
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptical bio-sensing of DNA methylation analysis: an overview of recent progress and future prospects
Mina Adampourezare*ab,
Mohammad Hasanzadeh *bc and
Farzad Seidid
*bc and
Farzad Seidid
aDepartment of Biology, Faculty of Natural Science, University of Tabriz, Tabriz, Iran. E-mail: m.adampour@tabrizu.ac.ir
bPharmaceutical Analysis Research Center, Tabriz University of Medical Sciences, Tabriz, Iran. E-mail: hasanzadehm@tbzmed.ac.ir
cNutrition Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
dJiangsu Co-Innovation Center for Efficient Processing and Utilization of Forest Resources and International Innovation Center for Forest Chemicals and Materials, Nanjing Forestry University, Nanjing 210037, China
First published on 9th September 2022
Abstract
DNA methylation as one of the most important epigenetic modifications has a critical role in regulating gene expression and drug resistance in treating diseases such as cancer. Therefore, the detection of DNA methylation in the early stages of cancer plays an essential role in disease diagnosis. The majority of routine methods to detect DNA methylation are very tedious and costly. Therefore, designing easy and sensitive methods to detect DNA methylation directly and without the need for molecular methods is a hot topic issue in bioscience. Here we provide an overview on the optical biosensors (including fluorescence, FRET, SERs, colorimetric) that have been applied to detect the DNA methylation. In addition, various types of labeled and label-free reactions along with the application of molecular methods and optical biosensors have been surveyed. Also, the effect of nanomaterials on the sensitivity of detection methods is discussed. Furthermore, a comprehensive overview of the advantages and disadvantages of each method are provided. Finally, the use of microfluidic devices in the evaluation of DNA methylation and DNA damage analysis based on smartphone detection has been discussed.
1. Introduction
DNA methylation in the 5′ carbon of cytosine nucleotides is an important epigenetic modification that has crucial roles in gene expression, embryonic development,1 cancer, and neurobiological diseases such as Alzheimer's disease.2 This important epigenetic modification is a post-replication change that causes a change in the chromosome without changing its sequence. Important consequences of epigenetic modification include controlling genetic expression,3 genomic imprinting4 and cellular differentiation silencing of the X chromosome.5,6There are two main types of changes in the DNA methylation pattern: one is global hypomethylation, which usually targets non-coding regions and can cause chromosomal instability and aberrant expression of genes, and the other is hypermethylation of CpG islands in the promoter regions of genes, which causes deactivation of these genes.7
The most important genes that are silenced by hypermethylation in the promoter region include tumor suppressor genes, tumor invasion and spread suppressor genes, DNA repair genes, hormone receptor genes, and angiogenesis inhibitor genes.7 The global hypomethylation at the CpG rich region leads to weak transcription suppression of genes such as oncogenes (tumorigenic genes) or retrotransposons (Fig. 1).7
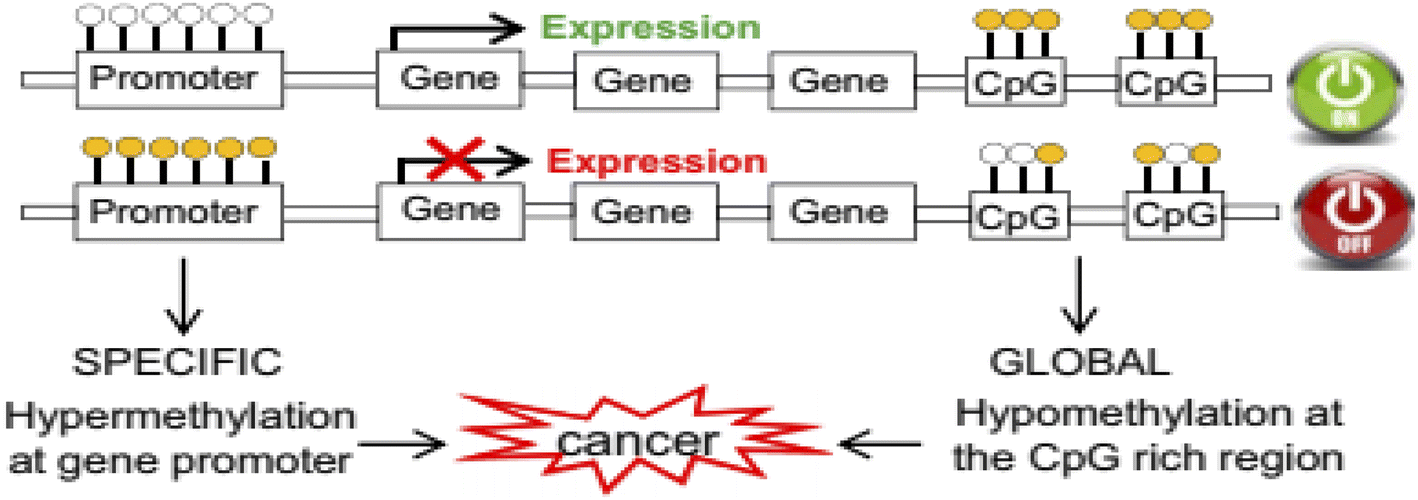 | ||
| Fig. 1 Rearrangement of the DNA methylation patterns associated with tumorigenesis.7 | ||
Since cytosine and methyl cytosine have similar Watson–Crick's behavior, conventional sequencing cannot be used to detect methyl cytosine. Recently, DNA methylation was detected by various methods including bisulfite genomic sequencing,8 the use of restriction enzymes,9 and the use of anti-methyl cytosine antibodies.9 One of the most important benefits of bisulfite treatment is deamination of non-methylated cytosine and to convert it to uracil (U).10 Limitation of bisulfite treatment are DNA degradation, time-consuming, incomplete conversion of non-methylated cytosine to uracil, require expensive instruments, and high cost of sequencing.11–14
Enzymatic/chemical hydrolysis based methods such as high performance liquid chromatography (HPLC)15 and high performance capillary electrophoresis (HPCE)16 need, lengthy experimental processes, abundant DNA samples, and expensive instruments. A major limitation of DNA restriction enzyme digestion based methods such as PCR (MSRE-PCR),17 methylation-specific multiplex ligation and dependent probe amplification (MS-MLPA)18 applied for the site-specific detection of DNA methylation is restriction of specific enzyme identification sequences. Therefore, information about the methylation status of the entire genome cannot be obtained. The major advantages of bisulfite treatment methods such as methylation sensitive high resolution melting (MS-HRM),19 combined bisulfite restriction analysis (COBRA),20 methylation specific PCR (MS-PCR),21 methlight,22 bead arrays (Illumina),23 and pyrosequencing24,25 is converting methylation changes to base changes (unmethylated cytosine to uracil and finally thymine). The major disadvantages of this method are DNA degradation due to high temperature and prolonged incubation with chemicals, incomplete conversion of unmethylated cytosine, time-consuming, need to use of expensive equipment, and high cost of sequencing services.26–28 Affinity enrichment technique using DNA methyl binding domain (MBD) protein are appropriate for high efficiency analysis but time-consuming and requiring antibodies and instrument are the main limitations of this method. Isotope-labeled S-adenosylmethionine (SAM) and immune reactions focus on direct exposure detection of DNA methylation using its dynamics assay. Time-consuming and too costly materials are the disadvantages of these methods.29
Therefore, highly sensitive, accurate, and quantitative assay methods of DNA methylation would be valuable for genetic disease diagnosis. In recent years, scientists have tried to develop various methods to determine methylation, which one of them is the use of optical biosensors.30
Optical biosensors are powerful detection and analysis tools that measure changes in the optical properties including absorbing, reflecting, and emitting specific light wavelengths in response to a connecting event between a receiver and target molecules.31,32 An optical biosensor consists of a compact analytical tool that contains a bio-recognition sensing element integrated with an optical transducer system. The main purpose of an optical biosensor is to generate a signal that is proportional to the concentration of a measured substance (analyte).
These biosensors can be classified into five distinct classes based on the nature of the transfer mode including fluorescence, surface plasmon resonance (SPR), surface-enhanced Raman scattering (SERS), colorimetry and luminescence.32,33 The following subsections concentrate on the applied optical biosensors in the recent researches for DNA methylation detection by different transduction events. In addition to the use of biosensors, the use of nanomaterials improves the quality and sensitivity of detection. In accordance with this, with the development of nanotechnology, new nanomaterials have been designed and synthesized such as gold nanoparticles,34 carbon nanomaterials,35 semiconductor quantum dots,36 and metal nanoclusters.37 Nanomaterials have some attractive optical properties due to their unique size and shape and may act as optical emitters.38–40 In addition, bioactivation of nanomaterials through physical adsorption, electrostatic and/or covalent coupling, makes them a powerful tool for making suitable biosensors.40,41 In this review, critical role of labelled and label free optical methods on the photonic biosensing of DNA methylation are surveyed. Also, important role of advanced nanomaterials on the optical biosensing with emphasize on the key role of detection method are investigated.
2. Evaluation of DNA methylation using fluorescence method
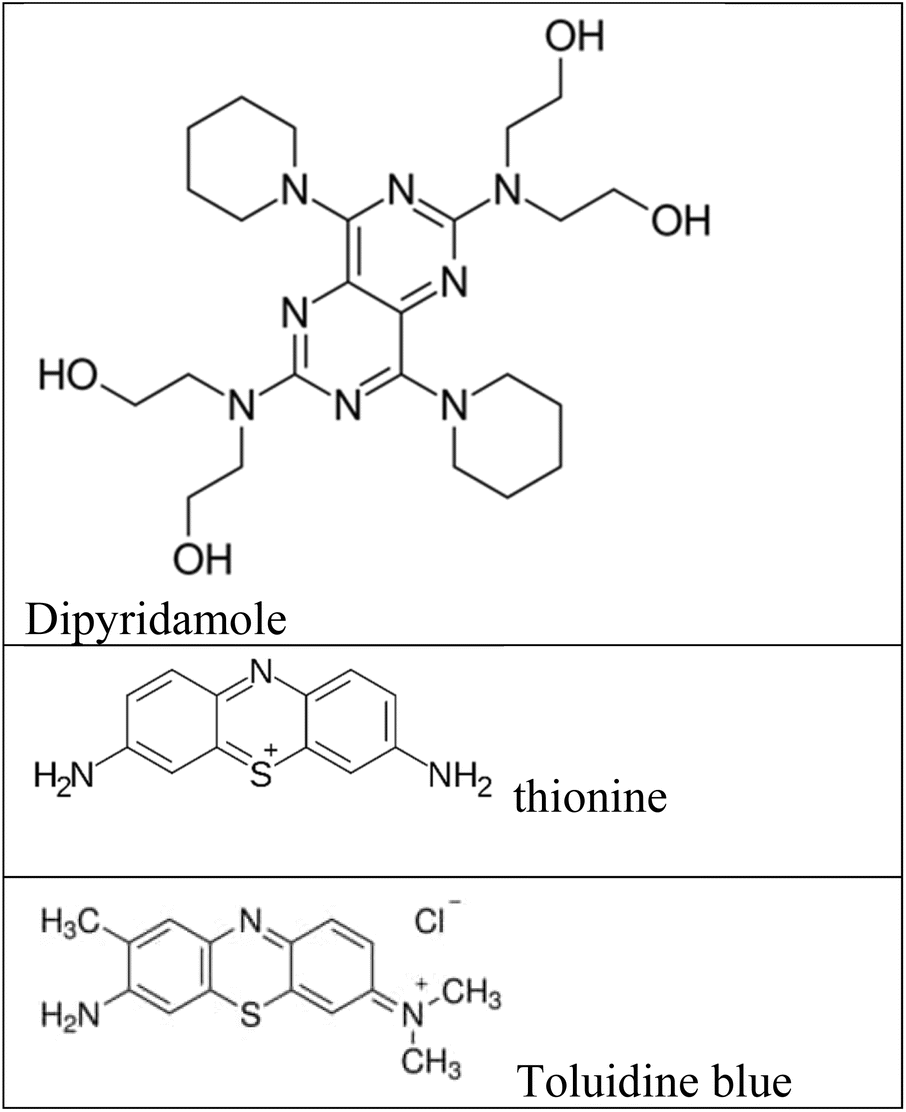
Fluorescence assays dominate some fields of biosensing because of high intrinsic sensitivity of this optical technique and the possibility of integration with receptors with high selectivity. In fluorescence detection, a specific wavelength of electromagnetic radiation excites fluorophore molecules and an optical transducer detects the intensity of shifted and emitted light.32,42 Three different approaches to bioassay target molecules using fluorescence methods including labeled, label free, and fluorescence resonance energy transfer (FRET) methods.The label free method is direct assay method of target molecules before and after the binding events. The labeled method is indirect detection method of target molecules by adding fluorescent labeling reagents such as organic dyes and nanoparticles (quantum dots, dyedoped silica nanoparticles).33,43 For example, a fluorometric nanobiosensor was used to detect DNA methylation based on the use of gold coated magnetic nanomaterials to stabilize DNA probe (SH–(CH2)6–5′-CCG TCG AAA ACC CGC CGA-3′).44 In this study, dipyridamole was used as a fluorescence optical probe and was able to distinguish methylated sequences from unmethylated sequences. The detection limit for methylated and unmethylated sequences was 3.1 × 10−16 M and 1.2 × 10−16 M, respectively (Fig. 2A). Fluorometric nanobiosensors based on carbon materials and organic dyes of thionine45 (Fig. 2B) and toluidine blue46 were developed to detect DNA methylation. These optical probes were used as a platform as well as fluorophore and were able to distinguish methylated sequences from unmethylated sequences. The designed nanobiosensor had a detection limit of 1 ZM.
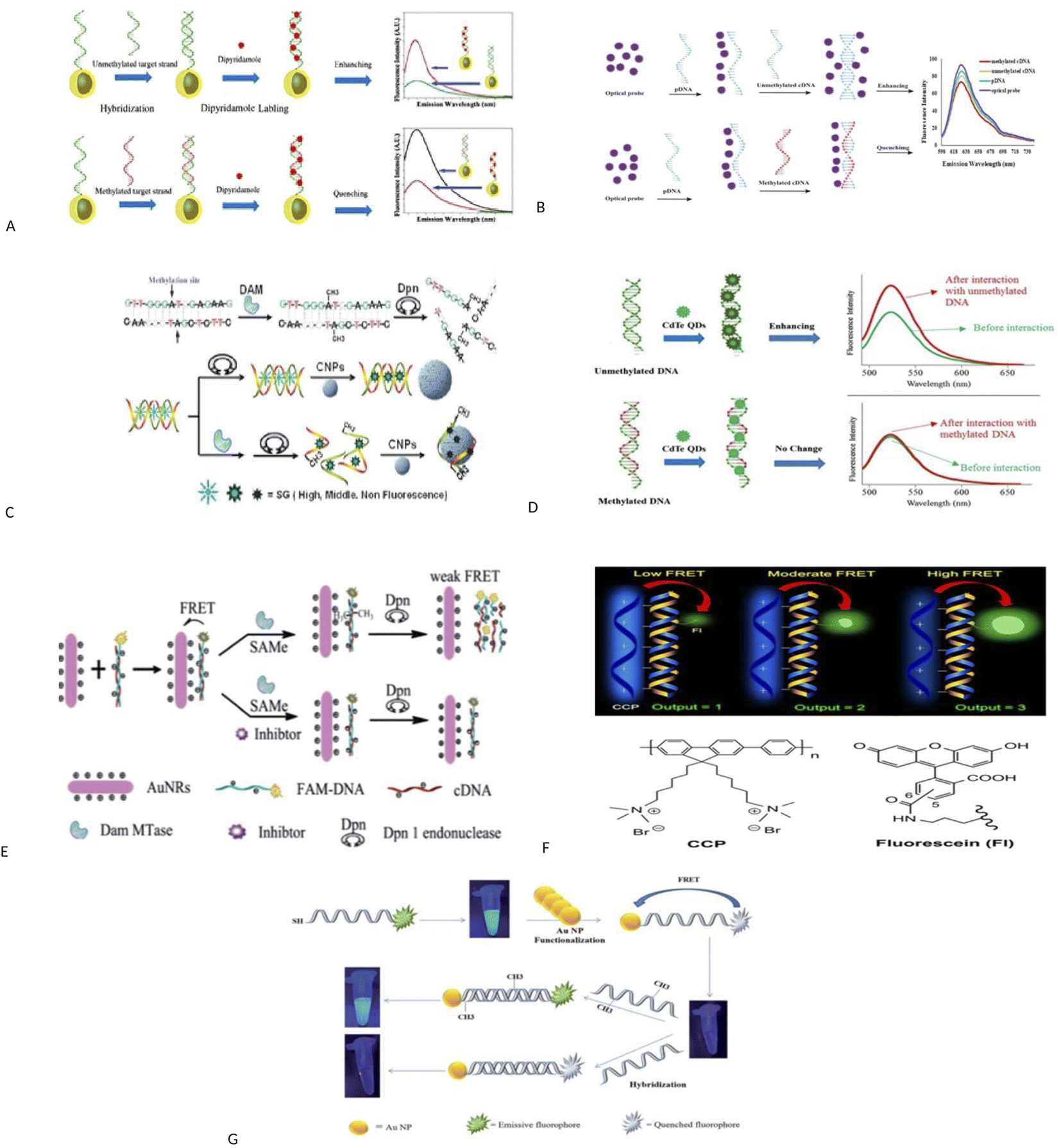 | ||
| Fig. 2 (A) Schematic representation of the bio-assay for DNA methylation detection.44 (B) Schematic diagram of nanobiosensor preparation steps for DNA methylation assay.45 (C) Schematic description for label-free fluorescence assay of DNA methylation based on CNPs, SG and enzyme–linkage reactions.48 (D) Schematic representation of direct detection of DNA methylation by CdTe QDs.49 (E) A schematic illustration of the ultrasensitive detection of DNA methylation by gold-nanorods-based fluorescence resonance energy transfer (FRET) assay.52 (F) Principle of Methylation Level Analysis of Cancer-Related Genes Using CCP-Based FRET Technique, together with the Chemical Structures of CCP and Fluorescein Used in the Detection.53 (G) Schematic representation of a FRET based approach for the detection of methylated DNA.54 (H) Detection procedures of cytosine methylation in DNA by the FRET probe based on UCNPs and AuNRs.55 | ||
A label free and sensitive fluorometric nanobiosensor based on quantum dot graphene was developed by Rafiei and et al.47 Graphene quantum dot (GQD) had a strong affinity for double-stranded DNA and was intercalated within double-stranded DNA in a major groove. This nanobiosensor quenched the fluorescence when attached to methylated sequences, but increased the fluorescence intensity when attached to unmethylated sequences. It has been assumed that the intercalation of GQD to double-stranded methylated DNA, DNA conformation changes from form B to form A, but in double-stranded unmethylated DNA, DNA conformation does not change and this change in conformation causes an increase or decrease in fluorescence intensity. The nanobiosensor had a detection limit of 7.3 × 10−11 M. Indeed, research group of Xiangyuan48 developed a label free and sensitive fluorometric nanobiosensor based on DNA intercalator dye, endonuclease and carbon nanomaterials for detecting DNA methylation and evaluating DNA methyltransferase activity. In this study, the basis of the work was designed in such a way that single-stranded DNA was absorbed on the surface of carbon nanomaterials using π–π bonds, but double-stranded DNA did not have this feature. Also, when organic dyes were attached to carbon nanomaterials, their fluorescence was quenched, and SYBR Green I (SG) were more inclined to double-stranded DNA than single-stranded DNA. In this study, double-stranded DNAs were methylated by DNA methyltransferase enzyme and then were cleaved by methylation-sensitive restriction endonuclease. Single-stranded DNAs were obtained, and the SG was released and placed on carbon nanomaterials, and its fluorescence was quenched. Double-stranded DNAs without methyl groups were not cleaved by methylation-sensitive restriction endonuclease and thus their fluorescence intensity was higher. The nanobiosensor had a detection limit of 7.3 × 10−11 M (Fig. 2C).
Interestingly, a fluorometric nanobiosensor was built based on the capped CdTe quantum dots as a fluorophore probe.49 This optical probe tended to have double-stranded DNA sequences and increased the fluorescence intensity in interaction with unmethylated sequences, but no change in fluorescence intensity was observed in interaction with methylated sequences. The different mobility in electrophoresis assay was confirmed the results of the spectroscopic study. The detection limit for unmethylated DNA was 6.2 × 10−11 M (Fig. 2D). The third strategy for fluorescence evaluation is fluorescence resonance energy transfer (FRET). The mechanism of FRET, a donor fluorophore excites and transfers its excitation energy to a nearby acceptor chromophore having a similar resonance frequency in a non-radiative (radiationless) fashion and the distance-dependent (approximately 1–10 nm) through dipole–dipole interactions.50–52
FRET is a spectroscopic technique that is widely applied in various biological and medical fields such as excavating biomolecules conformational changes, assaying enzyme activity and intermolecular interactions, and nucleic acid analysis.51 Various nanomaterial compositions have been made by the FRET mechanism for use as nanosensors. FRET is a distance-dependent energy transduction method that energy transfers from a donor molecule to an acceptor molecule. Due to its sensitivity to distance, molecular interactions are assayed by FRET.51
For example, a fluorescence method based on FRET assay was designed by using UCNPs and gold nanorods (AuNRs) as a fluorescence quencher for detection of DNA methylation and to evaluate DNA methyltransferase activity.52 In this method, the probe DNA was attached to a fluorescent material (FAM) and hybridized with its complement DNA. The resulted dsDNA was methylated by DNA methyltransferase and was cleaved by Dpn. The fluorescence measurements were performed after the addition of gold nanorods. This method is able to exhibit methyltransferase activity with a low limit of detection 0.25 U mL−1 (Fig. 2E). In another study, DNA methylation assay was performed based on FRET using cationic conjugated polymer in RASSF1A, OPCML, and HOXA9 promoters as ovarian cancer markers.53 After digestion of genomic DNA by methylation-sensitive restriction endonuclease and then PCR amplification of digested productions, the methylation level is detected through FRET arising from incorporation CCP and fluorescein into the PCR products. The duration was about 20 h (Fig. 2F).
The FRET assay method also was applied to detect DNA methylation and to evaluate DNA methyltransferase activity using gold nanoparticles based on fluorescence retrieval of FAM-labeled DNA with gold nanoparticles (AuNPs).54 In this study, one end of the DNA probes was attached to FAM fluorophore and the other end was modified with thiol groups (6′-FAM–5′-TCCGGTTCCCGACCCGGACTCCGCAAAAAA-3′-SH). The thiol groups at the end of the probe were functionalized with AuNPs, which brings the FAM fluorophores close to the AuNPs. This caused the overlap of the FAM fluorescence emission and the absorption spectrum of the AuNPs and triggered a FRET event and caused the fluorescence quenching of FAM fluorophore.
Then, FAM-labeled DNA was hybridized with its complementary sequences. Hybridization of the FAM-labeled DNA with its complementary DNA created a specific CpG locus for the activity of M.SssI methyltransferase enzyme. The methyltransferase caused the methylation of DNA at specific CpG sites and converted quenched FAM fluorophore into an emissive fluorophore. Conversely, no change in fluorescence signal was observed in unmethylated sequences. In this study, detection limit of 0.14 U mL−1 for M.SssI methyltransferase enzyme and 2.2 pM for methylated targets were resulted (Fig. 2G). In another study, methylation of cytosine in specific sequences in DNA was detected using FRET between up-conversion nanoparticles (UCNPs) and gold nanorods (AuNRs) and ssDNA probe (biotin-5′-ATACCmGGTCTAAA-3′-S-S).55 In this method, methylated single-stranded DNA, which contained biotin at one end and thiol group at the other, was hybridized to methylated single-stranded DNA (5′-AGACCmGGTAT-3′) and unmethylated single-stranded DNA (3′-TATGGCCAGA-5′). The resulting double-stranded DNAs were located adjacent to UCNPs and AuNRs. The UCNPs were interacted with biotin and the gold nanoparticles were attached to thiol group and placed close to each other and their fluorescence was quenched. Then, the resulted double-stranded DNAs were exposed to HpaII restriction enzyme. Two-stranded sequences containing one methyl group were cleaved with HpaII and the fluorescence signal was recovered, but the sequences containing the two methyl groups were not cleaved and the fluorescence signal did not change. Sequence containing the two methyl groups were cleaved by MspI enzyme and the fluorescence signal was recovered. Duration was 2 h (Fig. 2H).
Importantly, novel technology of MS-qFRET, methylation specific quantum dot FRET, was used to evaluate DNA methylation.56 In this method, first, bisulfite treatment and conversion of cytosine to uracil was done. Then PCR was done using Cy5-dCTP as an acceptor and methylation specific primers that forward primers contain a biotin label for post-PCR conjugation to quantum dots as a donor. In this method, DNAs containing methylcytosine were amplified, but DNAs containing unmethylated cytosine were not amplified. Amplified PCR product end contains biotin, which conjugated to quantum dot by biotin-streptavidin affinity. Upon excitation at 488 nm, QD emission is recorded at 605 nm and Cy5 (FRET acceptor) at 670 nm (Fig. 3A).
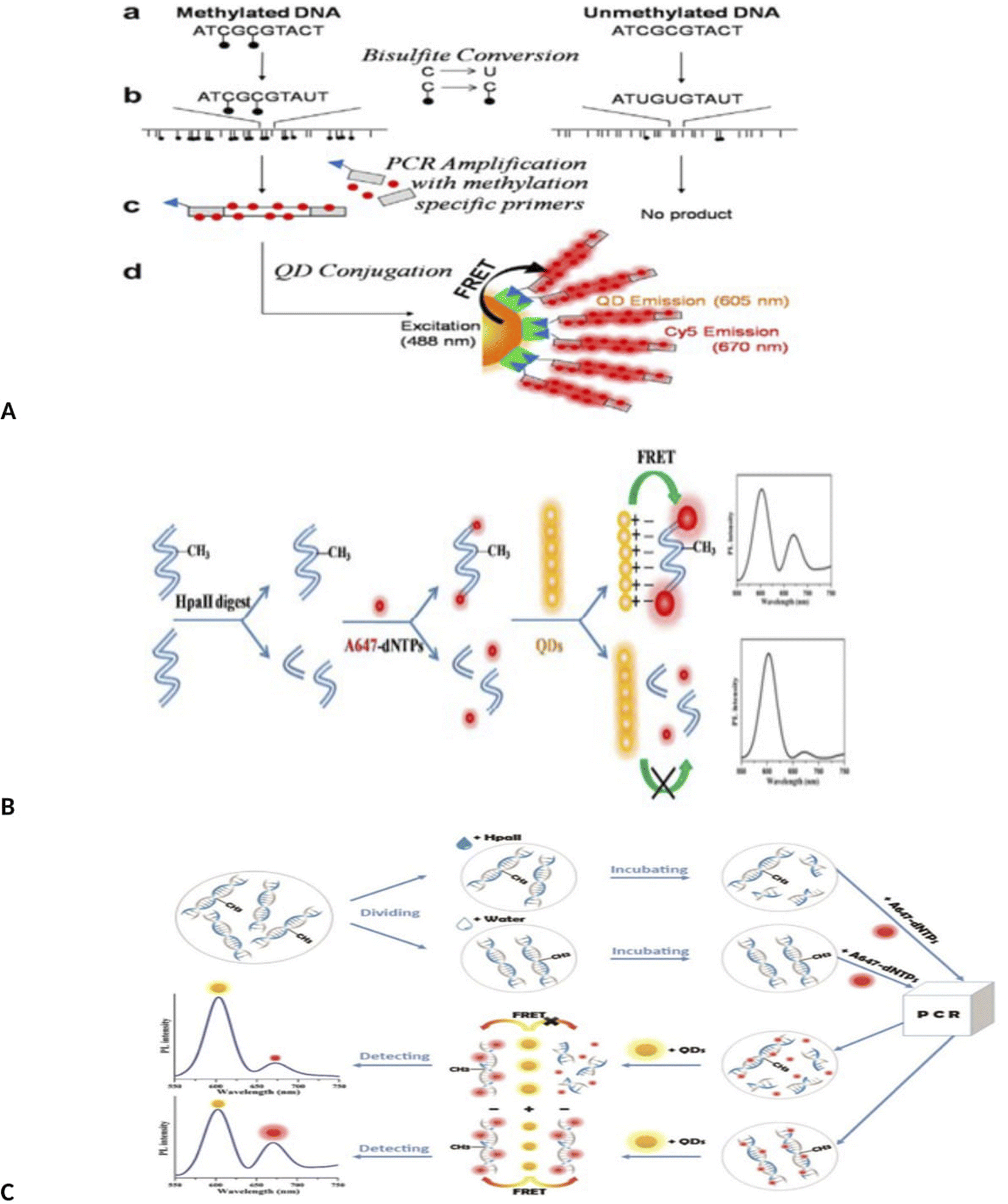 | ||
| Fig. 3 (A) Schematic depiction of identification of DNA methylation detection via MS-qFRET technology.56 (B) Schematic procedures for the detection of methylated DNA based on FRET using up-conversion nanoparticles (UCNPs) and intercalating dye.57 (C) Schematic of QD-FLP, illustrating the improved design. After bissulfite conversion and PCR amplification with modified primers, the FLP distinguishes the target amplicon, thereby eliminating background noise from the non-specific PCR amplicons.61 (D) Schematic illustration of the detection of DNA methylation based on QDs-FRET.62 (E) Schematic illustration of the detection of DNA methylation based on QDs-FRET.63 | ||
Interestingly, a MS-PCR, up-conversion nanoparticles (UCNPs) and intercalating-based FRET system also has been developed for the detection of DNA methylation of CDKN2Agene by Kim et al.57 Briefly, after bisulfite treatment of DNA and amplification with methylation-specific PCR with biotinylated forward primer (forward primer, 5′-biotin-TTATTAGAGGGTGGGGCGGATCGC-3′), amplified methylated DNA was hybridized with streptavidin-UCNPs and SYTOX Orange were intercalated to the streptavidin-UCNP. FRET was occurred between the UCNPs and SYTOX Orange and the intensity of the green fluorescence was decreased from the UCNPs as FRET donors (Fig. 3B). This method has a limit of detection as low as 0.1% (Fig. 3B).
MS-qFRET method58 developed based on MSP59 and the quantum dot fluorescence resonance energy transfer (QD-FRET) nanosensor technology,60 needs low concentrations of DNA. The disadvantages of this method are that MSP is dependent upon bisulfite conversion of extracted DNA and bisulfite conversion causes DNA damage.59 Also, formation of primer dimer and non-specific PCR products with dual biotin and Cy5 labels that can similarly attach to the QD surface, cause the FRET background.
A method similar to that used by Kim was also used to detect methylation by Keeleyi et al.61 The detection system consists of three main processes including bisulfite conversion, PCR and PCR amplicons conjugation to the surface of QDs (as donor) by FLPs (biotin functionalized DNA oligonucleotide probes) as acceptor for FRET detection. In this method, only sequences containing methyl cytosine were amplified by PCR and amplified products bound to the quantum dot surface by biotin–streptavidin binding. Due to overlap of the spectrum of the quantum dot and FLP, the fluorescence intensity decreased. Finally, QD-FRET signals were measured using nanodrop 3300 fluorospectrometer. In this study, the biological limitations of dysfunctional PCR and FRET signal arising from background noise generated using the MS-qFRET method was eliminated using FLP (Fig. 3C). In another study, quantum dots-based FRET method was used for DNA methylation assay of three tumor suppressor genes, PCDHGB6, HOXA9 and RASSF1A, in lung adenocarcinoma and adjacent nontumorous tissues (NT).62 In this study, DNA was extracted and divided into two parts. One part was placed in the vicinity of HpaII enzyme. This enzyme cleavages unmethylated sequences but does not cleavage methylated sites. The second part of the DNA was not in the vicinity of the enzyme. Both samples were amplified by PCR using 647-dNTP. In the first part, methylated DNA could be amplified since it was insensitive to cleavage by HpaII. Unmethylated DNA remains unamplified because it was cleaved by the HpaII enzyme. The resulting PCR products were mixed with amino-QDs dots. Positively charged amine QDs formed electrostatic interaction with negatively charged A647 labeled DNA. This compound could be detected by FRET. With the increase in the number of methyl groups, the intensity of the FRET signal was stronger. In the case of products not treated with the enzyme (second part of DNA), both methylated and unmethylated DNA were amplified, resulting in high-efficiency FRET signals. The limit detection was obtained up to 90% for cancer (up to 90% of sequences with methyl group were detected. The detection sensitivity of this method was 90% of the sequences containing the group) (Fig. 3D).
Similarly, a methylation-sensitive restriction enzyme, bisulfite free, quantum dots (QDs) based FRET method using A647 fluorophores (Alexa Fluor-647 (A647) fluorophores) was designed to assay the promoter methylation in NSCLC tissue samples and noninvasive bronchial brushing specimens.63 Using this method, the methylation level was determined by measuring the signal amplification during QDs-FRET. Using this method, DNA methylation was assessed in three genes (PCDHGB6, HOXA9 and RASSF1A) and in two sample groups (control and patient). This method was able to detect methylation with a high degree of sensitivity. One of the advantages of this method is not using bisulfite treatment and labeled primers (Fig. 3E).
3. Evaluation of DNA methylation using SPR method
SPR (superficial plasmon resonance) is one of the strongest analytical methods for biomolecular detection.64 In an SPR-based optical biosensor, the binding of the target analyte to the surface-connected receptor causes a positional alteration in refractive indicator on the sensor surface, which is quantitatively associated with an increase in the relative mass associated with the target absorption.65 SPR measures kinetic and affinity constants of bimolecular interactions in real-time and label-free fashion.66,67 Since this SPR has distinct advantages over radioactive or fluorescent labeling methods as this label may impair binding. Additionally, SPR is cost effective and measures directly binding constant and affinity; and consumes less reagents.68 In this subsection, some of SPR-based biosensing techniques were surveyed.For the example, a novel SPR-based biosensor optical was applied for detection of methylated CpG sites on specific DNA sequences in the promoter of the adenomatous polyposis coli (APC) gene using a dual detection mechanism with oligonucleotide probe hybridization and specific protein binding.69 In this report, first, the probe immobilized (5′-biotin-TGmCGGAGTGmCGGGTmCGGGAAGmCGG-3′) onto a SPR based biosensor chip captured complementary target DNA. Then, the recombinant methyl CpG binding domain (MBD) protein was added on the surface to recognize and bind to methylated CpG sites. Binding events increased the refractive index, and generated a detectable optical signal. The detection limit was around 5 pmol within 1 h for methylated APC promoter. The main disadvantage of this method is the use of specific proteins bound to methyl groups (methyl binding domain protein). One of the advantages of this technique is that it does not require bisulfite treatment.
A real-time, label-free, molecular inversion probes (MIPs), bisulfite treatment, circularized DNA probe and SPR method was applied for DNA methylation detection of En1 region DNA extracted from MCF7 cells and human whole genomic DNA.70,71 In this method, after genomic DNA extraction from cell lines and bisulfite treatment (conversion of unmethylated cytosines into uracile), the desired region of genomic DNA is identified by MIP and MIP binds to the DNA and a multi-play gap is created between the ends of the MIP identification, which represents the desired methylated region. This gap is filled by a polymerase and closed by a ligase, creating a circular DNA. This circular product is then converted into a single-stranded amplicon by asymmetric PCR, which is attached to a single-stranded oligonucleotide immobilized on SPR. In the final stage, detection is done by SPR. In the presence of methylated DNA compared to unmethylated DNA, an increase in the SPR signal was observed. The concentration range was from 0 to 400 nM with low detection limit 100 nM. Combined detection of SPR and EAP has three advantages including instant read-outs, cost-effective, ability to detect multiple samples simultaneously using multi-channel SPR and MIP multiplication capability (Fig. 4A).
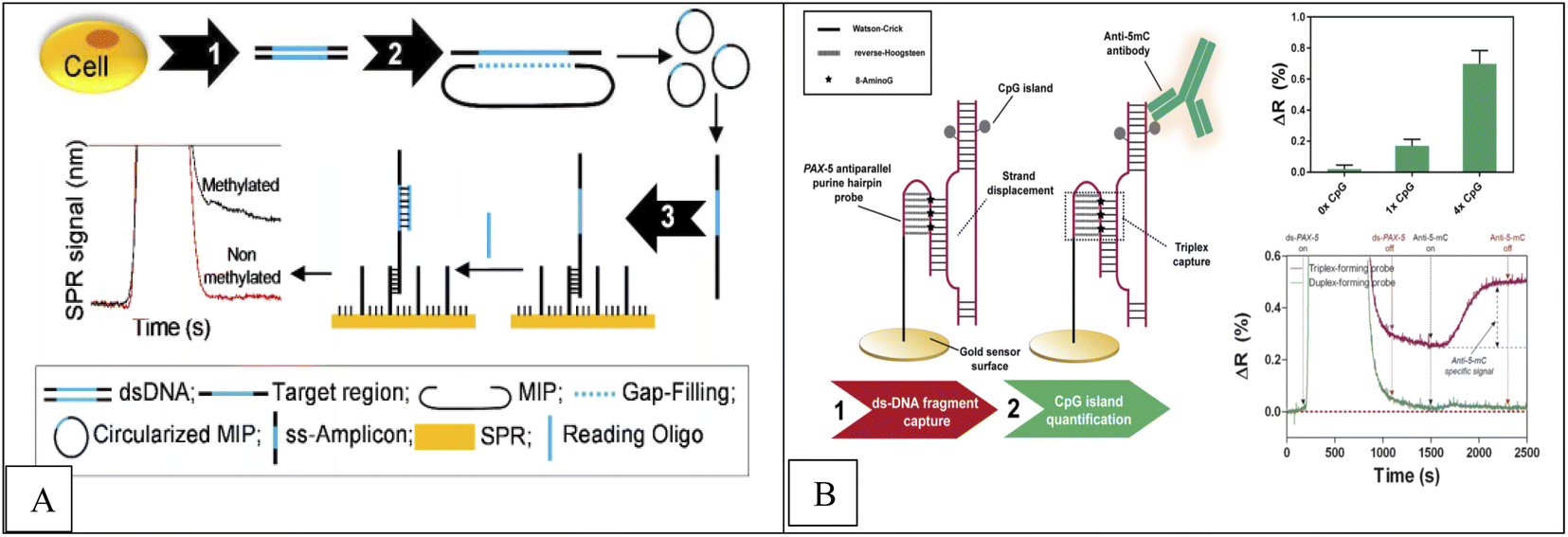 | ||
| Fig. 4 (A) Methodological approach for the label-free and real-time detection of regional DNA methylation based on MIP and SPR.70 (B) Scheme of the DNA methylation biosensor methodology.72 | ||
Huertas et al.72 reported a label-free, SPR-immunoassay using poly-purine reverse-Hoogsteen hairpin (PPRH) probes (DNA molecules composed of two mirrors symmetrical polypurine stretches and a poly-thymidine loop) to assess DNA methylation in PAX-5 gene fragment. Due to their structural properties, these oligonucleotides can form reverse-Hoogsteen hairpins and form Watson–Crick bonds with polypyrimidine double-stranded DNA targets to form three stranded (triplex) conformation.74 Finally, anti-methylcytosine antibody was used to detect methylcytosine, which specifically recognizes cytosine roots and binds specifically to methyl groups in cytosine roots, and a sensor signal proportional to the quantity of methyl groups is obtained (Fig. 4B). Similarly, Li et al.,74 reported a novel platform based on Au nanorod (AuNR) (as label), polymerization and nicking reactions, of enzyme cyclic signal enhancement methods, SPR technique using double stranded DNA (dsDNA) probe that was self-hybridized with a palindromic sequence of 5′-GATC-3′ for DNA methylation detection and adenine methylation (Dam) methyltransferase (MTase) activity assay. The concentration range was from 0.5 to 120 U mL−1 with a detection limit of 0.2 U mL−1.
Despite researches done at the use of SPR in the evaluation of DNA methylation, the methods used also have some limitations, such as the use of restriction enzymes and antibodies or the use of complex primer design methods. It is also necessary to use some traditional methods in combination with biosensor techniques. Therefore, designing new methods that can study methylation without using molecular methods19,20 is of interest to researchers.
4. Evaluation of DNA methylation using surface enhanced Raman scattering (SERS) method
SERS is a label-free method that has been extensively applied in the characterization of molecular monolayers, interfacial reactions and various biological surfaces.76–78 It is also considered as a physical phenomenon that happens on metal surfaces which can replace fluorescence-based detection methods.79–81 SERS is based on highly amplified Raman signals from molecules attached to nanometer sized metallic structures.82 SERS has significant beneficial over fluorescence, such as unique spectrum characteristics with massive informational content, narrow spectral bands for multiplied assays, and free from both photobleaching and self-quenching of fluorophores.82In contrast, the overlapping fluorescence emission spectra strongly restrict multiplexing. In addition, SERS also has comparable sensitivity yet improved photostability over fluorophores.79–81 Recently, Raman spectroscopy was applied for evaluation of DNA methylation.76,83–86 Raman spectroscopy has several advantages over methods based on fluorescence, because multiple sites of nucleoside methylation in DNA is detectable using SERs simultaneously.76,85 In addition, it does not require any chemical additives and minimizes chemical changes in the samples.83,87 Raman bands represent molecular fingerprints with narrow bandwidths, which makes it possible to identify multiple sites.76,83
Recently, SERS has been employed for DNA identification and single nucleotide polymorphism (SNP) analysis.88–92 For example, Hu and coworkers used single base extension reaction and SERs spectroscopy to detect DNA methylation using AuNPs-modified capture probe in tumor suppressor gene CDKN2/p16/MTS1 (p16) and a detection limit of 3 was obtained.76 In the other study, label-free method of Raman Spectroscopy was applied for detection of DNA methylation using synthesized sequences according to the promoter regions of cancer-related genes such as cadherin 1 (CDH1) and retinoic acid receptor beta (RARB).93
Ganesh et al. was applied SERs to assay the DNA methylation profile in cancer stem cells.94 Ouyang et al. applied laser wrapped graphene-Ag array for detection of methylated DNA and its oxidation derivatives (5-hydroxymethylcytosine and 5-carboxylcytosine) by SERS. In this method, first, the target DNAs are identified and captured from the global DNA by AuNPs functionalized with a specific antibody. Since methylated DNAs have a weak SER signal, the captured target DNAs are marked by dye SYBR Green I (SG) as Raman tag, which can specifically bind to double-stranded DNA and increase the SER signal. Also, for further SERS enhancement, Au-DNA conjugates are directly dipped onto the graphene wrapped Ag array. The detection limit 1.8 pmol L−1 and duration 60 min was obtained (Fig. 5A).95 Wang et al., applied SERS via ligase chain reaction (LCR) for DNA methylation detection. 10% changes in methylation can be detected by this method (Fig. 5B).96
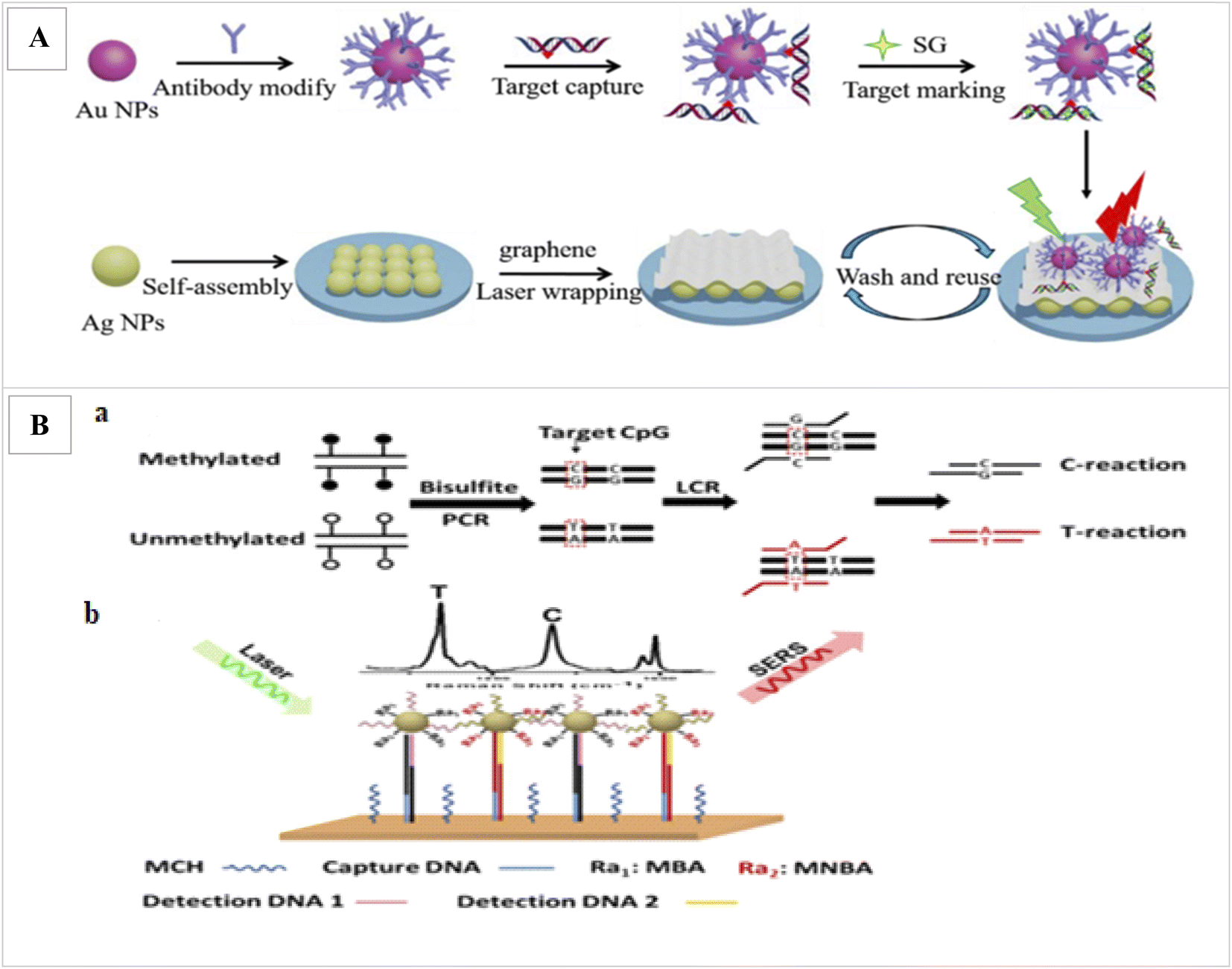 | ||
| Fig. 5 (A) Illustration of the proposed SERS strategy for detection of methylated DNA and its derivatives.95 (B) Schematic illustration for simultaneous C and T single DNA base change detection by LCR (a), and DNA methylation analysis via simultaneous SERS nanotags detection on a gold surface array (b).96 | ||
In this study, after bisulfite treatment of genomic DNA and conversion of unmethylated C to uracils and subsequently T after PCR, for the PCR amplification of a sequence containing a CpG of interest, LCR was applied to amplify a sequence containing a CpG of interest and to identify the C/T base change. In order binding LCR products onto the SERS array, one end of the LCR products contain the specific sequence for binding to DNA probe immobilized onto the SERS array and the other end contains a methylation state specific sequence to bind methylation-specific SERS nanotag. Finally, the methylation levels of captured LCR products were identified by methylation state-specific SERS nanotags. These nanotags were made from AuNPs functionalized with methylation specific DNA probes and Raman reporters. Final detection was performed by laser. Finally, to determine the level of methylation, the SER signals intensities produced from the LCR products of the C-reaction and T-reaction were compared.
However, the major limitation of clinical application of Raman spectroscopy is that the signals from single-stranded and double-stranded DNA are similar. To overcome those constraints, inculcated internal SERS nanotags were used to quantify DNA by Wang et al.85 This method provides real-time molecular information at high resolution without the use of labels. However, it travails from weak signal and restricted penetration depth, typically for optical based techniques.
5. Evaluation of DNA methylation using colorimetric method
Colorimetric nanosensors are a (semi) quantification method and popular in molecular diagnostics. The colorimetric method has various advantages, including the ability to visualize with naked-eye, cost effectiveness, simplicity, portability and a low limit of detection.97,98For the example, a colorimetric assay was applied to detect DNA methylation using methyl-binding domain (MBD) proteins for evaluation methylated prostate cancer biomarker in the urine. The method can detect 5% methylation differences in total genomic and gene-specific methylation and the limit of detection for 50 ng or less DNA input under 2 h was obtained.99 In this method, genomic DNA was first affected by enzymatic digestion and the resulting DNA fragments were enzymatically labeled with biotin to produce a DNA-biotin polymer. Then MBD protein conjugated with magnetic beads was used to select methylated DNA. Finally, methylated DNA was recognized by SA-HRP (streptavidin conjugated horse radish peroxidase) via the biotin/streptavidin interaction. Finally, methylation levels were visually evaluated via the HRP-mediated reduction of a chromophore (TMB substrate). In the second method, after enzymatic digestion of DNA, the resulting fragment was amplified by isothermal amplification using biotin–deoxynucleotides (dNTPs). Then, HRP/TMB colorimetric reaction was applied to indicate the presence of methylated regions (Fig. 6).
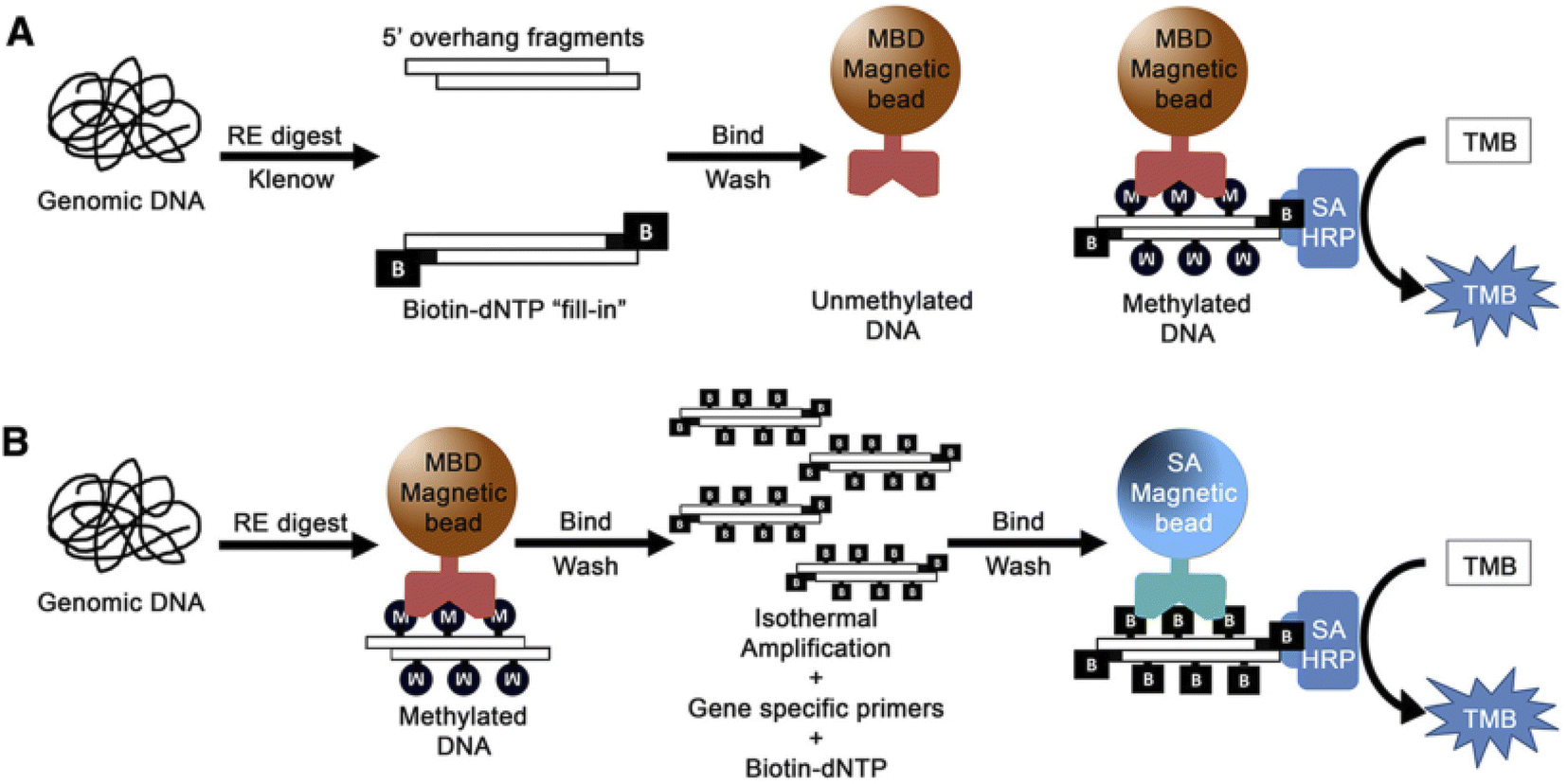 | ||
| Fig. 6 (A) Strategy for total genomic methylation. Genomic DNA. (B) Strategy for gene-specific methylation. Genomic DNA.99 | ||
A colorimetric nanosensor based on gold nanoparticle, sodium bisulfite treatment, PCR amplification and 5-aza-2′-deoxycytidine was designed for DNA methylation assay in nasopharyngeal carcinoma cells by Lin.100 Also, Li et al., designed a label-free and enzyme-free colorimetric method based on gold nanoparticle for DNA methylation assay in human serum samples without using complicated instruments or expensive biological enzymes or common organic dyes during the reaction. The employed strategy was the difference in electrostatic interaction of single-stranded DNA and double-stranded DNA against salt-induced aggregation of Au NPs. The method has a low detection limit of 8.47 nM.101 Interestingly, Geng et al., reported colorimetric method based on combining methylation-sensitive endonuclease digestion and hyperbranched rolling circle amplification (HRCA) method for DNA methylation analysis in p16/CDKN2 promoter of breast cancer samples. The concentration range was from 100 fM to 10 nM and duration was 3 h.102 The colorimetric method is generally low sensitivity and selectivity and is quantitative/semi-quantitative.97–100 The colorimetric methods are mostly based on the color change properties due to the accumulation of nanoparticles. On the other hand, the use of gold nanoparticles alone has less sensitivity and selectivity. Also, the increase in the size of gold nanoparticles due to their accumulation in solution and eventually the formation of sediment, the color of the suspension becomes colorless over time and prevents accurate quantification. In addition, false positive results due to non-specific accumulation of functional nanoparticles in complex biological samples limit their practical application.103 Therefore, the use of DNA amplification and endonuclease based digestion methods along with colorimetric methods can improve the sensitivity of the detection72,104–107 and amplify the detection signals.103,108–111
6. DNA methylation assay using microfluidic devices
Over the past two decades, microfluidics technologies have emerged as promising tools for a variety of biomedical applications.112,113 Microfluidic systems are small and miniature devices that have several advantages over traditional methods, including precise fluid volume control, low consumption of test samples and solutions, sample contamination reduction during transferring between each step, reaction time, no need for laboratory equipment and skilled operators.114–116 Therefore, these devices are very suitable tools for point-of-care (POC) diagnosis. Microfluidic systems are designed that the entire analytical process can be performed on a single chip. Many detection tools could be integrated into microfluidic systems such as electrochemical, colorimetric, optical biosensors, molecular techniques and etc.117According to different stages of methylation evaluation, microfluidic devices are classified into different types including microfluidic for bisulfite treatment,118 DNA extraction,119 methylcytosine assay based on bisulfite conversion,120–123 microfluidic based on restriction enzyme and bisulfite free,124,125 microfluidic for methyltransferase assay126,127 and morphological properties.128
7. Optical analysis systems of DNA damages using smartphones
Recently, smartphones and smartphone-based technology have developed rapidly, and smartphones have become a versatile multimedia device. Smartphones have several advantages, including portability, internet connection, increased battery and computing capacity, and a large number of sensors and applications.129–131 Therefore, these features make this device a laboratory level sensor and make it suitable for medical, biological analysis and the point of care diagnosis.132–134 Numerous types of optical and electrical biosensors have been integrated with smartphones, including smartphone-based colorimetry,135 smartphone-based FRET spectroscopy,136 smartphone-based fluorimetry137 and smartphone-based SPR.138In the colorimetric method integrated with smartphones, the reaction is first performed in aqueous medium or using paper-based systems such as strip tests or paper microfluidic devices. Then, changes in the color of the solution or paper substrates are detected by smartphone cameras. The smartphone analysis is performed by a app on a smartphone or an application on a computer after transferring data.139–148
Smartphones are used not only in colorimetric analysis but also in spectroscopic, fluorimetric and electrochemical analyzes. Smartphones are able to detect wavelengths and analyze the entire visible spectrum.149 The use of smartphone technology has also been proven in colorimetric and fluorometric analyzes of single and double strand DNA detection,150 DNA sequencing,151 quantitative DNA analysis using mobile PCR digital devices based on smartphones.152
For example, Lazaro153 and coworkers used smartphone-based detection method evaluate single-nucleotide variants detection in genes KRAS and NRAS in cultured cells and tissues biopsied from cancer patients. For this purpose, first isothermal amplification was done, then allele-specific hybridization chain reaction (AS-HCR) was done using allele specific probes immobilized on the chip in a microanalysis format. Despite the high ratio of wild type cells to mutant cells, AS-HCR was able to identify specific DNA variants. Finally, the data were detected with a smartphone array reader. The advantage of this method is that the analysis time using this method was 1 h compared to traditional methods that take 2 h. Secondly, AS-HCR was an isothermal method and inexpensive and instrument free. Also, the amount of contamination in this method is lower than conventional PCR method. The DNA concentration range was less than 103 copies and the detection limit was 0.7% (Fig. 7).
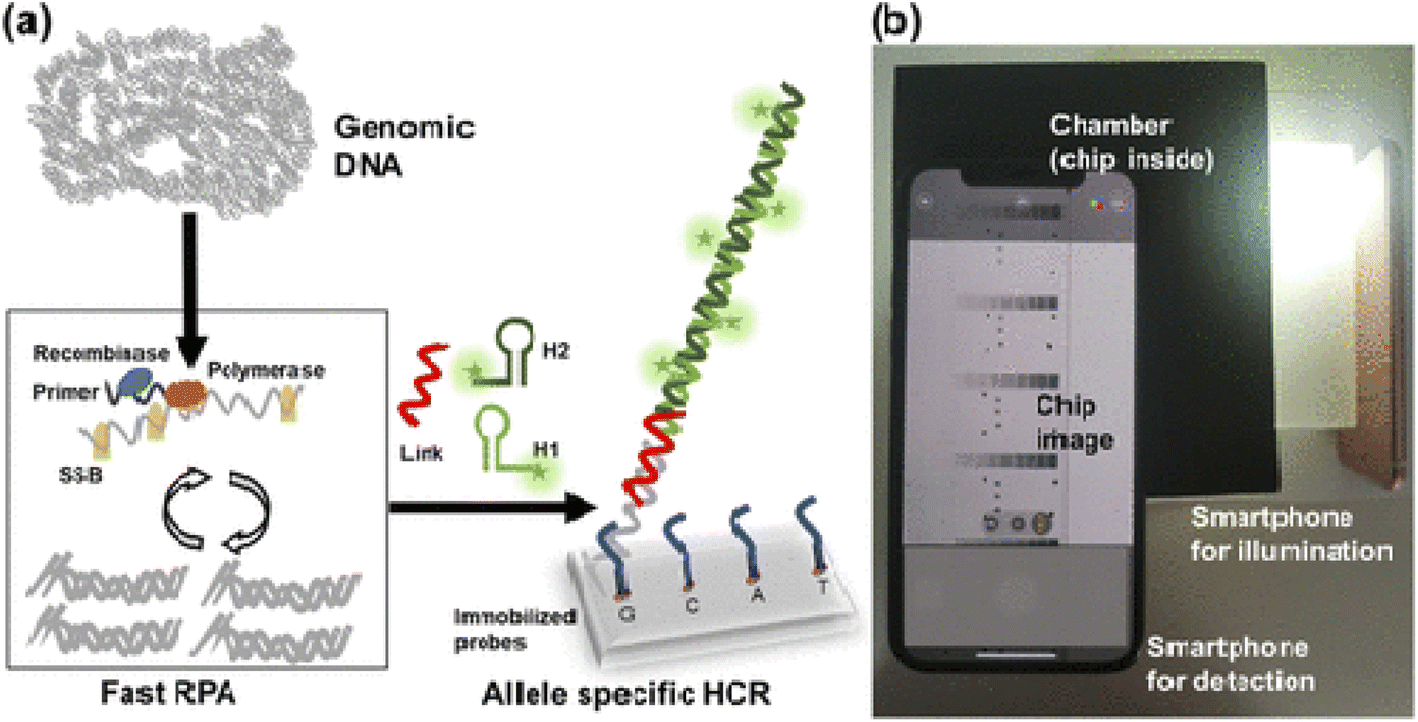 | ||
| Fig. 7 Scheme of genotyping by AS-HCR with colorimetric detection. (a) Mechanism of the AS-HCR method for identifying single-nucleotide variants from genomic DNA and (b) assembly for smartphone detection.153 | ||
Kuhnemund and et al.154 used a smartphone to detect single-nucleotide mutations and DNA sequencing based on selector probes, pad lock probe in situ and RCA to generate micron sized DNA coil, each of which could be labeled with fluorescent probes or quenched. In this method, a sequencing library was first generated from synthetic fragments of gene KRAS with two types of wild type and mutation 12 codon. Products of individual RCA could be identified and quantified by mobile phone based microscope. The result showed that using this method, very small amounts of variant sequences can be identified (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 ratio). This study was performed on biopsy specimens of colon cancer. The results of the smartphone analysis were consistent with the results of the PCR-based sequencing analysis (Fig. 8).
:1000 ratio). This study was performed on biopsy specimens of colon cancer. The results of the smartphone analysis were consistent with the results of the PCR-based sequencing analysis (Fig. 8).
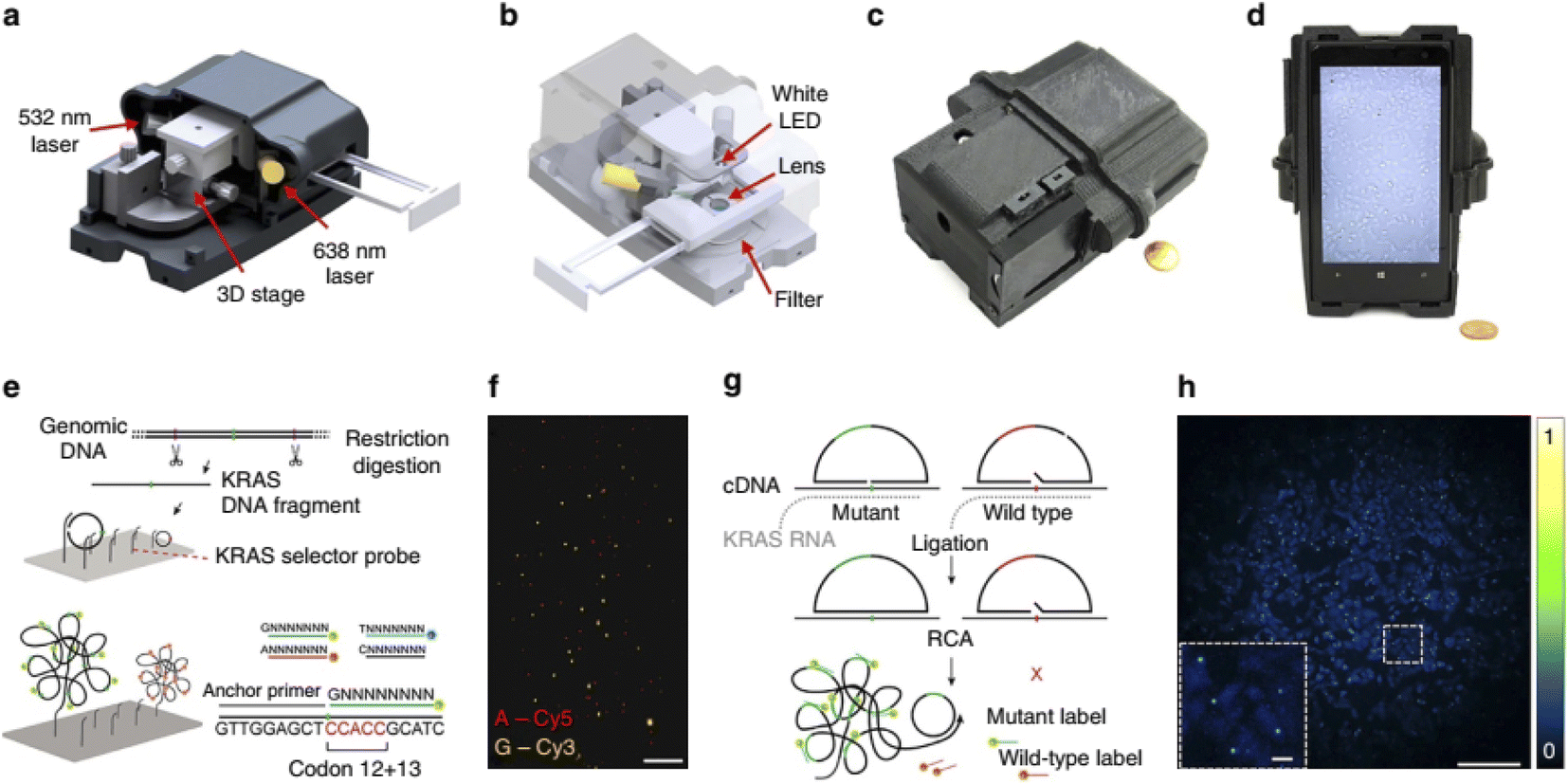 | ||
| Fig. 8 Multimodal mobile microscopy device and schematics of RCA assays. (a and b) 3D schematic illustration of the inner structure and the optical design of the mobile-phone-based microscopy platform. (c and d) Photographs of the mobile-phone-based microscope from different viewing perspectives. Mobile phone screen of (d) shows a bright-field image of fixated A549 cells captured by the phone. (e) DNA sequencing sample preparation scheme: genomic DNA is restriction digested and the KRAS DNA fragment selectively circularized on KRAS selector probes attached to slides. The DNA fragments are ligated and amplified on the slide, and the RCA products sequenced by unchained SBL chemistry.15,20 DNA sequencing reactions are then imaged through our mobile phone microscope. (f) Dual-colour mobile phone microscope image of a targeted SBL reaction of KRAS codon 12 in genomic DNA extracted from A427 cells which are heterogeneous for a KRAS codon 12 mutation. RCPs are either stained with Cy3 corresponding to base G (KRAS wild type), or Cy5 corresponding to base A (KRAS mutant). Scale bar, 50 mm. (g) Schematic diagram of in situ point mutation detection assay through padlock probes and RCA. KRAS mRNA is converted to cDNA, which is targeted by single-base-discriminating padlock probes. Mutant specific padlock probes are ligated and amplified through RCA. Wild-type-specific probes do not ligate on mutated KRAS cDNA and generate no RCP. (h) A full field of view image of the A549 cell line with in situ RCA detected codon 12 point mutations, imaged with our mobile phone fluorescence microscope. Scale bar, 200 mm (full field of view); Scale bar, 20 mm (inset).154 | ||
Mai and et al.155 applied an enzyme-free colorimetric biosensor based on catalytic hairpin assembly amplification and G-quadruplex DNAzyme to assess BRCA1 gene. Colorimetric readout was recorded using a smartphone camera and a UV-vis spectrophotometer. The detection limit was 10 pM in a concentration range of 50 pM to 200 nM. This method can detect target DNA at a concentration of 0.2 nM. This colorimetric platform can be selected to detect BRCA1 gene in serum samples. To complete our discussion in this review, Table 1 summarizes recent reports on the optical bio-sensing of DNA methylation and DNA damage analysis.45–112,156
| Detection method | Biomarker | Clinical sample | Linear range | LOD | Nanomaterial | Sequences of detection probes | Ref. |
|---|---|---|---|---|---|---|---|
| Fluorescence | Adenomatous polyposis coli gene | Plasma | Un-methylated DNA: 1.6 × 10−15–6.6 × 10−13 M | 1.2 × 10−16 M | Fe3O4/Au core/shell, dipyridamole | SH-(CH2)6-5′-CCG TCG AAA ACC CGC CGA-3′ | 45 |
| Methylated DNA: 3.2 × 10−15–8.0 × 10−13 M | 3.1 × 10−16 M | ||||||
| Fluorescence and UV-Vis | Adenomatous polyposis coli gene | Plasma | 10−21–0.1 × 10−12 M | 1 zM | Carbon materials and an organic dye (thionine) | TCCGCTTCCCGACCCGCACTCCGC | 46 |
| Fluorescence and UV-Vis | Adenomatous polyposis coli gene | Plasma | 10−21–0.1 × 10−12 M | 1 zM | Carbon materials and an organic dye (toluidine blue) | TCCGCTTCCCGACCCGCACTCCGC | 47 |
| Fluorescence | 7.3 × 10−11 M | Graphene quantum dot and SYBER green as dye reporter | 48 | ||||
| Fluorescence | Adenomatous polyposis coli | — | 1.0 × 10−10 to 1.0 × 10−6 M | 6.2 × 10−11 M | CdTe quantum dots | 5′-TCCGCTTCCCGACCCGCACTCCGC-3′ | 50 |
| FRET | — | — | 40 nM | 0.25 U mL−1 | Gold nanorods | FAM-5′-CCTTTTGATCATTTT-FAM-3′ | 53 |
| FRET | RASSF1A, OPCML, and HOXA9 promoters | Ovarian cancer | 85.7% | Cationic conjugated polymer (CCP, poly{(1,4-phenylene)-2,7-[9,9-bis(6′-N,N,N-trimethyl ammonium)-hexyl fluorene]dibromide}) | RASSF1A | 54 | |
| F:5′-GGAGGCGCTGAAGTCGG-3′ | |||||||
| R:5′-GCCCAGCGGGTGCCA-3 | |||||||
| OPCML | |||||||
| 5′-GCCAGTGTCAGTTTTCAGTTTG-3′ | |||||||
| 5′-ATCCCTGACCGCCACTTT-3′ | |||||||
| HOXA9 | |||||||
| 5′-TGGACTCGTTCCTGCTGG-3′ | |||||||
| 5′-TGGTGGTGATGGTGGTGGTA-3′ | |||||||
| FRET | P53 tumor suppressor gene promoter | Human serum sample | 5 pM to 100 pM | 2.2 pM | Gold nanoparticle | 6′-FAM-5′-TCCGGTTCCCGACCCGGACTCCGCAAAAAA-3′-SH | 55 |
| FRET | — | 0–100 nM | — | Up-conversion nanoparticles (UCNPs) and gold nanorods (AuNRs) | Probe: biotin-5′-ATACCmGGTCTAAA-3′-S-S methylated single-stranded DNA (target): 5′-AGACCmGGTAT-3′ unmethylated single-stranded DNA (target): 3′-TATGGCCAGA-5′ | 40 | |
| MS-qFRET | p15INK4B | Cell lines, clinical samples from patients with acute myeloid leukemia. | — | — | Quantum dots | Primer sequences for p15INK4B: Methylated sense 5′-GGTTTTTTATTTTGTTAGAGCGAGGC-3′ | 57 |
| Methylated anti-sense 5′-TAACCGCAAAATACGAACGCG-3′ | |||||||
| Unmethylated sense 5′-GGTTGGTTTTTTATTTTGTTAGAGTGAGGT-3′ | |||||||
| Unmethylated anti-sense 5′-AACCACTCTA ACCACAAAATAC | |||||||
| AAACACA-3′ | |||||||
| MS-UC-FRET | CDKN2Agene | — | — | 0.1% | Up-conversion nanoparticles (UCNPs), SYTOX orange dye | 5′-biotin-TTATTAGAGGGTGGGGCGGATCGC-3′ | 58 |
| Detection method | Biomarker | Clinical sample | Linear range | LOD | Nanomaterial | Sequences of detection probe | Ref. |
|---|---|---|---|---|---|---|---|
| FRET | CDKN2A, TFPf2, CHFR | Synthesized sequence | — | — | Quantum dots, FRET linker probes (FLPs) | CDKN2A | 63 |
| Sense: TTATTAGAGGGTGGGGCGGATCGC | |||||||
| Anti-sense: GACCCCGAACCGCGACCGTAA | |||||||
| FLP: CTACCTACTCTCCCCCTCTCCRCAACCRCC | |||||||
| TFPf2 | |||||||
| Sense: GTTCGTTGGGTAAGGCGTTC | |||||||
| Anti-sense: CATAAAACGAACACCCGAACCG | |||||||
| FLP: ACCGCGCACCTCCTCCCGCCAA | |||||||
| CHFR | |||||||
| Sense: GTTATTTTCGTGATTCGTAGGCGAC | |||||||
| Anti-sense: CGAAACCGAAAATAACCCGCG | |||||||
| FLP: CGCTCGACCATCTTTAATCCTAACCAAACGACTTC | |||||||
| FRET | PCDHGB6, HOXA9 and RASSF1A | Lung adenocarcinoma and adjacent nontumorous tissues | — | Up to 90% | Amino-CdSe/CdS/ZnS QDs | PCDHGB6 | 64 |
| GATGTACACCTGCATTTTCG | |||||||
| CGTTCGCTCGGGTTCTCGCT | |||||||
| HOXA9 | |||||||
| CCAACGGGTGAGAATAAAC | |||||||
| AAAAACTACAAGTGGCATGA | |||||||
| RASSF1A | |||||||
| AAGATCACGGTCCAGCCTC | |||||||
| CTTCGTCCCCTCCTCACAC | |||||||
| PCDHGB6, HOXA9 and RASSF1A | NSCLC tissue samples and noninvasive bronchial brushing specimens | — | Sensitivity of 92% (AUC1/40.977, Po0.001) and 80% (AUC1/40.907, Po0.001) | Quantum dots | — | 64 | |
| SPR | Adenomatous polyposis coli | Synthesized sequence | — | 5 pmol | — | 5′-biotin-CTGmCGGAGTGmCGGGTmCGGGAAGmCGG-3′ | 70 |
| SPR | — | MCF7 cell | 0 to 400 nM | 100 nM | — | — | 71 |
| SPR | — | — | 0.5 to 120 U/mL | 0.2 U/mL | Au nanorod | — | 75 |
| SERS | CDKN2/p16/MTS1 (p16) | Synthesized sequence | — | 3 pM | AuNPs | 5′-SH-C6-TAC CTA CTC TAC CCC TCTCC-3′ | 76 |
| SERS | Cancer-related genes cadherin 1 (CDH1) and retinoic acid receptor beta (RARB) | Synthesized sequence | — | — | — | CDH1 | 93 |
| Methylated probe (CMP): 5′-TAA TTT TAG GTT AGA GGG TTA TmCG mCG -3′ | |||||||
| Unmethylated probe (CUMP): 5′-TAA TTT TAG GTT AGA GGG TTA TTG TG-3′ | |||||||
| Methylated target (CMT): 5′-CG CGA TAA CCC TCT AAC CTA AAA TTA-3′ | |||||||
| Unmethylated target (CUMT): 5′-CA CAA TAA CCC TCT AAC CTA AAA TTA-3′ | |||||||
| RARB | |||||||
| Methylated probe (RMP): 5′-GGT TAG TAG TTmCGGG TAG GGT TTA TmC-3′ | |||||||
| Unmethylated probe (RUMP): 5′- GGT TAG TAG TTTGGG TAG GGT TTA TT-3′ | |||||||
| Methylated target (RMT): 5′-GAT AAA CCC TAC CCG AAC TAC TAA CC-3′ | |||||||
| Unmethylated target (RUMT): 5′-AAT AAA CCC TAC CCA AAC TAC TAA CC-3′ | |||||||
| SERS | Real cell sample | — | 1.8 pmol L-1 | Wrapped graphene-Ag array, AuNPs | — | 95 |
| Detection method | Biomarker | Clinical sample | Linear range | LOD | Nanomaterial | Sequences of detection probe | Ref. |
|---|---|---|---|---|---|---|---|
| SERS | MDA-MB-231, MDAMB-468 and HCC1937 cell lines, | Synthetic targets, breast cancer cell lines and a serum-derived DNA sample | — | 0.5 pM | AuNPs, 4-mercapto-3-nitro benzoic acid (MNBA), 4-mercaptobenzoic acid (MBA) | Capture probe HS-AGTTGTGCAGGTGGT | 96 |
| SERS detection probe 1 HS-CAGATCGTCATGTTC | |||||||
| SERS detection probe 2 HS-TCTGCACCAATGTAC | |||||||
| Colorimetry | GSPTP1 gene | Urine | — | 5% methylation differences | MBD magnetic beads, SA-HRP | Forward primer: AACCCCCTTATCCCTCCGTCGTGTGGCTTTTAC | 99 |
| Reverse primer: AAACAGGTTCCTCCGAAGATTTCACACAACACT | |||||||
| Colorimetry | CpG islands of microRNA 9–1 | Cancer cell line | — | — | Gold nanoparticle | No reported | 100 |
| Colorimetry | — | Human serum | 20 to 120 nM | 0.13% | Gold nanoparticle | No reported | 101 |
| Colorimetry | p16/CDKN2 promoter | Breast cancer samples | 100 fM to 10 nM | 93 fM | 3,3′,5,5′-Tetramethylbenzidine | Methylated DNA: CGAGCTGCCTGGAGTTGCGTTCCAGGCGTmCGGCmCC | 102 |
| CTGGGCCGTCACCGCG | |||||||
| Unmethylated DNA: CGAGCTGCCTGGAGTTGCGTTCCAGGCGTCCGGCC | |||||||
| CCTGGGCCGTCACCGCG | |||||||
| Circular template: CACGCGATCCGCCCCACCCTCCGCGGTGACGGCCCAG | |||||||
| GAATTCGTGTAACTACACGAATTCCAACCGCCGAACG | |||||||
| Primer: ACCAAGAGCAACTACACGAATTC | |||||||
| Capture probe: HOOC-AACCGTCTTCCAAGAGACCTTCTCCAGGCAGCTC G | |||||||
| Biotinylated probe: GCGATCCGCCCCA-biotin |
8. Conclusion and future perspective
In conclusion, DNA methylation is an epigenetic and post-replication change that is common to all vertebrates. Studies have shown that loss of DNA methylation interferes with tissue homeostasis. Loss of DNA methylation causes abnormal activation of gene expression and is associated with in vivo differentiation of adult stem cells.156–160 Due to the limitations of routine methods in the detection of DNA methylation in the clinic diagnosis, designing sensitive methods for evaluating DNA methylation in all cell lines and in all cell sections is essential. In the meantime, it seems that the design and development of optical nanobiosensors can help in the detection of DNA methylation at a low cost and in a short time to diagnose diseases related to methylation. The use of nanomaterials in these biosensors will make these devices more sensitive and practical in point-of-care early diagnosis. Early diagnosis will help to survive patients. Successful development of biosensors to diagnose various diseases will require finding the appropriate financing to move technology from research to commercial product realization. Given the importance of microfluidic systems in assessing DNA modification as well as the integration of smartphones with biosensors, it is expected that in the very near future we will see the development of these technologies and their use in medicine and clinical and personalized medical diagnoses.In summary, recent progresses on the epigenetic researches using optical biosensing of DNA methylation were surveyed. Many examples of the optical biosensors have been reported for extraction and bisulfite treatment, DNA methylation detection based photonic devices/methods using microfluidic and smartphone-based techniques. Also, assay of methyltranseferase activity using photonic measurement by was performed. Many nanomaterials have been reported to increase the sensitivity of microfluidic and lateral flow tests, such as carbon nanotubes, quantum dots, and metal nanoparticles that allow highly sensitive and rapid measurements in small volumes of materials. Using a combination of nanoparticles and other optical based amplification methods integrated with microfluid and smartphone, in the future these tests will be used with high sensitivity to detect DNA methylation. There are also some problems in using these tests, including non-specific and non-selective adsorption when using real samples. We hope that the relevant problems will be overcome by increasing progress in improving these tests.
In this review, labeled and unlabeled methods for evaluating DNA methylation using fluorimetric, SERS, SPR, FRET, and colorimetric methods and evaluating nucleotide changes in DNA sequence based on mobile phones and integrating these methods into microfluidic systems have been reviewed and many examples are given. In this sensing process, different types of nanomaterials, including GQD, QD, magnetic nanomaterials, cationic conjugated polymer, MBD magnetic beads, Au nanorod, AuNPs, CdTe, UCNPs, wrapped Ag array, as well as organic dyes as chromophores and fluorophores have been used. Therefore, the use of a combination of these methods and the use of different nanomaterials can increase the detection sensitivity and by reducing the analysis time, it enables the evaluation in low volume samples. In real biological samples, due to the complexity of the cellular environment and the large size of the genome, first, it is necessary to extract DNA, then amplify the desired region of the genome by PCR. The toxicity of nanomaterials used in synthesis probes can affect the structure of DNA in real biological samples and should be further studied and clarified.
It is hoped that in the future, with the advancement of science and technology and the use of high-sensitivity nanomaterials and eliminating the disadvantages of these methods, microfluidic methods can be routinely used to detection of DNA methylation and early stage diagnosis of cancer.
Surface Plasmon Resonance (SPR) based biosensors are simple and fast and label-free detection assays which is used to detect different types of biomolecules without using any fluorophores and chromophores. Also, it measures directly binding constant and affinity; and consumes less reagents.69 Surface-enhanced Raman spectroscopy (SERS) is sensitive to any changes in conformation of the biomolecules. Low detection limit, interference-free high, and selectivity are several desirable properties. In this method, a wide range of excitation wavelengths from NIR to red excitations can be used, which reduces photo damaging to biological samples. The colorimetric methods are very simple and is based on color changes due to the reaction of nanomaterials with the desired analyte that can be seen with the naked eye. Color changes due to the reaction of nanomaterials with the desired analyte can be observed. Low sensitivity and selectivity and semi-quantitative are the major disadvantages of this method. Decrease in the color intensity of the solution containing the sample with the passage of time and false positive results due to non-specific accumulation of functional nanoparticles in complex biological samples limit their practical application.112 In fluorescence resonance energy transfer (FRET), energy transfers from an excited donor to an atomic structurally appropriate acceptor fluorophore. FRET provides indirect information about the various biomedical actions like conformation changes. FRET is highly sensitive to the distance between acceptor and donor (about 10 nm) which makes it a very sensitive technology for the detection of near-field interaction between molecules. Fluorescence assays has high intrinsic sensitivity and can be integrated with receptors with high selectivity. In fluorescence detection, a specific wavelength of electromagnetic radiation excites fluorophore molecules and an optical transducer detects the intensity of shifted and emitted light.32,42
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Researchers gratefully acknowledge the University of Tabriz and Tabriz University of Medical Sciences for their cooperation.References
- J. A. Yoder, C. P. Walsh and T. H. Bestor, Cytosine methylation and the ecology of intragenomic parasites, Trends Genet., 1997, 13, 335–340 CrossRef CAS PubMed.
- S. Ledoux, J. Nalbantoglu and N. R. Cashman, Amyloid precursor protein gene expression in neural cell lines: influence of DNA cytosine methylation, Mol. Brain Res., 1994, 24, 140–144 CrossRef CAS.
- P. A. Jones, DNA methylation errors and cancer, Cancer Res., 1996, 56, 2463–2467 CAS.
- M. Monk, Epigenetic programming of differential gene expression in development and evolution, Dev. Genet., 1995, 17, 188–197 CrossRef CAS PubMed.
- R. M. Rivera and L. B. Bennett, Epigenetics in humans: An overview, Curr. Opin. Endocrinol., Diabetes Obes., 2010, 17, 493–499 CrossRef PubMed.
- A. Portela and M. Esteller, Epigenetic modifications and human disease, Nat. Biotechnol., 2010, 28, 1057–1068 CrossRef CAS PubMed.
- L. Krejcova, L. Richtera, D. Hynek and J. Labuda, DNA methylation: Bisulphite modification and analysis, Nat. Protoc., 2006, 1, 2353–2364 CrossRef PubMed.
- H. Jin, Y. Ma, Q. Shen and X. Wang, Methylation: from DNA, RNA and histones to diseases and treatment, In Tech Open Access Publisher, 2013, vol. 6, pp. 137–152 Search PubMed.
- S. Clark, A. Statham, C. Stirzaker, P. L. Molloy and M. Frommer, DNA methylation: Bisulphite modification and analysis, Nat. Protoc., 2006, 1, 2353–2364 CrossRef CAS PubMed.
- Z. Xiong and P. W. Laird, COBRA: a sensitive and quantitative DNA methylation assay, Nucleic Acids Res., 1997, 25, 2532–2534 CrossRef CAS PubMed.
- J. Tost and I. G. Gut, DNA methylation analysis by pyrosequencing, Nat. Protoc., 2007, 2, 2265–2275 CrossRef CAS PubMed.
- M. L. Gonzalgo and P. A. Jones, Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE), Nucleic Acids Res., 1997, 25, 2529–2531 CrossRef CAS.
- T. K. Wojdacz and A. Dobrovic, Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation, Nucleic Acids Res., 2007, 35, 41 CrossRef.
- K. M. Armstrong, E. N. Bermingham, S. A. Bassett, B. P. Treloar, N. C. Roy and M. P. Barnett, Global DNA methylation measurement by HPLC using low amounts of DNA, Biotechnol. J., 2011, 6, 113–117 CrossRef CAS PubMed.
- M. F. Fraga, R. Rodriguez and M. J. Canal, Rapid quantification of DNA methylation by high performance capillary electrophoresis, Electrophoresis, 2000, 21, 2990–2994 CrossRef CAS.
- E. M. Southern, Detection of specific sequences among DNA fragments separated by gel electrophoresis, J. Mol. Biol., 1975, 98, 503–517 CrossRef CAS PubMed.
- A. O. Nygren, N. Ameziane, H. M. Duarte, R. N. Vijzelaar, Q. Waisfisz and C. J. Hess, Methylation-Specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences, Nucleic Acids Res., 2005, 33, e128 CrossRef.
- T. K. Wojdacz and A. Dobrovic, Methylation-sensitive high resolution melting (MSHRM): a new approach for sensitive and high-throughput assessment of methylation, Nucleic Acids Res., 2007, 35, e41 CrossRef PubMed.
- Z. Xiong and P. W. Laird, COBRA: a sensitive and quantitative DNA methylation assay, Nucleic Acids Res., 1997, 25, 2532–2534 CrossRef CAS PubMed.
- J. G. Herman, J. R. Graff, S. Myohanen, B. D. Nelkin and S. B. Baylin, Methylationspecific PCR: a novel PCR assay for methylation status of CpG islands, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9821–9826 CrossRef CAS.
- C. A. Eads, K. D. Danenberg and K. Kawakami, MethyLight: a high-throughput assay to measure DNA methylation, Nucleic Acids Res., 2000, 28, 32e-0 CrossRef.
- M. Bibikova, Z. Lin, L. Zhou, E. Chudin, E. W. Garcia and B. Wu, High-throughput DNA methylation profiling using universal bead arrays, Genome Res., 2006, 16, 383–393 CrossRef CAS PubMed.
- S. E. Cottrell, J. E. Distler and N. S. Goodman, A real-time PCR assay for DNAmethylation using methylation-specific blockers, Nucleic Acids Res., 2004, 32, 10e CrossRef PubMed.
- J. Tost and I. G. Nat, DNA methylation analysis by pyrosequencing, Nat. Protoc., 2007, 2, 2265–2275 CrossRef CAS.
- H. K. Komori, S. A. LaMere, A. Torkamani, G. T. Hart, S. Kotsopoulos and J. Warner, Application of microdroplet PCR for large-scale targeted bisulfite sequencing, Genome Res., 2011, 21, 1738–1745 CrossRef CAS PubMed.
- Y. Zhang, V. Bailey, C. M. Puleo, H. Easwaran, E. Griffiths and J. G. Herman, DNA methylation analysis on a droplet-in-oil PCR array, Lab Chip, 2009, 9, 1059–1064 RSC.
- M. Kalofonou and C. Toumazou, Semiconductor technology for early detection of DNA methylation for cancer: From concept to practice, Sens. Actuators, B, 2013, 178, 572–580 CrossRef CAS.
- S. P. Stabler and R. H. Allen, Quantification of serum and urinary Sadenosylmethionine and S-adenosylhomocysteine by stable-isotope-dilution liquid chromatography-mass spectrometry, Clin. Chem., 2004, 50, 365–372 CrossRef CAS.
- R. Narayanaswamy and O. S. Wolfbeis, Optical Sensors: Industrial, environmental and diagnostic applications, Springer, New York, 2004 Search PubMed.
- X. Fan, I. M. White, S. I. Shopova, H. Zhu, J. D. Suter and Y. Sun, Sensitive optical biosensors for unlabeled targets: A review, Anal. Chim. Acta, 2008, 620, 8–26 CrossRef CAS PubMed.
- M. N. Velasco-Garcia, Optical biosensors for probing at the cellular level: A review of recent progress and future prospects, Semin. Cell Dev. Biol., 2009, 20, 27–33 CrossRef CAS PubMed.
- I. E. Tothill, Biosensors for cancer markers diagnosis, Semin. Cell Dev. Biol., 2009, 20, 55–62 CrossRef CAS PubMed.
- R. A. Sperling, R. Rivera Gil, F. Zhang, M. Zanella and W. J. Parak, Biological applications of gold nanoparticles, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- D. Jariwala, D. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Carbon nanomaterials for electronics, optoelectronics, photovoltaics, and sensing, Chem. Soc. Rev., 2013, 42, 2824–2860 RSC.
- X. Gao, Y. Cui, R. M. Levenson, L. W. Chung and S. Nie, In vivo cancer targeting and imaging with semiconductor quantum dots, Nat. Biotechnol., 2004, 22, 969–976 CrossRef CAS PubMed.
- J. P. Wilcoxon and B. L. Abrams, Synthesis, structure and properties of metal nanoclusters, Chem. Soc. Rev., 2006, 35, 1162–1194 RSC.
- J. Lei and H. Ju, Signal amplification using functional nanomaterials for biosensing, Chem. Soc. Rev., 2012, 41, 2122–2134 RSC.
- M. Holzinger, M. Le Goff and S. Cosnier, Nanomaterials for biosensing applications: a review, Front Chem., 2014, 2, 63 Search PubMed.
- H. Ju, C-dots assisted synthesis of gold nanoparticles as labels to catalyze copper deposition for ultrasensitive electrochemical sensing of proteins, Sci. China Chem., 2011, 54, 1202–1217 CrossRef CAS.
- I. Willner and B. Willner, Biomolecule-Based Nanomaterials and Nanostructures, Nano Lett., 2010, 10, 3805–3815 CrossRef CAS PubMed.
- J. Yao, M. Yang and Y. Duan, Chemistry, biology, and medicine of fluorescent nanomaterials and related systems: new insights into biosensing, bioimaging, genomics, diagnostics, and therapy, Chem. Rev., 2014, 114, 6130–6178 CrossRef CAS PubMed.
- B. Li, Q. Yu and Y. Duan, Fluorescent labels in biosensors for pathogen detection, Crit. Rev. Biotechnol., 2015, 35, 82–93 CrossRef CAS PubMed.
- W. Zhong, Nanomaterials in fluorescence-based biosensing, Anal. Bioanal. Chem., 2009, 394, 47–59 CrossRef CAS PubMed.
- M. Dadmehr, M. Hosseini, S. Hosseinkhani, M. R. Ganjali, M. Khoobi, H. Behzadi, H. Hamedani and R. Sheikhnejad, DNA methylation detection by a novel fluorimetric nanobiosensor for early cancer diagnosis, Biosens. Bioelectron., 2014, 60, 35–44 CrossRef CAS.
- M. Adampourezare, G. H. Dehghan, M. Hasanzadeh and M. A. Hosseinpoure Feizi, Identification of DNA methylation by novel optical genosensing: A new platform in epigenetic study using biomedical analysis, J. Mol. Recognit., 2021, 2938 Search PubMed.
- M. Adampourezare, M. Hasanzadeh, G. Dehghan, M. A. Hosseinpourefeizi and F. Seidi, An innovative fluorometric bioanalysis strategy towards recognition of DNA methylation using opto-active polymer: A new platform for DNA damage studies by genosensor technology, J. Mol. Recognit., 2022 Search PubMed.
- S. Rafiei, M. Dadmehr, M. Hosseini, H. Ahmadzade Kermani and M. R. Ganjali, A fluorometric study on the effect of DNA methylation on DNA interaction with graphene quantum dots, Methods Appl. Fluoresc., 2019, 7(2) CAS.
- X. Ouyang, J. Liu, J. Li and R. Yang, A carbon nanoparticle-based low-background biosensing platform for sensitive and label-free fluorescent assay of DNA methylation, Chem. Commun., 2012, 48, 88–90 RSC.
- M. Hosseini, F. Khaki, E. Shokri, H. Khabbaz, M. Dadmehr, M. R. Ganjali, M. Feizabadi and D. Ajloo, Study on the interaction of the CpG alternating DNA with CdTe quantum dots, J. Fluoresc., 2017, 27(6), 2059–2068 CrossRef CAS PubMed.
- G. Chen, F. Song, X. Xiong and X. Peng, Fluorescent nanosensors based on fluorescence resonance energy transfer (FRET), Ind. Eng. Chem. Res., 2013, 52, 11228–11245 CrossRef CAS.
- T. D. Martins, A. C. Ribeiro, D. L. Dias, H. P. Cavalcante, H. S. Camargo, P. A. Costa Filho, in State of the Art in Biosensors, New Insights on Optical Biosensors: Techniques, Construction and Application, ed. T. Rinken, Intech Open Access Publisher, 2013, DOI:10.5772/52330.
- G. L. Wang, H. Q. Luo and N. B. Li, Gold nanorods-based FRET assay for ultrasensitive detection of DNA methylation and DNA methyltransferase activity, Analyst, 2014, 139, 4572 RSC.
- J. Zhang, B. Xing, B. Song, F. Zhang, C. Nie, L. Jiao, L. Liu, F. Lv and S. Wang, Associated analysis of DNA methylation for cancer detection using CCP-based FRET Technique, Anal. Chem., 2014, 86, 346–350 CrossRef CAS.
- M. A. Karimi, M. Dadmehr, M. Hosseini, B. Korouzhdehi and F. Oroojalian, Sensitive detection of methylated DNA and methyltransferase activity based on the lighting up of FAM-labeled DNA quenched fluorescence by gold nanoparticles, RSC Adv., 2014, 9, 12063–12069 RSC.
- M. Wu, X. Wang, K. Wang and Z. Guo, Sequence-specific detection of cytosine methylation in DNA via FRET mechanism between upconversion nanoparticles and gold nanorods, Chem. Commun., 2016, 52, 8377–8380 RSC.
- B. J Bailey, B. M. P. Keeley, C. R. Razavi, E. Griffiths, H. E. Carraway and T. H. Wang, DNA methylation detection using MS-qFRET, a quantum dot-based nanoassay, Methods, 2010, 52, 237–241 CrossRef PubMed.
- S. Kim, S. H. Hwang, S. Gyeong, M. K. Lee, C. H. Lee and S. Jun, Upconversion Nanoparticle-Based Forster Resonance Energy Transfer for Detecting DNA Methylation, Sensors, 2016, 16, 1259 CrossRef PubMed.
- V. J. Bailey, H. Easwaran, Y. Zhang, E. Griifiths, S. Beliknsy and G. Herman, DNA methylation detection using MS-qFRET a quantum dot-based nanoassay, Genome Res., 2009, 19, 1455–1461 CrossRef CAS.
- J. G. Herman, J. R. Graff, S. Myohanen, B. D. Nelkin and S. B. Baylin, Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9821–9826 CrossRef CAS.
- C. Y. Zhang, H. C. Yeh, M. T. Kuroki and T. H. Wang, Single-quantum-dot-based DNA nanosensor, Nat. Mater., 2005, 4, 826–831 CrossRef CAS PubMed.
- B. Keeleyi, Y. Zhang, Y. Zhang, A. Starki, T. H. Wang, Quantum dot FRET linker probes for highly sensitive DNA methylation detection, The International Conference Centre Birmingham, 2012, 2, pp. 20–23 Search PubMed.
- Y. Ma, H. Zhang, F. Liu, Z. Wu, S. Luc, Q. Jin, J. Zhao and X. Zhong, Highly sensitive detection of DNA methylation level by using quantum dots-based FRET method, Nanoscale, 2015, 7(41), 17547–17555 RSC.
- Y. Ma, Y. Bai, H. Mao, Q. Hong and D. Yang, A panel of promoter methylation markers for invasive and noninvasive early detection of NSCLC using a quantum dots-based FRET approach, Biosens. Bioelectron., 2016, 85, 641–648 CrossRef CAS.
- L. G. Carrascosa, A. Calle and L. M. Lechuga, Label-free detection of DNA mutations by SPR: application to the early detection of inherited breast cancer, Anal. Bioanal. Chem., 2009, 393, 1173–1182 CrossRef CAS PubMed.
- J. Homola, Surface plasmon resonance sensors for detection of chemical and biological species, Chem. Rev., 2008, 108, 462–493 CrossRef CAS PubMed.
- K. D. Kihm, S. Cheon, J. S. Park, H. J. Kim, J. S. Lee, I. T. Kim and H. J. Yi, Surface plasmon resonance (SPR) reflectance imaging: Far-Field recognition of near-field phenomena, Opt. Lasers Eng., 2012, 50, 64–73 CrossRef.
- P. Singh, SPR biosensors: Historical perspectives and current challenges, Sens. Actuators, B, 2016, 229, 110–130 CrossRef CAS.
- J. A. Maynard, N. C. Lindquist, J. N. Sutherland and A. Lesuffleur, Next generation SPR technology of membrane-bound proteins for ligand screening and biomarker discovery, Anal. Chem., 2009, 81, 2854–2859 CrossRef PubMed.
- S. Pan, J. Xu, Y. Shu, F. Wang and W. Xia, Double recognition of oligonucleotide and protein in the detection of DNA methylation with surface plasmon resonance biosensors, Biosens. Bioelectron., 2010, 26, 850–853 CrossRef CAS PubMed.
- L. G. Carrascosa, A. A. Sina, R. Palanisamy and B. Sepulveda, Molecular inversion probe-based SPR biosensing for specific, label-free and real-time detection of regional DNA methylation, Chem. Commun., 2014, 50, 3585–3588 RSC.
- C. Liang, Y. Chu, S. Cheng, H. Wu, T. Kajiyama, H. Kambara and G. Zhou, Multiplex loop-mediated isothermal amplification detection by sequence-based barcodes coupled with nicking endonucleasemediated pyrosequencing, Anal. Chem., 2012, 84(8), 3758–3763 CrossRef CAS PubMed.
- S. H. Cesar, A. Avino, C. Kurachi, A. Pique, J. Sandoval, R. Eritja, M. Esteller and L. M. Lechuga, Label-free DNA-methylation detection by direct dsDNA fragment screening using poly-purine hairpins, Biosens. Bioelectron., 2018, 120, 47–54 CrossRef PubMed.
- S. Coma, V. Noe, R. Eritja and C. J. Ciudad, Strand displacement of double-stranded DNA by triplex-forming antiparallel purine-hairpins, Oligonucleotides, 2005, 15, 269–283 CrossRef CAS PubMed.
- X. Li, T. Song and X. Guo, DNA methylation detection with end-to-end nanorod assemblyenhanced surface plasmon resonance, Analyst, 2015,(140), 6230–6233 RSC.
- J. Hu and C. Y. Zhang, Single base extension reaction-based surface enhanced Raman spectroscopy for DNA methylation assay, Biosens. Bioelectron., 2012, 31, 451–457 CrossRef CAS PubMed.
- R. Liu, S. Zhu, M. Si, Z. Liu and D. Zhang, Surface-enhanced Raman scattering-based approach for DNA detection at low concentrations via polyvinyl alcohol-protected silver grasslike patterns, J. Raman Spectrosc., 2012, 43, 370–379 CrossRef CAS.
- A. Talari, C. Evans, I. Holen, R. Coleman and I. U. Rehman, Raman spectroscopic analysis differentiates between breast cancer cell lines, J. Raman Spectrosc., 2015, 46, 421–427 CrossRef CAS.
- S. Schlücker, SERS Microscopy: Nanoparticle probes and biomedical applications, ChemPhysChem, 2009, 10, 1344 CrossRef.
- B. H. Jun, G. Kim, M. S. Noh, H. Kang, Y. K. Kim, M. H. Cho, D. H. Jeong and Y. S. Lee, Surface-enhanced Raman scattering-active nanostructures and strategies for bioassays, Nanomedicine, 2012, 6, 1463 CrossRef PubMed.
- Y. Wang and S. Schlucker, Rational design and synthesis of SERS labels, Analyst, 2013, 138, 2224 RSC.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. Dasari and M. S. Feld, Single molecule detection using surface-enhanced raman scattering (SERS), Phys. Rev. Lett., 1997, 78(9), 1667–1670 CrossRef CAS.
- Y. Wang, E. J. Wee and M. Trau, Accurate and sensitive total genomic DNA methylation analysis from sub-nanogram input with embedded SERS nanotags, Chem. Commun., 2016, 52, 3560–3563 RSC.
- A. Barhoumi and N. J. Halas, Detecting chemically modified DNA bases using surface enhanced Raman spectroscopy, J. Phys. Chem. Lett., 2011, 2, 3118–3123 CrossRef CAS PubMed.
- Y. Wang, E. J. Wee and M. Trau, Highly sensitive DNA methylation analysis at CpG resolution by surface-enhanced Raman scattering via ligase chain reaction, Chem. Commun., 2015, 51, 10953–10956 RSC.
- L. Guerrini, Z. Krpetic, D. van Lierop, R. A. Alvarez-Puebla and D. Graham, Direct surface-enhanced Raman scattering analysis of DNA duplexes, Angew. Chem., Int. Ed. Engl., 2015, 54, 1144–1148 CrossRef CAS PubMed.
- Z. Zhou, X. Han, G. G. Huang and Y. Ozaki, Label-free detection of binary mixtures of proteins using surface-enhanced Raman scattering, J. Raman Spectrosc., 2012, 43, 706–711 CrossRef CAS.
- G. Braun, S. J. Lee, M. Dante, T. Q. Nguyen, M. Moskovits and N. Reich, Surface-enhanced raman spectroscopy for DNA detection by Nanoparticle Assembly onto Smooth Metal Films, J. Am. Chem. Soc., 2007, 129(20), 6378–6379 CrossRef CAS PubMed.
- Y. W. Cao, R. C. Jin and C. A. Mirkin, Nanoparticles with raman spectroscopic fingerprints for DNA and RNA detection, Science, 2002, 297(5586), 1536–1540 CrossRef CAS PubMed.
- K. Faulds, W. E. Smith and D. Graham, Evaluation of surface-enhanced resonance raman scattering for quantitative DNA analysis, Anal. Chem., 2004, 76(2), 412–417 CrossRef CAS PubMed.
- J. Hu and C. Y. Zhang, Sensitive detection of nucleic acids with rolling circle amplification and surface-enhanced raman scattering spectroscopy, Anal. Chem., 2010, 82(21), 8991–8997 CrossRef CAS PubMed.
- N. R. Isola, D. Stokes and T. Vo-Dinh, Surface-Enhanced Raman Gene Probe for HIV Detection, Anal. Chem., 1998, 70(7), 1352–1356 CrossRef CAS PubMed.
- J. Kim, H. J. Park, J. H. Kim, B. Chang and H. K. Park, Label free detection for a DNA methylation assay using raman spectroscopy, Chin. Med. J., 2017, 130(6), 1961–1967 CrossRef CAS.
- S. Ganesh, K. Venkatakrishnan and B. Tan, Quantum scale organic semiconductors for SERS detection of DNA methylation and gene expression, Nat. Commun., 2020, 11, 1135 CrossRef CAS.
- L. Ouyang, Y. Hu, L. Zhu, G. J. Cheng and J. Irudayaraj, A reusable laser wrapped graphene-Ag array-based SERS sensor for trace detection of genomic DNA methylation, Biosens. Bioelectron., 2016, 15(92), 755–762 Search PubMed.
- Y. Wang, E. Wee and M. Trau, Highly sensitive DNA methylation analysis at CpG resolution by surface-enhanced raman scattering via ligase chain reaction, Chem. Commun., 2013, 1–3 Search PubMed.
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Fluorescent and colorimetric sensors for detection of lead, cadmium, and mercury ions, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- D. Vilela, M. C. Gonzalez and A. Escarpa, Sensing colorimetric approaches based on gold and silver nanoparticles aggregation: Chemical creativity behind the assay, A review, Anal. Chim. Acta, 2012, 751, 24–43 CrossRef CAS PubMed.
- E. Wee, T. Ngo and M. Trau, Colorimetric detection of both total genomic and loci-specific DNA methylation from limited DNA inputs, Clin. Epigenet., 2015, 7, 65 CrossRef.
- Y. Z. Lin and P. L. Chang, Colorimetric determination of DNA methylation based on the strength of the hydrophobic interactions between DNA and gold nanoparticles, ACS Appl. Mater. Interfaces, 2013, 5(22), 12045–12051 CrossRef CAS PubMed.
- Z. M. Li, T. Pi, X. L. Yan, X. M. Tang, R. H. Deng and X. J. Zheng, Label-free and enzyme-free one-step rapid colorimetric detection of DNA methylation based on unmodified gold nanoparticles, Spectrochim. Acta, Part A, 2020, 238, 118375 CrossRef CAS.
- Y. Geng, J. Wu, L. Shao, F. Yan and H. Ju, Sensitive colorimetric biosensing for methylation analysis of p16/CDKN2 promoter with hyperbranched rolling circle amplification, Biosens. Bioelectron., 2014, 61, 593–597 CrossRef CAS PubMed.
- L. Guo, Y. Xu, A. R. Ferhan, G. Chen and D. H. Kim, Oriented Gold Nanoparticle Aggregation for Colorimetric Sensors with Surprisingly High Analytical Figures of Merit, J. Am. Chem. Soc., 2013, 135, 12338–12345 CrossRef CAS PubMed.
- Y. Zhang, J. Hu and C.-y. Zhang, Sensitive detection of transcription factors by isothermal exponential amplification-based colorimetric assay, Anal. Chem., 2012, 84(21), 9544–9549 CrossRef CAS PubMed.
- Y. Zhao, F. Chen, Y. Wu, Y. Dong and C. Fan, Highly sensitive fluorescence assay of DNA methyltransferase activity via methylation-sensitive cleavage coupled with nicking enzyme assisted signal amplification, Biosens. Bioelectron., 2013, 42, 56–61 CrossRef CAS.
- L. Cui, G. Ke, W. Y. Zhang and C. J. Yang, A universal platform for sensitive and selective colorimetric DNA detection based on Exo III assisted signal amplification, Biosens. Bioelectron., 2011, 26(5), 2796–2800 CrossRef CAS.
- W. Xu, X. Xue, T. Li, H. Zeng and X. Liu, Ultrasensitive and selective colorimetric DNA detection by nicking endonuclease assisted nanoparticle amplification, Angew. Chem., Int. Ed., 2009, 48(37), 6849–6852 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Triggered amplification by hybridization chain reaction, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(43), 15275–15278 CrossRef CAS.
- C. Park, Y. Song, K. Jang, C. H. Choi and S. Na, Target switching catalytic hairpin assembly and gold nanoparticle colorimetric for EGFR mutant detection, Sens. Actuators, B, 2018, 261, 497–504 CrossRef CAS.
- B. Li, Y. Jiang, X. Chen and A. D. Ellington, Probing spatial organization of DNA strands using enzyme-free hairpin assembly circuits, J. Am. Chem. Soc., 2012, 134(34), 13918–13921 CrossRef CAS PubMed.
- A. X. Zheng, J. Li, J. R. Wang, X. R. Song, G. N. Chen and H. H. Yang, Enzyme-free signal amplification in the DNAzyme sensor via target-catalyzed hairpin assembly, Chem. Commun., 2012, 48(25), 3112–3114 RSC.
- B. D’angelo, E. Benedetti and A. Cimini, MiRNAs: a puzzling tool in cancer diagnostics and therapy, Anticancer Res., 2016, 36, 5571–5576 CrossRef PubMed.
- W. Kwang, Multidisciplinary Role of Microfluidics for Biomedical and Diagnostic Applications: Biomedical Microfluidic Devices, Micromachines, 2017, 8(12), 343 CrossRef.
- V. Gubala, L. F. Harris, A. J. Ricco, M. X. Tan and D. E. Williams, Point of care diagnostics: status and future, Anal. Chem., 2011, 84, 487–515 CrossRef PubMed.
- G. Gauglitz, Point-of-care platforms, Annu. Rev. Anal. Chem., 2014, 7, 297–315 CrossRef CAS.
- B. Srinivasan and S. Tung, development and applications of portable biosensors, J. Lab. Autom., 2015, 20(4), 365–389 CrossRef CAS PubMed.
- M. Adampourezare, G. Dehghan, M. Hasanzadeh and M. A. Hosseinpoure Feizi, Application of lateral flow and microfluidic bio-assay and biosensing towards identification of DNA methylation and cancer detection: Recent progress and challenges in biomedicine, Biomed. Pharmacother., 2021, 141, 111845 CrossRef CAS PubMed.
- B. Keeley, A. Stark, T. R. Pisanic, R. Kwak and Y. Zhang, Extraction and processing of circulating DNA from large sample volumes using methylation on beads for the detection of rare epigenetic events, Clin. Chim. Acta, 2013, 425, 169–175 CrossRef CAS.
- Y. Shin, A. P. Perera, C. C. Wong and M. K. Park, Solid phase nucleic acid extraction technique in a microfluidic chip using a novel non-chaotropic agent: dimethyl adipimidate, Lab Chip, 2014, 14, 359–368 RSC.
- J. Yoon, M. K. Park, T. Y. Lee, Y.-J. Yoon and Y. Shin, LoMA-B: a simple and versatile lab-on-a-chip system based on single-channel bisulfite conversion for DNA methylation analysis, Lab Chip, 2015, 15, 3530–3539 RSC.
- M. Koo Kevin, J. H. Wee Eugene, R. Sakandar, J. A. Muhammad Shiddiky and M. Trau, Microdevices for detecting locus-specific DNA methylation at CpG resolution, Biosens. Bioelectron., 2014, 56, 278–285 CrossRef CAS.
- M. Christine, D. Giammanco, S. Li, R. Pisanic II and T. J. Wang, Multilayer microfluidic array for highly efficient sample loading and digital melt analysis of DNA methylation, Lab Chip, 2019, 19, 444–451 RSC.
- R. Kurita, H. Yanagisawa, K. Yoshioka and O. Niwa, On-chip sequence-specific immunochemical epigenomic analysis utilizing outward-turned cytosine in a DNA bulge with handheld surface plasmon resonance equipment, Anal. Chem., 2015, 87, 11581–11586 CrossRef CAS.
- T. Yoon Lee, Y. Shin and M. Kyoung Park, A simple, low-cost, and rapid device for a DNA methylation-specific amplification/detection system using a flexible plastic and silicon complex, Lab Chip, 2014, 14, 4220–4229 RSC.
- C. H. Wang, H. C. Lai, T. M. Liou, K. F. Hsu, C. Y. Chou and G. B. Lee, A DNA methylation assay for detection of ovarian cancer cells using a HpaII/MspI digestion-based PCR assay in an integrated microfluidic system, Microfluid. Nanofluid., 2013, 15, 575–585 CrossRef CAS.
- F. Lyko, R. Brown and J. Natl, DNA methyltransferase inhibitors and the development of epigenetic cancer therapies, J. Natl. Cancer Inst., 2005, 97, 1498–1506 CrossRef CAS PubMed.
- M. Ronen, D. Avrahami and D. Gerber, A sensitive microfluidic platform for a high throughput DNA methylation assay, Lab Chip, 2014, 14, 2354–2362 RSC.
- D. Onoshima, N. Kawakita, D. Takeshita, H. Niioka, H. Yukawa and J. Miyake, Measurement of DNA length changes upon CpG hypermethylation by microfluidic molecular stretching, Cell Med., 2017, 9, 61–66 CrossRef PubMed.
- M. Mohammadniaei, H. V. Nguyen, M. V. Tieu and M. H. Lee, 2D materials in development of electrochemical point-of-care cancer screening devices, Micromachines, 2019, 10(10), 662 CrossRef PubMed.
- D. Ji, N. Xu, Z. Liu, Z. Shi, S. S. Low, J. Liu, C. Cheng, J. Zhu, T. Zhang and H. Xu, Smartphone-based differential pulse amperometry system for real-time monitoring of levodopa with carbon nanotubes and gold nanoparticles modified screen-printing electrodes, Biosens. Bioelectron., 2019, 129, 216e223 Search PubMed.
- D. Ji, Z. Liu, L. Liu, S. S. Low, Y. Lu, X. Yu, L. Zhu, C. Li and Q. Liu, Smartphone-based integrated voltammetry system for simultaneous detection of ascorbic acid, dopamine, and uric acid with graphene and gold nanoparticles modified screen-printed electrodes, Biosens. Bioelectron., 2018, 119, 55e62 CrossRef PubMed.
- R. Uthoff, B. Song, M. Maarouf, V. Shi and R. Liang, Pointof-care, multispectral, smartphone-based dermascopes for dermal lesion screening and erythema monitoring, J. Biomed. Opt., 2020, 25, 1–21 Search PubMed.
- L.-J. Wang, Y.-C. Chang, R. Sun and L. Li, A multichannel smartphone optical biosensor for high-throughput pointof-care diagnostics, Biosens. Bioelectron., 2017, 87, 686–692 CrossRef CAS PubMed.
- S. Wang, X. Zhao, I. Khimji, R. Akbas, W. Qiu, D. Edwards, D. W. Cramer, B. Ye and U. Demirci, Integration of cell phone imaging with microchip ELISA to detect ovarian cancer HE4 biomarker in urine at the point-of-care, Lab Chip, 2011, 11, 3411–3418 RSC.
- T. H. Wu, C. C. Chang, J. Vaillant, A. Bruyant and C. W. Lin, DNA biosensor combining single-wavelength colorimetry and a digital lock-in amplifier within a smartphone, Lab Chip, 2016, 16, 4527–4533 RSC.
- C. Severi, N. Melnychuk and A. S. Klymchenko, Smartphone-assisted detection of nucleic acids by light-harvesting FRET-based nanoprobe, Biosens. Bioelectron., 2020, 168, 112515 CrossRef CAS PubMed.
- C. Vietz, M. L. Schütte, Q. Wei, L. Richter and B. Lalkens, Benchmarking Smartphone Fluorescence-Based Microscopy with DNA Origami Nanobeads: Reducing the Gap toward Single-Molecule Sensitivity, ACS Omega, 2019, 4, 637–642 CrossRef CAS PubMed.
- A. Baba, K. Shinboa and K. Kato, A smartphone-based surface plasmon resonance platform Chutiparn Lertvachirapaiboon, Anal. Methods, 2018, 10, 4732 RSC.
- E. Petryayeva and W. R. Algar, Multiplexed homogeneous assays of proteolytic activity using a smartphone and quantum dots, Anal. Chem., 2014, 86, 3195–3202 CrossRef CAS PubMed.
- T. San Park, C. Baynes, S. I. Cho and J. Y. Yoon, Paper microfluidics for red wine tasting, RSC Adv., 2014, 4, 24356–24362 RSC.
- Y. Wang, S. Wang, N. Dong, W. Kang, K. Li and Z. Nie, Titanium carbide mxenes mediated in situ reduction allows label-free and visualized nanoplasmonic sensing of silver ions, Anal. Chem., 2020, 92, 4623–4629 CrossRef CAS.
- T. Liu, S. Zhang, W. Liu, S. Zhao, Z. Lu, Y. Wang, G. Wang, P. Zou, X. Wang, Q. Zhao and H. Rao, Smartphone based platform for ratiometric fluorometric and colorimetric determination H2O2 and glucose, Sens. Actuators, B, 2020, 305, 127524 CrossRef CAS.
- W. Xu, S. Lu, Y. Chen, T. Zhao, Y. Jiang, Y. Wang and X. Chen, Simultaneous color sensing of O2 and pH using a smartphone, Sens. Actuators, B, 2015, 220, 326–330 CrossRef CAS.
- S. K. Vashist, T. van Oordt, E. M. Schneider, R. Zengerle, F. von Stetten and J. H. Luong, A smartphone-based colorimetric reader for bioanalytical applications using the screen-based bottom illumination provided by gadgets, Biosens. Bioelectron., 2015, 67, 248–255 CrossRef CAS PubMed.
- W. Chen, F. Cao, W. Zheng, Y. Tian, Y. Xianyu, P. Xu, W. Zhang, Z. Wang, K. Deng and X. Jiang, Detection of the nanomolar level of total Cr(iii) and (vi) by functionalized gold nanoparticles and a smartphone with the assistance of theoretical calculation models, Nanoscale, 2015, 7, 2042–2049 RSC.
- N. Lopez-Ruiz, V. F. Curto, M. M. Erenas, F. Benito-Lopez, D. Diamond, A. J. Palma and L. F. Capitan-Vallvey, Smartphone-based simultaneous pH and nitrite colorimetric determination for paper microfluidic devices, Anal. Chem., 2014, 86, 9554–9562 CrossRef CAS PubMed.
- L. Shen, J. A. Hagen and I. Papautsky, Point-of-care colorimetric detection with a smartphone, Lab Chip, 2012, 12, 4240–4243 RSC.
- A. W. Martinez, S. T. Phillips, E. Carrilho, S. W. Thomas, H. Sindi and G. M. Whitesides, Simple telemedicine for developing regions: camera phones and paper-based microfluidic devices for real-time, off-site diagnosis, Anal. Chem., 2008, 80, 3699–3707 CrossRef CAS PubMed.
- R. Zhao, T. Shen, T. Lang and B. Cao, Visible smartphone spectrometer based on the transmission grating, 16th International Conference on Optical Communications and Networks (ICOCN), Wuzhen, China, 2017, pp. 1–3 Search PubMed.
- T. Heng, W. Chia-Chen Chang, J. Vaillant, A. Bruyant and C. Wann, DNA biosensor combining single-wavelength colorimetry and digital Lock-in Amplifier within a Smartphone, Lab Chip, 2016, 1–20 Search PubMed.
- A. Palatnick, B. Zhou, E. Ghedin and M. C. Schatz, iGenomics: Comprehensive DNA sequence analysis on your Smartphone, GigaScience, 2020, 9, 1–12 CrossRef CAS PubMed.
- T. Goua, J. Hua, W. W. Xiong, D. Shufang, Z. Weibo and F. Ying, Smartphone-based mobile digital PCR device for DNA quantitative analysis with high accuracy, Biosens. Bioelectron., 2018, 120, 144–152 CrossRef.
- A. Lazaro, A. Maquieira and L. A. Tortajada-Genaro, Discrimination of single-nucleotide variants based on an allele specific hybridization chain reaction and smartphone detection, ACS Sens., 2022, 7, 758–765 CrossRef CAS PubMed.
- M. K. hnemund, Q. Wei, E. Darai, Y. Wang, I. Hernández-Neuta, Z. Yang, D. Tseng, A. Ahlford, L. Mathot, T. Sjöblom, A. Ozcan and M. Nilsson, Targeted DNA sequencing and in situ mutation analysis using mobile phone microscopy, Nat. Commun., 2017, 8, 13913 CrossRef.
- C. Y. Mai, Y. F. Lai and L. Zou, Smartphone-assisted colorimetric detection of BRCA-1 gene based on catalytic hairpin assembly amplification and G-quadruplex DNAzyme, Chin. J. Anal. Chem., 2021, 49, 41–46 Search PubMed.
- R. Eiges, M. Schuldiner, M. Drukker, O. Yanuka, J. Itskovitz- ldor and N. Benvenisty, Establishment of human embryonic stem cell-transfected clones carrying a marker for undifferentiated cells, Curr. Biol., 2001, 11(7), 514–518 CrossRef CAS PubMed.
- C. Bock, I. Beerman, W. H. Lien, Z. Smith and H. G. Boyle, DNA methylation dynamics during in vivo differentiation of blood and skin stem cells, Mol. Cell, 2012, 47, 633–647 CrossRef CAS PubMed.
- A. Bird, DNA methylation patterns and epigenetic memory, Genes Dev., 2012, 16, 6–21 CrossRef.
- R. Lister, M. Pelizzola, R. H. Dowen, R. D. Hawkins, G. Hon, J. Tonti-Filippini, J. R. Nery, L. Lee, Z. Ye and Q. M. Ngo, Human DNA methylomes at base resolution show widespread epigenomic differences, Nature, 2009, 462, 315–322 CrossRef CAS PubMed.
- R. Guerrero-Preston, C. Michailidi, L. Marchionni, C. R. Pickering and M. Frederick, Key tumor suppressor genes inactivated by greater promoter methylation and somatic mutations in head and neck cancer, Epigenetics, 2014, 9(7), 1031–1046 CrossRef PubMed.
- B. Sharma, R. R. Frontiera, A.-I. Henry, E. Ringe and R. P. Van Duyne, SERS: Materials, applications, and the future, Mater. Today, 2012, 15, 16–25 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |