production under hydrogen-free atmosphere over bimetallic catalyst CoMo supported on activated carbon, Fuel Process. Technol., 2020, 208 Search PubMed.
- S. J. Patil and P. D. Vaidya, Production of Hydrotreated Jatropha Oil Using Co–Mo and Ni–Mo Catalysts and Its Blending with Petroleum Diesel, Energy Fuels, 2018, 32(2), 1812–1821 CrossRef CAS.
- A. E. Coumans and E. J. M. Hensen, A real support effect on the hydrodeoxygenation of methyl oleate by sulfided NiMo catalysts, Catal. Today, 2017, 298, 181–189 CrossRef CAS.
- K. Wu, et al., Preparation of hydrophobic MoS2, NiS2-MoS2 and CoS2-MoS2 for catalytic hydrodeoxygenation of lignin-derived phenols, Mol. Catal., 2019, 477 Search PubMed.
- C.-C. Tran, D. Akmach and S. Kaliaguine, Hydrodeoxygenation of vegetable oils over biochar supported bimetallic carbides for producing renewable diesel under mild conditions, Green Chem., 2020, 22(19), 6424–6436 RSC.
- F. Wang, et al., Hydrotreatment of vegetable oil for green diesel over activated carbon supported molybdenum carbide catalyst, Fuel, 2018, 216, 738–746 CrossRef CAS.
- M. Alvarez-Galvan, J. Campos-Martin and J. Fierro, Transition Metal Phosphides for the Catalytic Hydrodeoxygenation of Waste Oils into Green Diesel, Catalysts, 2019, 9(3), 242–249 CrossRef.
- M. de Oliveira Camargo, et al., Green diesel production by solvent-free deoxygenation of oleic acid over nickel phosphide bifunctional catalysts: Effect of the support, Fuel, 2020, 281 Search PubMed.
- L. K. H. Pham, et al., Formation and activity of activated carbon supported Ni2P catalysts for atmospheric deoxygenation of waste cooking oil, Fuel Process. Technol., 2019, 185, 117–125 CrossRef CAS.
- N. Kochaputi, et al., Catalytic Behaviors of Supported Cu, Ni, and Co Phosphide Catalysts for Deoxygenation of Oleic Acid, Catalysts, 2019, 9(715), 1–12 Search PubMed.
- S. Rakmae, et al., Defining nickel phosphides supported on sodium mordenite for hydrodeoxygenation of palm oil, Fuel Process. Technol., 2020, 198 Search PubMed.
- K. N. Papageridis, et al., Effect of operating parameters on the selective catalytic deoxygenation of palm oil to produce renewable diesel over Ni supported on Al2O3, ZrO2 and SiO2 catalysts, Fuel Process. Technol., 2020, 209 Search PubMed.
- M. B. Griffin, et al., An investigation into support cooperativity for the deoxygenation of guaiacol over nanoparticle Ni and Rh2P, Catal. Sci. Technol., 2017, 7(14), 2954–2966 RSC.
- L. M. Raymundo, et al., Deoxygenation of Biomass Pyrolysis Vapors via in Situ and ex Situ Thermal and Biochar Promoted Upgrading, Energy Fuels, 2019, 33(3), 2197–2207 CrossRef CAS.
- M. Safa Gamal, et al., Solvent-free catalytic deoxygenation of palm fatty acid distillate over cobalt and manganese supported on activated carbon originating from waste coconut shell, J. Anal. Appl. Pyrolysis, 2019, 144 Search PubMed.
- W. N. Adira Wan Khalit, et al., Development of bimetallic nickel-based catalysts supported on activated carbon for green fuel production, RSC Adv., 2020, 10(61), 37218–37232 RSC.
- P. Roy, et al., Performance of biochar assisted catalysts during hydroprocessing of non-edible vegetable oil: Effect of transition metal source on catalytic activity, Energy Convers. Manage., 2022, 252 Search PubMed.
- T. Thangadurai and C. T. Tye, Performance of Activated Carbon Supported Cobalt Oxides and Iron Oxide Catalysts in Catalytic Cracking of Waste Cooking Oil, Period. Polytech., Chem. Eng., 2021, 65(3), 350–360 CrossRef CAS.
- F. Marrakchi, et al., Mesoporous-activated carbon prepared from chitosan flakes via single-step sodium hydroxide activation for the adsorption of methylene blue, Int. J. Biol. Macromol., 2017, 98, 233–239 CrossRef CAS PubMed.
- M. Dizbay-Onat, U. K. Vaidya and C. T. Lungu, Preparation of industrial sisal fiber waste derived activated carbon by chemical activation and effects of carbonization parameters on surface characteristics, Ind. Crops Prod., 2017, 95, 583–590 CrossRef CAS.
- N. Kaewtrakulchai, K. Faungnawakij and A. Eiad-Ua, Parametric Study on Microwave-Assisted Pyrolysis Combined KOH Activation of Oil Palm Male Flowers Derived Nanoporous Carbons, Materials, 2020, 13(2876), 1–16 Search PubMed.
- S. Li, et al., Preparation and characterization of super activated carbon produced from gulfweed by KOH activation, Microporous Mesoporous Mater., 2017, 243, 291–300 CrossRef CAS.
- N. Asikin-Mijan, et al., Production of renewable diesel from Jatropha curcas oil via pyrolytic-deoxygenation over various multi-wall carbon nanotube-based catalysts, Process Saf. Environ. Prot., 2020, 142, 336–349 CrossRef CAS.
- E. W. Yihunu, et al., Preparation, characterization and cost analysis of activated biochar and hydrochar derived from agricultural waste: a comparative study, SN Appl. Sci., 2019, 1, 1–8 Search PubMed.
- A. Zubrik, et al., Preparation of chemically activated carbon from waste biomass by single-stage and two-stage pyrolysis, J. Cleaner Prod., 2017, 143, 643–653 CrossRef CAS.
- Q.-L. Gao, et al., Biomass-derived mesoporous carbons materials coated by α-Mn3O4 with ultrafast zinc-ion diffusion ability as cathode for aqueous zinc ion batteries, Electrochim. Acta, 2020, 335 Search PubMed.
- Z. Asadi Zeidabadi, et al., Synthesis, characterization and evaluation of biochar from agricultural waste biomass for use in building materials, Constr. Build. Mater., 2018, 181, 301–308 CrossRef CAS.
- N. Dejang, O. Somprasit and S. Chindaruksa, A Preparation of Activated Carbon from Macadamia Shell by Microwave Irradiation Activation, Energy Procedia, 2015, 79, 727–732 CrossRef CAS.
- A. Abdelaal, et al., The impact of pyrolysis conditions on orange peel biochar physicochemical properties for sandy soil, Waste Manage. Res., 2021, 39(7), 995–1004 CrossRef CAS PubMed.
- J. K. Ratan, M. Kaur and B. Adiraju, Synthesis of activated carbon from agricultural waste using a simple method: Characterization, parametric and isotherms study, Mater. Today: Proc., 2018, 5(2), 3334–3345 CAS.
- M. N. Mahamad, M. A. A. Zaini and Z. A. Zakaria, Preparation and characterization of activated carbon from pineapple waste biomass for dye removal, Int. Biodeterior. Biodegrad., 2015, 102, 274–280 CrossRef CAS.
- A. Hai, et al., Date seeds biomass-derived activated carbon for efficient removal of NaCl from saline solution, Process Saf. Environ. Prot., 2019, 129, 103–111 CrossRef CAS.
- F. Meng and D. Wang, Effects of vacuum freeze drying pretreatment on biomass and biochar properties, Renewable Energy, 2020, 155, 1–9 CrossRef CAS.
- Y. Shen, et al., Activated carbons synthesized from unaltered and pelletized biomass wastes for bio-tar adsorption in different phases, Renewable Energy, 2020, 146, 1700–1709 CrossRef CAS.
- E. M. Mistar, T. Alfatah and M. D. Supardan, Synthesis and characterization of activated carbon from Bambusa vulgaris striata using two-step KOH activation, J. Mater. Res. Technol., 2020, 9(3), 6278–6286 CrossRef CAS.
- D. Li, et al., Preparation of porous carbons with high low-pressure CO 2 uptake by KOH activation of rice husk char, Fuel, 2015, 139, 68–70 CrossRef CAS.
- A. Toprak and T. Kopac, Carbon Dioxide Adsorption Using High Surface Area Activated Carbons from Local Coals Modified by KOH, NaOH and ZnCl2 Agents, Int. J. Chem. React. Eng., 2017, 15(3), 1–16 Search PubMed.
- N. K. Kalagatur, et al., Application of Activated Carbon Derived from Seed Shells of Jatropha curcas for Decontamination of Zearalenone Mycotoxin, Front. Pharmacol., 2017, 8, 760 CrossRef PubMed.
- P. Salimi, et al., Magnetic biochar obtained through catalytic pyrolysis of macroalgae: A promising anode material for Li-ion batteries, Renewable Energy, 2019, 140, 704–714 CrossRef CAS.
- P. N. Bhandari, et al., Synthesis and evaluation of biochar-derived catalysts for removal of toluene (model tar) from biomass-generated producer gas, Renewable Energy, 2014, 66, 346–353 CrossRef CAS.
- M. T. H. Siddiqui, et al., Synthesis and optimization of chitosan supported magnetic carbon bio-nanocomposites and bio-oil production by solvothermal carbonization co-precipitation for advanced energy applications, Renewable Energy, 2021, 178, 587–599 CrossRef CAS.
- M. F. Kamaruzaman, Y. H. Taufiq-Yap and D. Derawi, Green diesel production from palm fatty acid distillate over SBA-15-supported nickel, cobalt, and nickel/cobalt catalysts, Biomass Bioenergy, 2020, 134 Search PubMed.
- C. Guo, et al., Novel inexpensive transition metal phosphide catalysts for upgrading of pyrolysis oil via hydrodeoxygenation, AIChE J., 2016, 62(10), 3664–3672 CrossRef CAS.
- S. S. Lam, et al., Activated Carbon for Catalyst Support from Microwave Pyrolysis of Orange Peel, Waste Biomass Valorization, 2016, 8(6), 2109–2119 CrossRef.
- S. Thongkumkoon, et al., Catalytic activity of trimetallic sulfided Re-Ni-Mo/γ-Al2O3 toward deoxygenation of palm feedstocks, Renewable Energy, 2019, 140, 111–123 CrossRef CAS.
- A. S. Al-Fatesh, et al., CO2 reforming of CH4: Effect of Gd as promoter for Ni supported over MCM-41 as catalyst, Renewable Energy, 2019, 140, 658–667 CrossRef CAS.
- W. Xie, Y. Han and H. Wang, Magnetic Fe3O4/MCM-41 composite-supported sodium silicate as heterogeneous catalysts for biodiesel production, Renewable Energy, 2018, 125, 675–681 CrossRef CAS.
- X. Liu, et al., Improvement of hydrodeoxygenation stability of nickel phosphide based catalysts by silica modification as structural promoter, Fuel, 2017, 204, 144–151 CrossRef CAS.
- J. Masud, et al., Iron phosphide nanoparticles as an efficient electrocatalyst for the OER in alkaline solution, J. Mater. Chem. A, 2016, 4(25), 9750–9754 RSC.
- M. Usman, et al., Highly selective and stable hydrogenation of heavy aromatic-naphthalene over transition metal phosphides, Sci. China: Chem., 2014, 58(4), 738–746 CrossRef.
- M. C. Alvarez-Galvan, et al., Metal phosphide catalysts for the hydrotreatment of non-edible vegetable oils, Catal. Today, 2018, 302, 242–249 CrossRef CAS.
- Z. Pan, et al., Deoxygenation of methyl laurate to hydrocarbons on silica-supported Ni-Mo phosphides: Effect of calcination temperatures of precursor, J. Energy Chem., 2015, 24(1), 77–86 CrossRef.
- R. S. R. M. Hafriz, et al., Effect of Ni/Malaysian dolomite catalyst synthesis technique on deoxygenation reaction activity of waste cooking oil, Renewable Energy, 2021, 178, 128–143 CrossRef CAS.
- K. N. Papageridis, et al., Promoting effect of CaO-MgO mixed oxide on Ni/γ-Al2O3 catalyst for selective catalytic deoxygenation of palm oil, Renewable Energy, 2020, 162, 1793–1810 CrossRef CAS.
- H. Xin, et al., Production of high-grade diesel from palmitic acid over activated carbon-supported nickel phosphide catalysts, Appl. Catal., B, 2016, 187, 375–385 CrossRef CAS.
- Y. Yang, et al., Transition Metal Phosphide Nanoparticles Supported on SBA-15 as Highly Selective Hydrodeoxygenation Catalysts for the Production of Advanced Biofuels, J. Nanosci. Nanotechnol., 2015, 15(9), 6642–6650 CrossRef CAS PubMed.
- P. Liu, et al., Controlled synthesis of nickel phosphide nanoparticles with pure-phase Ni2P and Ni12P5 for hydrogenation of nitrobenzene, React. Kinet., Mech. Catal., 2018, 126(1), 453–461 CrossRef.
- D. Xiong, et al., Facile synthesis of iron phosphide nanorods for efficient and durable electrochemical oxygen evolution, Chem. Commun., 2016, 52(56), 8711–8714 RSC.
- K. C. Kwon, et al., Catalytic deoxygenation of liquid biomass for hydrocarbon fuels, Renewable Energy, 2011, 36(3), 907–915 CrossRef CAS.
- C.-y. Liu, et al., Hydrodeoxygenation of fatty acid methyl esters and isomerization of products over NiP/SAPO-11 catalysts, J. Fuel Chem. Technol., 2016, 44(10), 1211–1216 CrossRef CAS.
- N. Kaewtrakulchai, et al., Palm Oil Conversion to Bio-Jet and Green Diesel Fuels over Cobalt Phosphide on Porous Carbons Derived from Palm Male Flowers, Catalysts, 2020, 10(694), 1–18 Search PubMed.
- N. Srihanun, et al., Biofuels of Green Diesel–Kerosene–Gasoline Production from Palm Oil: Effect of Palladium Cooperated with Second Metal on Hydrocracking Reaction, Catalysts, 2020, 10(241), 1–13 Search PubMed.
- W. Zhou, et al., The Deoxygenation Pathways of Palmitic Acid into Hydrocarbons on Silica-Supported Ni12P5 and Ni2P Catalysts, Catalysts, 2018, 8(153), 1–20 Search PubMed.
- P. Bui, et al., Studies of the synthesis of transition metal phosphides and their activity in the hydrodeoxygenation of a biofuel model compound, J. Catal., 2012, 294, 184–198 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |