
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot two-step catalytic synthesis of 6-amino-2-pyridone-3,5-dicarbonitriles enabling anti-cancer bioactivity†
Lynden G. Nicely‡
a,
Ruturajsinh M. Vala‡ b,
Dipti B. Upadhyayb,
Joaquina Nogalesa,
Celestine Chic,
Sourav Banerjee*a and
Hitendra M. Patel*b
b,
Dipti B. Upadhyayb,
Joaquina Nogalesa,
Celestine Chic,
Sourav Banerjee*a and
Hitendra M. Patel*b
aDepartment of Cellular and Systems Medicine, School of Medicine, University of Dundee, Dundee DD1 9SY, UK. E-mail: s.y.banerjee@dundee.ac.uk
bDepartment of Chemistry, Sardar Patel University, Vallabh Vidyanagar 388120, Gujarat, India. E-mail: hm_patel@spuvvn.edu
cDepartment of Medical Biochemistry and Microbiology, Uppsala University, SE-75123 Uppsala, Sweden
First published on 24th August 2022
Abstract
We report a one-pot two-step synthesis of a bioactive 6-amino-2-pyridone-3,5-dicarbonitrile derivative using natural product catalysts betaine and guanidine carbonate. Anti-cancer bioactivity was observed in specific molecules within the library of 16 derivatives. Out of the compounds, 5o had the most potent anti-cancer activity against glioblastoma cells and was selected for further study. Compound 5o showed anti-cancer properties against liver, breast, lung cancers as well as primary patient-derived glioblastoma cell lines. Furthermore, 5o in combination with specific clinically relevant small molecule inhibitors induced enhanced cytotoxicity in glioblastoma cells. Through our current work, we establish a promising 6-amino-2-pyridone-3,5-dicarbonitrile based lead compound with anti-cancer activity either on its own or in combination with specific clinically relevant small molecule kinase and proteasome inhibitors.
1. Introduction
Establishing novel and cost-effective strategies to synthesise unique bioactive chemical scaffolds is essential for future development of cancer therapeutics. For some forms of cancers like glioblastoma (stage IV brain cancer),1 there are very limited options for patients and is categorised as a major ‘unmet need’. Various blood–brain-barrier (BBB) penetrant small molecules have either undergone or are currently undergoing clinical evaluations like PI-103,2 buparlisib,3 abemaciclib,4 bozitinib,5 marizomib,6 nilotinib,7 osimertinib8 and others. However, maintaining a large and diverse pipeline of novel cancer therapeutics remains the need of the hour.Previously, we had successfully established a synthetic strategy of 6-amino-2-pyridone-3,5-dicarbonitriles9 using piperidine acetate as a catalyst. To further establish a non-hazardous synthetic approach for 6-amino-2-pyridone-3,5-dicarbonitriles, we decided to utilise a natural product catalyst. The main advantages of natural product catalysts are that they require milder reaction conditions and do not contain any heavy metal, making them non-toxic. In our previous work, we synthesised pyrazolo[3,4-b]quinolinones using the natural product pyridine-2-carboxylic acid10 and spiro-heterocycles by using the natural product betaine based deep-eutectic solvents.11,12 Herein, we explored the catalytical activity of pyridine-2-carboxylic acid and betaine for the synthesis of 6-amino-2-pyridone-3,5-dicarbonitriles. We also explored the catalytical activity of guanidine carbonate for the reaction. As a catalyst, guanidine carbonate is used in the methanolysis of triacylglycerols13 and synthesis of graphitic carbon nitride.14 It is rarely used to catalyse one-pot synthesis; however, it has been used as a substrate in one-pot multicomponent synthesis.15–17 We explored the catalytical activity of guanidine carbonate in one-pot synthesis of 6-amino-2-pyridone-3,5-dicarbonitriles. Consequently, we observed anti-cancer cytotoxicity of the lead compound 5a and attempted to establish similar biological activity of the derivatives of 5a to establish the best derivative for future medicinal chemistry efforts. Although the current series of 16 derivatives were not predicted to be blood–brain barrier penetrant, we explored the potential of combining a 6-amino-2-pyridone-3,5-dicarbonitrile with clinically relevant BBB-penetrant small molecules18–22 to establish potency for future medicinal chemistry efforts to optimise a BBB-penetrant drug.
2. Results and discussion
2.1 Chemistry
To synthesise 6-amino-2-pyridone-3,5-dicarbonitrile, we followed the same protocol used in our previously reported work.9 p-Tolylidenemalononitrile 3a synthesised by reaction of p-tolualdehyde 1a and malononitrile 2. Some natural products like pyridine-2-carboxylic acid, betaine, guanidine hydrochloride and guanidine carbonate are used as a catalyst for the one-pot reaction. At room temperature, 1a and 2 were mixed with 10 mol% catalyst and stirred. When betaine or guanidine carbonate was used, the reaction mixture solidified within 5 minutes in a round bottom flask, however, the reaction was still incomplete, therefore, we added 1 ml of methanol to the reaction mixture and stirred it again for 10 minutes. Aldehyde was completely used but, guanidine carbonate produced 3a with water-soluble impurities. Whereas betaine produced 3a as a single product. Thus, betaine is a suitable catalyst for the first step.After completion of the first step with betaine catalyst, N-benzyl-2-cyanoacetamide 4a was added to the reaction mixture with 1 ml methanol. The reaction mixture was refluxed with pyridine-2-carboxylic acid, betaine, guanidine hydrochloride or guanidine carbonate catalyst. Betaine did not convert 3a to 5a completely in 5 minutes. However, guanidine carbonate efficiently catalysed this step and produced desired product 5a in 10 minutes. Thus, the one-pot two-step process is more suitable than the multi-component procedure for the synthesis of 6-amino-2-pyridone-3,5-dicarbonitrile derivatives using two catalysts: betaine for the first step and guanidine carbonate for the second step (Fig. 1 & Table 1).
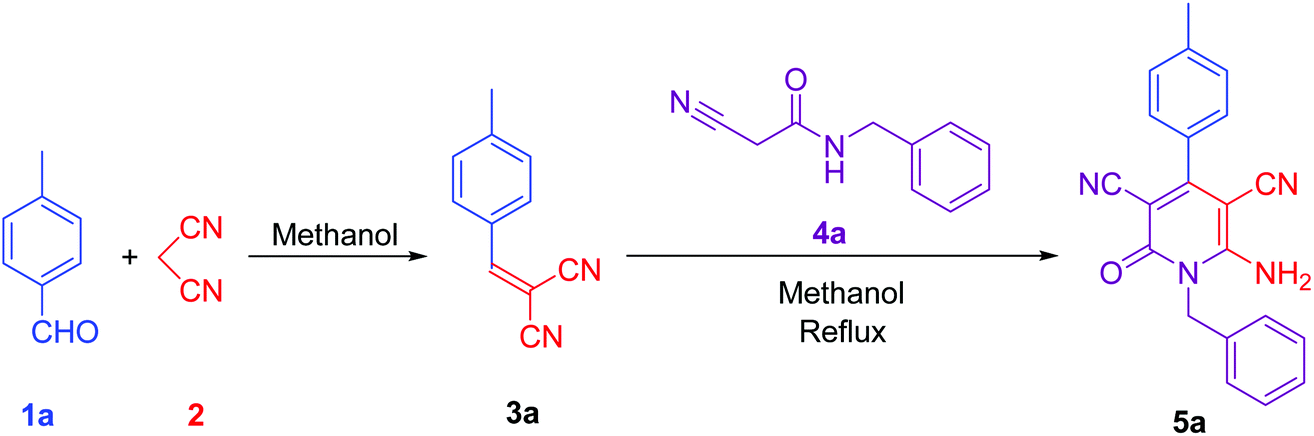 | ||
| Fig. 1 Synthesis of lead compound. Reaction condition: 2 mmol p-tolualdehyde, 2 mmol mole malononitrile, 2 mmol N-benzyl-2-cyanoacetamide in methanol solvent. | ||
| Entry | Catalyst (10 mol%) | Reaction time of first step | Conversion relative to aldehyde | Reaction time of second step | Conversion relative to 3a |
|---|---|---|---|---|---|
| 1 | Pyridine-2-carboxylic acid | 30 min | Incomplete | 1 h | Incomplete |
| 2 | Betaine | 15 min | 100% | 1 h | Incomplete |
| 3 | Guanidine hydrochloride | 30 min | Incomplete | 1 h | Incomplete |
| 4 | Guanidine carbonate | 10 min | 100% | 10 min | 100% |
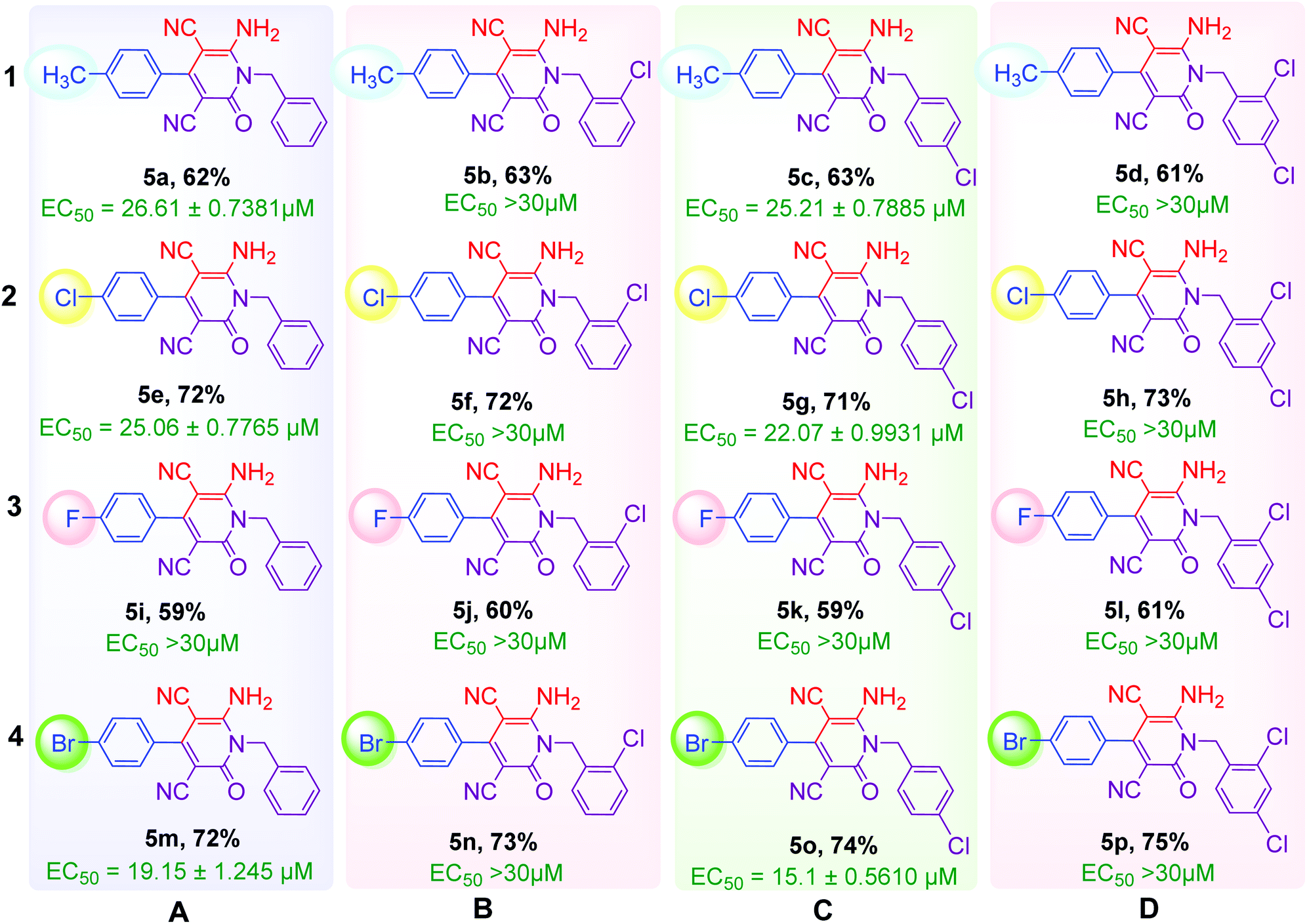
As shown in Table 2, sixteen 6-amino-2-pyridone-3,5-dicarbonitriles 5(a–p) were synthesised via a one-pot two-step reaction. 5(a–p) were characterised by 1H NMR, 13C{1H} NMR and HRMS analysis. Some of the products were also analysed by DEPT-135 or HSQC analysis. HSQC spectra confirmed the presence of benzyl group (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2Ph), aromatic rings and methyl group in case of 5a (Fig. 2A). DEPT-135 spectra confirmed the presence of different types of carbons (Fig. 2B). Due to C–F coupling singles of C-16, C-4, C-9 and C-10 are observed as a doublet with coupling constant (J) as 247.77, 21.38, 8.80 and 2.52 Hz, respectively.
H2Ph), aromatic rings and methyl group in case of 5a (Fig. 2A). DEPT-135 spectra confirmed the presence of different types of carbons (Fig. 2B). Due to C–F coupling singles of C-16, C-4, C-9 and C-10 are observed as a doublet with coupling constant (J) as 247.77, 21.38, 8.80 and 2.52 Hz, respectively.

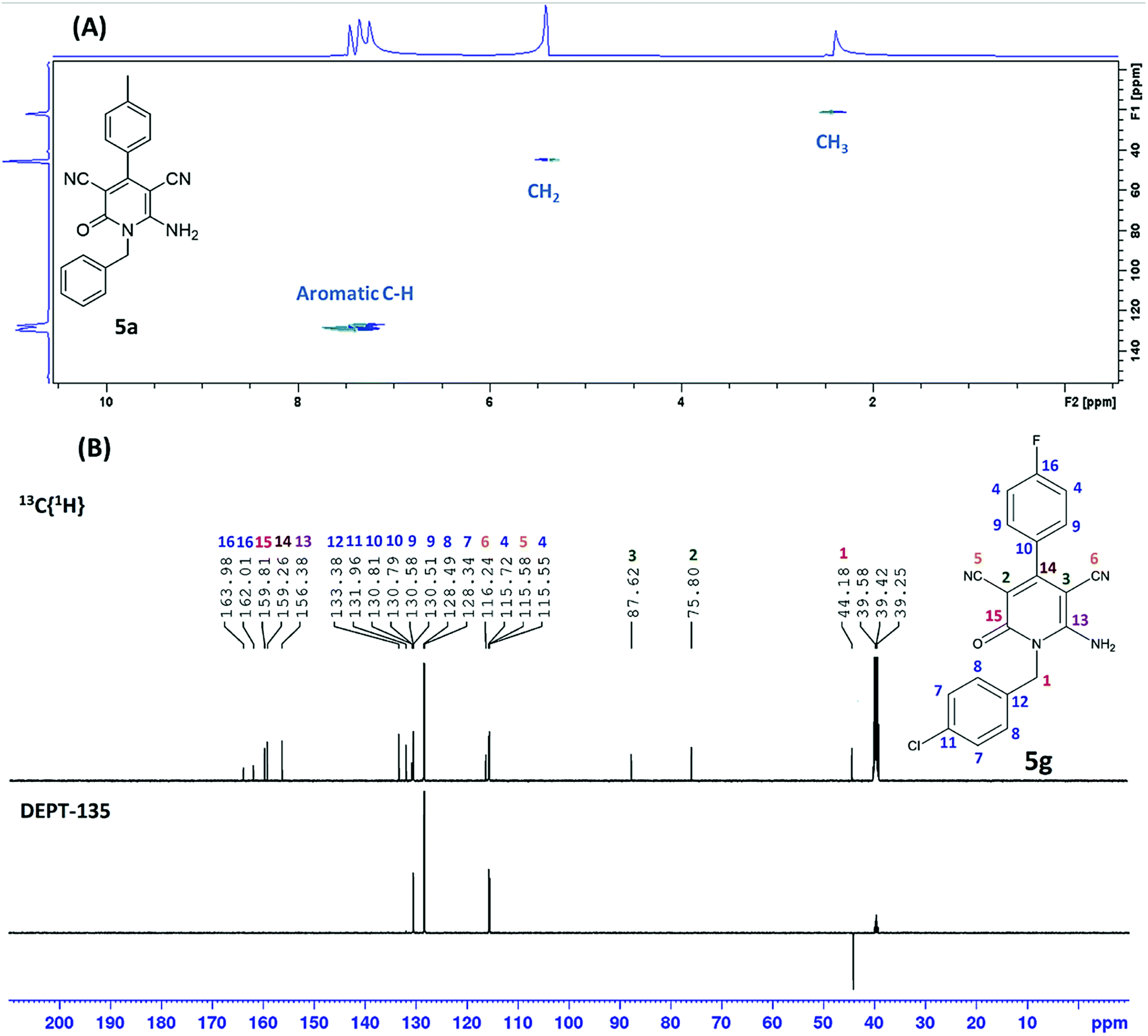 | ||
| Fig. 2 (A) HSQC spectrum of 5a, (B) DEPT-135 spectrum of 5g. | ||
We reported the synthesis of intermediate aryledinemalononitrile by betaine catalyst for the first time. However, other betaine base catalysts are reported for different heterocyclic reactions. We also tried the synthesis of aryledinemalononitrile by the solvent-free grinding method in the presence of betaine catalyst. Betaine successfully catalysed this room-temperature synthesis and produced intermediate 3 within 10 minutes. However, this solvent-free grinding method did not carry out the second step and hence expected product was not synthesised. All subsequent derivatives were synthesised using the one-pot two-step synthesis in the round bottom flask.
2.2 Biology
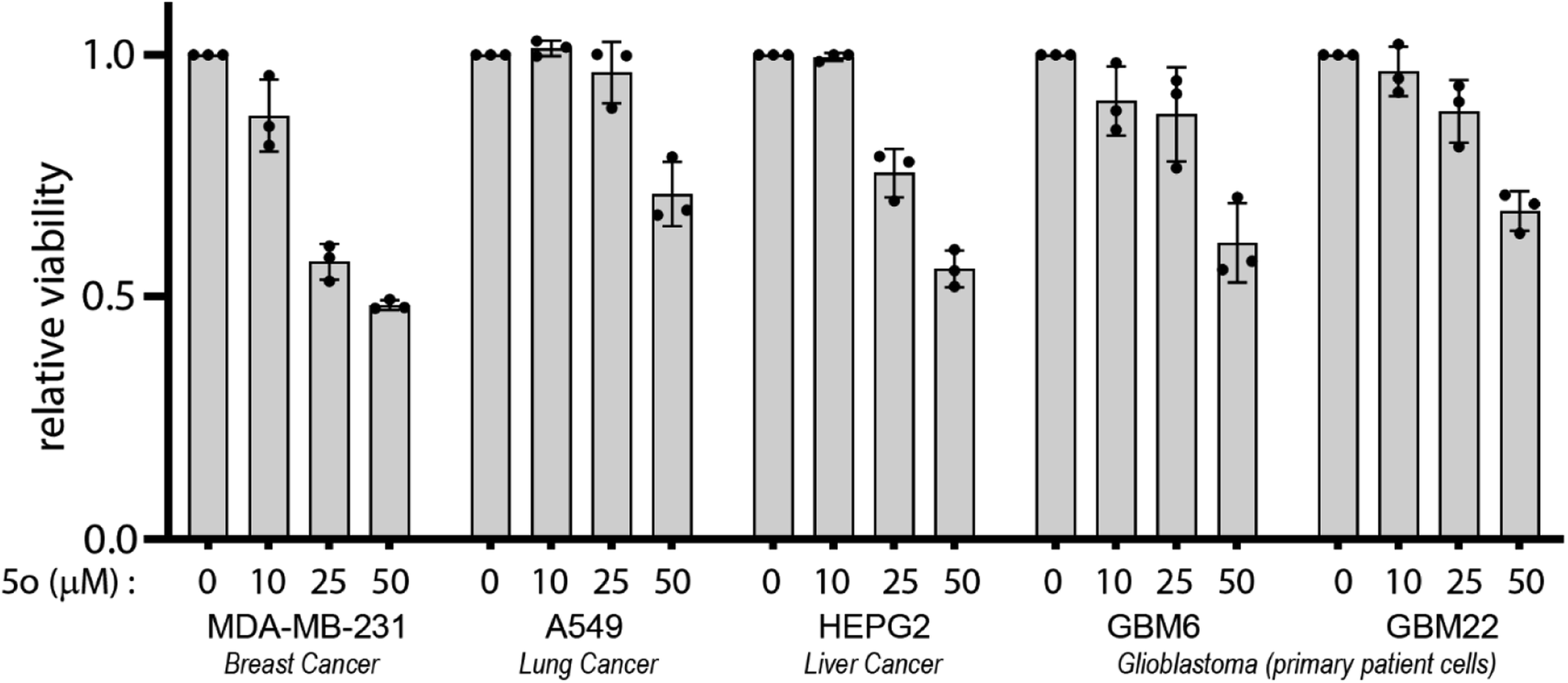 | ||
| Fig. 3 5o exhibits anti-cancer activity in diverse cancer cell lines. Indicated cell lines were treated with various doses of 5o for 72 hours and their viability was measured using CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay kit. Viability of DMSO-treated cells was used as control. Data are represented as fold viability of DMSO-treated control for each cell line with n = 3 biological replicates. | ||
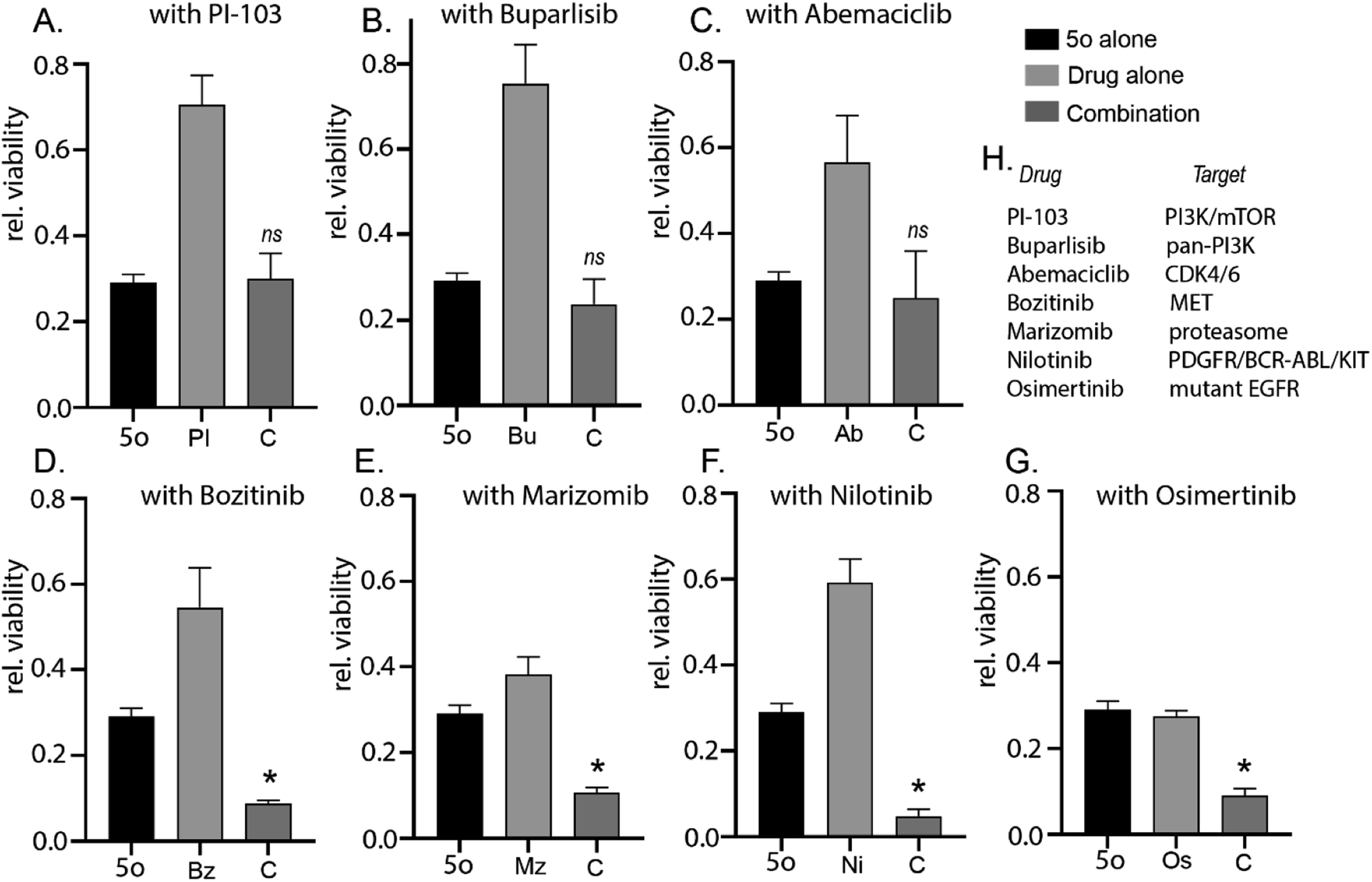 | ||
| Fig. 4 5o induces enhanced cytotoxicity in combination with specific clinically relevant brain-penetrant small molecule inhibitors. GL261 cells were treated with 5o alone (30 μM) or clinically relevant brain-penetrant drugs alone (A) PI-103 100 nm, (B) Buparlisib 500 nm, (C) Abemaciclib 1 μM, (D) Bozitinib 7 μM, (E) Marizomib 400 nM, (F) Nilotinib 5 μM, (G) Osimertinib 2 μM, or a combination of both for 72 hours and cell viability was analysed by CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay kit. Viability of DMSO-treated cells was used as control. Data are represented as fold viability of DMSO-treated control for each cell line (* indicates statistical significance compared to each single compound treatment; one-way ANOVA with Dunnett's multiple comparison; ns: not significant). Same dataset for 5o was used across all combination studies from (A–G). (H) Table lists the cellular targets of the respective clinically relevant brain-penetrant drugs. | ||
3. Conclusion
A series of novel 6-amino-2-pyridone-3,5-dicarbonitrile derivatives were synthesised using a one-pot two-step scheme using natural product catalysts. Out of the library we identified 5o with the most potent anti-cancer activity in a diverse set of cancer cell lines including primary patient-derived cells. Furthermore, in combination with brain-penetrant small molecule inhibitors targeting receptor tyrosine kinases and proteasome, 5o induced potent cytotoxicity which attests for the further medicinal chemistry and development of the 6-amino-2-pyridone-3,5-dicarbonitrile backbone. Therefore, this study establishes a simple yet novel synthetic scheme which will allow for the development of future clinically relevant anti-cancer molecules which can be used either as a monotherapeutic option or used in combination with specific molecules targeting diverse cancer signalling pathways.4. Experimental procedure
4.1 Chemistry
4.1.2.1 6-Amino-1-benzyl-2-oxo-4-p-tolyl-1,2-dihydropyridine-3,5-dicarbonitrile (5a). White solid (0.423 g, 62%), mp: 292–294 °C; 1H NMR (600 MHz, DMSO-d6) δ: 2.42 (s, 3H, CH3), 5.39 (s, 2H, CH2), 7.28 and 7.49 (ABq, J = 8.1 Hz, 2H, Ar–H), 7.32 (t, J = 7.2 Hz, 1H, Ar–H), 7.39 (d, J = 5.4 Hz, 3H, Ar–H), 8.46 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 21.5, 45.2, 76.1, 88.0, 116.4, 117.0, 127.0, 127.9, 128.5, 129.0, 129.6, 132.2, 135.0, 140.7, 157.1, 160.0, 161.35; MS (ESI-TOF) m/z calcd For C21H16N4O (M + H)+: 341.1402, found: 341.1420.
4.1.2.2. 6-Amino-1-(2-chlorobenzyl)-2-oxo-4-p-tolyl-1,2-dihydropyridine-3,5-dicarbonitrile (5b). White solid (0.473 g, 63%), mp: 292–294 °C; 1H NMR (600 MHz, DMSO-d6) δ: 2.40 (s, 3H, CH3), 5.28 (s, 2H, CH2), 6.91 (d, J = 6 Hz, 1H, Ar–H), 7.31–7.39 (m, 4H, Ar–H), 7.47–7.54 (m, 3H, Ar–H), 8.53 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 21.5, 44.8, 76.3, 87.8, 116.5, 117.0, 125.8, 127.9, 128.5, 129.3, 129.6, 130.0, 132.2, 132.4, 132.7, 140.7, 157.4, 159.8, 161.6; MS (ESI-TOF) m/z calcd for C21H15ClN4O (M + H)+: 375.1013, found: 375.1028.
4.1.2.3 6-Amino-1-(4-chlorobenzyl)-2-oxo-4-p-tolyl-1,2-dihydropyridine-3,5-dicarbonitrile (5c). White solid (0.472 g, 63%), mp: 250–252 °C; 1H NMR (600 MHz, DMSO-d6) δ: 2.40 (s, 3H, CH3), 5.35 (s, 2H, CH2), 7.31 (d, J = 8.4 Hz, 2H, Ar–H), 7.38 (d, J = 7.8 Hz, 2H, Ar–H), 7.45 (dd, J = 8.4, 13.8 Hz, 2H, Ar–H), 8.46 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 21.5, 44.8, 76.2, 88.0, 116.3, 117.0, 128.5, 128.9, 129.1, 129.6, 132.10, 132.6, 134.0, 140.7, 157.0, 160.0, 161.4; MS (ESI-TOF) m/z calcd for C21H15ClN4O (M + H)+: 375.1013, found: 375.0982.
4.1.2.4 6-Amino-1-(2,4-dichlorobenzyl)-2-oxo-4-p-tolyl-1,2-dihydropyridine-3,5-dicarbonitrile (5d). White solid (0.499 g, 61%), mp: 314–316 °C; 1H NMR (600 MHz, DMSO-d6) δ: 2.42 (s, 3H, CH3), 5.23 (s, 2H, CH2), 6.98 (d, J = 7.8 Hz, 1H, Ar–H), 7.39–7.46 (m, 5H, Ar–H), 7.73 (s, 1H, Ar–H), 8.58 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 21.5, 44.6, 76.4, 87.8, 116.4, 116.9, 127.3, 127.9, 128.5, 129.4, 129.6, 131.7, 132.2, 133.0, 133.6, 140.7, 157.4, 159.7, 161.6; MS (ESI-TOF) m/z calcd for C21H14Cl2N4O (M + H)+: 409.0623, found: 409.0844.
4.1.2.5 6-Amino-1-benzyl-4-(4-chlorophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5e). White solid (0.519 g, 72%), mp: 272–274 °C; 1H NMR (500 MHz, DMSO-d6) δ: 5.35 (s, 2H, CH2), 7.25 (d, J = 7.5 Hz, 2H, Ar–H), 7.31 (t, J = 7.25, 1H, Ar–H), 7.38 (t, J = 7.25, 2H, Ar–H), 7.60 (dd, J = 2.0, 6.5 Hz, 2H, Ar–H), 7.66 (dd, J = 2.0, 6.5 Hz, 2H, Ar–H), 8.49 (s, 2H, NH2); 13C{1H} NMR (125 MHz, DMSO-d6) δ: 44.6, 75.6, 87.4, 115.5, 116.2, 126.4, 127.3, 128.4, 128.7, 129.9, 133.3, 134.3, 135.1, 156.4, 159.2, 159.5; MS (ESI-TOF) m/z calcd for C20H13ClN4O (M + H)+: 361.0856, found: 361.0871.
4.1.2.6 6-Amino-1-(2-chlorobenzyl)-4-(4-chlorophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5f). White solid (0.569 g, 72%), mp: 284–286 °C; 1H NMR (600 MHz, DMSO-d6) δ: 5.28 (s, 2H, CH2), 6.93 (d, J = 7.8 Hz, 1H, Ar–H), 7.39 (dt, J = 7.2, 22.8 Hz, 2H, Ar–H), 7.55–7.56 (m, 1H, Ar–H), 7.62 (d, J = 8.4 Hz, 1H, Ar–H), 7.68 (d, J = 9.0 Hz, 1H, Ar–H), 8.61 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 44.8, 76.3, 87.9, 116.2, 116.7, 125.8, 127.8, 129.3, 130.0, 130.5, 132.2, 132.7, 134.0, 135.7, 157.8, 159.6, 160.4; MS (ESI-TOF) m/z calcd for C20H12Cl2N4O (M + H)+: 395.0466, found: 395.0511.
4.1.2.7 6-Amino-1-(4-chlorobenzyl)-4-(4-chlorophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5g). White solid (0.561 g, 71%), mp: 244–246 °C; 1H NMR (600 MHz, DMSO-d6) δ: 5.32 (s, 2H, CH2), 7.29 (d, J = 8.4 Hz, 2H, Ar–H), 7.44 (d, J = 8.4 Hz, 2H, Ar–H), 7.59 (d, J = 8.4 Hz, 2H, Ar–H), 7.66 (d, J = 9.0 Hz, 2H, Ar–H), 8.51 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 44.8, 76.2, 88.1, 116.1, 116.7, 128.9, 129.1, 129.3, 130.5, 132.6, 133.9, 133.9, 135.7, 157.0, 159.8, 160.2; MS (ESI-TOF) m/z calcd for C20H12Cl2N4O (M + H)+: 395.0466, found: 395.0476.
4.1.2.8 6-Amino-4-(4-chlorophenyl)-1-(2,4-dichlorobenzyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5h). White solid (0.627 g, 73%), mp: 292–294 °C (d); 1H NMR (600 MHz, DMSO-d6) δ: 5.23 (s, 2H, CH2), 6.99 (d, J = 8.4 Hz, 1H, Ar–H), 7.39 (dd, J = 1.5, 8.1 Hz, 1H, Ar–H), 7.61 (d, J = 8.4 Hz, 2H, Ar–H), 7.68 (d, J = 8.4 Hz, 2H, Ar–H), 7.73 (d, J = 1.2 Hz, 1H, Ar–H), 8.62 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 44.7, 76.4, 87.9, 116.2, 116.7, 127.3, 127.9, 129.3, 129.5, 130.5, 131.6, 133.0, 133.7, 133.9, 135.7, 157.4, 159.6, 160.4; MS (ESI-TOF) m/z calcd for C20H11Cl3N4O (M + H)+: 429.0077, found: 429.0107.
4.1.2.9 6-Amino-1-benzyl-4-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5i). White solid (0.406 g, 59%), mp: 228–230 °C; 1H NMR (500 MHz, DMSO-d6) δ: 5.35 (s, 2H, CH2), 7.25 (d, J = 9 Hz, 2H, Ar–H), 7.30–7.36 (m, 1H, Ar–H), 7.37–7.44 (m, 4H, Ar–H), 7.63–7.66 (m, 2H, Ar–H), 8.47 (s, 2H, NH2); 13C{1H} NMR (125 MHz, DMSO-d6) δ: 44.6, 75.7, 87.6, 115.5, 115.6, 115.7, 116.3, 126.4, 127.3, 128.4, 130.5, 130.6, 130.8, 130.9, 134.3, 156.4, 159.3, 159.8, 162.0, 164.0; MS (ESI-TOF) m/z calcd for C20H13FN4O (M + H)+: 345.1152, found: 345.1145.
4.1.2.10 6-Amino-1-(2-chlorobenzyl)-4-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5j). White solid (0.454 g, 60%), mp: 298–300 °C; 1H NMR (600 MHz, DMSO-d6) δ: 5.27 (s, 2H, CH2), 6.92 (d, J = 7.8 Hz, 1H, Ar–H), 7.32–7.38 (m, 2H, Ar–H), 7.45 (t, J = 8.7 Hz, 2H, Ar–H), 7.55 (d, J = 7.2 Hz, 1H, Ar–H), 7.65 (dd, J = 5.7, 8.1 Hz, 2H, Ar–H), 8.58 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 44.7, 76.5, 88.0, 116.2, 116.3, 116.3, 116.8, 125.8, 127.9, 129.3, 130.0, 131.1, 131.2, 131.5, 132.2, 132.6, 157.4, 159.6, 160.6, 162.8, 164.4; MS (ESI-TOF) m/z calcd for C20H12ClFN4O (M + H)+: 379.0762, found: 379.0756.
4.1.2.11 6-Amino-1-(4-chlorobenzyl)-4-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5k). White solid (0.447 g, 59%), mp: 130–132 °C; 1H NMR (500 MHz, DMSO-d6) δ: 5.32 (s, 2H, CH2), 7.28 (d, J = 10.2 Hz, 2H, Ar–H), 7.41–7.45 (m, 4H, Ar–H), 7.61–7.64 (m, 2H, Ar–H), 8.48 (s, 2H, NH2); 13C{1H} NMR (125 MHz, DMSO-d6) δ: 44.2, 75.8, 87.6, 115.6, 115.6, 115.7, 116.2, 128.3, 128.5, 130.5, 130.6, 130.8, 130.8, 132.0, 133.4, 156.4, 159.3, 159.8, 162.0, 164.0; MS (ESI-TOF) m/z calcd for C20H12ClFN4O (M + H)+: 379.0762, found: 379.0768.
4.1.2.12 6-Amino-1-(2,4-dichlorobenzyl)-4-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5l). White solid (0.504 g, 61%), mp: 278–280 °C; 1H NMR (600 MHz, DMSO-d6) δ: 5.22 (s, 2H, CH2), 6.98 (d, J = 8.4 Hz, 1H, Ar–H), 7.39 (d, J = 7.8 Hz, 1H, Ar–H), 7.45 (t, J = 8.4 Hz, 2H, Ar–H), 7.63–7.66 (m, 2H, Ar–H), 7.73 (s, 1H, Ar–H), 8.59 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 44.6, 76.5, 88.0, 116.2, 116.2, 116.3, 116.8, 127.3, 127.9, 129.5, 131.1, 131.1, 131.4, 131.6, 133.0, 133.6, 157.8, 159.6, 160.6, 162.8, 164.4; MS (ESI-TOF) m/z calcd for C20H11Cl2FN4O (M + H)+: 413.0372, found: 413.0603.
4.1.2.13 6-Amino-1-benzyl-4-(4-bromophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5m). White solid (0.584 g, 72%), mp: 280–282 °C; 1H NMR (500 MHz, DMSO-d6) δ: 5.34 (s, 2H, CH2), 7.25 (d, J = 7.5 Hz, 2H, Ar–H), 7.31 (t, J = 7.5 Hz, 1H, Ar–H), 7.37–7.40 (m, 3H, Ar–H), 7.52–7.54 (m, 2H, Ar–H), 7.79–7.81 (m, 2H, Ar–H), 8.49 (s, 2H, NH2); 13C{1H} NMR (125 MHz, DMSO-d6) δ: 44.6, 75.5, 87.4, 115.5, 116.2, 123.8, 126.4, 127.3, 128.4, 130.1, 131.6, 133.7, 134.2, 156.4, 159.2, 159.6; MS (ESI-TOF) m/z calcd for C20H13BrN4O (M + H)+: 405.0351, found: 405.0569.
4.1.2.14 6-Amino-4-(4-bromophenyl)-1-(2-chlorobenzyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5n). White solid (0.642 g 73%), mp: 290–292 °C; 1H NMR (600 MHz, DMSO-d6) δ: 5.27 (s, 2H, CH2), 6.93 (d, J = 7.2 Hz, 1H, Ar–H), 7.35 (dt, J = 7.5, 23.4 Hz, 2H, Ar–H), 7.55 (dd, J = 4.5, 6.9 Hz, 3H, Ar–H), 7.82 (d, J = 7.8 Hz, 2H, Ar–H), 8.61 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 44.8, 76.3, 87.8, 116.2, 116.7, 124.4, 125.8, 127.9, 129.3, 130.0, 130.5, 130.7, 132.2, 132.6, 134.4, 157.4, 159.6, 160.4; MS (ESI-TOF) m/z calcd for C20H12BrClN4O (M + H)+: 438.9961, found: 439.0189.
4.1.2.15 6-Amino-4-(4-bromophenyl)-1-(4-chlorobenzyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5o). White solid (0.650 g, 74%), mp: 260–262 °C; 1H NMR (600 MHz, DMSO-d6) δ: 5.32 (s, 2H, CH2), 7.28 (d, J = 10.2 Hz, 2H, Ar–H), 7.41–7.45 (m, 4H, Ar–H), 7.61–7.64 (m, 2H, Ar–H), 8.48 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 44.8, 76.2, 88.0, 116.1, 116.7, 124.5, 128.9, 129.1, 130.7, 132.2, 132.6, 133.9, 134.2, 157.0, 159.8, 160.2; MS (ESI-TOF) m/z calcd for C20H12BrClN4O (M + H)+: 438.9961, found: 439.0183.
4.1.2.16 6-Amino-4-(4-bromophenyl)-1-(2,4-dichlorobenzyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (5p). White solid (0.714 g, 75%), mp: 284–286 °C; 1H NMR (600 MHz, DMSO-d6) δ: 5.22 (s, 2H, CH2), 6.98 (d, J = 8.4 Hz, 1H, Ar–H), 7.39 (dd, J = 1.8, 8.4 Hz, 1H, Ar–H), 7.53 (d, J = 8.4 Hz, 2H, Ar–H), 7.73 (d, J = 1.8 Hz, 1H, Ar–H), 7.82 (d, J = 8.4 Hz, 2H, Ar–H), 8.61 (s, 2H, NH2); 13C{1H} NMR (151 MHz, DMSO-d6) δ: 44.7, 76.3, 87.8, 116.2, 116.7, 124.5, 127.3, 127.9, 129.5, 130.7, 131.6, 132.2, 133.0, 133.6, 134.3, 157.4, 159.6, 160.5; MS (ESI-TOF) m/z calcd for C20H11BrCl2N4O (M + H)+: 472.9572, found: 472.9563.
4.2 Cell based assays
5000–8000 cells were plated per well in 96-well plate. 24 hours post-plating, the appropriate drug was added to the cells in triplicate at varying concentrations with a DMSO control. 72 hours after treatment, cell viability was then determined using CellTiter 96® AQueous Non-Radioactive Cell proliferation assay, adhering to manufacturer instructions. Absorbance was measured using a Tecan multi-well plate reader and data was represented relative to DMSO treated control.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
SB is funded by the Cancer Research UK EDDPMA-May21\100005, Tenovus Scotland T21-05, and Royal Society RGS\R2\212056. The authors thank the Department of Chemistry, Sardar Patel University, Vallabh Vidyanagar for valuable support. The authors are thankful to the Biomolecular NMR center Uppsala, which is funded by the Department of Chemistry-BMC and the Disciplinary Domain of Medicine and Pharmacy. The graphical abstract was generated in part using Biorender.References
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. B. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer and R. O. Mirimanoff, N. Engl. J. Med., 2005, 352, 987–996 CrossRef CAS PubMed.
- T. Bagci-Onder, H. Wakimoto, M. Anderegg, C. Cameron and K. Shah, Cancer Res., 2011, 71, 154–163 CrossRef CAS PubMed.
- P. Y. Wen, M. Touat, B. M. Alexander, I. K. Mellinghoff, S. Ramkissoon, C. S. McCluskey, K. Pelton, S. Haidar, S. S. Basu, S. C. Gaffey, L. E. Brown, J. E. Martinez-Ledesma, S. Wu, J. Kim, W. Wei, M. A. Park, J. T. Huse, J. G. Kuhn, M. L. Rinne, H. Colman, N. Y. R. Agar, A. M. Omuro, L. M. DeAngelis, M. R. Gilbert, J. F. de Groot, T. F. Cloughesy, A. S. Chi, T. M. Roberts, J. J. Zhao, E. Q. Lee, L. Nayak, J. R. Heath, L. L. Horky, T. T. Batchelor, R. Beroukhim, S. M. Chang, A. H. Ligon, I. F. Dunn, D. Koul, G. S. Young, M. D. Prados, D. A. Reardon, W. K. A. Yung and K. L. Ligon, J. Clin. Oncol., 2019, 37, 741–750 CrossRef CAS PubMed.
- T. J. Raub, G. N. Wishart, P. Kulanthaivel, B. A. Staton, R. T. Ajamie, G. A. Sawada, L. M. Gelbert, H. E. Shannon, C. Sanchez-Martinez and A. De Dios, Drug Metab. Dispos., 2015, 43, 1360–1371 CrossRef CAS PubMed.
- H. Hu, Q. Mu, Z. Bao, Y. Chen, Y. Liu, J. Chen, K. Wang, Z. Wang, Y. Nam, B. Jiang, J. K. Sa, H. J. Cho, N. G. Her, C. Zhang, Z. Zhao, Y. Zhang, F. Zeng, F. Wu, X. Kang, Y. Liu, Z. Qian, Z. Wang, R. Huang, Q. Wang, W. Zhang, X. Qiu, W. Li, D. H. Nam, X. Fan, J. Wang and T. Jiang, Cell, 2018, 175, 1665–1678 CrossRef CAS PubMed.
- D. A. Bota, W. Mason, S. Kesari, R. Magge, B. Winograd, I. Elias, S. D. Reich, N. Levin, M. Trikha and A. Desjardins, Neurooncol. Adv., 2021, 3, vdab142 Search PubMed.
- S. Vairy, G. Le Teuff, F. Bautista, E. De Carli, A. I. Bertozzi, A. Pagnier, F. Fouyssac, K. Nysom, I. Aerts, P. Leblond, F. Millot, C. Berger, S. Canale, A. Paci, V. Poinsignon, A. Chevance, M. Ezzalfani, D. Vidaud, A. Di Giannatale, R. Hladun-Alvaro, F. M. Petit, G. Vassal, B. Geoerger, M. C. Le Deley and J. Grill, Neurooncol. Adv., 2020, 2, vdaa075 Search PubMed.
- G. Chagoya, S. G. Kwatra, C. W. Nanni, C. M. Roberts, S. M. Phillips, S. Nullmeyergh, S. P. Gilmore, I. Spasojevic, D. L. Corcoran, C. C. Young, K. V. Ballman, R. Ramakrishna, D. A. Cross, J. M. Markert, M. Lim, M. R. Gilbert, G. J. Lesser and M. M. Kwatra, Oncotarget, 2020, 11, 2074–2082 CrossRef PubMed.
- R. M. Vala, D. M. Patel, M. G. Sharma and H. M. Patel, RSC Adv., 2019, 9, 28886–28893 RSC.
- M. G. Sharma, R. M. Vala and H. M. Patel, RSC Adv., 2020, 10, 35499–35504 RSC.
- D. M. Patel, H. J. Patel, J. M. Padrón and H. M. Patel, RSC Adv., 2020, 10, 19600–19609 RSC.
- D. M. Patel and H. M. Patel, ACS Sustainable Chem. Eng., 2019, 7, 18667–18676 CrossRef CAS.
- S. Peter and E. Weidner, Eur. J. Lipid Sci. Technol., 2007, 109, 11–16 CrossRef CAS.
- X. Zhang, C. Jia and J. Liu, Int. J. Hydrogen Energy, 2021, 46, 19939–19947 CrossRef CAS.
- M. Zakeri, M. M. Nasef and E. Abouzari-Lotf, J. Mol. Liq., 2014, 199, 267–274 CrossRef CAS.
- Q. Zhuang, H. Han, S. Wang, S. Tu and L. Rong, Synth. Commun., 2009, 39, 516–522 CrossRef CAS.
- L. Rong, H. Han, L. Gao, Y. Dai, M. Cao and S. Tu, Synth. Commun., 2010, 40, 504–509 CrossRef CAS.
- H. M. Patel, Green Sustain. Chem., 2015, 5, 137–144 CrossRef CAS.
- H. M. Patel, D. P. Rajani, M. G. Sharma and H. G. Bhatt, Lett. Drug Des. Discovery, 2019, 16, 119–126 CrossRef CAS.
- M. G. Sharma, J. Pandya, D. M. Patel, R. M. Vala, V. Ramkumar, R. Subramanian, V. K. Gupta, R. L. Gardas, A. Dhanasekaran and H. M. Patel, Polycyclic Aromat. Compd., 2021, 41, 1495–1505 CrossRef CAS.
- R. M. Vala, M. G. Sharma, D. M. Patel, A. Puerta, J. M. Padrón, V. Ramkumar, R. L. Gardas and H. M. Patel, Arch. Pharm., 2021, 354, 2000466 CrossRef CAS PubMed.
- V. Tandon, R. M. Vala, A. Chen, R. L. Sah, H. M. Patel, M. C. Pirrung and S. Banerjee, Biosci. Rep., 2022, 42, BSR20212721 CrossRef CAS PubMed.
- S. Banerjee, T. Wei, J. Wang, J. J. Lee, H. L. Gutierrez, O. Chapman, S. E. Wiley, J. E. Mayfield, V. Tandon, E. F. Juarez, L. Chavez, R. Liang, R. L. Sah, C. Costello, J. P. Mesirov, L. de la Vega, K. L. Cooper, J. E. Dixon, J. Xiao and X. Lei, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 24881–24891 CrossRef CAS PubMed.
- V. S. Tagliabracci, S. E. Wiley, X. Guo, L. N. Kinch, E. Durrant, J. Wen, J. Xiao, J. Cui, K. B. Nguyen, J. L. Engel, J. J. Coon, N. Grishin, L. A. Pinna, D. J. Pagliarini and J. E. Dixon, Cell, 2015, 161, 1619–1632 CrossRef CAS PubMed.
- K. M. Al-Zaydi, Ultrason. Sonochem., 2009, 16, 805–809 CrossRef CAS PubMed.
- R. M. Vala, V. Tandon, L. G. Nicely, L. Guo, Y. Gu, S. Banerjee and H. M. Patel, ChemRxiv, 2021 DOI:10.26434/chemrxiv.14774835.v1 , preprint.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03579k |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |